





Abstract
The traditional view of avian evolution places ratites and tinamous at the base of the phylogenetic tree of modern birds (Neornithes). In contrast, most recent molecular studies suggest that neognathous perching birds (Passeriformes) compose the oldest lineage of modern birds. Here, we report significant molecular support for the traditional view of neognath monophyly based on sequence analyses of nuclear and mitochondrial DNA (4.4 kb) from every modern avian order. Phylogenetic analyses further show that the ducks and gallinaceous birds are each other's closest relatives and together form the basal lineage of neognathous birds. To investigate why other molecular studies sampling fewer orders have reached different conclusions regarding neognath monophyly, we performed jackknife analyses on our mitochondrial data. Those analyses indicated taxon-sampling effects when basal galloanserine birds were included in combination with sparse taxon sampling. Our phylogenetic results suggest that the earliest neornithines were heavy-bodied, ground-dwelling, nonmarine birds. This inference, coupled with a fossil bias toward marine environments, provides a possible explanation for the large gap in the early fossil record of birds.
Introduction
The order of appearance of the major lineages of birds is important for understanding the evolution of their complex morphologies and behaviors, yet the earliest branching pattern among modern avian orders remains unresolved. The traditional classification of modern birds, based primarily on their differing palatal anatomies, groups together ratites and tinamous in the infraclass Palaeognathae and all other birds in the Neognathae (Pycraft 1900 ; Cracraft 1988 ; Cracraft and Mindell 1989 ). However, molecular studies have not yielded consistent results for the higher-level relationships of birds. Studies of nuclear genes and proteins have largely supported neognath monophyly, whereas studies of mitochondrial genes have supported neognath paraphyly. The nuclear genes and proteins that have been studied include transferrin and serum albumin (Ho et al. 1976 ; Prager et al. 1976 ), alpha crystallins (Stapel et al. 1984 ; Caspers et al. 1997 ), and the recombination-activating gene 1 (Groth and Barrowclough 1999 ). A DNA-DNA hybridization study of all avian orders indicated that galloanserine birds (including ducks and fowl) are closer to palaeognaths than to other neognaths, a result that was considered to reflect differences in maturation time (Sibley and Ahlquist 1990 ). Otherwise, the DNA-DNA hybridization results are similar to those of the nuclear gene studies in placing the perching birds, Passeriformes, together with other neognath avian orders.
Mitochondrial DNA sequence studies have instead supported a phylogenetic tree of avian orders that differs considerably from the traditional classification. Those studies of mitochondrial protein-coding genes found that the palaeognath and galloanserine birds occupy derived positions within Neognathae, whereas Passeriformes occupy the most basal position among all modern birds (Mindell et al. 1997 ; Härlid, Janke, and Arnason 1998 ; Härlid and Arnason 1999 ; Mindell et al. 1999 ). Some morphological support for a basal Passeriformes and a derived position of the galloanserine birds also has been proposed (Woodbury 1998 ).
With such disparate results from mitochondrial and nuclear genes, and from morphology, it is clear that additional data are needed to better understand the early history of modern birds. Of the previous molecular studies, those with broad taxonomic sampling (Ho et al. 1976 ; Prager et al. 1976 ; Sibley and Ahlquist 1990 ) did not address the statistical significance of their findings, and those reporting statistical significance (Mindell et al. 1997 ; Härlid, Janke, and Arnason 1998 ; Groth and Barrowclough 1999 ; Härlid and Arnason 1999 ; Mindell et al. 1999 ) did not sample all avian orders. Therefore, in this study, we attempted to do both by sampling a large number of nucleotide sites in all avian orders.
Materials and Methods
Sequencing
The complete mitochondrial 12S rRNA, tRNA-Valine, and 16S rRNA genes were sequenced for 41 taxa using a standard protocol (Hedges and Sibley 1994 ; Hedges et al. 1995 ; van Tuinen, Sibley, and Hedges 1998 ). A partial sequence from one ratite (Apteryx australis) was obtained from GenBank (accession numbers U76043, U76063, and X67626) and completed through sequencing of a 400-bp fragment. Previously described ratite and tinamou sequences (van Tuinen, Sibley, and Hedges 1998 ) were added to the data set, and corresponding sequences for Gallus gallus (Galliformes), Anas platyrhynchos (Anseriformes), Alligator mississipiensis, and Trachemys scripta were obtained from GenBank (accession numbers X52392, L16770, Y13113, and L28077, respectively) in order to include one or more representatives of all avian orders and two outgroup taxa (table 1 ). For some taxa, partial sequences obtained in earlier studies (Hedges and Sibley 1994 ; Hedges et al. 1995 ) were extended. To improve the resolution of the earliest avian divergences, 32 of the 48 taxa (representatives of all avian orders) as well as an outgroup (A. mississipiensis) also were sequenced for the more conserved nuclear 18S rRNA gene using the same polymerase chain reaction protocol with nine primers. The turtle 18S rRNA sequence (T. scripta) was obtained previously (accession number M59398). The galliform sequence is a composite of fowl (G. gallus) mitochondrial ribosomal genes and the closely related quail (Coturnix pectoralis) nuclear ribosomal gene because of an anomalous phylogenetic position in the 18S rRNA–based tree for G. gallus. Sequences were aligned using CLUSTAL X (Thompson, Higgins, and Gibson 1994 ), and the basal nodes resulting from this alignment were confirmed with the alignment obtained with the divide-and-conquer method (Stoye 1998 ; http://www.bibiserv.techfak.uni-bielefeld.de/dca). Alignments and a list of primers used in this study are available from the authors. The new sequences have been deposited in GenBank (accession numbers AF173561–AF173638).
Phylogenetic Analyses
For phylogenetic analysis, the neighbor-joining method was performed in both MEGA (Kumar, Tamura, and Nei 1993 ) and PAUP 4.0 (Swofford 1998 ), using a Kimura (1980) two-parameter distance and the complete-deletion option. To test the earliest branching patterns, either all substitutions (nuclear) or transversions only (mitochondrial, combined) were used, depending on the type of analysis. Topological effects of compositional bias were tested by performing separate phylogenetic analyses using the Tamura-Nei distance (Kumar, Tamura, and Nei 1993 ), resulting in effects only on the topology of the unresolved parts of the avian tree (within Neoaves, the cohort of neognaths excluding the galloanserine orders). Chi-square analyses of separate base frequencies, including only variable sites, show no significant deviation from stationarity among all taxa. Nonetheless, we employed log determinant distances to correct for heterogeneous base composition (Swofford 1998 ) in conjunction with the neighbor-joining method. Topological effects were again observed only among the neoavian birds. For analysis of the combined data, resulting neighbor-joining trees were compared with those obtained by maximum-parsimony and maximum-likelihood methods (Swofford 1998 ). For subsequent analyses, only neighbor-joining analyses were employed, considering the sizes of the data sets and the computational time involved. Separate analyses were performed on the full alignment from CLUSTAL X (liberal alignment) and on a more conservative data set in which hypervariable regions (549 bp total) were removed after manual adjustment of the liberal alignment. The bootstrap method with 2,000 replications was replications was used to assess the confidence of individual nodes in the phylogenetic trees. Additionally, significance of interior branches resulting from transversion-only analyses was tested using PHYLTEST (Kumar 1996 ). Tests for alternative phylogenetic signal were performed using SPECTRUM 2.0 (Hendy and Penny 1993 ; 20 taxa, including 2 outgroup, 5 palaeognath, 3 galloanserine, 1 passeriform, and 9 remaining neoavian taxa).
The analyses of cytochrome b sequences obtained from GenBank were performed using Poisson-corrected distances from sequences of the complete gene (379 amino acids) and included 70 taxa representing the following bird orders (Sibley and Ahlquist 1990 ): Anseriformes (L08385), Bucerotiformes (U89190), Ciconiiformes (U74347, U74348, U74351, U74352, U83305, U83311, U83312, U83314, U83316, AF076044, AF076052, AF076054, AF076062, AF076076, AF076090, AF076094), Coliiformes (U89173, U89175), Coraciiformes (U89184, U89186, U89188), Craciformes (L08384, AF068190), Cuculiformes (U89197, U89198), Galliformes (L08377, L08381, AF013763, AF028791, AF028795, AF028798, AF028802, AF068192, AF068193, Gruiformes (U11060, U27543), Passeriformes (U15206, U25736, U25738, U77331, U77336, X74251, X74256, Y16885, AF081958, AF081959, AF082007), Piciformes (U89192, U89193), Psittaciformes (U89176, U89178, U89179), Strigiformes (U89171, U89172, U89194), Struthioniformes (U76050, U76051, U76052, U76054, U76055), Tinamiformes (U76053, U76056), Trochiliformes (U89180), Trogoniformes (U89201, U89202), and Upupiformes (U89189). To test the sensitivity of the ingroup topology to choice of outgroup, alligators (Y13113) and turtles (AF039066) were used in combination and separately as outgroup.
Jackknife Analyses
The effect of sampling different numbers of taxa (avian orders) was explored through separate jackknife analyses of the mitochondrial data set and the complete data set (nuclear and mitochondrial rRNA sequences). In each case, a fixed number of palaeognath and neognath taxa were sampled 100 times randomly, without replacement, and a neighbor-joining tree was constructed using the Kimura two-parameter and transversions-only distance. In all cases, the outgroup (alligators + turtles) was included and the tree was scored as supporting or not supporting neognath monophyly. Taxon-specific jackknife analyses were performed by substituting one of the variable (randomly chosen) neognath taxa for a galliform (Gallus/Coturnix), anseriform (Anas), or passeriform (Tyrannus) in each jackknife sample. In a separate jackknife analysis (40 iterations because of extensive computational time), a measure of phylogenetic signal (tRASA; Lyons-Weiler, Hoelzer, and Tausch 1996 ) for each jackknifed data set was calculated and averaged.
Results
Phylogenetic analyses of the complete data set significantly resolve the earliest branches in modern birds and indicate that all orders of neognathous birds form a monophyletic group (fig. 1 ). These results are supported by analyses of the separate nuclear and mitochondrial data sets regardless of the tree-building method (neighbor-joining, maximum-parsimony, or maximum-likelihood) or the choice of outgroup employed. Furthermore, the mitochondrial ribosomal genes and the combined data set indicate that the palaeognaths are monophyletic and that the galloanserine clade (ducks and gallinaceous birds) is a sister group to all remaining neognaths (fig. 1 ). The conserved nuclear sequences alone are not sufficient to resolve the monophyly of Galloanserae but indicate that ducks, fowl, and cracids are basal neognaths. All of these results are robust using the most liberal and the most conserved versions of the alignments (fig. 1 ; Materials and Methods), do not change with choice of alignment method (Thompson, Higgins, and Gibson 1994 ; Stoye 1998 ), and are unaffected by sequentially removing constant sites from analysis of the complete data set.
The largest single molecular study to date of any major group of vertebrates is Sibley and Ahlquist's (1990) DNA-DNA hybridization study of approximately 1,700 species of birds representing all extant orders. That study formed the basis of a revised classification of birds (table 1 ) and, although its methods have been debated, has been a point of reference for many evolutionary studies of birds (Sheldon and Bledsoe 1993 ). The correlation between the evolutionary distances calculated from pairwise comparison of the mitochondrial and nuclear sequences obtained in this study and the corresponding distances calculated from DNA hybridization melting curves (fig. 2 ) indicates broad agreement and provides support for the continued usefulness of the DNA hybridization data set. Furthermore, the linear correlation of the mitochondrial sequence divergence with the hybridization distances suggests that transversion saturation does not affect our mitochondrial data set.
To further investigate the phylogenetic signal in our data, we performed spectral analyses on the complete data set using the conservative alignment obtained by CLUSTAL X (Thompson, Higgins, and Gibson 1994 ). A spectral analysis is independent of any tree-building method and quantifies the support and conflict for any grouping, thereby exploring the extent of alternative phylogenetic signals (Hendy and Penny 1993 ). Results indicate comparatively high levels (3.8 × 10−3) of support and low levels (0.0) of conflict for neognath monophyly. The alternative “monophyletic Palaeognathae-Galloanserae” and “Passeriformes basal” signals were each accompanied by lower support (1.7 × 10−4, 8.4 × 10−7) and higher conflict (0.4, 3.3). Similarly, low support-to-conflict ratios were found for other clusters that resulted in neognath paraphyly.
A jackknife analysis of our data sets shows that neognath monophyly was almost always supported in small or large subsets of the complete data set and in the mitochondrial data set when 20 or more orders were sampled. However, when fewer than 20 orders were included in the mitochondrial data set, neognath monophyly was not supported in as many as one quarter of the jackknife samples (table 2 ). To further explore this taxon-sampling problem, we examined those specific jackknife samples (n = 81; table 2 ) that resulted in neognath paraphyly and found that 91% (74 taxa) included a galloanserine taxon. Separate taxon-specific jackknife analyses were then performed by including a galliform, an anseriform, or a passeriform in each subset. The latter neognath order was tested because it was found to be basal among modern avian orders in recent mitochondrial studies (Mindell et al. 1997 ; Härlid, Janke, and Arnason 1998 ; Mindell et al. 1999 ; Härlid and Arnason 1999 ). As predicted from the first jackknife analysis, support for neognath monophyly was significantly lower when either a galliform or an anseriform taxon was included. However, support for neognath monophyly from the jackknife analysis using a passeriform was the same as that for randomly chosen neoavian taxa (table 2 ).
We also investigated taxon-sampling effects in a published data set (cytochrome b) that supported neognath paraphyly (Härlid, Janke, and Arnason 1998 ). Although the tree from that gene was considered to provide support for a basal position of the passeriform birds under broad taxon sampling, the analysis included only seven orders. Whereas some orders were broadly sampled, the unusual topological placement of Anseriformes and the resulting unconventional unrooted tree were probably caused by sparse sampling of the anseriform lineage. Changing the root to the traditional placement fails to yield a monophyletic Galloanserae and Neoaves due to the sister group relationship of the single anseriform lineage to Neoaves. Although the cytochrome b gene is relatively small (1,143 bp) and is not expected to robustly resolve the relationships of avian orders, we nonetheless performed a new analysis with additional sequences that have since become available. With 11 additional avian orders available in ENTREZ/GenBank release 112.0, we did not find statistical support for a basal position of Passeriformes. Furthermore, the tree supported neognath paraphyly if alligators + turtles were used as the root and neognath monophyly if the turtle alone was used as the root. This finding is concordant with our jackknife analysis showing support for neognath paraphyly only in the analyses of small subsets of taxa (table 2 ).
Discussion
These results suggest that the finding of neognath paraphyly in recent molecular studies (Mindell et al. 1997 ; Härlid, Janke, and Arnason 1998 ; Härlid and Arnason 1999 ; Mindell et al. 1999 ) was probably the result of insufficient taxon sampling. The reason why inclusion of galloanserine taxa in smaller samples of taxa increases the probability of neognath paraphyly could be related to their basal position among neognaths and the longer branch lengths of the neoavian orders and outgroup taxa relative to the galloanserine and palaeognath birds. The neoavian clade has a 30% longer branch length than the galloanserine clade and a 74% longer branch length than the palaeognath clade when examined using PHYLTEST. Therefore, the importance of the galloanserine taxa lies in their intermediate phylogenetic position and intermediate branch lengths.
These jackknife analyses also revealed increases in phylogenetic signal as taxon sampling was increased (table 2 ). Given that the tree-independent measure of signal we used (Lyons-Weiler, Hoelzer, and Tausch 1996 ) is known to decrease as branch length heterogeneity increases (Lyons-Weiler and Hoelzer 1997 ), this result provides further support for the hypothesis that a taxon-sampling problem is involved. In such cases, long-branch attraction may result, owing to sparse taxon sampling. When sufficient taxon sampling becomes available for a diversity of genes, it will be possible to test for gene-specific effects on phylogeny.
The unrooted trees are identical for the traditional Palaeognathae-Neognathae concept and for the recently found topology (Passeriformes basal) derived from the mitochondrial concatenated protein-coding genes (Mindell et al. 1997 ; Härlid and Arnason 1999 ; Mindell et al. 1999 ). Together with these jackknife data, this observation indicates that improper rooting can occur when using sparse taxon sampling. The unrooted tree from the cytochrome b sequences (Härlid, Janke, and Arnason 1998 ) is unconventional. Nonetheless, the reanalyses of this gene with additional taxa show that a taxon-sampling problem is probably involved. These reanalyses further point to the need for optimal outgroup usage in phylogenetic studies (Lyons-Weiler, Hoelzer, and Tausch 1998 ).
The group comprising the orders of neognath birds exclusive of Galloanserae was found by Sibley, Ahlquist, and Monroe (1988) based on DNA-DNA hybridization data and named Neoaves. In that study, Turniciformes were left unplaced, but otherwise the composition of the group is identical to that found here. Sibley and Ahlquist (1990) and Sibley and Monroe (1990) , as well as other authors (Kurochkin 1995 ; Rotthowe and Starck 1998 ), later used Neoaves in reference to all neognath birds including Galloanserae. Because the taxon Neoaves has been used differently, Groth and Barrowclough (1999) proposed a new name, “Plethornithae,” for the group originally named by Sibley, Ahlquist, and Monroe (1988) . However, definitions of taxonomic names frequently change, and this is usually not a reason to abandon a taxonomic name. Although the International Code of Zoological Nomenclature does not apply to these higher-level groupings, we follow standard taxonomic practice and use the name first applied to the group in question: Neoaves. Otherwise, we agree with Groth and Barrowclough (1999) in retaining the names Palaeognathae and Neognathae as infraclasses of the subclass Neornithes, and in treating the two remaining taxa (Galloanserae, and, in this case, Neoaves) as cohorts (fig. 1 ).
Inferring the order of appearance of the major lineages of birds permits a better understanding of the morphology and ecology of the earliest neornithine birds. The late Cretaceous avian fossils of neornithines (Chiappe 1995 ; Padian and Chiappe 1998 ), aside from those of a ducklike bird and a parrot (Stidham 1998 ), are mostly of marine taxa (Ciconiiformes, sensuSibley and Ahlquist 1990 ). These fossils led to the proposal that all orders of modern birds arose from a marine (shore bird) ancestor (Feduccia 1995 ). In contrast, the basal lineages identified in this study (tinamous, ratites, anseriforms, galliforms, and craciforms) include almost entirely terrestrial, nonmarine, ground-dwelling, and heavy-bodied birds. If our identification of the earliest neornithines is correct and the ancestral neornithines also shared these morphological and ecological traits, then the neornithine fossil record is biased, perhaps as a result of the greater chances of fossilization of aquatic, particularly marine, taxa (Benton 1997 ). Furthermore, this taphonomic bias could provide an explanation of the large gap in the neornithine fossil record as implied by molecular time estimates (Hedges et al. 1996 ; Cooper and Penny 1997 ; Härlid, Janke, and Arnason 1997, 1998 ; Waddell et al. 1999 ).
Shozo Youoyama, Reviewing Editor
Keywords: molecular avian phylogeny Palaeognathae-Neognathae Galloanserae Neoaves taxon sampling fossil record bias
Address for correspondence and reprints: S. Blair Hedges, Department of Biology, 208 Mueller Laboratory, Pennsylvania State University, University Park, Pennsylvania 16802. E-mail: sbh1@psu.edu.

Table 2 Jackknife Analysis Showing Effects of Taxon Sampling on Phylogeny Estimation
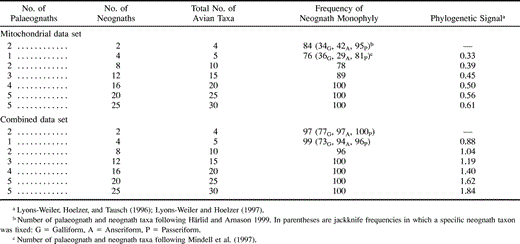
Table 2 Jackknife Analysis Showing Effects of Taxon Sampling on Phylogeny Estimation
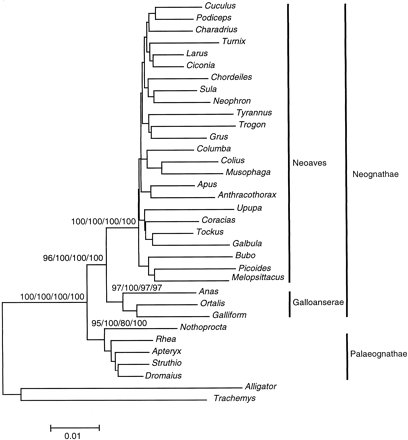
Fig. 1.—Phylogenetic tree of modern birds based on neighbor-joining analysis of nuclear and mitochondrial rRNA gene sequences (4.4 kb) of 32 avian taxa. The alligator and the turtle are used to root the tree. Confidence values of >95% are indicated on nodes (bootstrap using complete alignment/interior branch test using complete alignment/bootstrap using conserved alignment/interior branch test using conserved alignment). Names of infraclasses and cohorts within Neornithes are shown. Note the lack of resolution within Neoaves and within ratites
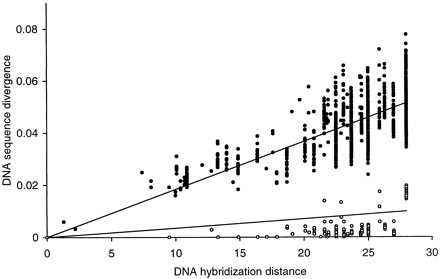
Fig. 2.—Correlation between DNA sequence divergence (this study) and DNA hybridization distances (Sibley and Ahlquist 1990 ) for multiple pairwise comparisons. For DNA sequence divergence, closed circles indicate mitochondrial data (Kimura transversion distance; r = 0.84), and open circles indicate nuclear rRNA data (Jukes-Cantor distance; r = 0.43). The DNA hybridization distances were calculated by averaging the ΔT50H values in Sibley and Ahlquist (1990). A similar correlation has been obtained for a smaller avian data set (Hedges and Sibley 1994 )
We thank Carla A. Hass, James Lyons-Weiler, Sonja J. Pyott, and Pat Shipman for comments and discussion; Shannon G. Kindl for laboratory assistance; and James Lyons-Weiler for assistance with the RASA program. This work was supported by private funds (C.G.S.), a grant from the Howard Hughes Medical Institute (Undergraduate Biological Sciences Education Program) to Penn State, a grant from the Netherlands Organization for International Cooperation in Higher Education (M.v.T.) and grants from NSF and NASA (S.B.H.). C.G.S. is deceased (April 12, 1998).
literature cited
Caspers, G.-J., D. UitdeWeert, J. Wattel, and W. W. DeJong.
Cooper, A., and D. Penny.
Cracraft, J.
Cracraft, J., and D. P. Mindell.
Groth, J. G., and G. F. Barrowclough.
Härlid, A., and U. Arnason.
Härlid, A., A. Janke, and U. Arnason.
———.
Hedges, S. B., P. H. Parker, C. G. Sibley, and S. Kumar.
Hedges, S. B., and C. G. Sibley.
Hedges, S. B., M. D. Simmons, M. A. M. van Dijk, G.-J. Caspers, W. W. deJong, and C. G. Sibley.
Ho, C. Y.-K., E. Prager, A. C. Wilson, D. T. Osuga, and R. E. Feeney.
Kimura, M.
Kumar, S., K. Tamura, and M. Nei.
Kurochkin, E. N.
Lyons-Weiler, J., and G. A. Hoelzer.
Lyons-Weiler, J., G. A. Hoelzer, and R. J. Tausch.
Mindell, D. P., M. D. Sorenson, D. E. Dimcheff, M. Hasegawa, J. C. Ast, and T. Yuri.
Mindell, D. P., M. D. Sorenson, C. J. Huddleston, H. C. Miranda Jr., A. Knight, S. J. Sawchuk, and T. Yuri.
Prager, E. M., A. C. Wilson, D. T. Osuga, and R. E. Feeney.
Pycraft, W. P.
Rotthowe, K., and J. M. Starck.
Sheldon, F. H., and A. H. Bledsoe.
Sibley, C. G., and J. Ahlquist.
Sibley, C. G., J. Ahlquist, and B. L. Monroe Jr.
Sibley, C. G., and B. L. Monroe Jr.
Stapel, S. O., J. A. Leunissen, M. Versteeg, J. Wattel, and W. deJong.
Swofford, D. L.
Thompson, J. D., D. G. Higgins, and T. J. Gibson.
van Tuinen, M., C. G. Sibley, and S. B. Hedges.
Waddell, P. J., Y. Cao, M. Hasegawa, and D. P. Mindell.

{kind=link}
{kind=link}