





Abstract
Species are commonly delimited on the basis of gaps in patterns of morphological variation, but there seems to be little recent work on methods to objectively assess such gaps. Here, we introduce a statistical approach that uses measurements of continuous morphological characters and geographic variation in those characters to (i) measure the strength of the evidence for the existence of a gap in morphological variation between two hypothesized species and (ii) examine if a gap in morphological variation between two hypothesized species can be explained by an alternative hypothesis of geographic variation within a species. This approach is based on recent developments in analyses of multivariate normal mixtures, estimates of multivariate tolerance regions, and principal coordinates of neighboring matrices. We demonstrate the application of the approach by examining previously proposed hypotheses of species limits in the plant genus Escallonia. We discuss the main features of the method, including potential limitations, in relation to other approaches that use gaps in morphological variation as a criterion for species delimitation. The method we propose can help strengthen the link between the theory and practice of species delimitation by increasing the transparency and consistency of taxonomic decisions based on morphology, thus contributing to integrative approaches for species delimitation that consider morphological and geographic data on an equal footing with other kinds of information.
Conventionally, species are recognized by seemingly discontinuous, nonoverlapping patterns of variation in characters of individuals from geographically circumscribed populations (Mayr 1942; Davis and Heywood 1963). However, identifying and delimiting species has been a controversial endeavor in systematics. Disagreements stem largely from the desire of biologists to provide an objective definition of species (Mayr 1982). Debates range from philosophical arguments as to what species are (ontology, e.g., Mishler and Donoghue 1982; Wilson 1999; Stamos 2003) to discussions on operational methods to delimit species (epistemology, e.g., Howard and Berlocher 1998; Sites and Marshall 2004). Underlying the controversy is the notion that understanding species boundaries is essential for elucidating the processes driving the origin and maintenance of biological diversity (Coyne and Orr 2004), as well as for basic and applied research in other areas of biology (Gaston 2000; Agapow et al. 2004; Padial and De La Riva 2006; Light et al. 2008). de Queiroz (1998), de Queiroz (2005) suggested that recognizing species as separately evolving segments of metapopulation-level lineages provides a context for integrating information from different sources for inferring lineage separation and thus for evaluating hypotheses about species boundaries. This pluralistic and unifying framework fosters the use and development of a wide range of methods for inferring species limits (de Queiroz 2007) including non–tree- and tree-based approaches (Sites and Marshall 2004). Non–tree-based approaches focus on gene flow assessments, such as crossability experiments to evaluate the level of pre- and postzygotic reproductive isolation (e.g., Ackermann et al. 2008). Tree-based approaches focus on properties of phylogenetic trees (Sites and Marshall 2004) and include the use of morphological or molecular data to evaluate reciprocal monophyly (e.g., Wiens and Penkrot 2002; Lehtonen 2008) and coalescent theory to model the relationship between gene trees and species history (e.g., Knowles and Carstens 2007; Cummings et al. 2008). Recent studies have combined non–tree- and tree-based approaches, using georeferenced occurrence records and spatially explicit environmental information to evaluate demographic exchangeability and gene flow between populations (e.g., Raxworthy et al. 2007; Stockman and Bond 2007; Bond and Stockman 2008). However, non–tree-based approaches to delimit species using morphological variation have received little attention recently (Wiens 2007, but see Wiens and Servedio 2000) despite substantial past interest (e.g., Sneath and Sokal 1973). This is surprising because many, perhaps most, species are still delimited based on statements made by museum/herbarium-based systematists about patterns of morphological variation (Luckow 1995; Futuyma 1998; Wiens and Servedio 2000).
How do systematists use information on morphological variation to decide what to call a species? Often, seemingly nonoverlapping patterns of morphological variation are used as a criterion for inferring species boundaries (Davis and Nixon 1992; Wiens and Servedio 2000; Sites and Marshall 2004). This operational criterion—gaps in the pattern of morphological variation—is based on the idea that morphological discontinuities suggest that some evolutionary force (e.g., lack of gene flow, natural selection) may prevent two distinct lineages from homogenizing (Wiens and Servedio 2000; Wiens and Penkrot 2002; Coyne and Orr 2004; Mallet 2008). Here, we focus on two issues that systematists contend with when using gaps in morphological variation as a criterion for inferring species limits.
Foremost, systematists need to weigh the strength of the evidence indicating a gap in morphological variation (or lack thereof). However, there seems to be little current work on how to do so. Various kinds of cluster analyses have been proposed to examine species limits using morphological data (Sneath and Sokal 1973), but algorithms used to define clusters lack a clear relationship to the operational criterion of a morphological gap (Dunn and Everitt 1982, p. 101–104) and respective underlying theory (Mayr 1992; de Queiroz and Good 1997; Wiens and Penkrot 2002). Clustering algorithms assume a hierarchical structure even though morphological variation at and below the species level is unlikely to be hierarchical and thus force data into groups irrespective of whether such groups exist in nature (Crisp and Weston 1993; de Queiroz and Good 1997). Some systematists have also tried to examine species limits using statistics that evaluate differences in central tendency (e.g., analysis of variance, discriminant analysis) among sets of samples (Luckow 1995; Henderson 2006); however, a difference in central tendency between samples is not the same as a gap between samples. In univariate or multivariate space, two samples may differ in central tendency in the absence of a gap separating two modes (McLachlan and Peel 2000; Ray and Lindsay 2005). Because the operational criterion of interest is a gap in morphology (see above), and not differences in central tendency (Luckow 1995, see also Patten and Unitt 2002), statistical tests focusing on measures of central tendency would not seem germane. Recently, a way to measure the strength of the evidence for the existence of a gap in qualitative or discrete characters between pairs of hypothesized species was developed based on statistical tolerance regions (Wiens and Servedio 2000). Here, we extend this approach to the case of quantitative continuous morphological characters using recent developments in statistics related to the topography of multivariate normal mixtures (Ray and Lindsay 2005) and estimates of multivariate tolerance regions (Krishnamoorthy and Mathew 1999; Krishnamoorthy and Mondal 2006).
A second issue that systematists confront is that the operational criterion of a morphological gap may require, contingent on species definitions (sensu de Queiroz 1998), that populations of the hypothesized species are sympatric (Mayr 1963; Futuyma 1998; Coyne and Orr 2004; Mallet 2008). Accordingly, systematists who try to evaluate species limits among nonsympatric populations may want to assess whether a morphological discontinuity interpreted as a species boundary can be explained by an alternative hypothesis of geographic variation within a single species (de Queiroz and Good 1997; de Queiroz 2007). Ideally, the alternative hypothesis would identify the factors thought to shape geographic variation in morphology and would be contrasted against a hypothesis of a species boundary using spatial data on such factors. In practice, often little is known about the factors determining morphological variation and their spatial variation, and systematists resort to variables derived from spatial coordinates to examine geographic patterns of morphological variation. For instance, Mantel tests (Mantel 1967; Sokal 1979; de Queiroz and Good 1997) can be used to model simple large-scale spatial trends, akin to those predicted in neutral traits according to models of isolation by distance (Wright 1943, Wright 1946, Wright 1969). Aside from isolation by distance, environmental differences can determine spatial variation in morphology within a species (e.g., local adaptation) potentially resulting in spatial patterns more complex than large-scale spatial trends, which are difficult to describe using Mantel approaches or trend surface polynomial regression (Gabriel and Sokal 1969; Legendre and Legendre 1998). Here, we show how recent advances in spatial ecology, particularly the development of spatial analysis by means of principal coordinates of neighbor matrices (Borcard and Legendre 2002, Dray et al. 2006, Griffith and Peres-Neto 2006), can be used to model complex spatial patterns of morphological variation, like those that can arise from spatially structured evolutionary forces acting on a single species and thus provide a powerful alternative hypothesis of geographic variation in morphology that does not require a species boundary.
Below, we first describe the conceptual basis of the approach that systematists can use to deal with both issues stated above. Next, we demonstrate the application of this approach by using herbarium data to evaluate previously proposed hypotheses (Sleumer 1968) about species limits in the plant genus Escallonia L.f (Escalloniaceae), a widespread group of neotropical shrubs and trees.
THE CONCEPTUAL BASIS
We emphasize that the approach we describe here is not designed to formulate hypotheses of species limits. Such hypotheses can be derived from previous taxonomic studies, as in our example data set (see below), or generated from the evaluation of other operational criteria (e.g., molecular phylogenetics or artificial crosses). Our method is designed to help systematists weigh the strength of the evidence supporting a previously proposed hypothesis about a species boundary. Thus, we consider taxa designated as species to be hypotheses of species as biological entities in nature (Hey et al. 2003; Wheeler 2004; Henderson 2005). Accordingly, we refer to taxa designated as species as “hypothesized species” and to the limits of those taxa as “hypothesis of species limits” or “hypothesis of species boundaries.”
Inferring Gaps in Morphology
Wiens and Servedio (2000) developed a statistical approach to evaluate hypotheses of species limits based on qualitative or discrete characters. Their approach was based on theory that emphasizes pairwise comparisons among hypothesized species, similar in this sense to approaches based on assessments of crossability (e.g., Rieseberg 2000; Dettman et al. 2003; Ackermann et al. 2008) that are necessarily based on pairwise comparisons. Here, we extend the approach developed by Wiens and Servedio (2000) to the case of quantitative continuous characters. In this case, we will assume that phenotypic variation arises from the effect of several genes and that there is random mating among the individuals in a geographic locality that belong to a single species. Under these assumptions, morphological variation is reasonably described by a normal distribution (Sokal and Rohlf 1995; Templeton 2006). Conversely, the hypothesis that there are two species in a geographic locality is supported when the distribution of morphological variation has two (or more, see below) modes (local maxima), so that there is a gap separating two approximately normal distributions (Futuyma 1998). Like the criterion based on gaps in the distribution of qualitative or discrete characters, the criterion based on gaps in the distribution of continuous characters assumes that bimodal (or multimodal) distributions do not result from polymorphisms, ontogenetic variation or phenotypic plasticity. Based on these assumptions, a hypothesis of species limits can be evaluated by assessing the number of modes in the distribution of continuous morphological characters of two hypothesized species, as we explain below.
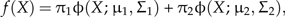
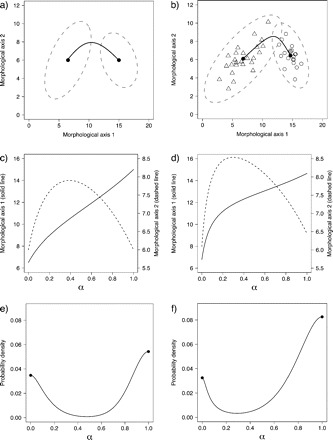
where π1 and π2 are the mixing proportions of the distributions describing the morphological variation of each of the two hypothesized species, φ(X;μ1,Σ1) is the pdf of the distribution describing the morphological variation of one species with mean μ1 and variance Σ1 and φ(X;μ2,Σ2) is the pdf of the distribution describing the morphological variation of the other species with mean μ2 and variance Σ2. For our purposes, both hypothesized species should be weighted equally, so π1 = π2 = 0.5. Hereafter, we use carets to denote sample-based estimates of population parameters. For instance, , and denote estimates of f(X), φ(X;μ1,Σ1) and φ(X;μ2,Σ2), respectively.
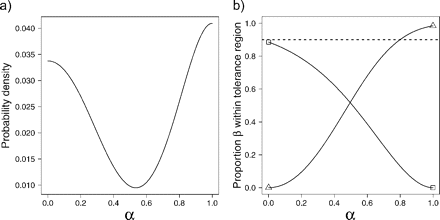
When X is univariate, a mixture of two normal distributions can have one or two modes (Robertson and Fryer 1969), and evidence for bimodality (i.e., two local maxima) can be detected by visually inspecting a plot of , as illustrated by a numerical example of f(X) (Fig. 1a), random samples from φ(X;μ1,Σ1) and φ(X;μ2,Σ2) (Fig. 1b) and the respective (Fig. 1c). Bimodality in can be regarded as a necessary, but not sufficient, condition to support the hypothesis that a sample of morphological measurements represents two species. Although bimodality implies that two local maxima are separated by a local minimum, the latter being the gap in morphological variation that suggests a species boundary, it does not directly inform how morphologically distinct two hypothesized species are. Following Wiens and Servedio (2000), one may select a priori a frequency cutoff below which overlap of phenotypic values between hypothesized species is considered rare enough as to suggest evolutionary isolation. Then, univariate tolerance regions for normal distributions (Natrella 1963) can be used to determine the degree of phenotypic overlap between hypothesized species. By example, the value in Figure 1b estimates with confidence γ = 0.95 the upper limit of the one-tailed tolerance region encompassing a proportion β = 0.9 of the hypothesized species represented by . Likewise, the value in Figure 1b estimates with confidence γ = 0.95 the lower limit of the one-tailed tolerance region encompassing a proportion β = 0.9 of the hypothesized species represented by . The confidence level γ is the frequency with which estimated tolerance regions encompass a proportion ≥ β of the sampled population: If we were to draw a large number of random samples from a given statistical population (φ(X;μ1,Σ1) or φ(X;μ2,Σ2)), a proportion γ of those random samples would yield estimated tolerance regions that contain a proportion ≥ β of the population from which the samples were drawn (Fig. 1d). For a given confidence level γ, no overlap between tolerance regions would indicate that there is a gap between φ(X;μ1,Σ1) and φ(X;μ2,Σ2), so that proportions ≥ β of the two distributions do not overlap (Fig. 1b). If the overlap does not exceed the a priori frequency cutoff, then the results would support the hypothesis that the sample of morphological measurements represents two species.
Numerical example of univariate normal mixtures and tolerance regions. a) pdf for the mixture of two distributions of a morphological variable (continuous line) and the corresponding statistical populations describing hypothesized species with parameters chosen for illustrative purposes (φ(X;μ1 = − 4,Σ1 = 2.25) dashed line, φ(X;μ2 = 3,Σ2 = 4) dotted lines). b) Simulated random samples from each of the two statistical populations in (a). Gray bars: , , n = 15; white bars: , 2 = 3.987, n = 30. Values and estimate with confidence γ = 0.95 the limits of one-tailed tolerance regions encompassing a proportion β = 0.9 of the hypothesized species represented by gray and white bars, respectively. c) pdf of the normal mixture (continuous line) and its components (dashed and dotted lines) estimated from the samples in (b). d) Distribution of 100,000 estimated tolerance limits for each of the two populations in (a). Each limit was calculated for γ = 0.95 and β = 0.9, from simulated samples as those in (b). A proportion 0.947 (∼γ) of the estimated limits for φ(X;μ1 = − 4,Σ1 = 2.25) (gray bars) were equal to or higher than a, the actual (as opposed to estimated) upper limit of the interval containing a proportion = 0.9 of the population. Likewise, a proportion 0.946 (∼γ) of the estimated limits for φ(X;μ2 = 3,Σ2 = 4) (white bars) were equal to or lower than b, the respective actual lower limit.
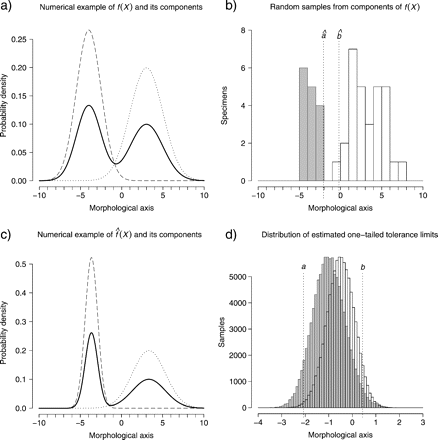
In contrast to the case above in which X is univariate, a mixture of two multivariate normal distributions can have more than two modes (Ray and Lindsay 2005). Detecting and characterizing evidence for more than one mode in multivariate space X involves inspecting a plot of along the ridgeline manifold. The ridgeline manifold is a curve guaranteed to include all the critical points (maxima, minima, and saddles) of (Fig. 2; Ray and Lindsay 2005). Points along the ridgeline manifold correspond to values of variable α that range from zero at the multivariate mean of one hypothesized species to one at multivariate mean of the other (Fig. 2c,d). Visual inspection can reveal local maxima and minima of along the ridgeline manifold (Fig. 2f). For the same reason described above, more than one mode in along the ridgeline manifold can be regarded as a necessary, but not sufficient, condition to support the hypothesis that the sample of morphological measurements represents two species. The frequency cutoff approach of Wiens and Servedio (2000) can also be applied when X is multivariate by examining overlap of ellipsoidal tolerance regions for multivariate normal distributions (Krishnamoorthy and Mathew 1999; Krishnamoorthy and Mondal 2006). Given a confidence level γ, one can calculate various ellipsoids defining tolerance regions for one of the distributions, for example, , using various β values. Each of these ellipsoids shares a single point along the ridgeline manifold (corresponding to a single value of α) with an ellipsoid defining a tolerance region for the other distribution, (Fig. 3a, Ray and Lindsay 2005). A plot of the proportions β covered by these ellipsoids for several points along the ridgeline manifold (corresponding to several values of α) reveals the estimated phenotypic overlap between the hypothesized species (Fig. 3b). If the overlap does not exceed the frequency cutoff selected a priori, then there would be empirical support for the hypothesis that the sample of morphological measurements represents two species.
Numerical example of multivariate normal mixtures and tolerance regions. a) Two multivariate normal distributions describing hypothesized species with parameters chosen for illustrative purposes 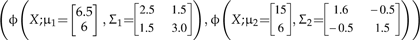 are represented by ellipses delineating regions including a proportion to 0.95 of each statistical population and the respective multivariate (closed circles). The black continuous line that joins both multivariate means is the ridgeline manifold. b) Simulated random samples from each of the statistical populations in (a). Triangles:
are represented by ellipses delineating regions including a proportion to 0.95 of each statistical population and the respective multivariate (closed circles). The black continuous line that joins both multivariate means is the ridgeline manifold. b) Simulated random samples from each of the statistical populations in (a). Triangles: 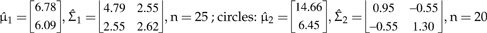 Estimated (as opposed to actual) multivariate means are shown as closed circles and the estimated ridgeline manifold as a black continuous line. The dashed lines delimit tolerance regions for proportion β = 0.9 and confidence level γ = 0.95. c) Points along the ridgeline manifold in (a) correspond to values of variable α that range from 0 to 1 and map onto morphological space as shown by the two ordinates representing morphological Axes 1 and 2. d) Relationship between α and morphological Axes 1 and 2 as in (c) but based on estimated values from the random samples in (b). e) pdf evaluated at various points corresponding to values of α along the ridgeline manifold in (a). f) Estimated pdf evaluated at various points corresponding to values of α along the ridgeline manifold in (b).
Estimated (as opposed to actual) multivariate means are shown as closed circles and the estimated ridgeline manifold as a black continuous line. The dashed lines delimit tolerance regions for proportion β = 0.9 and confidence level γ = 0.95. c) Points along the ridgeline manifold in (a) correspond to values of variable α that range from 0 to 1 and map onto morphological space as shown by the two ordinates representing morphological Axes 1 and 2. d) Relationship between α and morphological Axes 1 and 2 as in (c) but based on estimated values from the random samples in (b). e) pdf evaluated at various points corresponding to values of α along the ridgeline manifold in (a). f) Estimated pdf evaluated at various points corresponding to values of α along the ridgeline manifold in (b).
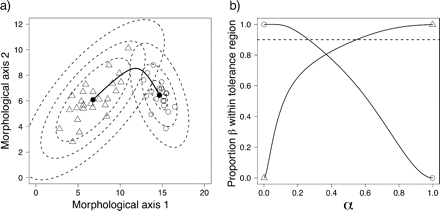
Numerical example of multivariate normal mixtures and tolerance regions. a) Estimated multivariate means (closed circles) and ridgeline manifold (black continuous line) based on simulated random samples (same as in Fig. 2b) from the two hypothesized species in Figure 2a. Dashed lines delimit three elliptical tolerance regions for each sample. These six tolerance regions form three pairs, each pair sharing a single point in the ridgeline manifold (corresponding to a single value of α). b) The continuous lines show, for different values of α along the ridgeline manifold (in the abscissa), the estimated proportion β (in the ordinate) covered with confidence γ = 0.95 by the two elliptical tolerance regions sharing a single point in the ridgeline manifold. Open triangles mark the ends of the line representing one hypothesized species and open circles for the other. The horizontal dotted line marks proportion β = 0.9.
Gaps versus Geographic Variation in Morphology
Phenotypic discontinuities can result from geographic differentiation within a single species (e.g., McGowen et al. 2001; Stenström et al. 2002; Olson et al. 2004). Therefore, under some species definitions (sensu de Queiroz 1998), it is often useful to examine whether a morphological discontinuity can be explained solely by geographic variation within a lineage, without having to invoke a boundary between species (de Queiroz and Good 1997; de Queiroz 2007). To this end, geographic variation in morphology can be modeled using principal coordinates of neighbor matrices (Borcard and Legendre 2002; Dray et al. 2006; Griffith and Peres-Neto 2006). This approach uses the geographic coordinates of the collection localities of individual specimens on which morphological variables were measured to construct a matrix of geographic distances. Principal coordinate analysis of this distance matrix yields eigenvectors, known as spatial eigenvectors, which are useful to describe complex patterns of spatial variation in morphology, and can be used as explanatory variables in regression models to directly confront a hypothesis of species limits against the alternative hypothesis of geographic variation within a species, as explained next.
Multiple regression models can be used when morphological space X is univariate. A first step in this analysis involves accounting for spatial linear trends in morphology by regressing the morphological variable on the geographic coordinates of the specimens in the sample (see Borcard and Legendre 2002; Borcard et al. 2004). When a statistically significant spatial linear trend is detected in this first step, the response variable in the following step would be the residuals yielded after extracting the spatial linear trend. Otherwise, the response variable in the following step would be the original morphological variable. The next step involves a second multiple regression in which spatial eigenvectors enter as explanatory variables to determine whether variation in morphology can be modeled as geographic variation within a single species (Fig. 4). A dummy variable (coding one of the hypothesized species as zero and the other as one (Draper and Smith 1998)) also enters as explanatory variable to determine whether a significant amount of variation in morphology can be explained by the hypothesis of a species boundary (Fig. 4). Further independent variables in this second multiple regression are interaction terms between each spatial eigenvector and the dummy variable, constructed by multiplying each spatial eigenvector by the dummy variable. These interaction terms also represent the hypothesis of a species boundary. For instance, the interaction term between the dummy variable and the first spatial eigenvector represents the idea that the relationship between the continuous morphological character (the response variable) and the first spatial eigenvector differs between both hypothesized species. The hypothesis of species limits would be supported if the analysis yields at least one statistically significant regression coefficient for the dummy variable or any of its interactions with spatial eigenvectors. In that case, one can conclude that variables representing the hypothesis of a species limit (the dummy variable or any of its interactions) accounted for variation in morphology that was not captured by spatial variables (spatial eigenvectors) representing an alternative hypothesis of geographic variation within a species. This result would considerably strengthen the evidence supporting the hypothesis of species limits because the spatial eigenvectors have remarkable power to model a wide range of spatial structures (Borcard and Legendre 2002; Dray et al. 2006; Griffith and Peres-Neto 2006).
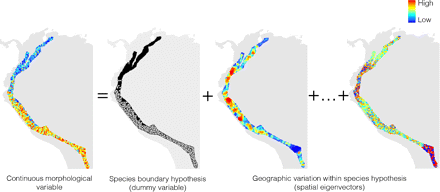
Illustration of how a multiple linear regression can be used to determine whether a gap in morphology can be explained by a hypothesis of a species boundary or by a hypothesis of geographic variation within a single species. The map on the left of the equal sign shows the geographic pattern of morphological variation measured on specimens of two hypothesized species. The hypothesis of a species boundary assigns the specimens measured to one (white circles) or the other (black circles) hypothesized species. The hypothesis of geographic variation in morphology within a single species is modeled with spatial eigenvectors derived from a principle coordinate analysis of a matrix of geographic distances of the collection localities of the specimens measured. Only two spatial eigenvectors are shown here, corresponding to eigenvalues of rank 1 (left) and 1001 (right).
When X is multivariate, redundancy analysis (RDA, Rao 1964) can be used to evaluate a hypothesis of species limits against the alternative hypothesis of geographic variation within a species. RDA is an extension of multiple regression that allows regressing several response variables (instead of just one response variable in multiple regression) on several independent variables (ter Braak 1995; Legendre and Legendre 1998). The analysis proceeds in two steps that are similar to those used when X is univariate. First, one runs an RDA in which the multivariate morphological measurements are the response variables and the geographic coordinates of the specimens in the sample are the only independent variables. If a statistically significant spatial linear trend is detected in this first step, the response variables in the next step would be the (multivariate) residuals around the linear trend, otherwise the response variables would be the original multivariate morphological measurements. The second step in the analysis involves an RDA in which the spatial eigenvectors are independent variables representing the hypothesis of geographic variation within a species, and as before, a dummy variable and its interactions with spatial eigenvectors represent the hypothesis of species limits. A statistically significant coefficient for any of the two latter independent variables provides support for the hypothesis of a species limit and indicates that a multivariate morphological discontinuity hypothesized to delimit two species cannot be entirely explained by a hypothesis of geographic variation within a species.
EMPIRICAL EXAMPLES
Study System
To illustrate the application of our approach, we used 2 examples from an ongoing systematic study on the evolution and diversification of the plant genus Escallonia. The complete data set examined here consists of 6 of the 39 taxa designated as species in the only available monograph of the genus (Sleumer 1968), taken here as hypotheses of species in nature: Escallonia micrantha, Escallonia millegrana, Escallonia discolor, Escallonia piurensis, Escallonia resinosa, and Escallonia schreiteri. Example A includes Escallonia millegrana and Escallonia micrantha, a pair of morphologically similar taxa for which species limits are uncertain. Molecular phylogenies based on nuclear loci show that individuals sampled from both these taxa form a monophyletic group well isolated from all other taxa within Escallonia (Zapata 2010). Example B includes a group of four taxa that are morphologically similar in reproductive characters (i.e., inflorescence type and petal, ovary, and calyx shapes) but differ in vegetative morphology (i.e., leaf shape). All individuals sampled from these four taxa belong to a single well-supported clade in the aforementioned molecular phylogenies (Zapata 2010). Examples A and B illustrate different outcomes that may result from using the methods developed here.
Materials and Methods
We used a total of 85 herbarium specimens to represent the geographic range of morphological and ecological variation in the six taxa included in Examples A and B (Fig. 5). Specimens were assigned to each taxon after detailed studies of morphology, using a dichotomous key (Sleumer 1968), and carrying out comparative analyses using collections available from more than 17 herbaria including many of the same specimens that Sleumer studied. For each specimen, latitude–longitude coordinates were obtained from herbarium labels, high resolution maps, and online gazetteers (Guralnick et al. 2006). We characterized variation in 6 vegetative and 21 reproductive continuous characters ( Supplementary Data available from http://www.sysbio.oxfordjournals.org/), selected after careful study of specimens and literature review (Kausel 1953; Sleumer 1968), that describe the form of leaves (e.g., petiole length, lamina length, and width), inflorescences (length of main axis and first secondary axis), and flowers (e.g., ovary length and width, calyx tube length, calyx lobe length and width, and petal length and width). We measured only mature leaves and flowers in all specimens. Vegetative characters were measured using a standard metric ruler on dried specimens, and floral characters were measured using a digital caliper (Digimatic CD-6” CS; Mitutoyo Japan) or a micrometer eyepiece on a stereoscopic dissecting microscope (SMZ645; Nikon USA) on flowers that were previously rehydrated. All measurements were recorded from three different structures for each specimen whenever possible and then averaged for each specimen.
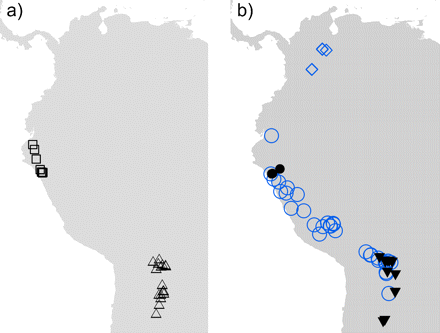
Outline of Northwestern South America and collection localities for the herbarium specimens included in the morphological samples of hypothesized Escallonia species. a) Example A, □ = Escalloniamicrantha, ▵ = Escalloniamillegrana. b) Example B: = Escalloniadiscolor, • = Escalloniapiurensis, circ = Escalloniaresinosa, = Escalloniaschreiteri.
For each of the two examples (A and B), we used measurements of continuous characters to derive orthogonal morphological axes using principal component analysis (PCA) on a correlation matrix. The first two resulting principal components, PC1 and PC2, captured 48.8% and 59.6% of the morphological variation in the samples for Examples A and B, respectively, and we used them to define the multivariate morphological space X for further analyses. In particular, we used the multivariate spaces defined by PC1 and PC2 to estimate the ridgeline manifold (Fig. 2b) and corresponding pdf (Fig. 2f) for each pair of taxa (i.e., hypothesized species). When the pdf along the ridgeline manifold did not exhibit more than one mode, we stopped the analyses because the first necessary condition to support the hypothesis of a species boundary was not met (see Inferring Gaps in Morphology section). Otherwise, we performed further analyses to estimate how morphologically distinct the two taxa were. In particular, we calculated a series of ellipsoid tolerance regions for each taxon (Fig. 3a) and estimated the proportions β covered by each of these ellipsoids (Fig. 3b) using a fixed confidence level γ = 0.95. Following Wiens and Servedio (2000), we used 0.1 as an a priori frequency cutoff below which overlap of phenotypic values between hypothesized species was considered rare enough to suggest lineage isolation. Therefore, if the overlap of tolerance regions for two taxa calculated with γ = 0.95 did not exceed the frequency cutoff, we considered the two taxa sufficiently distinct to support the hypothesis of a species limit.
When taxa were morphologically distinct according to the frequency cutoff described above, we used RDA to ask whether the morphological discontinuity could be explained by a hypothesis of geographic variation within a single species. First, the values of PC1 and PC2 defining X were evaluated for spatial linear trends using the geographic coordinates of each specimen as explanatory variables (see Borcard et al. 2004). After detrending, we used the multivariate residuals as response variables in a second RDA. Among the explanatory variables in the second RDA, we used the spatial eigenvectors associated with positive eigenvalues (see Borcard and Legendre 2002; Dray et al. 2006; Griffith and Peres-Neto 2006) to represent the hypothesis of geographic variation within a single species. We used the geographic coordinates of the specimens to derive the spatial eigenvectors following the description in Dormann et al. (2007). All other explanatory variables in the second RDA represented the hypothesis of a species limit, including a dummy variable coding for each putative species and its interactions with the spatial eigenvectors. Only explanatory variables that were not highly correlated were retained in the RDA models. We assessed the statistical significance of the regression coefficient for each explanatory variable through permutation (Legendre and Legendre 1998). We used a standard model simplification procedure (Crawley 2002) to find a minimal adequate model by removing nonsignificant terms that did not increase model explanatory power. We measured model explanatory power as the “adjusted redundancy statistic,” which is the RDA equivalent of the adjusted regression coefficient of determination (Peres-Neto et al. 2006). All statistical analyses were carried out in the R environment (R Development Core Team 2009) using the packages ellipse (Murdoch and Chow 2007), labdsv (Roberts 2007), mvtnorm (Genz et al. 2009), spdep (Bivand et al. 2009), vegan (Oksanen et al. 2009), and our scripts ( Supplementary Data).
Results
Example A.—We measured 11 specimens assigned to E. micrantha and 17 assigned to E. millegrana. These samples separated clearly in multivariate morphological space (Fig. 6a). A plot of the estimated pdf along the ridgeline manifold revealed two local maxima, one at α = 0, the estimated mean of the distribution describing E. millegrana, and the other at α = 1, the estimated mean of the distribution describing E. micrantha (Fig. 7a). This implied bimodality in , and thus, it fulfilled the first necessary condition to suggest a morphological gap separating this pair of taxa (hypothesized species). However, tolerance regions covering proportions β < 0.9 of the distributions of both taxa overlapped (Fig. 7b), failing to support the hypothesis of a species boundary according to the a priori frequency cutoff. As we did not find enough evidence in these data to support the idea that E. micrantha and E. millegrana were separated by a gap in the morphological space defined by PC1 and PC2, we conducted no further analyses because it was not necessary to confront the hypothesis of a species boundary against an alternative hypothesis of geographic variation within a species.
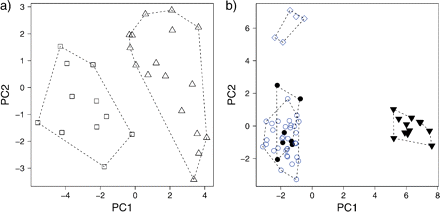
PCA describing the pattern of morphological variation among hypothesized Escallonia species. a) Example A, □ = Escalloniamicrantha, ▵ = Escalloniamillegrana. b) Example B: = Escalloniadiscolor, • = Escalloniapiurensis, circ = Escalloniaresinosa, = Escalloniaschreiteri. The dashed polygons correspond to the minimum convex hulls for the samples of each hypothesized species.
Inference of morphological gap between Escallonia species in Example A. a) Estimated pdf, , evaluated for various points along the ridgeline manifold (α) for the mixture of two normal distributions of morphological variation implied by the hypothesized species limit; b) estimated proportion β (in the ordinate) covered by the elliptical tolerance regions (γ = 0.95) sharing a single point at α in the ridgeline manifold (in the abscissa). Symbols mark the end of lines representing each species; □ = Escalloniamicrantha, ▵ = Escalloniamillegrana. The horizontal dotted line marks proportion β = 0.9.
Example B.—Only 7 specimens were available for E. piurensis and 5 for E. discolor, whereas 12 and 35 specimens were available for E. schreiteri and E. resinosa, respectively. Specimens assigned to E. discolor and E. schreiteri separated clearly from all other taxa in the morphological space defined by PC1 and PC2, whereas specimens assigned to E. piurensis and E. resinosa did not (Fig. 6b).
The plots of along the ridgeline manifold for E. schreiteri and each of the other taxa were all bimodal (Fig. 8a, c, e), suggesting a morphological gap between E. schreiteri and all other three taxa. The corresponding plots of the proportions covered by tolerance regions revealed that the phenotypic overlap was smaller than the frequency cutoff in all cases (Fig. 8b, d, f), indicating that E. schreiteri was sufficiently distinct from all other three taxa to support the hypothesis of a species limit. Therefore, we evaluated this hypothesis against an alternative of geographic variation within a species. Although the pattern of morphological variation in the pairwise comparison between E. schreiteri and E. resinosa was spatially structured and partly explained by spatial eigenvectors, a dummy variable representing a species boundary accounted for variation in morphology that was not captured by spatial variables (Table 1). In contrast, the morphological gaps separating E. schreiteri from E. discolor and E. schreiteri from E. piurensis were explained by geographic differentiation among allopatric populations (Fig. 5) within a single species. The relationship between PC1 and PC2 and the variables representing the hypotheses of species limits were not statistically significant for either of the latter pairwise comparisons (Table 1). Furthermore, the pattern of morphological variation in both pairwise comparisons corresponded to large-scale spatial trends (RDA for detrending yielded significant regression coefficients, not shown) rather than to more complex spatially structured patterns (Table 1, nonsignificant regression coefficients for spatial eigenvectors), suggesting simple geographic gradients in morphology in these taxa.
Results from RDA after accounting for spatial linear trend for three taxa (hypothesized species) pairs
| Taxa pair and adjusted redundancy statistic | Source of variation | df | Variance | F | P |
| Escallonia schreiteri | SEV1 | 1 | 0.017 | 0.640 | 0.48 |
| and | (0.016) | (0.684) | (0.43) | ||
| Escallonia resinosa | SEV2 | 1 | 0.462 | 18.150 | 0.01 |
| Ra2 = 0.88 | (0.463) | (19.147) | (0.01) | ||
| (Ra2 = 0.89) | SEV3 | 1 | 0.410 | 16.000 | 0.01 |
| (0.407) | (17.114) | (0.01) | |||
| SEV4 | 1 | 0.022 | 0.852 | 0.37 | |
| (0.022) | (0.9145) | (0.34) | |||
| SEV5 | 1 | 0.016 | 0.612 | 0.34 | |
| (0.016) | (0.657) | (0.40) | |||
| SEV6 | 1 | 0.091 | 3.570 | 0.03 | |
| (0.091) | (3.822) | (0.06) | |||
| SEV7 | 1 | 0.004 | 0.154 | 0.76 | |
| Dum. | 1 | 7.950 | 311.264 | 0.01 | |
| (7.926) | (333.089) | (0.01) | |||
| SEV1 * Dum. | 1 | 0.000 | 0.040 | 0.93 | |
| SEV2 * Dum. | 1 | 0.016 | 0.626 | 0.43 | |
| SEV3 * Dum. | 1 | 0.001 | 0.054 | 0.93 | |
| SEV4 * Dum. | 1 | 0.022 | 0.852 | 0.37 | |
| (0.022) | (0.9145) | (0.34) | |||
| SEV5 * Dum. | 1 | 0.022 | 0.880 | 0.35 | |
| SEV7 * Dum. | 1 | 0.043 | 1.710 | 0.16 | |
| Residual | 30 | 0.766 | |||
| (37) | (0.880) | ||||
| E. schreiteri | SEV1 | 1 | 0.036 | 0.250 | 0.73 |
| and | Dum. | 1 | 0.160 | 1.080 | 0.33 |
| Escallonia discolor | SEV1 * Dum. | 1 | 0.004 | 0.003 | 0.97 |
| Ra2 = 0 | Residual | 13 | 1.933 | ||
| E. schreiteri | SEV1 | 1 | 0.002 | 0.017 | 0.99 |
| and | Dum. | 1 | 0.002 | 0.019 | 0.96 |
| Escallonia piurensis | SEV1 * Dum. | 1 | 0.085 | 0.753 | 0.40 |
| Ra2 = 0 | Residual | 15 | 1.693 |
| Taxa pair and adjusted redundancy statistic | Source of variation | df | Variance | F | P |
| Escallonia schreiteri | SEV1 | 1 | 0.017 | 0.640 | 0.48 |
| and | (0.016) | (0.684) | (0.43) | ||
| Escallonia resinosa | SEV2 | 1 | 0.462 | 18.150 | 0.01 |
| Ra2 = 0.88 | (0.463) | (19.147) | (0.01) | ||
| (Ra2 = 0.89) | SEV3 | 1 | 0.410 | 16.000 | 0.01 |
| (0.407) | (17.114) | (0.01) | |||
| SEV4 | 1 | 0.022 | 0.852 | 0.37 | |
| (0.022) | (0.9145) | (0.34) | |||
| SEV5 | 1 | 0.016 | 0.612 | 0.34 | |
| (0.016) | (0.657) | (0.40) | |||
| SEV6 | 1 | 0.091 | 3.570 | 0.03 | |
| (0.091) | (3.822) | (0.06) | |||
| SEV7 | 1 | 0.004 | 0.154 | 0.76 | |
| Dum. | 1 | 7.950 | 311.264 | 0.01 | |
| (7.926) | (333.089) | (0.01) | |||
| SEV1 * Dum. | 1 | 0.000 | 0.040 | 0.93 | |
| SEV2 * Dum. | 1 | 0.016 | 0.626 | 0.43 | |
| SEV3 * Dum. | 1 | 0.001 | 0.054 | 0.93 | |
| SEV4 * Dum. | 1 | 0.022 | 0.852 | 0.37 | |
| (0.022) | (0.9145) | (0.34) | |||
| SEV5 * Dum. | 1 | 0.022 | 0.880 | 0.35 | |
| SEV7 * Dum. | 1 | 0.043 | 1.710 | 0.16 | |
| Residual | 30 | 0.766 | |||
| (37) | (0.880) | ||||
| E. schreiteri | SEV1 | 1 | 0.036 | 0.250 | 0.73 |
| and | Dum. | 1 | 0.160 | 1.080 | 0.33 |
| Escallonia discolor | SEV1 * Dum. | 1 | 0.004 | 0.003 | 0.97 |
| Ra2 = 0 | Residual | 13 | 1.933 | ||
| E. schreiteri | SEV1 | 1 | 0.002 | 0.017 | 0.99 |
| and | Dum. | 1 | 0.002 | 0.019 | 0.96 |
| Escallonia piurensis | SEV1 * Dum. | 1 | 0.085 | 0.753 | 0.40 |
| Ra2 = 0 | Residual | 15 | 1.693 |
Note: Taxa names and adjusted redundancy statistic (Ra2) for each RDA are shown in the first column. For each source of variation listed in the second column, the subsequent columns provide degrees of freedom (d.f.), variance, the F statistic, and the significance of the respective regression coefficient (P). Throughout the table values obtained after model simplification are shown in parentheses. Spatial eigenvectors (SEV) are listed in descending order according to their respective eigenvalues from SEV1 to SEVn. The dummy variable (Dum.) and its interactions with spatial eigenvectors (e.g., SEV1*Dum.) are listed afterwards. Rows corresponding to statistically significant (P < 0.05) regression coefficients are shown in bold font.
Results from RDA after accounting for spatial linear trend for three taxa (hypothesized species) pairs
| Taxa pair and adjusted redundancy statistic | Source of variation | df | Variance | F | P |
| Escallonia schreiteri | SEV1 | 1 | 0.017 | 0.640 | 0.48 |
| and | (0.016) | (0.684) | (0.43) | ||
| Escallonia resinosa | SEV2 | 1 | 0.462 | 18.150 | 0.01 |
| Ra2 = 0.88 | (0.463) | (19.147) | (0.01) | ||
| (Ra2 = 0.89) | SEV3 | 1 | 0.410 | 16.000 | 0.01 |
| (0.407) | (17.114) | (0.01) | |||
| SEV4 | 1 | 0.022 | 0.852 | 0.37 | |
| (0.022) | (0.9145) | (0.34) | |||
| SEV5 | 1 | 0.016 | 0.612 | 0.34 | |
| (0.016) | (0.657) | (0.40) | |||
| SEV6 | 1 | 0.091 | 3.570 | 0.03 | |
| (0.091) | (3.822) | (0.06) | |||
| SEV7 | 1 | 0.004 | 0.154 | 0.76 | |
| Dum. | 1 | 7.950 | 311.264 | 0.01 | |
| (7.926) | (333.089) | (0.01) | |||
| SEV1 * Dum. | 1 | 0.000 | 0.040 | 0.93 | |
| SEV2 * Dum. | 1 | 0.016 | 0.626 | 0.43 | |
| SEV3 * Dum. | 1 | 0.001 | 0.054 | 0.93 | |
| SEV4 * Dum. | 1 | 0.022 | 0.852 | 0.37 | |
| (0.022) | (0.9145) | (0.34) | |||
| SEV5 * Dum. | 1 | 0.022 | 0.880 | 0.35 | |
| SEV7 * Dum. | 1 | 0.043 | 1.710 | 0.16 | |
| Residual | 30 | 0.766 | |||
| (37) | (0.880) | ||||
| E. schreiteri | SEV1 | 1 | 0.036 | 0.250 | 0.73 |
| and | Dum. | 1 | 0.160 | 1.080 | 0.33 |
| Escallonia discolor | SEV1 * Dum. | 1 | 0.004 | 0.003 | 0.97 |
| Ra2 = 0 | Residual | 13 | 1.933 | ||
| E. schreiteri | SEV1 | 1 | 0.002 | 0.017 | 0.99 |
| and | Dum. | 1 | 0.002 | 0.019 | 0.96 |
| Escallonia piurensis | SEV1 * Dum. | 1 | 0.085 | 0.753 | 0.40 |
| Ra2 = 0 | Residual | 15 | 1.693 |
| Taxa pair and adjusted redundancy statistic | Source of variation | df | Variance | F | P |
| Escallonia schreiteri | SEV1 | 1 | 0.017 | 0.640 | 0.48 |
| and | (0.016) | (0.684) | (0.43) | ||
| Escallonia resinosa | SEV2 | 1 | 0.462 | 18.150 | 0.01 |
| Ra2 = 0.88 | (0.463) | (19.147) | (0.01) | ||
| (Ra2 = 0.89) | SEV3 | 1 | 0.410 | 16.000 | 0.01 |
| (0.407) | (17.114) | (0.01) | |||
| SEV4 | 1 | 0.022 | 0.852 | 0.37 | |
| (0.022) | (0.9145) | (0.34) | |||
| SEV5 | 1 | 0.016 | 0.612 | 0.34 | |
| (0.016) | (0.657) | (0.40) | |||
| SEV6 | 1 | 0.091 | 3.570 | 0.03 | |
| (0.091) | (3.822) | (0.06) | |||
| SEV7 | 1 | 0.004 | 0.154 | 0.76 | |
| Dum. | 1 | 7.950 | 311.264 | 0.01 | |
| (7.926) | (333.089) | (0.01) | |||
| SEV1 * Dum. | 1 | 0.000 | 0.040 | 0.93 | |
| SEV2 * Dum. | 1 | 0.016 | 0.626 | 0.43 | |
| SEV3 * Dum. | 1 | 0.001 | 0.054 | 0.93 | |
| SEV4 * Dum. | 1 | 0.022 | 0.852 | 0.37 | |
| (0.022) | (0.9145) | (0.34) | |||
| SEV5 * Dum. | 1 | 0.022 | 0.880 | 0.35 | |
| SEV7 * Dum. | 1 | 0.043 | 1.710 | 0.16 | |
| Residual | 30 | 0.766 | |||
| (37) | (0.880) | ||||
| E. schreiteri | SEV1 | 1 | 0.036 | 0.250 | 0.73 |
| and | Dum. | 1 | 0.160 | 1.080 | 0.33 |
| Escallonia discolor | SEV1 * Dum. | 1 | 0.004 | 0.003 | 0.97 |
| Ra2 = 0 | Residual | 13 | 1.933 | ||
| E. schreiteri | SEV1 | 1 | 0.002 | 0.017 | 0.99 |
| and | Dum. | 1 | 0.002 | 0.019 | 0.96 |
| Escallonia piurensis | SEV1 * Dum. | 1 | 0.085 | 0.753 | 0.40 |
| Ra2 = 0 | Residual | 15 | 1.693 |
Note: Taxa names and adjusted redundancy statistic (Ra2) for each RDA are shown in the first column. For each source of variation listed in the second column, the subsequent columns provide degrees of freedom (d.f.), variance, the F statistic, and the significance of the respective regression coefficient (P). Throughout the table values obtained after model simplification are shown in parentheses. Spatial eigenvectors (SEV) are listed in descending order according to their respective eigenvalues from SEV1 to SEVn. The dummy variable (Dum.) and its interactions with spatial eigenvectors (e.g., SEV1*Dum.) are listed afterwards. Rows corresponding to statistically significant (P < 0.05) regression coefficients are shown in bold font.
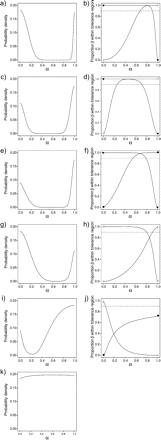
Inference of morphological gaps among Escallonia species in Example B. Each panel in the left column corresponds to the estimated pdf, , evaluated for various points along the ridgeline manifold (α) for the mixture of two normal distributions of morphological variation implied by the hypothesized species limits. Each panel in the right column corresponds to the estimated proportion β (in the ordinate) covered by the elliptical tolerance regions (γ = 0.95) sharing a single point at α in the ridgeline manifold (in the abscissa). In these latter panels, symbols mark the end of lines representing each species and the horizontal dotted line marks proportion β = 0.9. a–b) Escallonia discolor (◊) and Escallonia schreiteri (▾), c–d) Escallonia resinosa (○) and E. schreiteri (▾), e–f) Escallonia piurensis (•) and E. schreiteri (▾), g–h) E. discolor () and E. resinosa (○), i–j) E. piurensis (•) and E. discolor (◊), k) E. piurensis and E. resinosa.
The plots of along the ridgeline manifold for E. discolor and E. resinosa and E. discolor and E. piurensis were both bimodal (Fig. 8g, i), consistent with the idea that a morphological gap separates each of these pairs of taxa. However, for each of these pairwise comparisons, tolerance regions covering proportions β < 0.9 overlapped (Fig. 8h, j), failing to support the hypothesis of a species boundary. As we did not find enough evidence in these data to support the idea that E. discolor, E. resinosa, and E. piurensis were separated by morphological gaps, it was not necessary to conduct further analyses to evaluate the hypothesis of geographic variation within a species.
Finally, the plot along the ridgeline manifold for E. piurensis and E. resinosa was unimodal (Fig. 8k), thus not meeting the first necessary condition to support the hypothesis of a species boundary. Therefore, we conducted no further analyses for this pairwise comparison.
DISCUSSION
Numerous statistical methods have been developed recently to help systematists weigh the strength of the evidence related to different operational criteria to infer species boundaries (e.g., Wiens and Servedio 2000; Wiens and Penkrot 2002; Sites and Marshall 2004; Pons et al. 2006; Knowles and Carstens 2007; Raxworthy et al. 2007; O'Meara 2010). However, there is little recent work on how to measure the strength of the evidence related to the operational criterion of nonoverlapping patterns of variation in quantitative continuous morphological characters (Wiens 2007). Here, we presented a method that uses morphological and geographic data to formally assess whether there is enough evidence to indicate that a morphological gap supports a hypothesis of a species boundary and whether such a gap could be explained by an alternative hypothesis of geographic variation within a single species. This method, hereafter GCG (Gaps in Continuous characters across Geography), is based on recent developments in analyses of multivariate normal mixtures (Ray and Lindsay 2005), estimates of multivariate tolerance regions (Krishnamoorthy and Mathew 1999; Krishnamoorthy and Mondal 2006), and principal coordinates of neighboring matrices (Borcard and Legendre 2002; Borcard et al. 2004). We illustrated the application of GCG by using herbarium data to evaluate previously proposed hypotheses (Sleumer 1968) about species limits within two groups of morphologically similar taxa, both forming well-supported clades in molecular phylogenies of the neotropical plant genus Escallonia (Zapata 2010). Analysis of these empirical examples shows that GCG is readily applied to real-world data of the kind typically available to museum/herbarium-based systematists. Further work on groups where species boundaries are well-known a priori will be needed to assess the performance of GCG relative to alternatives.
Below, we discuss properties of GCG that may be important determinants of its performance and use those properties as a basis of comparison with other non–tree-based approaches (sensu Sites and Marshall 2004) that emphasize discontinuities in morphological variation (i.e., gaps) as a criterion to infer species limits: population aggregation analysis (Davis and Nixon 1992), hereafter PAA, and estimates of tolerance regions for discrete or qualitative characters (Wiens and Servedio 2000), hereafter GD (Gaps in Discrete characters). We do not further discuss approaches based on clustering analyses or statistical tests of differences in central tendency because they do not seem to be clearly related to the operational criterion of a gap in morphological variation (see introduction). Likewise, we do not discuss tree-based approaches (sensu Sites and Marshall 2004) to delimit species using morphological data (Baum and Donoghue 1995, Wiens and Penkrot 2002) because they are based on fundamentally different criteria and underlying theory (Sites and Marshall 2004) and thus are outside the scope of this paper.
In principle, hypotheses of species limits can be supported or rejected (Hey et al. 2003, Wheeler 2004, Henderson 2006); however, how a single empirical study employing GCG can support or reject a given hypothesis depends on the extent to which various assumptions are reasonably met, as well as other aspects of the study design and system. Like PAA and GD, GCG assumes that morphological discontinuities presented as evidence supporting hypotheses of species limits do not result from polymorphisms, ontogenetic variation, or phenotypic plasticity. GCG, but not PAA or GD, assumes that morphological variation is reasonably described by a normal distribution. When PAA, GD, or GCG fail to support a particular hypothesis, it is possible to argue that important morphological characters supporting the hypothesis were not included in the analysis. This argument, however, would not apply to hypotheses of species limits explicitly stated in terms of particular morphological characters, as may be the case for taxa described in formal statements about measures of dispersion of different characters (Wheeler 2004; Henderson 2005).
An important property of GCG is that it can be effectively applied to morphological spaces with any number of dimensions. For simplicity, we illustrated the implementation of this method using examples based on analyses in bivariate morphological spaces (the first two axes from PCA). However, all steps in the method (i.e., estimating the pdf along the ridgeline manifold, the tolerance regions and coefficients of the RDA) can be carried out using data in more dimensions. It is worth noting that the ridgeline manifold of the multivariate normal mixture of two taxa (i.e., two hypothesized species) is always one dimensional, regardless of the number of dimensions of the morphological space (Ray and Lindsay 2005). This implies a major reduction of dimensionality that readily allows visual inspection of key features of high-dimensional morphological spaces. Specifically, inspecting the pdf along the one-dimensional manifold (Fig. 2f) reveals gaps and modes that are otherwise difficult to visualize. Furthermore, using the one-dimensional manifold to compare the proportions covered by tolerance regions (Fig. 3b) simplifies the otherwise difficult task of assessing by visual inspection the overlap between high-dimensional ellipsoidal tolerance regions. Importantly, GCG accounts for within-taxa correlations between morphological dimensions (through covariance terms), unlike PAA and GD, and does not assume independence of the variables used to describe multivariate morphological space, in contrast to GD.
The distinction between morphological gaps among samples (i.e., groups of specimens representing particular hypothesized species) and morphological gaps among the populations from which the samples were derived is nontrivial and central to species delimitation (Wiens and Servedio 2000). In GCG, as well as in GD, support for a hypothesis of a species boundary is gauged according to the overlap of tolerance regions inferred from samples. The degree of overlap between tolerance regions is an estimate of morphological gaps between the populations from which the samples were derived. In contrast, in PAA species limits are drawn by gauging overlap of intervals defined by extreme sample values. We believe that focusing on tolerance regions is desirable because they attempt to describe the properties of the populations from which the samples were drawn. Hypothesis about species limits are about such populations (Hey et al. 2003; Wheeler 2004; Henderson 2005), not about samples.
Calculation of tolerance regions to infer morphological distinctiveness, as proposed here following GD, assumes that the observed sample is a random draw from a statistical population representing a hypothesized species. This assumption means that the probability of any individual being included in the sample is equal across all individuals in the statistical population and that the inclusion of one individual in the sample does not influence the probability of including any other individual (Zar 1999). The samples used in systematic studies are generally not random samples of hypothesized species. This is particularly exacerbated in museum/herbarium-based studies where species are often recognized from limited samples, which themselves frequently cover a small part of the hypothesized species' total geographic range and phenotypic variability (Davis and Heywood 1963; Henderson 2006). Even though PAA does not resort to formal inferences about statistical populations based on sample estimates (see above), error in estimates of species limits produced by this method can also result from nonrandom sampling of individuals and populations. Problems may arise, for instance, when bias geographic sampling fails to secure specimens from a zone of intergradation that bridge what may be mistakenly perceived as a morphological gap between allopatric populations (Davis and Nixon 1992). The mismatch between assumed and actual sampling strategies is not only an issue for methods emphasizing discontinuities in morphological variation as a criterion to infer species limits but is also common to virtually all approaches to infer species limits (see also Manel et al. 2003; Pons et al. 2006; Bond and Stockman 2008; Lohse 2009; Papadopoulou et al. 2009). Implementing formal sampling designs across large geographical scales may often be difficult, and future simulation work may help us understand the relationships between properties of different methods to delimit species and the procedures by which samples of hypothesized species are obtained. In the mean time, the mismatch between assumed and actual sampling strategies mandates caution in the interpretation of approaches for species delimitation.
Difficulties in the interpretation of results can arise when an analysis based on GCG fails to support a given hypothesis because often it is not possible to equate absence of evidence with evidence of absence, an issue common to all attempts to evaluate scientific hypotheses using statistical inferences (Gotelli and Ellison 2004). For instance, it is possible to interpret absence of evidence for a morphological gap as a lack of statistical power due to insufficient data. Eliminating this possibility may be difficult because there is not a way to formally determine what constitutes a large enough sample size without knowing the population parameters defining the distribution mixture corresponding to a pair of hypothesized species (i.e., the population means, variances and covariances). In this context, there is a noteworthy difference between overlap of tolerance regions, as used by GCG and GD, and overlap of intervals defined by extreme sample values used in PAA. Given random sampling of a population (e.g., φ(X;μ1,Σ1)) and fixed values of β and γ, tolerance regions become narrower as sample size increases ( Supplementary Data) because increased sample size results in increased precision in the estimate of the population distribution (i.e., increased precision of ). This implies higher statistical power to detect morphological discontinuities. In consequence, as sample size increases, GCG as well as GD may tend to recognize more morphological discontinuities. This contrasts with the tendency of the interval defined by extreme sample values to increase as sample size increases (Zar 1999; Supplementary Data) and the respective tendency of PAA to recognize less morphological discontinuities as sample size increases (Davis and Nixon 1992). Strictly speaking, these differences among methods occur under random sampling of statistical populations representing hypothesized species. However, random sampling is not common in systematic studies (see above).
PAA was explicitly designed as a method for the identification of phylogenetic species (sensu Nixon and Wheeler 1990, see Davis and Nixon 1992). In contrast, GCG and GD were designed to gauge the strength of the evidence indicating a discontinuity in morphological variation and, as such, they are not necessarily tied to any particular species definition (sensu de Queiroz 1998) because a gap in morphology can provide evidence of a species boundary under different species definitions, potentially yielding different results (Crisp and Weston 1993; Luckow 1995). For instance, when two taxa are considered sufficiently distinct to support the hypothesis of a species limit based on nonoverlapping tolerance regions calculated for some a priori values of γ and β, but morphological distinctiveness can be explained by geographic differentiation among allopatric populations, the phylogenetic (Cracraft 1983, Cracraft 1989; Nixon and Wheeler 1990) and biological (Mayr 1942) species definitions may lead to different conclusions about species limits (Cracraft 1992). Under the phylogenetic species definition, allopatric taxa that are morphologically distinct (such as taxa pairs E. schreiteri – E. discolor and E. schreiteri–E. piurensis in our empirical Example B) would be considered different species, whereas under the biological species definition, it is possible to regard allopatric taxa that are morphologically distinct as geographic variants of the same species. Thus, to yield consistent conclusions about species limits, results from the approach we presented here need to be interpreted in the light of an explicit species definition.
Beyond particular species definitions, recogniz-ing species as separately evolving segments of metapopulation-level lineages (de Queiroz 1998; de Queiroz 2005; de Queiroz 2007) implies that multiple operational criteria can be used to infer lineage separation, but no single one of them is necessary. Thus, a hypothesis of species limits that is not supported by evidence of morphological discontinuities can, nonetheless, correspond to a separation between lineages in nature, such as with morphologically convergent species and/or morphologically cryptic species (Whittall et al. 2004). Such hypotheses could be supported by other operational criteria, including pre- and postzygotic reproductive isolation, or differential exclusivity of gene genealogies. Therefore, support for a hypothesis of species limits may often be more easily interpreted than the absence of evidence supporting such hypothesis. This asymmetry might seem to be a problem if one requires that an inference from a single empirical study should be able to falsify a hypothesis of species boundaries. However, hypothesis-driven research does not need to adopt such a strict requirement. Support (or lack thereof) for a given scientific hypothesis can be judged in the light of multiple statistical inferences derived from several empirical studies, interpreted considering the nuances related to samples size and other aspects of study design (Bernays and Wege 1987; Hilborn and Mangel 1997; Gotelli and Ellison 2004 but see Krebs 2000). This approach recognizes that a single empirical study does not evaluate any hypothesis in isolation but rather bundles containing the hypothesis of interest along with background assumptions (Quine 1951; Duhem 1954).
The above issues regarding the relationship between GCG and species limits decisions involve major controversies about the ontology of species and the nature of the scientific method that are largely outside the scope of the present paper. Our focus here was on presenting an approach, explicitly based on theory about species limits, to weigh the strength of the evidence indicating a gap in morphological variation and assess whether such gap can be interpreted as geographic variation within a single species rather than as a species boundary. We hope that this approach will help to address concerns raised by several systematists (Davis and Heywood 1963; McDade 1995; Luckow 1995; Stevens 2000; Henderson 2006; Wiens 2007) regarding current practices to delimit species based on morphology. In particular, GCG can help strengthen the link between the theory and practice of species delimitation by increasing the transparency and consistency of taxonomic decisions based on morphology. There might be inevitable disagreements regarding decisions such as the relevance of a particular species definition (sensu de Queiroz 1998) or the a priori selection of a frequency cutoff below which overlap of phenotypic values between hypothesized species is considered rare enough as to suggest evolutionary isolation (Wiens and Servedio 2000). However, once those decisions are adopted, and given a set of explicit assumptions about the distribution of morphological variation and sampling of such a distribution, application of GCG to a particular data set provides an objective assessment of the strength of the evidence indicating whether a morphological discontinuity supports a particular hypothesis about species limits.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported in part by U.S. National Science Foundation (grant number OISE-0738118 to Peter F. Stevens and F.Z.); American Society of Plant Taxonomists, Idea Wild; and the Whitney R. Harris World Ecology Center at the University of Missouri–St. Louis.
We especially thank P.F. Stevens for insightful discussions on species delimitation. We thank D.W. Stevens and C.M. Taylor for discussions regarding species-level taxonomy. P.F. Stevens, E.A Kellogg, K.M. Olsen, P.C. Hoch, and C.D. Cadena provided helpful comments on drafts of this manuscript. We also thank R. DeBry, R. Glor, J. Sites, and two anonymous reviewers for constructive comments on earlier versions of this manuscript.
References
Author notes
Felipe Zapata and Iván Jiménez have contributed equally to this work.
Associate Editor: Richard Glor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}