





Abstract
Acrylamide (AA) is an animal carcinogen, neurotoxin, and reproductive toxin. AA is formed in baked and fried carbohydrate-rich foods. Metabolism of AA occurs via epoxidation to glycidamide (GA) or direct conjugation with glutathione. Using CYP2E1-null mice, recent studies in this laboratory demonstrated that induction of somatic and germ cell mutagenicity in AA-treated mice is dependent on CYP2E1. We hypothesized that AA metabolism to GA is a prerequisite for the induction of AA-induced mutagenicity. Current studies were undertaken to assess the role of CYP2E1 in the epoxidation of AA to GA and the formation of DNA and hemoglobin (HGB) adducts. AA was administered to CYP2E1-null or wild-type mice at 50 mg/kg ip. Mice were euthanized 6 h later and blood and tissues were collected. Using LC-ES/MS/MS, AA, GA, and DNA- and HGB-adducts were measured. While the plasma levels of AA and GA were 115 ± 14.0 and 1.7 ± 0.31 μM in CYP2E1-null mice, they were 0.84 ± 0.80 and 33.0 ± 6.3 μM in the plasma of AA-treated wild-type mice. Administration of AA to wild-type mice caused a large increase in N7-GA-Gua and N3-GA-Ade adducts in the liver, lung, and testes. While traces of N7-GA-Gua adducts were measured in the tissues of AA-treated CYP2E1-null mice, these levels were 52- to 66-fold lower than in wild-type mice. Significant elevation of both AA- and GA-HGB adducts was detected in AA-treated wild-type mice. In AA-treated CYP2E1-null mice, levels of AA-HGB adducts were roughly twice as high as those in wild-type mice. In conclusion, current work demonstrated that CYP2E1 is the primary enzyme responsible for the epoxidation of AA to GA, which leads to the formation of GA–DNA and HGB adducts.
Acrylamide (AA; CH2=CH–CO–NH2) is an important industrial chemical primarily used in the production of polyacrylamide and as a chemical intermediate in the synthesis of a variety of other chemicals. Polyacrylamide is used in the paper, textile and cosmetics industries, as a flocculent in the treatment of waste water, as a soil conditioner, and in ore processing and gel electrophoresis (Berger, 1995; LeQuesne, 1991; Friedman, 2003). AA is also present in cigarette smoke (Smith et al., 2001).
In recent years, AA was identified in baked and fried carbohydrate-rich foods such as French fries, potato chips, bread, and cereals (Rosen and Hellenas, 2002; Tareke et al., 2002; Weiss, 2002). AA can be formed in foods, especially carbohydrate-rich foods, during processing at very high temperatures via the Maillard browning reactions between amino acids and reducing sugars (Friedman, 2003; Rosen and Hellenas, 2002; Smith et al., 2001; Tareke et al., 2002). The discovery of AA in fried/baked carbohydrate-rich foods raised concerns that human exposure to AA, and potential hazardous effects are more widespread than originally thought. Human exposure may occur by ingestion of AA-containing foods, inhalation, or skin contact (Dearfield et al., 1995; IARC, 1994).
AA is neurotoxic, clastogenic, carcinogenic, and a reproductive toxicant in animals and was classified by IARC as a “probable human carcinogen” (Bull et al., 1984a,b; Damjanov et al., 1998; Dearfield et al., 1995; Ghanayem et al., 2005a; IARC, 1994; Tyl et al., 2000; Weiss, 2002).
It is currently established that AA is metabolized primarily in animals and humans via two competing pathways (Fig. 1): conjugation with reduced glutathione and oxidation to form the epoxide intermediate, glycidamide (GA) (Calleman et al., 1990; Ghanayem et al., 2000; Sumner et al., 1999; Wu et al., 1993). GA may subsequently undergo conjugation with reduced glutathione or hydrolysis catalyzed by epoxide hydrolase. Using 13C-NMR, comparative metabolite identification studies in CYP2E1-null and wild-type mice demonstrated that, in the absence of CYP2E1, mice did not excrete GA or metabolites derived from GA in the urine of 13C-AA-treated mice (Sumner et al., 1999). These studies concluded that functional CYP2E1 is required for the in vivo conversion of AA to GA. Subsequent studies in this laboratory showed protection against AA-induced male germ cell mutagenicity in CYP2E1-null compared with wild-type mice as determined via the dominant lethal mutation assay (Ghanayem et al., 2005a). It was concluded from these findings that CYP2E1-mediated oxidation of AA, presumably to GA, is required for the induction of male germ cell mutagenicity in mice treated with AA.
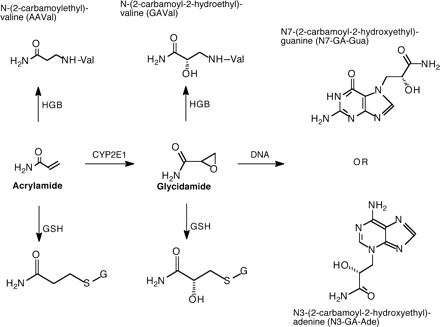
A proposed scheme of acrylamide (AA) metabolism showing the formation of glycidamide (GA), glutathione conjugates, and HGB and DNA adducts.
Epoxides are highly reactive, known to be potent mutagens and carcinogens, and interact with DNA to form adducts (Ghanayem et al., 2000; Hemminki, 1993; Melnick, 2002). DNA adducts play a significant role in the mutagenicity and carcinogenicity of epoxides and epoxide-forming chemicals (Ghanayem et al., 2000; Hemminki, 1993; Melnick, 2002). Formation of DNA adducts of GA (the reactive epoxide of AA) is well established, and it is currently suspected that formation of these adducts plays a role in the induction of mutagenicity, reproductive toxicity, and carcinogenicity by AA (Friedman, 2003; Gamboa da Costa et al., 2003; Ghanayem et al., 2005a; Segerback et al., 1995).
Both AA and GA can bind to the N-terminal of globin to form hemoglobin (HGB) adducts, which were detected in the blood of animals and humans exposed to AA (Bergmark, 1997; Bergmark et al., 1991, 1993; Sumner et al., 2003). HGB adducts are considered valuable markers of human exposure to chemicals and constitute a good measure of the internal dose. It is currently hypothesized that GA is responsible for the reproductive toxicity, mutagenicity, and carcinogenicity of AA (Friedman, 2003; Gamboa da Costa et al., 2003; Ghanayem et al., 2005a; Segerback et al., 1995). Although the proposition that metabolism of AA to GA is mediated by CYP2E1 has been around for sometime, no studies were available that directly proved this hypothesis. Using CYP2E1-null mice, the overall objectives of the current work were to assess the metabolic and molecular basis of AA-induced mutagenicity, reproductive toxicity, and carcinogenicity. In particular, the current studies were designed to (1) compare the metabolism of AA to GA using CYP2E1-null and wild-type mice and (2) compare the formation of DNA adducts in target organs of AA-induced effects (liver, lung, and testes) and HGB adducts in the blood of CYP2E1-null and wild-type mice.
MATERIALS AND METHODS
Chemicals.
AA (>99.5% pure) was manufactured by Fluka Chemie GmbH and purchased from Sigma-Aldrich (Milwaukee, WI). All other chemicals were of the highest commercially available purity.
Animals and treatments.
Male wild type (CYP2E1+/+; WT) and CYP2E1-null (CYP2E1−/−; KO) mice (derived from 129Sv mice and crossed to C57BL/6N2), were 3–4 months old and ranged in weight from 23 to 29 g, were first produced at the National Cancer Institute, Bethesda, MD (Lee et al., 1996), and were rederived and bred at Charles River Laboratories (Wilmington, DE) as previously described (Hoffler et al., 2003). Animals were housed in an animal facility with a 12-h light-dark cycle and fed NIH #31 diet. Both food and water were available ad libitum throughout the experiments. All animal care and experimentation were conducted according to NIH guidelines (U.S. Department of Health and Human Services, 1986).
AA dosing solutions were freshly prepared and administered by intraperitoneal injection (ip) to groups of CYP2E1-null and wild-type male mice at 0 or 50 mg/5 ml saline/kg. Dosing solutions were prepared in normal saline (0.9%) at a dose volume of 5 ml saline/kg. Control mice received 5 ml saline/kg ip. Six h after dosing, mice were euthanized with CO2/O2; blood was collected via cardiac puncture using heparinized syringes and was immediately processed to separate the plasma for AA and GA measurements and the RBCs for HGB adducts determinations. Livers, lungs, and testes were removed and immediately frozen using liquid nitrogen and stored at −80°C for DNA adducts measurements at a later time.
LC-ES/MS/MS analysis of AA and GA in plasma.
Analyses of AA and GA in plasma collected at sacrifice (6 h after dosing) were performed by using a high-throughput LC-ES/MS/MS method as previously described (Twaddle et al., 2004b). Briefly, labeled internal standards (13C3-AA and 13C3-GA) were added to each thawed plasma sample (10–100 ml); then samples were purified using solid phase extraction in 96-well plates and analyzed using LC-ES/MS/MS in the multiple reaction monitoring mode for 13C-labeled and unlabeled AA and GA transitions. In addition to the inherent specificity that comes from adding labeled internal standards for AA and GA to every plasma sample prior to sample preparation and analysis, further quality control measures were performed during every sample set, including the analysis of blank and spiked plasma samples, blank injections, and injections of authentic standards. The limit of quantification was 0.01 μM for AA, 0.1 μM for GA, and the intra- and inter-assay precision was 2–4% relative standard deviation for AA and GA spiked at 1 μM in blank plasma.
LC-ES/MS/MS analysis of GA-derived DNA adducts.
Livers, lungs, and testes collected at sacrifice were analyzed for GA-derived DNA adducts from AA-treated and control mice using the same total exposure time as in our previously published procedure (Gamboa da Costa et al., 2003). Briefly, tissue DNA was isolated and purified using Blood & Cell Culture Maxi kits (Qiagen Co., Valencia, CA), and 100-μg aliquots were subjected to neutral thermal hydrolysis to release N7-(2-carbamoyl-2-hydroxyethyl)guanine (N7-GA-Gua) and N3-(2-carbamoyl-2-hydroxyethyl)adenine (N3-GA-Ade) adducts, which were quantified by using 15N-labeled N7-GA-Gua and N3-GA-Ade internal standards and LC-ES/MS/MS in the multiple reaction monitoring mode. The limit of quantification was approximately 1.5 adducts in 108 nucleotides for N3-GA-Ade and 0.2 adducts in 108 nucleotides for N7-GA-Gua, and the intra- and inter-assay precision was 2–6% relative standard deviation for GA-modified DNA standards (18–1500 adducts in 108 nucleotides).
LC-ES/MS/MS analysis of AA- and GA-valine adducts in hemoglobin.
Six h after dosing, animals were anesthetized with CO2 and blood was collected via cardiac puncture in a 3-ml syringe coated with heparin. After collection, the blood was transferred to a 15-ml screw cap plastic tube, gently mixed, and placed on ice. Blood was centrifuged for 20 min at 3000 rpm to separate red blood cells from plasma. Using a glass Pasteur pipette, the plasma was carefully removed and transferred to a separate tube. The red blood cell pellet was washed three times with an equal volume of isotonic saline (0.9%). After removing the last saline wash, tubes containing the red blood cell pellets were capped and stored at −20°C until globin isolation. Globin was isolated from washed red blood cells (Mowrer et al., 1986) and derivatized as previously described (Fennell et al., 2003). N-(2-carbamoylethyl)valine (AAVal-PTH) and N-(2-carbamoyl-2-hydroxyethyl)valine (GAVal-PTH) adducts, derived from AA and GA, respectively, were measured in isolated globin samples using LC-ES/MS/MS in the multiple reaction monitoring mode as previously described (Fennell et al., 2003).
Statistical analyses.
Group mean comparisons were performed using Student's t-test two-tailed assuming equal variances. Values were considered statistically significant at p ≤ 0.05.
RESULTS
Administration of AA to CYP2E1-null and wild-type mice produced measurable levels of AA and GA in plasma when measured at 6 h after ip dosing with 50 mg/5 ml saline/kg (Fig. 2). This time point was selected to correspond to that used in our previous studies of DNA adducts and toxicokinetics of AA and GA (Doerge et al., 2005a,b; Gamboa da Costa et al., 2003; Twaddle et al., 2004b). The mean plasma concentration of AA in wild type mice was negligible (137-fold lower) compared to the levels found in the plasma of CYP2E1-null mice. In contrast, the mean GA concentration in the plasma of wild-type mice was significantly higher (19-fold) when compared to the levels in the plasma of CYP2E1-null mice (Fig. 2). Regardless of the genotype, the plasma levels of AA and GA in untreated mice were below the detection limits of the methods used in the current studies.
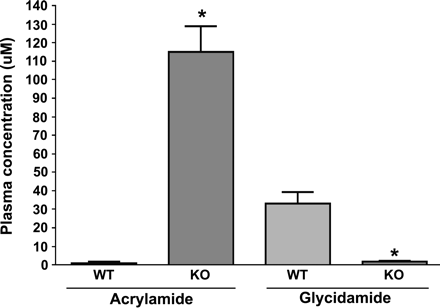
Comparison of acrylamide and glycidamide concentrations in the plasma of wild-type (WT) and CYP2E1-null (KO) mice at 6 h after treatment with a single ip dose of 50 mg acrylamide/kg body weight. Data are presented as mean ± SD of 3–5 animals. *Denotes values in KO mice that are significantly different from the corresponding WT mice at p ≤ 0.05.
Low levels of N7-GA-Gua adducts were detected in liver and testes from vehicle-treated wild type mice (9.3 ± 6.9; 31 ± 15; and 1.8 ± 0.3 adducts/108 nucleotides, respectively). In contrast, no N7-GA-Gua adducts were found in any of the tissues of vehicle-treated CYP2E1-null mice. The presence of GA–DNA adducts in tissues obtained from vehicle-treated mice is consistent with the presence of AA in the basal diet and is in agreement with what was previously described for an autoclaved rodent diet (Twaddle et al., 2004a).
Administration of AA to wild-type mice produced a significant increase in N7-GA-Gua and N3-GA-Ade adducts in all tissues tested (Fig. 3). Relatively minor differences in DNA adduct levels were found in the three tissues from AA-dosed wild-type mice, and this finding is in agreement with previously reported data in B6C3F1 mice (Doerge et al., 2005a; Gamboa da Costa et al., 2003). While traces of N7-GA-Gua adducts were detectable in the liver, testes, and lungs of CYP2E1-null mice treated with AA (Fig. 3), the levels of this adduct were much lower than those measured in wild-type mice (52- to 66-fold). N3-GA-Ade adducts were found in the liver, lung, and testes of wild type mice treated with AA at levels approximately 100-fold below those for the N7-GA-Gua. However, N3-GA-Ade adduct levels were below the detection limits in these tissues from similarly treated CYP2E1-null mice or untreated controls (Gamboa da Costa et al., 2003).
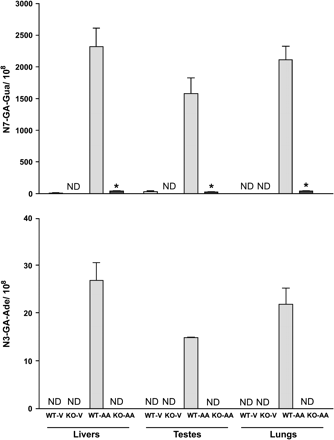
Comparison of glycidamide DNA adducts (N7-GA-Gua and N3-GA-Ade) in tissues (liver, testes, and lung) of wild type (WT) and CYP2E1-null (KO) mice at 6 h after treatment with a single ip dose of 50 mg acrylamide/kg body weight. Data are presented as the number of adducts in 108 nucleotides (mean ± SD of 3–5 animals). *Denotes values that are significantly different from the corresponding WT mice at p ≤ 0.05. ND = not detectable; V = Vehicle; AA = acrylamide; GA = glycidamide.
The concentration of the AAVal-PTH adducts from vehicle-treated mice were low but detectable in mice of both genotypes, ranging from 14 to 62 fmol/mg globin (Fig. 4). However, while GAVal-PTH adducts were low in vehicle-treated wild-type mice (ranging from nondetectable to 75 fmol/mg globin), they were not detectable in untreated CYP2E1-null mice. Significant elevation of both the AAVal-PTH and GAVal-PTH adducts was seen in wild-type mice 6 h after treatment with 50 mg AA /kg body weight, and the ratio of GAVal-PTH to AAVal-PTH was approximately 1.70. In CYP2E1-null mice treated with the same dose of AA, the levels of AAVal-PTH were approximately double those measured in wild-type mice (Fig. 4). The amount of GAVal-PTH in AA-treated CYP2E1-null mice was low, but significantly higher than in vehicle-treated CYP2E-null mice, and was approximately 33-fold lower than that found in AA-treated wild-type mice (Fig. 4). Overall, while the ratio of GAVal-PTH:AAVal-PTH in AA-treated wild-type mice was approximately 1.7, this ratio was approximately 0.02 in AA-treated CYP2E1-null mice.
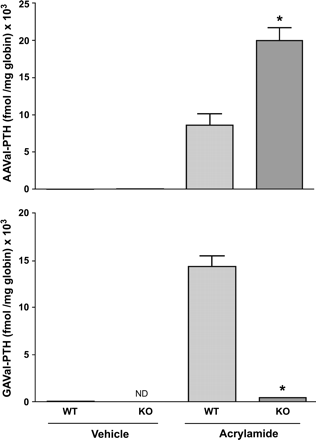
Comparison of AAVal-PTH (acrylamide HGB adducts) and GAVal-PTH (glycidamide HGB adducts) in wild type (WT) and CYP2E1-null (KO) mice at 6 h after treatment with a single ip dose of 50 mg acrylamide/kg body weight. Data are presented as mean ± SD of n = 3–5 animals. *Denotes values that are significantly different from the corresponding WT mice at p ≤ 0.05. ND = not detectable; KO = acrylamide; GA = glycidamide.
DISCUSSION
AA is a high-production-volume industrial chemical, which is heavily used in the manufacturing of polyacrylamide products. AA-derived polymers and copolymers are used in cosmetic, paper, and textiles manufacturing, in wastewater treatment, in biomedical research laboratories for gel electrophoresis, and in soil conditioning. The recent discovery of AA in baked and fried carbohydrate-rich foods such as French-fries, potato chips, bread, and cereals (Rosen and Hellenas, 2002; Tareke et al., 2002; Weiss, 2002) heightened concerns over the potential hazardous effects of this chemical to humans. This concern was compounded by the fact that AA is a known animal carcinogen, a germ cell mutagen, and a neurotoxicant (Friedman, 2003; Ghanayem et al., 2005a; IARC, 1994; Tyl and Friedman, 2003).
It is well established that AA metabolism proceeds primarily via two competing pathways (Fig. 1). Direct conjugation of AA with reduced glutathione results in the formation of a glutathione adduct, the degradation of which leads to the formation and urinary excretion of the mercapturic acid, N-acetyl-S-(2-carbamoylethyl) cysteine. This mercapturic acid was found in urine of mice as well as humans (Fennell et al., 2005; Sumner et al., 1999; Wu et al., 1993). The second major pathway involves oxidation of AA, leading to the formation of the epoxide intermediate, GA. GA-derived metabolites have been found in the urine of rodents and humans exposed to AA (Fennell et al., 2005; Sumner et al., 1999). Metabolism of GA may involve hydration via epoxide hydrolases or conjugation with reduced glutathione. Mercapturic acids originating from the GA-glutathione conjugate were found in human and mice urine after exposure to AA (Fennell et al., 2005; Sumner et al., 1999; Wu et al., 1993).
The presence of GA and its metabolites in addition to mercapturic acids derived from AA-glutathione conjugates in rodent and human urine clearly indicates that both rodents and humans oxidize AA to GA. Furthermore, the percentage of an AA dose metabolized via each of the two pathways appears to vary as a function of dose, species, and route of administration. Quantification of urinary metabolites using 13C-NMR demonstrated that no GA or metabolites originating from GA were detectable in the urine of CYP2E1-null mice treated with AA at 50 mg/kg (Sumner et al., 1999). This led to the conclusion that CYP2E1 is the principal enzyme responsible for the epoxidation of AA. Despite this observation, no data are currently available which directly assessed the role of CYP2E1 in the conversion of AA to GA in vivo. Epoxides and epoxide-forming chemicals are highly reactive with DNA, and most are genotoxic and carcinogenic (Ghanayem et al., 2000; Hemminki, 1993; Koskinen and Plna, 2000; Melnick, 2002). Recent studies in this laboratory using CYP2E1-null mice demonstrated that AA administration at 50 mg/kg (ip) induced male germ cell mutagenicity that required functional CYP2E1 (Ghanayem et al., 2005a). Our current hypothesis centers on the premise that GA is responsible for AA-induced genotoxicity, carcinogenicity, and reproductive toxicity. As part of a comprehensive investigation to test this hypothesis and assess the human health risks of AA, the current studies were undertaken to characterize the mechanism(s) of action of AA, to assess the role of CYP2E1 in the formation of GA, the epoxide intermediate of AA, and to quantify the formation of HGB and DNA adducts in CYP2E1-null and wild-type mice. Our choice of the route, dose, and time point was based on a number of earlier studies that used 50 mg/kg and intended to facilitate comparisons with these studies and to explain the relationships of GA formation with the mutagenicity and carcinogenicity of AA (Ghanayem et al., 2005a,b; Holland et al., 1999; Shelby et al., 1986; Sumner et al., 1999; Twaddle et al., 2004b).
As expected, the mean plasma concentration of AA in CYP2E1-null mice was 137-fold higher than the levels measured in wild-type mice (Fig. 2). In contrast, significant formation of GA was detected only in wild-type mice in comparison to traces of this metabolite formed in CYP2E1-null mice. This clearly demonstrated that, in the absence of a functional CYP2E1 enzyme, elimination of AA is diminished concomitant with a major reduction in GA formation. These results are consistent with previous findings in B6C3F1 mice that received AA at 50 mg/kg, which showed that the kinetics of GA formation (t1/2 = 0.77 h) and AA elimination (t1/2 = 0.73 h) were essentially identical (Twaddle et al., 2004b). These results, in conjunction with our recent findings that demonstrated the absence of germ and somatic cell mutagenicity in male CYP2E1-null mice treated with AA (Ghanayem et al., 2005a,b), support the hypothesis that GA is the ultimate toxic metabolite on the male reproductive system. The detection of trace amounts of GA as well as its HGB and DNA adducts in CYP2E1-null mice indicated that pathways other that CYP2E1 may lead to the formation of traces of GA, but that these pathways contribute less than 2% to the total GA–DNA adducts formed. This result differs from our earlier study, in which analysis of urinary metabolites showed no evidence of the presence of GA or GA-derived urinary metabolites in CYP2E1-null mice (Sumner et al., 1999). This difference may be attributed to the fact that LC-ES/MS/MS technology for measuring DNA and HGB adducts is more sensitive than 13C-NMR used for measuring urinary metabolites.
HGB adducts were also measured in mice treated with AA. HGB adducts provide a measure of the internal dose of reactive compounds and may serve as markers of human exposure to environmental chemicals (Fennell et al., 2003; Schettgen et al., 2002). Further, the ratio of HGB adducts of AA and GA may provide an insight into the metabolism of AA. Consistent with the measured concentrations of AA and GA in the plasma of mice of both genotypes, the amount of AA-derived HGB adducts (AAVal-PTH) in wild-type mice was less than one-half of the amount found in CYP2E1-null mice. Furthermore, wild-type mice generated approximately 33-fold more GA-derived HGB adducts (GAVal-PTH) than CYP2E1-null mice. The ratio of GAVal-PTH:AAVal-PTH in CYP2E1-null mice was approximately 1:45. The approximate doubling of the amount of AAVal-PTH adducts formed in CYP2E1-null mice confirmed that inhibition of CYP2E1-mediated metabolism leads to an increase in the t1/2 of AA and its area-under-the time-concentration curve (AUC) and, subsequently, the elevated formation of the amounts of AAVal-PTH adducts.
Differences in levels of GA-derived DNA adducts in the tissues of AA-treated mice and HGB adducts are consistent with the formation of this epoxide intermediate in mice in differing amounts which depend on the respective genotype. Administration of AA to wild-type mice produced a large increase in N7-GA-Gua and N3-GA-Ade adducts in the DNA of all tissues tested (lung, liver, and testes). The similarity of DNA adduct levels measured in all three tissues from AA-dosed wild-type mice is consistent with previously reported results in B6C3F1 mice (Gamboa da Costa et al., 2003). This finding is also consistent with the extensive tissue distribution of highly water-soluble compounds like AA and GA in mice (volume of distribution 0.6–0.7 l/kg) (Doerge et al., 2005a). While N7-GA-Gua adducts were also found in all organs obtained from CYP2E1-null mice treated with AA, the levels of this adduct were negligible compared to those found in wild-type mice (52- to 66-fold lower). No N3-GA-Ade adducts were detected in AA-treated CYP2E1-null mice, presumably because this minor adduct is formed at levels approximately 100-fold below those for the N7-GA-Gua (Gamboa da Costa et al., 2003; Doerge et al., 2005a). Traces of N7-GA-Gua but not the N3-GA-Ade adducts were detectable in the livers and testes obtained from vehicle-treated wild-type mice (Fig. 3). Both N7-GA-Gua and N3-GA-Ade were found at significantly higher levels in the lung, liver, and testes of vehicle-treated wild-type mice compared to traces of these adducts in CYP2E1-null mice (Fig. 3). The presence of the N7-GA-Gua adducts in tissues from untreated mice is in agreement with the previously reported presence of AA in autoclaved rodents diet (Twaddle et al., 2004a). Formation of GA–DNA adducts in vehicle-treated mice, resulting from basal diet consumption, suggests that intake of AA-containing foods such as French fries may lead to the formation of such adducts in humans.
In conclusion, these results clearly confirm that functional CYP2E1 is required for AA epoxidation to GA and subsequent formation of GA–DNA adducts. Traces of GA were found in the plasma, and DNA adducts derived from GA were found in tissues from CYP2E1-null mice treated with AA, which suggested that minor pathway(s), other than CYP2E1, may be functional in the absence of CYP2E1 enzyme. However, this pathway is negligible and may not be biologically significant, especially in light of the fact that male germ cell mutagenicity was absent in CYP2E1-null mice treated with AA (Ghanayem et al., 2005a) and the fact that AA was not mutagenic in CYP2E1-null versus wild-type mice using the micronuclei and COMET assays (Ghanayem et al., 2005b). Further, current work showed significant formation of GA–DNA adducts in the lung, liver, and testes of genetically intact wild-type mice with only traces detectable in CYP2E1-null mice (Fig. 3). While short-term 8-week exposure studies demonstrated that AA is carcinogenic in the lung and skin of genetically intact mice (IARC, 1994; Bull et al., 1984a,b), no long-term studies that assessed AA carcinogenicity in mice in a comprehensive fashion are available. However, N-methylolacrylamide is apparently converted to AA and GA in rodents (Fennell et al., 2003) and is carcinogenic in multiple organs from a 2-yr bioassay in B6C3F1 mice including liver and lung (Bucher et al., 1990). In rats, long-term administration of AA resulted in increased incidence of thyroid follicular-cell tumors and peritesticular mesotheliomas in male rats and mammary tumors and thyroid tumors in females (IARC, 1994; Johnson et al., 1986; Friedman et al., 1995). Whether formation of GA–DNA adducts (Fig. 3) is related to AA carcinogenesis remains to be established; however, it is logical to speculate that there is an association between formation of DNA adducts of GA and carcinogenicity of AA in the lung of genetically intact mice (Bull et al., 1984a,b; IARC, 1994). The relationship between GA–DNA adducts in the liver and testes and carcinogenicity in mice is currently unclear, especially in the absence of long-term studies in this animal species. However, a direct role for parent AA in DNA adduct formation can be eliminated based on minimal reactivity (Gamboa da Costa et al., 2003) as opposed to protein adduct formation, which is facile for both AA and GA through reaction with amine and sulfhydryl nucleophiles (Friedman, 2003). Although there is no direct evidence that formation of DNA adducts of GA in the testes of wild-type mice is involved in germ cell mutagenicity, the results of the current studies suggest that modification of DNA by GA may be involved. Earlier reports suggested that binding of GA and/or AA with spermatid protamines leads to dominant lethal mutations (Gamboa da Costa et al., 2004; Sega, 1991; Sega et al., 1989). While interaction with spermatid protamines remains a possibility, current results further confirmed that GA, and not AA, is responsible for AA-induced dominant lethal mutations. It is therefore clear that AA-induced male germ cell mutagenicity in wild-type mice is caused by GA through subsequent interaction with DNA in the testes/sperm or with spermatid protamines. Finally, work using CYP2E1-null mice may prove valuable in the assessment of the role of GA and GA–DNA adducts in AA-induced carcinogenicity and neurotoxicity.
We sincerely thank Dr. Michael Shelby and Kristine Witt for their insightful comments and suggestions. We also thank Dr. Frank Gonzalez for providing us animals to start the colony of CYP2E1-null and wild-type mice. Furthermore, we express our thanks to Dr. Mohammed Nayeem and Akef Rahman for their help in preparing the figures. Conflict of interest: none declared.
References
Berger, A. R., and Schaumburg, H. H. (
Bergmark, E. (
Bergmark, E., Calleman, C. J., and Costa, L. G. (
Bergmark, E., Calleman, C. J., He, F., and Costa, L. G. (
Bucher, J. R., Huff, J., Haseman, J. K., Eustis, S. L., Peters, A., and Toft, J. D. (
Bull, R. J., Robinson, M., Laurie, R. D., Stoner, G. D., Greisiger, E., Meier, J. R., and Stober, J. (
Bull, R. J., Robinson, M., and Stober, J. A. (
Calleman, C. J., Bergmark, E., and Costa, L. G. (
Damjanov, I., and Friedman, M. A. (
Dearfield, K. L., Douglas, G. R., Ehling, U. H., Moore, M. M., Sega, G. A., and Brusick, D. J. (
Doerge, D. R., Gamboa da Costa, G., McDaniel, L. P., Churchwell, M. I. Twaddle, N. C., and Beland, F. A. (
Doerge, D. R., Young, J. F., McDaniel, L. P., Twaddle, N. C., and Churchwell, M. I. (
Fennell, T. R., Snyder, R. W., Krol, W. L., and Sumner, S. C. (
Fennell, T. R., Sumner, S. C., Snyder, R. W., Burgess, J., Spicer, R., Bridson, W. E., and Friedman, M. A. (
Friedman, M. A., Dulak, L. H., and Stedham, M. A. (
Friedman, M. (
Gamboa da Costa, G., Churchwell, M. I., Hamilton, L. P., Beland, F. A., Marques, M. M., and Doerge, D. R. (
Ghanayem, B. I., Witt, K. L., El-Hadri, L., Hoffler, U., Kissling, G. E., Shelby, M. D., and Bishop, J. B. (
Ghanayem, B. I., Witt, K. L., Kissling, G. E., Tice, R. R., and Recio, L. (
Ghanayem, B. I., Wang, H., and Sumner, S. (
Hoffler, U., El-Masri, H., and Ghanayem, B. I. (
Holland, N., Ahlborn, T., Turteltaub, K., Mrkee, C., Moore, D., 2nd, Wyrobek, A. J., and Smith, M. T. (
Johnson, K. A., Gorzinski, S. J., Bonder, K. M., Campbell, R. A., Wolf, C. H., Friedman, M. A., and Mast, R. W. (
Koskinen, M., and Plna, K. (
Lee, S., Buters, J., Pineau, T., Fernadex-Salguero, P., and Gonzalez, F. (
Melnick, R. L. (
Mowrer, J., Tornqvist, M., Jensen, S., and Ehrenberg, L. (
Rosen, J., and Hellenas, K. E. (
Schettgen, T., Broding, H. C., Angerer, J., and Drexler, H. (
Segerback, D., Calleman, C. J., Schroeder, J. L., Costa, L. G., and Faustman, E. M. (
Sega, G. (
Sega, G., Alcota, R., Tancongco, C., and Brimer, P. (
Shelby, M., Cain, K., Hughes, L., Braden, L., and Generoso, W. (
Smith, C. J., Perfetti, T. A., Rumple, M. A., Rodgman, A., and Doolite, D. J. (
Sumner, S. C., Fennell, T. R., Moore, T. A., Chanas, B., Gonzalez, F., and Ghanayem, B. I. (
Sumner, S. C., Williams, C. C., Snyder, R. W., Krol, W. L., Asgharian, B., and Fennell, T. R. (
Tareke, E., Rydberg, P., Karlsson, P., Eriksson, S., and Tornqvist, M. (
Tyl, R. W., and Friedman, M. A. (
Tyl, R. W., Marr, M. C., Myers, C. B., Ross, W. P., and Friedman, M. A. (
Twaddle, N. C., Churchwell, M. C., McDaniel, L. P., and Doerge, D. R. (
Twaddle, N. C., Hamilton, L. P., Gamboa da Costa, G., Churchwell, M. I., Beland, F. A., and Doerge, D. R. (
U.S. Department of Health and Human Services (
Author notes
*Laboratory of Pharmacology and Chemistry, National Toxicology Program, NIEHS/NIH, Research Triangle Park, North Carolina 27709; †NCTR, Jefferson, AR 72079; and ‡RTI International, Research Triangle Park, North Carolina 27709

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments