Mitophagy, Mitochondrial Homeostasis, and Cell Fate
 Kaili Ma
Kaili Ma Guo Chen
Guo Chen Wenhui Li
Wenhui Li Oliver Kepp
Oliver Kepp Yushan Zhu
Yushan Zhu Quan Chen
Quan Chen- 1State Key Laboratory of Medicinal Chemical Biology, College of Life Sciences, Nankai University, Tianjin, China
- 2Suzhou Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
- 3Gustave Roussy Cancer Campus, Villejuif, France
- 4INSERM, UMR 1138, Centre de Recherche des Cordeliers, Sorbonne Université, Université de Paris, Paris, France
Mitochondria are highly plastic and dynamic organelles that have graded responses to the changing cellular, environmental, and developmental cues. Mitochondria undergo constant mitochondrial fission and fusion, mitochondrial biogenesis, and mitophagy, which coordinately control mitochondrial morphology, quantity, quality, turnover, and inheritance. Mitophagy is a cellular process that selectively removes the aged and damaged mitochondria via the specific sequestration and engulfment of mitochondria for subsequent lysosomal degradation. It plays a pivotal role in reinstating cellular homeostasis in normal physiology and conditions of stress. Damaged mitochondria may either instigate innate immunity through the overproduction of ROS or the release of mtDNA, or trigger cell death through the release of cytochrome c and other apoptogenic factors when mitochondria damage is beyond repair. Distinct molecular machineries and signaling pathways are found to regulate these mitochondrial dynamics and behaviors. It is less clear how mitochondrial behaviors are coordinated at molecular levels. BCL2 family proteins interact within family members to regulate mitochondrial outer membrane permeabilization and apoptosis. They were also described as global regulators of mitochondrial homeostasis and mitochondrial fate through their interaction with distinct partners including Drp1, mitofusins, PGAM5, and even LC3 that involved mitochondrial dynamics and behaviors. In this review, we summarize recent findings on molecular pathways governing mitophagy and its coordination with other mitochondrial behaviors, which together determine cellular fate.
Introduction
Mitochondria are organelles that govern energy transformation and ATP production through the tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OXPHOS). Moreover, mitochondria control redox homeostasis, Ca2+ signaling, iron metabolism, innate immunity, and apoptotic cell death (Zorov et al., 2014; Zong et al., 2016; Paul et al., 2017; Pathak and Trebak, 2018). Mitochondria are both the major source and the main targets of reactive oxygen species (ROS). Under homeostatic conditions, mitochondrial ROS serve as retrograde signaling molecules for cell growth (Diebold and Chandel, 2016). However, in conditions of stress or aging, mitochondrial ROS elicit oxidative damage to mitochondrial proteins, lipids, and DNA (mtDNA), causing the malfunction of mitochondria. Dysfunctional mitochondria may produce even more ROS via vicious cycle that further amplify the release of ROS and mtDNA into the cytosol, which in turn can act as instigators of inflammation (Nakahira et al., 2011; Zhou et al., 2011). Non-reparable and severe damage of mitochondria leads to the release from the intermembrane space into the cytosol of cytochrome c and other pro-death factors (Sinha et al., 2013) altogether triggering apoptosis, a specific form of programmed cell death. This process is governed by the BCL2 protein family that integrates apoptotic signals and controls mitochondrial outer membrane permeabilization (MOMP).
Mitochondria are highly dynamic organelles that undergo continuous fission and fusion, constant turnover through mitochondrial biogenesis and mitophagy to maintain mitochondrial morphology, homeostasis, and inheritance. When facing bioenergetic or oxidative challenges, mitochondria exhibit a graded response that involves changes in their morphology and dynamics through the activation of distinct molecular machineries that regulate mitochondrial fission, fusion, mitophagy, and mitochondrial biogenesis. Mitochondrial fission and fusion are tightly regulated by a complex protein machinery involving dynamin 1 like (DNM1L better known as Drp1), mitosfusin 1 (MFN1), mitosfusin 2 (MFN2), and Optic atrophy protein 1 (OPA1) in mammalian cells. Mitochondrial fission was found to contribute to mitochondrial apoptosis and was also suggested to be a prerequisite for mitophagy, while mitochondrial fusion is linked to an increase in mitochondrial metabolism. How these molecular machineries sense cellular stresses and how these complex mitochondrial behaviors are coordinated at the molecular level remains elusive. It is important to address these questions, as mitochondrial dynamics and homeostasis are tightly linked with cellular physiology and eventually cell fate.
John Lemasters first termed selective mitochondrial autophagy as “mitophagy” (Lemasters, 2005). Mitophagy is a process that selectively sequesters damaged or depolarized mitochondria into double-membraned autophagosomes for subsequent lysosomal degradation. The removal of damaged or unwanted mitochondria, mitophagy was found to be essential for maintaining cellular fitness. Both ubiquitin- and receptor-mediated mitophagy pathways have been described and extensively studied. Intriguingly, BCL2 family proteins were reported to participate in both mitochondrial dynamics and mitophagic processes, which puts them in the center of mitochondrial homeostasis. We recently have shown that PGAM5, a mitochondrial phosphatase, serves as a molecular switch for determining mitochondrial fate (apoptosis or mitophagy) by dephosphorylating BCL-xL, a key apoptosis inhibitor and FUNDC1, a mitophagy receptor. These results demonstrated the integration of stress signals and the coordinated execution of graded responses in response to mitochondrial stress conditions (Ma K. et al., 2019). Here, we provide a focused overview on the molecular mechanisms of mitophagy and its interplay with mitochondrial dynamics and behaviors, thus contributing to aging and aging-related diseases.
Molecular Regulation of Mitophagy
Mitophagy in Yeast
Electron microscopy has revealed that, in Saccharomyces cerevisiae, mitochondria can be specifically sequestered by autophagosomes, or be engulfed together with cytosolic material (Kissova et al., 2007). This process depends on the complete set of Atg-proteins such as Atg11, Atg17, and Atg29, as well as specific adaptor proteins (Farre et al., 2009). The mitochondrial outer membrane protein, Uth1, and mitochondrial protein phosphatase homolog, Aup1, have both been implicated in mitophagy (Kissova et al., 2007; Tal et al., 2007). Pioneering work from Ohsumi’s and Klionsky’s laboratories have identified that, Atg32, a mitochondria-anchored protein, is essential for mitophagy in yeast. It acts as a mitophagy-specific receptor and interacts with autophagy key proteins such as Atg8 via an Atg8 interacting motif (AIM) and Atg11 to recruit autophagosomes to mitochondria for their engulfment and final degradation (Kanki et al., 2009; Okamoto et al., 2009). Atg32 undergoes both transcriptional and post-translational regulation in response to mitophagy induction. Expression of Pichia pastoris Atg32 (PpAtg32, Atg32 homolog in P. pastoris) is highly suppressed in nutrient-rich media caused by the DNA-binding protein Ume6 and the histone deacetylase complex Sin3–Rpd3, which interact with the promoter region of the gene encoding PpAtg32 to repress its transcription (Aihara et al., 2014). Kang’s group provided evidence that the kinase CK2 could phosphorylate N-terminal cytosolic region of Atg32 at serine 114 and serine 119 to promote the Atg32–Atg11 interaction and further accelerate the mitophagic process (Kanki et al., 2013), but how CK2-dependent phosphorylation takes place during starvation remains elusive. The C-terminal intermembrane space domain of Atg32 was found to be proteolytically processed by inner membrane i-AAA (ATPases associated with various cellular activities) protease Yme1 during mitophagy induction (Wang et al., 2013).
Mitophagy in Mammalian System
It has become clear that the regulation of mitophagy in mammalian cells appears to be more complex. Thus, both ubiquitin-mediated and receptor-mediated pathways have been described to facilitate mitophagy in response to cellular, developmental, and environmental cues in mammalian systems.
Ubiquitin Pathways
In mammalian cells, the PTEN-induced putative kinase protein 1 (PINK1) and Parkin-mediated ubiquitination pathway is one of the most-studied mitophagy mechanisms so far. Two key factors, the serine/threonine kinase PINK1 and the E3 ubiquitin ligase Parkin, cooperatively sense cellular stress and mediate the removal of damaged mitochondria. Under physiological conditions with normal mitochondrial membrane potential, PINK1 is continuously imported into mitochondria where it is cleaved by the intramembrane protease presenilin associated rhomboid like (PARL), leading to its retro-translocation into the cytosol and rapid proteasomal degradation (Sekine and Youle, 2018). When mitochondrial membrane potential drops, PINK1 escapes from PARL-dependent cleavage and aggregates on the outer mitochondrial membrane to exert its pro-mitophagic function. Stabilized PINK1 phosphorylates both Parkin and ubiquitin (at Ser65) to promote ubiquitination of outer mitochondrial membrane proteins (Kane et al., 2014; Koyano et al., 2014). Phosphorylated ubiquitin binding to Parkin further unleashes Parkin from its autoinhibited state (Kazlauskaite et al., 2015). Activated Parkin appends ubiquitin moieties on specific mitochondrial outer membrane proteins such as MFN1, MFN2, FIS1, and translocase of outer mitochondrial membrane (TOMM) proteins, thus inducing their proteasomal degradation, which in turn promotes mitochondrial fission and mitophagy (Tanaka et al., 2010; Desai et al., 2018). The phosphatase and tensin homolog (PTEN)-long (PTEN-L) is able to dephosphorylate (Ser65 of) both ubiquitin and Parkin, which reduces the mitochondrial translocation of Parkin and negatively regulates mitophagy (Wang et al., 2018). On the other hand, the Parkin-mediated formation of ubiquitin chains on mitochondrial outer membrane proteins or even PINK1 itself can recruit ubiquitin-binding adaptor proteins such as optineurin (OPTN) and Calcium Binding And Coiled-Coil Domain 2 (CALCOCO2, better known as NDP52) onto mitochondrial surfaces, followed by the assembly of autophagy factors on Parkin and ubiquitin-marked mitochondria (Wong and Holzbaur, 2014; Lazarou et al., 2015). Ubiquitination of sperm mitochondria in both Caenorhabditis elegans and mammalian systems serves as “eat me” signal for their elimination by receptor-mediated mitochondrial degradation (Sutovsky et al., 1999; Molina et al., 2019). Both mitochondrial E3 ubiquitin protein ligase 1 (MUL1) and Parkin are necessary to remove paternal mitochondria from mouse embryos via mitophagy to ensure maternal mitochondrial inheritance (Rojansky et al., 2016).
Moreover, deubiquitinases play a crucial role in modulating the efficiency of PINK1 and Parkin-mediated mitophagy. Thus, ubiquitin-specific peptidase 8 (USP8) directly deubiquitinates Parkin and removes non-canonical Lys6-linked ubiquitin chains from Parkin, thereby promoting its translocation to depolarized mitochondria. In contrast to USP8 (Durcan et al., 2014), USP15 deubiquitinates the mitochondrial substrates of Parkin to inhibit mitophagy (Cornelissen et al., 2014). Recently, several deubiquitinases such as USP30, USP35, and USP33 were reported to antagonize Parkin-mediated ubiquitination and thus oppose Parkin-mediated mitophagy (Bingol et al., 2014; Wang et al., 2015; Niu et al., 2019). In addition, PINK1 and Parkin have been suggested to be required for mitochondria-derived vesicle (MDV)-dependent mitophagy such that vesicles budding from mitochondria under oxidative stress can be delivered to the lysosomes independent of LC3 (Soubannier et al., 2012; McLelland et al., 2014).
Mitophagy Receptor Pathway
Several mitophagy receptors have been identified in mammalian cells, significantly advancing the field of both mitochondrial and selective autophagy. Mitophagy receptors in mammalian cells are characterized by the presence of at least one LC3 interacting region (LIR) that can directly bind to the autophagy mediator LC3 to recruit autophagosomes to mitochondria.
BCL2 interacting protein 3 like (BNIP3L, better known as NIX) was identified as an essential mitophagy receptor for the autophagic clearance of mitochondria during the maturation of erythroid cells (Sandoval et al., 2008). Recently, the phosphorylation of the LIR domain of NIX was shown to further enhance the affinity of the interaction between NIX and LC3 (Rogov et al., 2017). Moreover, BCL2 interacting protein 3 (BNIP3), a homolog of NIX, was found to mediate mitophagy in conditions of hypoxia (Quinsay et al., 2010).
We have discovered that FUNDC1 acts as an important mitophagy receptor, whose function is regulated by its phosphorylation state (Liu et al., 2012). Structural analysis revealed the functional importance of the close proximity of Tyr18 of FUNDC1 with Asp19 of LC3. Consistently, phosphorylation of Tyr18 of FUNDC1 via SRC proto-oncogene, non-receptor tyrosine kinase (SRC) kinase significantly weakens its binding affinity for LC3 due to electrostatic repulsion in vitro (Kuang et al., 2016). The dephosphorylation (of Ser13) of FUNDC1 can promote mitophagy by recruiting Drp1 while dissociating it from OPA1, thus inducing mitochondrial fission (Chen et al., 2016).
Other mitophagy receptors have been reported such as BCL2 Like 13 (BCL2L13) (the functional homolog of ATG32 in mammals) (Otsu et al., 2015), FKBP prolyl isomerase 8 (FKBP8) (Bhujabal et al., 2017), NLR family member X1 (NLRX1) (Zhang Y. et al., 2019), autophagy and Beclin 1 regulator 1 AMBRA1 (Strappazzon et al., 2015), as well as the mitochondria inner membrane protein prohibitin 2 (PHB2) (Wei et al., 2017). All of them were found to interact with LC3 via the conserved LIR motif to mediate mitophagy when mitochondria become damaged. However, the molecular regulation and their cooperation in response to mitochondrial stresses are not completely understood. Moreover, mitophagy receptors are not limited to proteins, as certain types of lipids such as cardiolipin and ceramide have been reported to interact with LC3 and to mediate mitophagy (Sentelle et al., 2012; Chu et al., 2013).
The Interplay Between Mitochondrial Dynamics and Mitophagy
Distinct molecular machineries have been identified to regulate mitochondrial fission and fusion. In mammalian cells, the GTPase MFN1, MFN2, and OPA1 mediate the fusion of the outer and inner membranes of mitochondria, respectively. Mitochondrial fission is regulated by Drp1 that normally resides in the cytosol and is recruited to mitochondria by mitochondrial fission factors such as FIS1, MFF, MIEF1, or MIEF2 (Mishra and Chan, 2014). ER tubules, which are in contact with mitochondria, play an active role in the initial step of mitochondrial division and mediate mitochondrial constriction before Drp1 recruitment (Friedman et al., 2011). At the final step of mitochondrial division, the Drp1-mediated constriction promotes dynamin-2 (DNM2) assembly, which can induce membrane fission to complete division (Lee et al., 2016). In response to bioenergetic crisis and oxidative stress, these mediators of mitochondrial dynamics are posttranslationally modified to fine-tune their activities. Phosphorylation of Drp1 by protein kinase A (PKA, also known as cAMP-dependent protein kinase) at Ser637 (Chang and Blackstone, 2007) and Ser656 (Cribbs and Strack, 2007) inhibits Drp1, resulting in mitochondrial elongation, while dephosphorylation of Drp1 at Ser65 by the calcium-dependent protein phosphatase calcineurin or by protein phosphatase 2A (PP2A) enhances mitochondrial fragmentation (Cribbs and Strack, 2007). Another report suggested that Drp1 is phosphorylated at Ser616 by the cyclin-dependent kinase 1 (CDK1)/cyclin B complex during mitosis (Taguchi et al., 2007; Marsboom et al., 2012). The phosphorylation of the Drp1 receptor MFF by energy-sensing adenosine monophosphate (AMP)-activated protein kinase (AMPK) results in the recruitment of Drp1 and final mitochondrial fragmentation (Toyama et al., 2016). Other Drp1 modifications include S-nitrosylation (Cho et al., 2009) and ubiquitination by MARCH5 to mediate mitochondrial division (Karbowski et al., 2007) or by Parkin to inhibit mitochondrial fission (Wang et al., 2011).
The mitochondrial fusion molecule MFN1 can be phosphorylated by extracellular regulated kinase (ERK) at Thr562 to inhibit fusion (Pyakurel et al., 2015), ubiquitinated by MARCH5 (Park et al., 2014), and deubiquitinated by USP30 (Yue et al., 2014) to regulate protein stability and fusion activity, while MFN2 can be phosphorylated by mitogen-activated protein kinase 8 (MAPK8 better known as JNK) at Ser27 and ubiquitinated for degradation by HUWE1 (Leboucher et al., 2012), Parkin (Gegg et al., 2010), and MARCH5 (Sugiura et al., 2013), and deubiquitinated by USP30 (Yue et al., 2014). During mitophagy, MFN2 also functions as a mitochondrial receptor for the PINK1-dependent recruitment of Parkin. PINK1 phosphorylates MFN2 at Thr111 and Ser442 to promote the recruitment of Parkin to depolarized mitochondria (Chen and Dorn, 2013). OPA1 can be proteolytically processed by mitochondria-resident proteases, including YME1-like 1 ATPase (YME1L) (Griparic et al., 2007) and zinc metallopeptidase (OMA1) (Head et al., 2009), in response to intra-mitochondrial signals, to regulate fusion of the inner mitochondrial membrane.
It was suggested that mitochondrial fission is necessary for mitochondrial degradation by mitophagy because fission enables the separation of depolarized mitochondria from the mitochondrial network and allows their engulfment by autophagosomes. Mitochondrial stress-induced mitophagy is accompanied by enhanced mitochondrial fission. The inhibition of mitochondrial fission processes by overexpression of dominant negative Drp1K38A or knockdown of FIS1 decreases mitophagy and leads to the accumulation of oxidized mitochondrial proteins (Twig et al., 2008). In agreement with this, mitophagic players were found to regulate mitochondrial dynamics. Thus, Parkin is able to ubiquitinate MFN1 and MFN2 to promote their degradation, leading to increased fragmentation of mitochondria (Gegg et al., 2010). Our early work showed that Parkin also ubiquitinates and degrades Drp1 (Wang et al., 2011). This may be counterintuitive, as degradation of Drp1 prevents mitochondrial fragmentation. It is possible that under homeostatic conditions, Parkin monitors the molecular status of Drp1 to prevent mitochondrial fragmentation, and upon stress conditions, Parkin translocates to mitochondria to promote mitochondrial fragmentation and mitophagy.
Mitophagy receptors such as FUNDC1 and BNIP3 were found to promote mitochondrial fission in response to stress (Landes et al., 2010; Chen et al., 2016). FUNDC1 directly interacts and recruits Drp1 toward mitochondria for mitochondrial fission. Interestingly, FUNDC1 is a transmembrane protein with a motif that faces the mitochondrial intermembrane space and directly interacts with OPA1 to promote mitochondrial fission.
It was noted that mitochondrial fission is necessary, but not sufficient for mitophagy. Reports suggested that Drp1-mediated mitochondrial fission was dispensable for mitophagy (Song et al., 2015; Yamashita et al., 2016). We have found that mitochondrial targeting of the LIR-containing cytosolic portion of FUNDC1 is sufficient to induce mitophagy even in the absence of mitochondrial fragmentation, when phosphorylation of Tyr18 is blocked (Kuang et al., 2016). Recently, by using structure illumination microscopy (SR-SIM), Xian et al. (2019) observed that the overexpression of the SNARE protein syntaxin 17 (STX17) initiated mitophagy in FIS1-depleted cells but not in other mitochondria dynamic factors-silenced cells. They further demonstrated that FIS1 negatively regulated STX17 by inhibiting its trafficking to mitochondria-associated membranes (MAMs) and mitochondria independent of mitochondrial dynamics (Xian et al., 2019). In summary, a sensitive reaction to various types of stress mitochondrial fragmentation at early stages facilitates segregation and clearance of dysfunctional mitochondria from the mitochondrial network for maintaining mitochondrial and cellular homeostasis.
Mitochondrial Dynamics and Cell Death
Mitochondria in mammalian cells sense apoptotic stress, mainly through BCL2 and its family proteins, ultimately leading to MOMP and the subsequent release of cytochrome c and other apoptogenic factors for the activation of the caspase cascade governing apoptotic cellular disintegration. The BCL2 protein family is composed of antiapoptotic molecules including BCL2, BCL-xL, MCL1, and proapoptotic molecules such as BCL2 associated X (BAX), BCL2 antagonist/killer 1 (BAK), and BH-3-only subfamily proteins such as such as BIM, BAD, NOXA, and BID (Doerflinger et al., 2015). In healthy cells, BAX and BAK1 are blocked by antiapoptotic proteins such as BCL2, BCL-xL, and MCL1, which contain four BH motifs (BH1–4). The BH3-only proteins can induce apoptosis by direct interaction with BAX and BAK or by binding to antiapoptotic members and thus neutralizing the inhibitory sequestration of BAX and BAK (Chen et al., 2005; Chipuk et al., 2010). The antiapoptotic protein BCL-xL interacts with BAX to continuously retro-translocate mitochondrial BAX into the cytosol and keep it from integrating into the mitochondrial outer membrane (Edlich et al., 2011). In apoptotic cells, BAX and BAK oligomerization triggers MOMP and initiates the caspase cascade ultimately leading to cell death (Tait and Green, 2010).
Emerging evidence indicates that the mechanisms governing mitochondrial dynamics are also involved in the regulation of apoptotic processes. Inhibition of mitochondrial fission reduces cytochrome c release and apoptosis (Frank et al., 2001; Cereghetti et al., 2010). On the contrary, the dephosphorylation of Drp1 at Ser637 by the phosphatase calcineurin promotes Drp1-mediated mitochondrial fragmentation and leads to apoptosis (Cereghetti et al., 2010). Drp1-dependent mitochondrial fission through MIEF2 facilitates apoptotic cristae remodeling during the early phase of intrinsic apoptosis (Otera et al., 2016), and moreover, Drp1 can stimulate truncated Bid (tBID)-induced Bax oligomerization and cytochrome c release by promoting tethering and hemifusion of membranes. Dephosphorylation of Drp1 by the mitochondrial phosphatase PGAM5 can facilitate necroptosis by enhancing mitochondrial fission (Wang et al., 2012). However, other reports have shown that blocking mitochondrial fission can just delay but does not block apoptosis (Parone et al., 2006; Rolland and Conradt, 2010; Clerc et al., 2014).
On the other hand, BCL2 family members can affect the morphology of mitochondria. In healthy cells, BAX and BAK are required for mitochondrial fusion (Karbowski et al., 2006). Mitochondria are fragmented and have less network continuity in cells lacking BAX and BAK. The interaction between BAX and MFN2 activates the assembly of the MFN2 complex and changes its membrane mobility and distribution. However, the activation of pro-apoptotic BAX and BAK promotes the fragmentation of the mitochondrial network during apoptosis (Autret and Martin, 2009; Montessuit et al., 2010), which is not inhibited by the expression of BCL-xL, MCL1, or other members of the BCL2 subfamily (Sheridan et al., 2008). BAX and BAK form foci that colocalize with ectopic MFN2 and Drp1 at the sites of mitochondrial division to promote mitochondrial fission during apoptosis (Karbowski et al., 2002). Furthermore, BCL-w, an antiapoptotic BCL2 family member, was proposed to regulate mitochondrial fission in Purkinje cell dendrites (Liu and Shio, 2008). BCL-xL overexpression induces the remodeling of the mitochondrial network by altering the relative rates of mitochondrial fusion and fission (Delivani et al., 2006; Li et al., 2008). In neuronal cells, overexpression Bcl-xL can increase the rates of both fission and fusion and mitochondrial biomass (Berman et al., 2009). Other studies found that, in hippocampal neurons, BCL-xL increases synapse dynamics and the localization of mitochondria to synapses and vesicle clusters via a Drp1-dependent manner (Li et al., 2013). Alternatively BCL-xL can interact with Drp1 to function in mitochondrial fission during neuronal development (Li et al., 2008). Although the mechanisms of action require further clarification, these findings demonstrate that the BCL2 protein family indeed orchestrates mitochondrial morphology and apoptosis.
Mitophagy and Cell Death
The BCL2 family was initially recognized for their function in apoptosis, and is now widely proven to also have other roles in cellular function involving mitochondrial dynamics, autophagy/mitophagy, and cellular metabolism. Early studies have shown that the antiapoptotic protein BCL2 can interact with Beclin 1 (BECN1) to inhibit autophagy (Pattingre et al., 2005). Further analysis reveals that depending on its phosphorylation status, BCL2 has dual roles in regulating autophagy and apoptosis. It suggests that initial JNK1-mediated BCL2 phosphorylation may promote cellular survival by disrupting BCL2–BECN1 complexes and activating autophagy (Wei et al., 2008a). At a point when autophagy is no longer able to maintain survival, the phosphorylation of BCL2 serves to inactivate its antiapoptotic function for progression of regulated cell death (Wei et al., 2008b). Parkin-dependent mitophagy is antagonized by BCL-xL and MCL1 in a BECN1-independent manner. Specifically, BCL2 and BCL-xL suppress Parkin translocation to depolarized mitochondria, while BH3-only proteins (or BH3-only mimetics) can promote this process (Hollville et al., 2014).
Several mitophagy receptors including BNIP3, NIX, and BCL2L13 belong to the BCL2 family (Novak et al., 2010; Hanna et al., 2012; Murakawa et al., 2015), highlighting an intrinsic link of mitophagy with apoptosis. Apparently, these BCL2 family proteins have dual roles in both apoptosis and mitophagy. For example, BNIP3 and NIX can directly interact with antiapoptotic BCL2 or BCL-xL, which antagonizes the activation of proapoptotic BAX and BAK, to promote apoptosis (Imazu et al., 1999; Dorn, 2010). As discussed above, NIX also induces mitophagy via its interaction with LC-3, and enhanced interaction of BNIP3 with Atg8 family members promotes pro-survival mitophagy prior to cytochrome c release and apoptosis (Zhu et al., 2013). It was also found that the mitochondrial fragmentation is a prerequisite for BNIP3-induced mitophagy in cardiac myocytes, and dominant negative Drp1K38E mutant, or MFN1 overexpression inhibit BNIP3-induced mitochondrial division and mitophagy (Lee et al., 2011). Similar to NIX, BNIP3 induces the disintegration of elongated mitochondria into numerous spherical particles, accompanied by the recruitment of Drp1 to fragmented mitochondria in adult myocytes (Lee et al., 2011). Moreover, BNIP3 can directly interact with OPA1, promote the disassembly of OPA1 oligomers, and thus antagonize its fusion activity in HeLa cells (Landes et al., 2010). Thus, BCL2 family proteins act as general regulators of mitochondrial dynamics and homeostasis, in addition to their role in apoptosis-associated mitochondrial permeabilization.
Mitophagy was suggested to play a protective role in stress-induced cell death and early studies showed that Parkin strongly inhibits the translocation of BAX to mitochondria, thus preventing apoptosis (Darios et al., 2003; Johnson et al., 2012). Further studies revealed that Parkin is able to directly ubiquitinate the apoptotic effector proteins such as BAX and BAK, and the ubiquitination of BAK by Parkin impairs its activation and the formation of oligomers to suppress errant apoptosis (Bernardini et al., 2019). Parkin suppression of BAX-dependent apoptosis will allow the effective clearance of apoptotic mitochondria to limit their potential pro-inflammatory effect (Bernardini et al., 2019). Parkin suppression of apoptosis is likely the cellular context and apoptosis inducer dependent. Studies from Seamus Martin’s laboratory showed that upon mitochondrial depolarization, the BCL2 family member MCL1 underwent rapid PINK1- and Parkin-dependent polyubiquitination and degradation, which sensitized cells toward apoptosis via opening of the BAX and BAK-dependent pathway. Knockdown of BAX is able to suppress Parkin-dependent apoptosis in HeLa cells (Carroll et al., 2014). It was also reported that NIX-mediated mitophagy protects glioblastoma cells against hypoxia (Jung et al., 2019). Furthermore, abrogating NIX- and FUNDC1-mediated mitophagy during adult cardiac progenitor cells (CPCs), differentiation leads to increased susceptibility to cell death (Lampert et al., 2019).
In addition, there is certain evidence showing that mitophagy plays an accelerative role in programmed cell death. Thus, LC3–ceramide interactions provoked by ceramide treatment induce mitophagy and can progress to autophagic cell death in human cancer cells (Sentelle et al., 2012). Moreover, inhibition of mitophagy and mitochondrial fission reduces cigarette smoke-induced necroptosis in mice epithelial cells in vitro, and in chronic obstructive pulmonary disease (COPD) in vivo (Mizumura et al., 2014). Furthermore, in hippocampal neural stem cells deprived of insulin, Parkin-mediated mitophagy is necessary for autophagy-dependent cell death (Park et al., 2019). Moreover, drug-induced mitochondrial dysfunction and heme oxygenase 1 (HMOX1) overactivation synergize to trigger lethal mitophagy in glioma cells, which is significantly blocked by silencing of the mitophagy receptors BNIP3 and NIX (Meyer et al., 2018).
We have found that BCL-xL, but not BCL2, strongly suppresses FUNDC1-mediated mitophagy. BCL-xL interacts with and inhibits the mitochondrial Ser/Thr phosphatase PGAM5 to prevent the dephosphorylation of FUNDC1 (at Ser13), thus further blocking hypoxia-induced mitophagy (Wu et al., 2014). The functions of PGAM5 not only are limited to the induction of mitophagy but also involve the regulation of mitochondrial homeostasis (Figure 1). PGAM5 exists in an equilibrium between a dimeric and a multimeric state, which is sensitive to oxidative stress. Dimeric PGAM5 binds with and dephosphorylates BCL-xL in mitotically arrested cells, thus exerting its antiapoptotic function in vitro and in vivo. Mitochondrial oxidative stress enhances the multimerization of PGAM5, resulting in its dissociation from BCL-xL. Liberated multimeric PGAM5 dephosphorylates FUNDC1 to initiate mitochondrial fission and mitophagy. When FUNDC1-mediated mitophagy is blocked by the microtubule inhibitor vinblastine, PGAM5 dephosphorylates FUNDC1 and mediates mitochondrial fission that aggravates vinblastine-induced cell death (Ma K. et al., 2019).
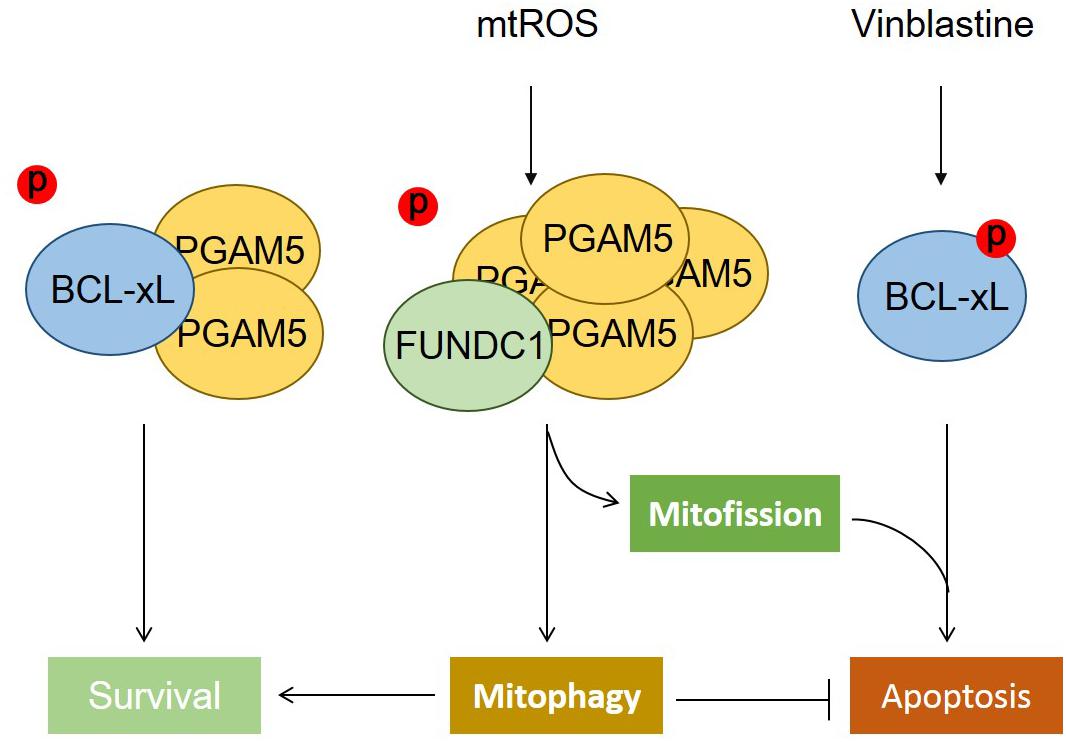
Figure 1. The phosphatase PGAM5 regulates mitochondrial fate. The phosphatase PGAM5 is a dimeric protein that can bind with and dephosphorylate BCL-xL at Ser62, which increases its antiapoptotic function and thus inhibits apoptotic cell death. Mitochondrial oxidative stress causes the transformation of dimeric PGAM5 into a multimeric state that fails to bind with BCL-xL, but instead interacts with and dephosphorylates FUNDC1 at Ser13 to mediate mitochondrial fission and mitophagy. The dephosphorylation of FUNDC1 cooperates with the phosphorylation of BCL-xL, aggravating cell death when mitophagy is blocked.
Mitophagy Is Balanced With Mitochondrial Biogenesis for Mitochondria Homeostasis
Mitophagy is balanced with mitochondrial biogenesis, together defining mitochondrial turnover. Mitochondrial biogenesis is a cellular process in which “new” mitochondria are produced, depending on the cooperation of nuclear and mitochondrial genome (Zhang and Xu, 2016). Mitochondrial biogenesis preserves mitochondrial function and cellular homeostasis (Rasbach and Schnellmann, 2007; Miwa et al., 2008; Gottlieb and Carreira, 2010), while mitophagy mitigates the source of oxidative stress that reduces the risk of apoptosis (Hickson-Bick et al., 2008). The crosstalk between mitophagy and mitochondrial biogenesis also allows cells to undergo metabolic reprogramming during development and differentiation. Similar to mitophagy, mitochondrial biogenesis is highly variable and tightly regulated in response to diverse stimuli such as energy demand, cell cycle, and intracellular stress (Zhang and Xu, 2016). Many cellular signaling pathways converge on the regulation of both mitophagy and mitochondrial biogenesis such as the mammalian target of rapamycin (mTOR), which regulates cellular growth and energy homeostasis in conditions of nutrient stress (Dibble and Cantley, 2015; Vyas et al., 2016). The mTOR signal pathway can transcriptionally and translationally regulate mitochondrial biogenesis. Thus, mTOR controls mitochondrial function through the modulation of PPARG coactivator 1 alpha (PGC1α) transcriptional activity (Cunningham et al., 2007) and ablation of PPARGC1B (PGC1β) is associated with the constitutive activation of mTORC1 (Camacho et al., 2012). At the transcriptional level, it can mediate the activation of PGC1α, which is a key transcriptional co-activator regulating mitochondrial biogenesis via its interaction with a variety of transcription factors (Morita et al., 2015). Similar to mTOR, the hypoxia-inducible factor 1 subunit alpha (HIF1A) signaling in response to limited oxygen availability can impinge on mitochondrial biogenesis, mitophagy, and mitochondrial metabolism through regulation of PGC1α (LaGory et al., 2015).
mTORC1 phosphorylates UNC-51 like autophagy activating kinase 1 (ULK1) and 2 (ULK2) and disrupts the interaction between ULK1 and protein kinase AMP (AMPK), leading to the inhibition of autophagy and mitophagy when nutrient levels are sufficient. Under nutrient starvation, ATP depletion leads to serine/threonine kinase 11 (LKB1)-mediated AMPK activation (Garcia and Shaw, 2017), which in turn leads to the phosphorylation of ULK1 to initiate autophagy and mitophagy (Kim et al., 2011; Tian et al., 2015). Tuberous sclerosis complex (TSC1/2) is an inhibitor of the mTOR signaling pathway. In TSC1/2-deficient neurons, axonal and global mitophagy is impaired and mitochondrial homeostasis can be restored by blocking mTORC1 (Ebrahimi-Fakhari et al., 2016). Moreover, TSC2-deficient cells exhibit constitutive mTOR activation, impaired autophagic flux, and accumulation of damaged mitochondria, which associates with reduced PINK1 expression and Parkin mitochondrial translocation to mitochondria. These data link mTOR signaling to PINK1-Parkin-mediated mitophagy (Bartolome et al., 2017).
Hypoxia-induced mtROS can activate HIFs leading to the upregulation of target genes, including the mitophagy receptor BNIP3 (Bell et al., 2007; Chourasia and Macleod, 2015). As a feedback mechanism, BNIP3-mediated mitophagy reduces the generation of mtROS, which in turn can stabilize HIF1A (Chourasia and Macleod, 2015). Nevertheless, ROS can also activate JNK–PGC1α signaling pathway to promote mitochondrial biogenesis and the expression of genes involved in OXPHOS (Chae et al., 2013). Increased mtROS was also found to promote cellular proliferation by activating NF-κB (Guha et al., 2010). Recently, we found that a hypoxic microenvironment can induce siah E3 ubiquitin protein ligase 2 (SIAH2)-dependent ubiquitination and subsequent proteasomal degradation of nuclear respiratory factor 1 (NRF1), a transcription factor crucial for mitochondrial biogenesis. In conditions of cancer, this signaling axis alters the level of mitochondrial biogenesis and is involved in metabolic adaptations finally maintaining tumor progression (Ma B. et al., 2019).
Mitochondrial Quality Control in Immune Responses
The innate and adaptive immune systems are able to sense pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) arising from exogenous clues including bacteria, virus, fungi, and parasites, as well as endogenous entities such as cancer cells and to mount defensive immune responses. Mitochondria have emerged as central organelles contributing to immune response at multiple levels, such as adaptations of mitochondrial metabolism, dynamics, biogenesis, and mitophagic turnover. Mitochondrial DNA (mtDNA), once released into the cytoplasm, acts as intrinsic DAMP, which can be sensed by toll-like receptor 9 (TLR9) and triggers nuclear factor kappa B (NF-κB) signaling in human polymorphonuclear neutrophils (Zhang et al., 2010). Moreover, mtDNA activates the NLRP3 inflammasome, which in turn boosts the production of cytokines such as IL18 and IL1β induces pyroptosis in immune cells (Liu et al., 2018). Furthermore, cellular mtDNA activates the STING pathway via cGAS and leads to the expression of IRF3-dependent genes such as type I interferons and participates to antiviral immune responses (West et al., 2015). Interestingly, live-cell lattice light-sheet microscopy observed mouse embryonic fibroblasts result has shown that BAK/BAX form macropores after activation and allowed mitochondrial matrix components, including the mtDNA releasing into the cytosol (McArthur et al., 2018). In addition, evidence has shown that mtROS enhances the NLRP3 inflammasome activation and upregulates NF-κB signaling. Mitophagy counteracts chronic inflammation via the elimination of damaged mitochondria, which are the major sources of mtDNA and ROS.
Parkin-mediated mitophagy restrains excess ROS and cytosolic mtDNA, and inhibits NLRP3 inflammasome activity in macrophages and favors tissue repair via a NF-κB and sequestosome 1 (SQSTM1, better known as p62)-dependent mitophagic pathway (Zhong et al., 2016). Intriguingly, Parkin is cleaved by caspase-1 to limit mitophagy and resultant excess inflammation (Yu et al., 2014). Defective mitophagy leads to the upregulation mRNA levels of inflammasome-related proteins in primary hepatocytes under palmitic acid treatment and in a murine model of NASH (Zhang N. P. et al., 2019). Defects in mitophagy arising from deletion, mutation, or silencing of mitophagy receptors, such as NIX, Parkin, and p62, lead to mitochondrial dysfunctions and have been linked to inflammasome activation and cancer (Drake et al., 2017). Likewise, ablation of FUNDC1 causes the inhibition of mitophagy and increases the accumulation of dysfunctional mitochondria, which in turn results in inflammasome activation and inflammatory responses that can promote hepatocyte tumorigenesis in vivo (Li et al., 2019). Listeria monocytogenes can induce mitophagy in macrophages to evade host immune response. Mechanistically, Nod-like receptor (NLR) family member X1 (NLRX1), a novel mitophagy receptor located at the mitochondria, directly interacts with LC3 via its LIR motif, thus contributing to the induction of mitophagy for the elimination of ROS, and maintains the survival of L. monocytogenes (Zhang Y. et al., 2019). Furthermore, interleukin 10 (IL-10), an anti-inflammatory cytokine that promotes mitophagy, leads to a decrease in the activation of the NLRP3 inflammasome and the production of IL-1β in macrophages (Ip et al., 2017).
The mitochondrial outer membrane is a platform for MAVS-mediated innate immune responses, which are activated by the viral RNA sensors RIG-I-mediated signaling cascade culminating in the activation by NF-κB and IRF3 (Seth et al., 2005). Alternatively, MAVS can oligomerize upon sensing mtROS independent of RIG-I to facilitate the production of type I interferon (Buskiewicz et al., 2016). Intriguingly, MAVS has been found to contain a LIR motif and act as a potential receptor for mitophagy (Sun et al., 2016). Furthermore, the ubiquitination of MAVS by ring finger protein 34 (RNF34) causes NDP52-associated mitophagy to mitigate innate immune response upon viral infection (He et al., 2019).
BCL2 family regulated mitochondria-dependent cell death has also been reported to play an important role in innate and adaptive immune responses. One of the therapeutic strategies to Legionnaires’ disease is the pharmacological inhibition of BCL-xL. Inhibition of BCL-xL can induce the apoptosis of macrophages infected with virulent Legionella and thus abrogate Legionella replication and disease progression in mice (Speir et al., 2016). Additionally, it is well known that activated T cells will undergo cell death once the antigen has disappeared. This mechanism is triggered by the BCL-xL- and BCL2-mediated release of pro-apoptotic BAX and BAK or the fact that BCL-xL without its unstructured loop, which cannot bind to any form of BAX and BAK, binds BIM less well than wild-type BCL-xL and thus sensitizes T cells to the induction of regulated cell death (Liu et al., 2006). Overall, more and more evidence demonstrates that mitochondrial quality control mechanisms regulate essential functions in immune cell and are important in controlling immune responses. Mitophagy plays a protective role in cellular homeostasis to negatively regulate innate immune response, but a systematic drawing of this intricate relationship has not been completed yet due to its complexity.
Dysregulation of Mitochondrial Homeostasis in Aging and Aging-Related Diseases
Aging increases the risk for the onset of various chronic diseases often associated with the accumulation of mtDNA mutations, altered mitochondrial mass, compromised mitochondrial functions, chronic immune activation, and accelerated cell death. These pathogenic manifestations are likely due to dysregulated mitochondrial dynamics and mitochondrial quality control mechanisms, leading to the accumulation of dysfunctional mitochondria, which enhances both chronic immune activation (through the release of mtDNA and ROS) and mitochondrial apoptosis (through the liberation of apoptogenic factors). There is emerging evidence that compromised mitophagy causes aging, while enhancing mitophagy by caloric restriction and physical exercises increases healthy life span. Thus, the reduction of mitophagy has been suggested as areas on for the accumulation of mitochondria in aged C. elegans (Palikaras et al., 2015), and the induction of mitophagy results in life span extension in this model (Ryu et al., 2016). Overexpression of the Drosophila PGC-1 homolog (dPGC-1/spargel) increases mitochondrial activity, and intestinal stem cell (ISC) lineage-specific expression of dPGC-1 leads to an extended life span in Drosophila melanogaster (Rera et al., 2011). Physical exercise caused the activation of AMPK that leads to ULK1 phosphorylation and enhanced mitophagy in skeletal muscle, which in turn promotes mitochondrial turnover and improves general health in murine models (Laker et al., 2017).
Mitochondrial dysfunction is a common pathogenic factor for neurodegenerative disorders. Mitochondria supply ATP, generate mtROS, and regulate calcium homeostasis, all of which affect neuronal cell physiology. Aberrations in mitochondrial ROS and Ca2+ homeostasis have been implicated in Parkinson’s disease (PD) (Ludtmann and Abramov, 2018). Both elevated cytosolic Ca2+ levels and mitochondrial ROS are pathological hallmarks of PD. Thus, PINK-1 deficiency in midbrain neurons leads to mitochondrial Ca2+ overload in response to dopamine, which further promotes ROS production and neuronal cell death (Gandhi et al., 2009). The loss of mitochondrial fission factor (MFF) increases mitochondrial size and mitochondrial Ca2+ uptake during neurotransmission, thus affecting neurotransmitter release and neuronal fitness (Lewis et al., 2018). Production of mtROS caused by damaged mitochondria in mice microglia promotes the secretion of pro-inflammatory cytokines and results in neurodegeneration (von Bernhardi et al., 2015). Furthermore, the accumulation of Ca2+ and ROS in the mitochondria triggers mitochondrial permeability transition pore (mPTP) opening, subsequently releasing cytochrome c and other pro-apoptotic intermembrane space proteins into the cytosol (Hunter and Haworth, 1979). It needs to be noted that BCL2 family proteins are involved in the regulation of Ca2+ dynamics of the ER and the mitochondria (Vervliet et al., 2016). For instance, a fraction of NIX is localized at the conjunction between mitochondria and the ER to regulate ER and mitochondrial Ca2+ homeostasis (Diwan et al., 2009). In addition, the upregulation of NIX in cardiac hypertrophy was associated with the apoptotic death of cardiomyocytes (Yussman et al., 2002). Altogether, mitochondrial quality control appears crucial for protecting neurons from damage and death.
It has been well established that compromised mitophagy contributes to the pathogenesis of Parkinson’s disease, and enlarged or swollen mitochondria have been observed in several disease models and in the brains of Parkinsonian patients. Mutations in PINK1 and Parkin are involved in rare familial cases of Parkinson’s disease (PD) (Kitada et al., 1998; Valente et al., 2004). PGAM5 deficiency disables PINK1-mediated mitophagy in vitro and causes a Parkinson’s-like phenotypes in mice model (Lu et al., 2014; Sekine et al., 2016). It is still puzzling that Parkin knockout mouse does not completely recapitulate PD phenotype. When Parkin knockout mouse was crossed with mouse that harbors high mtDNA mutation, the accumulation of mutated mtDNA in neuronal cells was observed, but these mice does not have much increase of mitochondrial mass (Gautier et al., 2008; Stevens et al., 2015; Pinto et al., 2018). Studies using the recently developed mitophagy reporter mice and Drosophila also show that mitophagy is rather constitutive and is minimally impacted by loss of PINK1 or Parkin (McWilliams et al., 2016; Whitworth and Pallanck, 2017; Lee et al., 2018; McWilliams et al., 2018), suggesting that additional factors are required in the absence of PINK1 or Parkin. Mitophagy has been implicated with disease progression in Alzheimer’s disease (Witte et al., 2009; Wilhelmus et al., 2011). Thus, Alzheimer’s disease phenotype-related accumulation of mutant amyloid beta precursor protein (APP) induces Parkin-dependent mitophagy in cultured human neurons and in the brain of Alzheimer’s patients (Ye et al., 2015). Recent studies showed that Tau pathology, another hallmark of Alzheimer’s disease, impairs mitophagy by inhibiting Parkin translocation to mitochondria (Cummins et al., 2019). These studies indicate that insufficient mitophagy might be the cause for the accumulation of damaged mitochondria in Alzheimer’s disease-affected neurons. Conversely, there are arguments that mitophagy improves the neuropathology of Alzheimer’s disease and reverses cognitive deficits in animal models (Kerr et al., 2017).
Moreover, recent studies have suggested that the loss of mitophagy regulators is closely linked to cardiovascular disease. Thus, Parkin-mediated mitophagy is required for the metabolic transition in the perinatal murine heart (Gong et al., 2015). Deletion of PINK1, Parkin, or other mitophagy receptors such as FUNDC1, BNIP3, or NIX leads to the accumulation of dysfunctional mitochondria and results in various heart defects involved in exacerbated ischemia/reperfusion injury and cardiomyopathy (Dorn, 2010; Kubli et al., 2013; Zhang et al., 2016). Consistently, impaired PINK1 and Parkin-mediated mitophagy affected by Parkin deficiency or mutations in MFN2 results in the retention of fetal cardiac mitochondria, reduced oxidative metabolism, heart failure, and premature death (Chen and Dorn, 2013; Gong et al., 2015), highlighting that mitophagy underlies mitochondrial plasticity and metabolic transitioning in developing cardiomyocytes.
Summary and Future Perspectives
Mitochondria are highly plastic organelles that adapt to cellular and environmental stress and developmental cues by changes in their morphology and their overall mass. Changes in the mitochondrial behaviors are regulated by distinct but interlinked molecular machineries that control mitochondrial dynamics (fission, fusion) and mitochondrial homeostasis (mainly through biogenesis and mitophagy), collectively allowing the graded response to stress (Figure 2). BCL2 family proteins, and in particular BCL-xL, act as global regulators of mitochondrial homeostasis and quality control through their interaction with various partners including DRP1, MFN1/2, and PGAM5, that are tightly controlled by reversible phosphorylation, acetylation, and ubiquitination, thereby modulating mitochondrial behaviors and cell fate. This is further exemplified by our recent finding that the mitochondrial phosphatase, PGAM5 exists in an equilibrium between a dimeric and a multimeric state to dephosphorylate FUNDC1 and BCL-xL, respectively, to switch on/off mitophagy and apoptosis. Further research is needed to explore the (patho-) physiological roles of this molecular switch in response to environmental and cellular stresses.
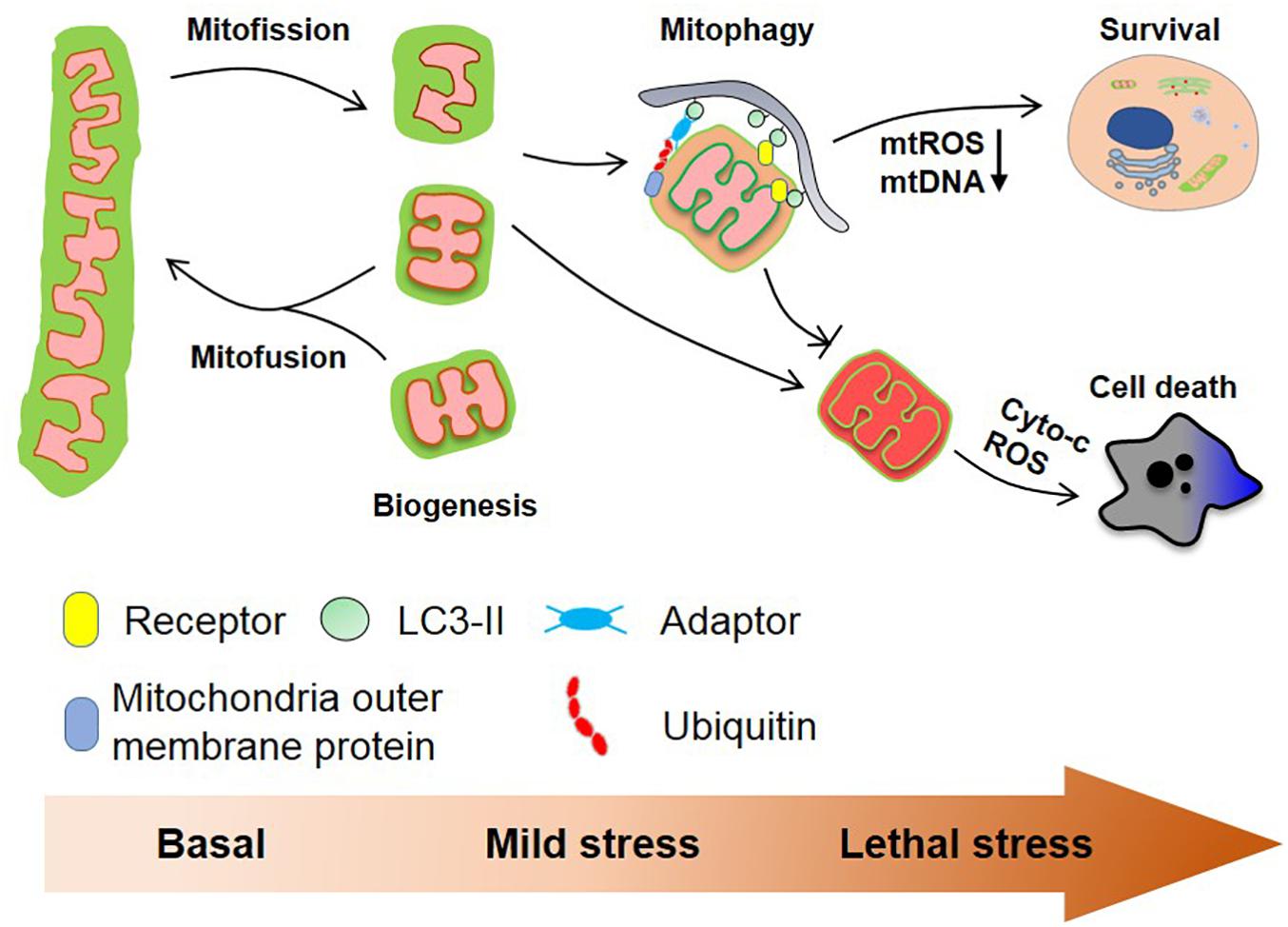
Figure 2. Mitochondrial homeostasis and cell fate. Mitochondria are dynamic organelles that constantly divide and fuse in healthy cells and mitochondrial biogenesis and mitophagy cooperate to maintain mitochondrial quality. Mitochondrial fission usually is a prerequisite for ubiquitin- and receptor-mediated mitophagy facilitating the removal of aged and damaged mitochondria. Severe stress-induced mitochondrial damage can lead to mitochondria outer membrane permeabilization (MOMP) that in turn triggers apoptosis. The clearance of depolarized mitochondria by mitophagy (before MOMP occurs) mitigates this process. Besides, mitophagy reduces the production of mtROS and the cytosolic secretion of mtDNA, which further prevents excess immune response.
By removing the damaged and unwanted mitochondria, mitophagy is essential for mitochondrial quality control and homeostasis. As discussed above, almost all mitophagy players including both receptor-dependent pathway and PINK1/Parkin pathway are found to regulate mitochondrial dynamics and apoptosis. Furthermore, mitophagy not only governs the mitochondrial quality and quantity, but also controls mitochondrial dynamics and behaviors. Enhanced mitophagy and mitochondrial turnover contributes to increased mitochondrial function and cellular activity. Conversely, the inhibition of mitophagy leads to accelerated aging and the manifestation of aging-associated diseases. Moreover, the accumulation of dysfunctional mitochondria, and the associated release of mtDNA, the overproduction of mtROS, and mitochondria-controlled apoptosis result in a chronic state of immune activation, which is also the common etiology for aging-associated neurodegenerative disease. We further suggest that targeting mitophagy is an important strategy to fight aging and aging-associated disease, which needs to be further explored in the future.
Author Contributions
KM, GC, QC, and WL wrote the manuscript. OK and YZ revised the manuscript. All authors provided intellectual input and read the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (31520103904, 31790404, and 91754114) to QC and YZ, the Ministry of Science and Technology of China (2016YFA0500201), and the Chinese Academy of Sciences Key project of Frontier Science (QYZDJSSW-SMC004) to QC.
Conflict of Interest
OK is one of the co-founders of Samsara Therapeutics, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aihara, M., Jin, X., Kurihara, Y., Yoshida, Y., Matsushima, Y., Oku, M., et al. (2014). Tor and the Sin3-Rpd3 complex regulate expression of the mitophagy receptor protein Atg32 in yeast. J. Cell Sci. 127(Pt 14), 3184–3196. doi: 10.1242/jcs.153254
Autret, A., and Martin, S. J. (2009). Emerging role for members of the Bcl-2 family in mitochondrial morphogenesis. Mol. Cell 36, 355–363. doi: 10.1016/j.molcel.2009.10.011
Bartolome, A., Garcia-Aguilar, A., Asahara, S. I., Kido, Y., Guillen, C., Pajvani, U. B., et al. (2017). MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol. Cell Biol. 37:e00441-17. doi: 10.1128/MCB.00441-17
Bell, E. L., Klimova, T. A., Eisenbart, J., Moraes, C. T., Murphy, M. P., Budinger, G. R., et al. (2007). The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 177, 1029–1036. doi: 10.1083/jcb.200609074
Berman, S. B., Chen, Y. B., Qi, B., McCaffery, J. M., Rucker, E. B. III, Goebbels, S., et al. (2009). Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J. Cell Biol. 184, 707–719. doi: 10.1083/jcb.200809060
Bernardini, J. P., Brouwer, J. M., Tan, I. K., Sandow, J. J., Huang, S., Stafford, C. A., et al. (2019). Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J. 38:e99916. doi: 10.15252/embj.201899916
Bhujabal, Z., Birgisdottir, A. B., Sjottem, E., Brenne, H. B., Overvatn, A., Habisov, S., et al. (2017). FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 18, 947–961. doi: 10.15252/embr.201643147
Bingol, B., Tea, J. S., Phu, L., Reichelt, M., Bakalarski, C. E., Song, Q., et al. (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375. doi: 10.1038/nature13418
Buskiewicz, I. A., Montgomery, T., Yasewicz, E. C., Huber, S. A., Murphy, M. P., Hartley, R. C., et al. (2016). Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci. Signal. 9:ra115. doi: 10.1126/scisignal.aaf1933
Camacho, A., Rodriguez-Cuenca, S., Blount, M., Prieur, X., Barbarroja, N., Fuller, M., et al. (2012). Ablation of PGC1 beta prevents mTOR dependent endoplasmic reticulum stress response. Exp. Neurol. 237, 396–406. doi: 10.1016/j.expneurol.2012.06.031
Carroll, R. G., Hollville, E., and Martin, S. J. (2014). Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 9, 1538–1553. doi: 10.1016/j.celrep.2014.10.046
Cereghetti, G. M., Costa, V., and Scorrano, L. (2010). Inhibition of Drp1-dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 17, 1785–1794. doi: 10.1038/cdd.2010.61
Chae, S., Ahn, B. Y., Byun, K., Cho, Y. M., Yu, M. H., Lee, B., et al. (2013). A systems approach for decoding mitochondrial retrograde signaling pathways. Sci. Signal. 6:rs4. doi: 10.1126/scisignal.2003266
Chang, C. R., and Blackstone, C. (2007). Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282, 21583–21587. doi: 10.1074/jbc.C700083200
Chen, L., Willis, S. N., Wei, A., Smith, B. J., Fletcher, J. I., Hinds, M. G., et al. (2005). Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17, 393–403. doi: 10.1016/j.molcel.2004.12.030
Chen, M., Chen, Z., Wang, Y., Tan, Z., Zhu, C., Li, Y., et al. (2016). Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12, 689–702. doi: 10.1080/15548627.2016.1151580
Chen, Y., and Dorn, G. W. II (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Chipuk, J. E., Moldoveanu, T., Llambi, F., Parsons, M. J., and Green, D. R. (2010). The BCL-2 family reunion. Mol. Cell 37, 299–310. doi: 10.1016/j.molcel.2010.01.025
Cho, D. H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., et al. (2009). S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105. doi: 10.1126/science.1171091
Chourasia, A. H., and Macleod, K. F. (2015). Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 11, 1937–1938. doi: 10.1080/15548627.2015.1085136
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. doi: 10.1038/ncb2837
Clerc, P., Ge, S. X., Hwang, H., Waddell, J., Roelofs, B. A., Karbowski, M., et al. (2014). Drp1 is dispensable for apoptotic cytochrome c release in primed MCF10A and fibroblast cells but affects Bcl-2 antagonist-induced respiratory changes. Br. J. Pharmacol. 171, 1988–1999. doi: 10.1111/bph.12515
Cornelissen, T., Haddad, D., Wauters, F., Van Humbeeck, C., Mandemakers, W., Koentjoro, B., et al. (2014). The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 23, 5227–5242. doi: 10.1093/hmg/ddu244
Cribbs, J. T., and Strack, S. (2007). Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944. doi: 10.1038/sj.embor.7401062
Cummins, N., Tweedie, A., Zuryn, S., Bertran-Gonzalez, J., and Gotz, J. (2019). Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 38:e99360. doi: 10.15252/embj.201899360
Cunningham, J. T., Rodgers, J. T., Arlow, D. H., Vazquez, F., Mootha, V. K., and Puigserver, P. (2007). mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450, 736–740. doi: 10.1038/nature06322
Darios, F., Corti, O., Lucking, C. B., Hampe, C., Muriel, M. P., Abbas, N., et al. (2003). Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum. Mol. Genet. 12, 517–526. doi: 10.1093/hmg/ddg044
Delivani, P., Adrain, C., Taylor, R. C., Duriez, P. J., and Martin, S. J. (2006). Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol. Cell 21, 761–773. doi: 10.1016/j.molcel.2006.01.034
Desai, S., Juncker, M., and Kim, C. (2018). Regulation of mitophagy by the ubiquitin pathway in neurodegenerative diseases. Exp. Biol. Med. 243, 554–562. doi: 10.1177/1535370217752351
Dibble, C. C., and Cantley, L. C. (2015). Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25, 545–555. doi: 10.1016/j.tcb.2015.06.002
Diebold, L., and Chandel, N. S. (2016). Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 100, 86–93. doi: 10.1016/j.freeradbiomed.2016.04.198
Diwan, A., Matkovich, S. J., Yuan, Q., Zhao, W., Yatani, A., Brown, J. H., et al. (2009). Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J. Clin. Invest. 119, 203–212. doi: 10.1172/JCI36445
Doerflinger, M., Glab, J. A., and Puthalakath, H. (2015). BH3-only proteins: a 20-year stock-take. FEBS J. 282, 1006–1016. doi: 10.1111/febs.13190
Dorn, G. W. II (2010). Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J. Cardiovasc. Transl. Res. 3, 374–383. doi: 10.1007/s12265-010-9174-x
Drake, L. E., Springer, M. Z., Poole, L. P., Kim, C. J., and Macleod, K. F. (2017). Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 47, 110–124. doi: 10.1016/j.semcancer.2017.04.008
Durcan, T. M., Tang, M. Y., Perusse, J. R., Dashti, E. A., Aguileta, M. A., McLelland, G. L., et al. (2014). USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 33, 2473–2491. doi: 10.15252/embj.201489729
Ebrahimi-Fakhari, D., Saffari, A., Wahlster, L., Di Nardo, A., Turner, D., and Lewis, T. L. Jr., et al. (2016). Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. 17, 1053–1070. doi: 10.1016/j.celrep.2016.09.054
Edlich, F., Banerjee, S., Suzuki, M., Cleland, M. M., Arnoult, D., Wang, C., et al. (2011). Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145, 104–116. doi: 10.1016/j.cell.2011.02.034
Farre, J. C., Krick, R., Subramani, S., and Thumm, M. (2009). Turnover of organelles by autophagy in yeast. Curr. Opin. Cell Biol. 21, 522–530. doi: 10.1016/j.ceb.2009.04.015
Frank, S., Gaume, B., Bergmann-Leitner, E. S., Leitner, W. W., Robert, E. G., Catez, F., et al. (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1, 515–525. doi: 10.1016/s1534-5807(01)00055-7
Friedman, J. R., Lackner, L. L., West, M., DiBenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., et al. (2009). PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 33, 627–638. doi: 10.1016/j.molcel.2009.02.013
Garcia, D., and Shaw, R. J. (2017). AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800. doi: 10.1016/j.molcel.2017.05.032
Gautier, C. A., Kitada, T., and Shen, J. (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 105, 11364–11369. doi: 10.1073/pnas.0802076105
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H., and Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi: 10.1093/hmg/ddq419
Gong, G., Song, M., Csordas, G., Kelly, D. P., Matkovich, S. J., and Dorn, G. W. II (2015). Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350:aad2459. doi: 10.1126/science.aad2459
Gottlieb, R. A., and Carreira, R. S. (2010). Autophagy in health and disease. 5. Mitophagy as a way of life. Am. J. Physiol. Cell Physiol. 299, C203–C210. doi: 10.1152/ajpcell.00097.2010
Griparic, L., Kanazawa, T., and van der Bliek, A. M. (2007). Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 178, 757–764. doi: 10.1083/jcb.200704112
Guha, M., Fang, J. K., Monks, R., Birnbaum, M. J., and Avadhani, N. G. (2010). Activation of Akt is essential for the propagation of mitochondrial respiratory stress signaling and activation of the transcriptional coactivator heterogeneous ribonucleoprotein A2. Mol. Biol. Cell 21, 3578–3589. doi: 10.1091/mbc.E10-03-0192
Hanna, R. A., Quinsay, M. N., Orogo, A. M., Giang, K., Rikka, S., and Gustafsson, A. B. (2012). Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 287, 19094–19104. doi: 10.1074/jbc.M111.322933
He, X., Zhu, Y., Zhang, Y., Geng, Y., Gong, J., Geng, J., et al. (2019). RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. EMBO J. 38:e100978. doi: 10.15252/embj.2018100978
Head, B., Griparic, L., Amiri, M., Gandre-Babbe, S., and van der Bliek, A. M. (2009). Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 187, 959–966. doi: 10.1083/jcb.200906083
Hickson-Bick, D. L., Jones, C., and Buja, L. M. (2008). Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J. Mol. Cell Cardiol. 44, 411–418. doi: 10.1016/j.yjmcc.2007.10.013
Hollville, E., Carroll, R. G., Cullen, S. P., and Martin, S. J. (2014). Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol. Cell 55, 451–466. doi: 10.1016/j.molcel.2014.06.001
Hunter, D. R., and Haworth, R. A. (1979). The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch. Biochem. Biophys. 195, 468–477. doi: 10.1016/0003-9861(79)90373-4
Imazu, T., Shimizu, S., Tagami, S., Matsushima, M., Nakamura, Y., Miki, T., et al. (1999). Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene 18, 4523–4529. doi: 10.1038/sj.onc.1202722
Ip, W. K. E., Hoshi, N., Shouval, D. S., Snapper, S., and Medzhitov, R. (2017). Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519. doi: 10.1126/science.aal3535
Johnson, B. N., Berger, A. K., Cortese, G. P., and Lavoie, M. J. (2012). The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc. Natl. Acad. Sci. U.S.A. 109, 6283–6288. doi: 10.1073/pnas.1113248109
Jung, J., Zhang, Y., Celiku, O., Zhang, W., Song, H., Williams, B. J., et al. (2019). Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res. 79, 5218–5232. doi: 10.1158/0008-5472.CAN-19-0198
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kanki, T., Kurihara, Y., Jin, X., Goda, T., Ono, Y., Aihara, M., et al. (2013). Casein kinase 2 is essential for mitophagy. EMBO Rep. 14, 788–794. doi: 10.1038/embor.2013.114
Kanki, T., Wang, K., Cao, Y., Baba, M., and Klionsky, D. J. (2009). Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell 17, 98–109. doi: 10.1016/j.devcel.2009.06.014
Karbowski, M., Lee, Y. J., Gaume, B., Jeong, S. Y., Frank, S., Nechushtan, A., et al. (2002). Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 159, 931–938. doi: 10.1083/jcb.200209124
Karbowski, M., Neutzner, A., and Youle, R. J. (2007). The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 178, 71–84. doi: 10.1083/jcb.200611064
Karbowski, M., Norris, K. L., Cleland, M. M., Jeong, S. Y., and Youle, R. J. (2006). Role of Bax and Bak in mitochondrial morphogenesis. Nature 443, 658–662. doi: 10.1038/nature05111
Kazlauskaite, A., Martinez-Torres, R. J., Wilkie, S., Kumar, A., Peltier, J., Gonzalez, A., et al. (2015). Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep. 16, 939–954. doi: 10.15252/embr.201540352
Kerr, J. S., Adriaanse, B. A., Greig, N. H., Mattson, M. P., Cader, M. Z., Bohr, V. A., et al. (2017). Mitophagy and Alzheimer’s Disease: cellular and Molecular Mechanisms. Trends Neurosci. 40, 151–166. doi: 10.1016/j.tins.2017.01.002
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kissova, I., Salin, B., Schaeffer, J., Bhatia, S., Manon, S., and Camougrand, N. (2007). Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 3, 329–336. doi: 10.4161/auto.4034
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Kuang, Y., Ma, K., Zhou, C., Ding, P., Zhu, Y., Chen, Q., et al. (2016). Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy 12, 2363–2373. doi: 10.1080/15548627.2016.1238552
Kubli, D. A., Zhang, X., Lee, Y., Hanna, R. A., Quinsay, M. N., Nguyen, C. K., et al. (2013). Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 288, 915–926. doi: 10.1074/jbc.M112.411363
LaGory, E. L., Wu, C., Taniguchi, C. M., Ding, C. C., Chi, J. T., von Eyben, R., et al. (2015). Suppression of PGC-1alpha is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep. 12, 116–127. doi: 10.1016/j.celrep.2015.06.006
Laker, R. C., Drake, J. C., Wilson, R. J., Lira, V. A., Lewellen, B. M., Ryall, K. A., et al. (2017). Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 8:548. doi: 10.1038/s41467-017-00520-9
Lampert, M. A., Orogo, A. M., Najor, R. H., Hammerling, B. C., Leon, L. J., Wang, B. J., et al. (2019). BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 15, 1182–1198. doi: 10.1080/15548627.2019.1580095
Landes, T., Emorine, L. J., Courilleau, D., Rojo, M., Belenguer, P., and Arnaune-Pelloquin, L. (2010). The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 11, 459–465. doi: 10.1038/embor.2010.50
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Leboucher, G. P., Tsai, Y. C., Yang, M., Shaw, K. C., Zhou, M., Veenstra, T. D., et al. (2012). Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 47, 547–557. doi: 10.1016/j.molcel.2012.05.041
Lee, J. E., Westrate, L. M., Wu, H., Page, C., and Voeltz, G. K. (2016). Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540, 139–143. doi: 10.1038/nature20555
Lee, J. J., Sanchez-Martinez, A., Zarate, A. M., Beninca, C., Mayor, U., Clague, M. J., et al. (2018). Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell Biol. 217, 1613–1622. doi: 10.1083/jcb.201801044
Lee, Y., Lee, H. Y., Hanna, R. A., and Gustafsson, A. B. (2011). Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 301, H1924–H1931. doi: 10.1152/ajpheart.00368.2011
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5. doi: 10.1089/rej.2005.8.3
Lewis, T. L. Jr., Kwon, S. K., Lee, A., Shaw, R., and Polleux, F. (2018). MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 9:5008. doi: 10.1038/s41467-018-07416-2
Li, H., Alavian, K. N., Lazrove, E., Mehta, N., Jones, A., Zhang, P., et al. (2013). A Bcl-xL-Drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat. Cell Biol. 15, 773–785. doi: 10.1038/ncb2791
Li, H., Chen, Y., Jones, A. F., Sanger, R. H., Collis, L. P., Flannery, R., et al. (2008). Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 2169–2174. doi: 10.1073/pnas.0711647105
Li, W., Li, Y., Siraj, S., Jin, H., Fan, Y., Yang, X., et al. (2019). FUN14 domain-containing 1-mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatology 69, 604–621. doi: 10.1002/hep.30191
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Liu, Q., Zhang, D., Hu, D., Zhou, X., and Zhou, Y. (2018). The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 103, 115–124. doi: 10.1016/j.molimm.2018.09.010
Liu, Q. A., and Shio, H. (2008). Mitochondrial morphogenesis, dendrite development, and synapse formation in cerebellum require both Bcl-w and the glutamate receptor delta2. PLoS Genet. 4:e1000097. doi: 10.1371/journal.pgen.1000097
Liu, X., Zhu, Y., Dai, S., White, J., Peyerl, F., Kappler, J. W., et al. (2006). Bcl-xl does not have to bind Bax to protect T cells from death. J. Exp. Med. 203, 2953–2961. doi: 10.1084/jem.20061151
Lu, W., Karuppagounder, S. S., Springer, D. A., Allen, M. D., Zheng, L., Chao, B., et al. (2014). Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson’s-like movement disorder. Nat. Commun. 5:4930. doi: 10.1038/ncomms5930
Ludtmann, M. H. R., and Abramov, A. Y. (2018). Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci. Lett. 663, 86–90. doi: 10.1016/j.neulet.2017.08.044
Ma, B., Cheng, H., Mu, C., Geng, G., Zhao, T., Luo, Q., et al. (2019). The SIAH2-NRF1 axis spatially regulates tumor microenvironment remodeling for tumor progression. Nat. Commun. 10“1034. doi: 10.1038/s41467-019-08618-y
Ma, K., Zhang, Z., Chang, R., Cheng, H., Mu, C., Zhao, T., et al. (2019). Dynamic PGAM5 multimers dephosphorylate BCL-xL or FUNDC1 to regulate mitochondrial and cellular fate. Cell Death Differ. 27, 1036–1051. doi: 10.1038/s41418-019-0396-4
Marsboom, G., Toth, P. T., Ryan, J. J., Hong, Z., Wu, X., Fang, Y. H., et al. (2012). Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 110, 1484–1497. doi: 10.1161/CIRCRESAHA.111.263848
McArthur, K., Whitehead, L. W., Heddleston, J. M., Li, L., Padman, B. S., Oorschot, V., et al. (2018). BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359: eaao6047. doi: 10.1126/science.aao6047
McLelland, G. L., Soubannier, V., Chen, C. X., McBride, H. M., and Fon, E. A. (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295. doi: 10.1002/embj.201385902
McWilliams, T. G., Prescott, A. R., Allen, G. F. G., Tamjar, J., Munson, M. J., Thomson, C., et al. (2016). mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 214, 333–345. doi: 10.1083/jcb.201603039
McWilliams, T. G., Prescott, A. R., Montava-Garriga, L., Ball, G., Singh, F., Barini, E., et al. (2018). Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 27, 439.e5–449.e5.
Meyer, N., Zielke, S., Michaelis, J. B., Linder, B., Warnsmann, V., Rakel, S., et al. (2018). AT 101 induces early mitochondrial dysfunction and HMOX1 (heme oxygenase 1) to trigger mitophagic cell death in glioma cells. Autophagy 14, 1693–1709. doi: 10.1080/15548627.2018.1476812
Mishra, P., and Chan, D. C. (2014). Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 15, 634–646. doi: 10.1038/nrm3877
Miwa, S., Lawless, C., and von Zglinicki, T. (2008). Mitochondrial turnover in liver is fast in vivo and is accelerated by dietary restriction: application of a simple dynamic model. Aging Cell 7, 920–923. doi: 10.1111/j.1474-9726.2008.00426.x
Mizumura, K., Cloonan, S. M., Nakahira, K., Bhashyam, A. R., Cervo, M., Kitada, T., et al. (2014). Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Invest. 124, 3987–4003. doi: 10.1172/JCI74985
Molina, P., Lim, Y., and Boyd, L. (2019). Ubiquitination is required for the initial removal of paternal organelles in C. elegans. Dev. Biol. 453, 168–179. doi: 10.1016/j.ydbio.2019.05.015
Montessuit, S., Somasekharan, S. P., Terrones, O., Lucken-Ardjomande, S., Herzig, S., Schwarzenbacher, R., et al. (2010). Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142, 889–901. doi: 10.1016/j.cell.2010.08.017
Morita, M., Gravel, S. P., Hulea, L., Larsson, O., Pollak, M., St-Pierre, J., et al. (2015). mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 14, 473–480. doi: 10.4161/15384101.2014.991572
Murakawa, T., Yamaguchi, O., Hashimoto, A., Hikoso, S., Takeda, T., Oka, T., et al. (2015). Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 6:7527. doi: 10.1038/ncomms8527
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
Niu, K., Fang, H., Chen, Z., Zhu, Y., Tan, Q., Wei, D., et al. (2019). USP33 deubiquitinates PRKN/parkin and antagonizes its role in mitophagy. Autophagy 16, 724–734. doi: 10.1080/15548627.2019.1656957
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
Okamoto, K., Kondo-Okamoto, N., and Ohsumi, Y. (2009). Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 17, 87–97. doi: 10.1016/j.devcel.2009.06.013
Otera, H., Miyata, N., Kuge, O., and Mihara, K. (2016). Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J. Cell Biol. 212, 531–544. doi: 10.1083/jcb.201508099
Otsu, K., Murakawa, T., and Yamaguchi, O. (2015). BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy 11, 1932–1933. doi: 10.1080/15548627.2015.1084459
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Park, H., Chung, K. M., An, H. K., Gim, J. E., Hong, J., Woo, H., et al. (2019). Parkin promotes mitophagic cell death in adult hippocampal neural stem cells following insulin withdrawal. Front. Mol. Neurosci. 12:46. doi: 10.3389/fnmol.2019.00046
Park, Y. Y., Nguyen, O. T., Kang, H., and Cho, H. (2014). MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 5:e1172. doi: 10.1038/cddis.2014.142
Parone, P. A., James, D. I., Da Cruz, S., Mattenberger, Y., Donze, O., Barja, F., et al. (2006). Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol. Cell Biol. 26, 7397–7408. doi: 10.1128/MCB.02282-05
Pathak, T., and Trebak, M. (2018). Mitochondrial Ca(2+) signaling. Pharmacol. Ther. 192, 112–123. doi: 10.1016/j.pharmthera.2018.07.001
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. doi: 10.1016/j.cell.2005.07.002
Paul, B. T., Manz, D. H., Torti, F. M., and Torti, S. V. (2017). Mitochondria and Iron: current questions. Expert Rev. Hematol. 10, 65–79. doi: 10.1080/17474086.2016.1268047
Pinto, M., Nissanka, N., and Moraes, C. T. (2018). Lack of parkin anticipates the phenotype and affects mitochondrial morphology and mtDNA Levels in a mouse model of Parkinsons disease. J. Neurosci. 38, 1042–1053. doi: 10.1523/JNEUROSCI.1384-17.2017
Pyakurel, A., Savoia, C., Hess, D., and Scorrano, L. (2015). Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell 58, 244–254. doi: 10.1016/j.molcel.2015.02.021
Quinsay, M. N., Thomas, R. L., Lee, Y., and Gustafsson, A. B. (2010). Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 6, 855–862. doi: 10.4161/auto.6.7.13005
Rasbach, K. A., and Schnellmann, R. G. (2007). Signaling of mitochondrial biogenesis following oxidant injury. J. Biol. Chem. 282, 2355–2362. doi: 10.1074/jbc.M608009200
Rera, M., Bahadorani, S., Cho, J., Koehler, C. L., Ulgherait, M., Hur, J. H., et al. (2011). Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab. 14, 623–634. doi: 10.1016/j.cmet.2011.09.013
Rogov, V. V., Suzuki, H., Marinkovic, M., Lang, V., Kato, R., Kawasaki, M., et al. (2017). Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 7:1131. doi: 10.1038/s41598-017-01258-6
Rojansky, R., Cha, M. Y., and Chan, D. C. (2016). Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. eLife 5:e17896. doi: 10.7554/eLife.17896
Rolland, S. G., and Conradt, B. (2010). New role of the BCL2 family of proteins in the regulation of mitochondrial dynamics. Curr. Opin. Cell Biol. 22, 852–858. doi: 10.1016/j.ceb.2010.07.014
Ryu, D., Mouchiroud, L., Andreux, P. A., Katsyuba, E., Moullan, N., Nicolet-Dit-Felix, A. A., et al. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22, 879–888. doi: 10.1038/nm.4132
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Sekine, S., Yao, A., Hattori, K., Sugawara, S., Naguro, I., Koike, M., et al. (2016). The ablation of mitochondrial protein phosphatase pgam5 confers resistance against metabolic stress. EBioMedicine 5, 82–92. doi: 10.1016/j.ebiom.2016.01.031
Sekine, S., and Youle, R. J. (2018). PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 16:2. doi: 10.1186/s12915-017-0470-7
Sentelle, R. D., Senkal, C. E., Jiang, W., Ponnusamy, S., Gencer, S., Selvam, S. P., et al. (2012). Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 8, 831–838. doi: 10.1038/nchembio.1059
Seth, R. B., Sun, L., Ea, C. K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. doi: 10.1016/j.cell.2005.08.012
Sheridan, C., Delivani, P., Cullen, S. P., and Martin, S. J. (2008). Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol. Cell 31, 570–585. doi: 10.1016/j.molcel.2008.08.002
Sinha, K., Das, J., Pal, P. B., and Sil, P. C. (2013). Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 87, 1157–1180. doi: 10.1007/s00204-013-1034-4
Song, M., Mihara, K., Chen, Y., Scorrano, L., and Dorn, G. W. II (2015). Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 21, 273–286. doi: 10.1016/j.cmet.2014.12.011
Soubannier, V., McLelland, G. L., Zunino, R., Braschi, E., Rippstein, P., Fon, E. A., et al. (2012). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22, 135–141. doi: 10.1016/j.cub.2011.11.057
Speir, M., Lawlor, K. E., Glaser, S. P., Abraham, G., Chow, S., Vogrin, A., et al. (2016). Eliminating Legionella by inhibiting BCL-XL to induce macrophage apoptosis. Nat. Microbiol. 1:15034. doi: 10.1038/nmicrobiol.2015.34
Stevens, D. A., Lee, Y., Kang, H. C., Lee, B. D., Lee, Y. I., Bower, A., et al. (2015). Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. U.S.A. 112, 11696–11701. doi: 10.1073/pnas.1500624112
Strappazzon, F., Nazio, F., Corrado, M., Cianfanelli, V., Romagnoli, A., Fimia, G. M., et al. (2015). AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 22, 419–432. doi: 10.1038/cdd.2014.139
Sugiura, A., Nagashima, S., Tokuyama, T., Amo, T., Matsuki, Y., Ishido, S., et al. (2013). MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol. Cell 51, 20–34. doi: 10.1016/j.molcel.2013.04.023
Sun, X., Sun, L., Zhao, Y., Li, Y., Lin, W., Chen, D., et al. (2016). MAVS maintains mitochondrial homeostasis via autophagy. Cell Discov. 2:16024. doi: 10.1038/celldisc.2016.24
Sutovsky, P., Moreno, R. D., Ramalho-Santos, J., Dominko, T., Simerly, C., and Schatten, G. (1999). Ubiquitin tag for sperm mitochondria. Nature 402, 371–372. doi: 10.1038/46466
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., and Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529. doi: 10.1074/jbc.M607279200
Tait, S. W., and Green, D. R. (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632. doi: 10.1038/nrm2952
Tal, R., Winter, G., Ecker, N., Klionsky, D. J., and Abeliovich, H. (2007). Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J. Biol. Chem. 282, 5617–5624. doi: 10.1074/jbc.M605940200
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D. F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Tian, W., Li, W., Chen, Y., Yan, Z., Huang, X., Zhuang, H., et al. (2015). Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 589, 1847–1854. doi: 10.1016/j.febslet.2015.05.020
Toyama, E. Q., Herzig, S., Courchet, J., Lewis, T. L. Jr., Loson, O. C., Hellberg, K., et al. (2016). Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. doi: 10.1126/science.aab4138
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Vervliet, T., Parys, J. B., and Bultynck, G. (2016). Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene 35, 5079–5092. doi: 10.1038/onc.2016.31
von Bernhardi, R., Eugenin-von Bernhardi, L., and Eugenin, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
Vyas, S., Zaganjor, E., and Haigis, M. C. (2016). Mitochondria and Cancer. Cell 166, 555–566. doi: 10.1016/j.cell.2016.07.002
Wang, H., Song, P., Du, L., Tian, W., Yue, W., Liu, M., et al. (2011). Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 286, 11649–11658. doi: 10.1074/jbc.M110.144238
Wang, K., Jin, M., Liu, X., and Klionsky, D. J. (2013). Proteolytic processing of Atg32 by the mitochondrial i-AAA protease Yme1 regulates mitophagy. Autophagy 9, 1828–1836. doi: 10.4161/auto.26281
Wang, L., Cho, Y. L., Tang, Y., Wang, J., Park, J. E., Wu, Y., et al. (2018). PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 28, 787–802. doi: 10.1038/s41422-018-0056-0
Wang, Y., Serricchio, M., Jauregui, M., Shanbhag, R., Stoltz, T., Di Paolo, C. T., et al. (2015). Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 11, 595–606. doi: 10.1080/15548627.2015.1034408
Wang, Z., Jiang, H., Chen, S., Du, F., and Wang, X. (2012). The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243. doi: 10.1016/j.cell.2011.11.030
Wei, Y., Chiang, W. C., Sumpter, R. Jr., Mishra, P., and Levine, B. (2017). Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168, 224.e10–238.e10. doi: 10.1016/j.cell.2016.11.042
Wei, Y., Pattingre, S., Sinha, S., Bassik, M., and Levine, B. (2008a). JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678–688. doi: 10.1016/j.molcel.2008.06.001
Wei, Y., Sinha, S., and Levine, B. (2008b). Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4, 949–951. doi: 10.4161/auto.6788
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., Lang, S. M., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. doi: 10.1038/nature14156
Whitworth, A. J., and Pallanck, L. J. (2017). PINK1/Parkin mitophagy and neurodegeneration-what do we really know in vivo? Curr. Opin. Genet. Dev. 44, 47–53. doi: 10.1016/j.gde.2017.01.016
Wilhelmus, M. M., van der Pol, S. M., Jansen, Q., Witte, M. E., van der Valk, P., Rozemuller, A. J., et al. (2011). Association of Parkinson disease-related protein PINK1 with Alzheimer disease and multiple sclerosis brain lesions. Free Radic. Biol. Med. 50, 469–476. doi: 10.1016/j.freeradbiomed.2010.11.033
Witte, M. E., Bol, J. G., Gerritsen, W. H., van der Valk, P., Drukarch, B., van Horssen, J., et al. (2009). Parkinson’s disease-associated parkin colocalizes with Alzheimer’s disease and multiple sclerosis brain lesions. Neurobiol. Dis. 36, 445–452. doi: 10.1016/j.nbd.2009.08.009
Wong, Y. C., and Holzbaur, E. L. (2014). Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. U.S.A. 111, E4439–E4448. doi: 10.1073/pnas.1405752111
Wu, H., Xue, D., Chen, G., Han, Z., Huang, L., Zhu, C., et al. (2014). The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy 10, 1712–1725. doi: 10.4161/auto.29568
Xian, H., Yang, Q., Xiao, L., Shen, H. M., and Liou, Y. C. (2019). STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun. 10:2059. doi: 10.1038/s41467-019-10096-1
Yamashita, S. I., Jin, X., Furukawa, K., Hamasaki, M., Nezu, A., Otera, H., et al. (2016). Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J. Cell Biol. 215, 649–665. doi: 10.1083/jcb.201605093
Ye, X., Sun, X., Starovoytov, V., and Cai, Q. (2015). Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 24, 2938–2951. doi: 10.1093/hmg/ddv056
Yu, J., Nagasu, H., Murakami, T., Hoang, H., Broderick, L., Hoffman, H. M., et al. (2014). Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. U.S.A. 111, 15514–15519. doi: 10.1073/pnas.1414859111
Yue, W., Chen, Z., Liu, H., Yan, C., Chen, M., Feng, D., et al. (2014). A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 24, 482–496. doi: 10.1038/cr.2014.20
Yussman, M. G., Toyokawa, T., Odley, A., Lynch, R. A., Wu, G., Colbert, M. C., et al. (2002). Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat. Med. 8, 725–730. doi: 10.1038/nm719
Zhang, N. P., Liu, X. J., Xie, L., Shen, X. Z., and Wu, J. (2019). Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab Invest 99, 749–763. doi: 10.1038/s41374-018-0177-6
Zhang, Q., Raoof, M., Chen, Y., Sumi, Y., Sursal, T., Junger, W., et al. (2010). Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107. doi: 10.1038/nature08780
Zhang, W., Ren, H., Xu, C., Zhu, C., Wu, H., Liu, D., et al. (2016). Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. eLife 5:e21407. doi: 10.7554/eLife.21407
Zhang, Y., and Xu, H. (2016). Translational regulation of mitochondrial biogenesis. Biochem. Soc. Trans. 44, 1717–1724. doi: 10.1042/BST20160071C
Zhang, Y., Yao, Y., Qiu, X., Wang, G., Hu, Z., Chen, S., et al. (2019). Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 20, 433–446. doi: 10.1038/s41590-019-0324-2
Zhong, Z., Umemura, A., Sanchez-Lopez, E., Liang, S., Shalapour, S., Wong, J., et al. (2016). NF-kappaB Restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910. doi: 10.1016/j.cell.2015.12.057
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. doi: 10.1038/nature09663
Zhu, Y., Massen, S., Terenzio, M., Lang, V., Chen-Lindner, S., Eils, R., et al. (2013). Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 288, 1099–1113. doi: 10.1074/jbc.M112.399345
Zong, W. X., Rabinowitz, J. D., and White, E. (2016). Mitochondria and Cancer. Mol. Cell 61, 667–676. doi: 10.1016/j.molcel.2016.02.011
Keywords: mitophagy, mitochondrial dynamics, mitochondrial apoptosis, cell fate, mitophagy receptors
Citation: Ma K, Chen G, Li W, Kepp O, Zhu Y and Chen Q (2020) Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 8:467. doi: 10.3389/fcell.2020.00467
Received: 01 February 2020; Accepted: 20 May 2020;
Published: 24 June 2020.
Edited by:
Yi Zhang, First Affiliated Hospital of Zhengzhou University, ChinaReviewed by:
Yansheng Feng, The University of Texas Health Science Center at San Antonio, United StatesAmalia M. Dolga, University of Groningen, Netherlands
Copyright © 2020 Ma, Chen, Li, Kepp, Zhu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yushan Zhu, zhuys@nankai.edu.cn; Quan Chen, chenq@ioz.ac.cn; chenq@nankai.edu.cn
†These authors have contributed equally to this work