 Pieter Ruytinx
Pieter Ruytinx Paul Proost
Paul Proost Jo Van Damme
Jo Van Damme Sofie Struyf
Sofie Struyf- Laboratory of Molecular Immunology, Department of Microbiology and Immunology, REGA Institute KU Leuven, Leuven, Belgium
Macrophages represent a heterogeneous cell population and are known to display a remarkable plasticity. In response to distinct micro-environmental stimuli, e.g., tumor stroma vs. infected tissue, they polarize into different cell subtypes. Originally, two subpopulations were defined: classically activated macrophages or M1, and alternatively activated macrophages or M2. Nowadays, the M1/M2 classification is considered as an oversimplified approach that does not adequately cover the total spectrum of macrophage phenotypes observed in vivo. Especially in pathological circumstances, macrophages behave as plastic cells modifying their expression and transcription profile along a continuous spectrum with M1 and M2 phenotypes as extremes. Here, we focus on the effect of chemokines on macrophage differentiation and polarization in physiological and pathological conditions. In particular, we discuss chemokine-induced macrophage polarization in inflammatory diseases, including obesity, cancer, and atherosclerosis.
Introduction
Monocytes arise in the bone marrow from hematopoietic stem cells (HSCs) and develop through a series of sequential differentiation stages. Common myeloid progenitor cells develop into granulocyte/macrophage colony forming units (GM-CFU), which in turn can commit to the macrophage colony-forming unit (M-CFU) or the granulocyte colony-forming unit (G-CFU). The M-CFU differentiates sequentially into monoblasts and promonocytes, which leave the bone marrow and enter the bloodstream, where they differentiate into mature monocytes (1). Mature monocytes represent about 10% of the leukocyte population in human peripheral blood and can circulate in the blood stream for up to 1–2 days before they undergo apoptosis. Alternatively, monocytes can migrate into the tissues and differentiate into specific macrophages (2). The major driver for the homeostatic control of monocyte/macrophage development is macrophage colony-stimulating factor (M-CSF), present in the blood circulation and produced by stromal cells in tissues (3–5). In inflammatory conditions, also other cytokines such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and the chemokine CXCL4 influence the differentiation and/or survival of mononuclear phagocytes (6–8).
In contrast to the classical model of macrophage development, where macrophages differentiate from circulating monocytes as described above, recent studies provided evidence that tissue-resident macrophages arise from yolk sac or fetal liver-derived progenitors (9). These tissue resident macrophages appear to have stem cell-like capacities as they persist independently of monocytes by self-renewal in situ (10). One of the major hallmarks of macrophages is their heterogeneity, which is reflected by their specialized function in a particular microenvironment. According to their tissue location, macrophages can take different names including microglia [central nervous system (CNS)], Kupffer cells (liver), alveolar macrophages (lung), osteoclasts (bone), histiocytes (spleen and connective tissue), Langerhans cells (skin), and tissue macrophages in the gut (11). Resident macrophages promote tissue homeostasis, whereas monocyte-derived macrophages primarily assist in host-defense. Moreover, macrophages recruited during and after embryogenesis co-exist in different organs (10, 12).
Besides their heterogeneity, macrophages are known to display remarkable plasticity. In response to different micro-environmental stimuli, a fully differentiated macrophage can adopt a polarized phenotype with specific functional characteristics. Traditionally, macrophages are subdivided into two subpopulations: the classically activated or M1 macrophages and the alternatively activated or M2 macrophages (13). M1 macrophages can be induced by the Th1 cytokines tumor necrosis factor (TNF)-α, interferon (IFN)-γ and bacterial components such as lipopolysaccharide (LPS). Activated M1 macrophages phagocytose and destroy microbes, eliminate tumor cells and present antigens to T cells to evoke an adaptive immune response. As such, they play an important role in protection against pathogens. The pro-inflammatory phenotype is characterized by the increased production of reactive nitrogen intermediates (RNI) and reactive oxygen species (ROS), which is essential for bacterial killing (14). In response to inflammatory mediators, M1 macrophages express the inducible nitric oxide synthase (iNOS), which uses L-arginine as a substrate to produce nitric oxide (NO) (15). Furthermore, classically activated macrophages release high levels of pro-inflammatory cytokines such as TNF-α, interleukin-6 (IL-6) and IL-1β to deal with infections and thereby promote Th1 responses (16).
M2 activation occurs in response to stimulation with IL-4, IL-10, and IL-13. These macrophages display high surface levels of scavenger, mannose and galactose type receptors involved in debris clearance. Furthermore, they show a more immunosuppressive phenotype characterized by decreased antigen presentation to T cells and production of cytokines that stimulate a Th2 response. In contrast to M1 macrophages, M2 macrophages constitutively express the enzyme Arginase 1 (ARG1), which hydrolyzes L-arginine to L-ornithine (13). L-ornithine is the main precursor for polyamines, essential for cell survival. Furthermore, L-ornithine can also be used as a building block to make proline and hydroxyproline, essential amino acids for the production of collagen, a crucial protein in tissue damage repair (17). As such, these macrophages are involved in long-term tissue repair, promote tumor growth and exert antiparasitic effects (18).
Nowadays the M1/M2 classification is considered as an oversimplified approach that does not fully cover the total spectrum of in vivo macrophage phenotypes. Especially, in pathological circumstances macrophages behave as plastic cells modifying in space and time their expression and transcription profile along a continuous spectrum, having M1 and M2 macrophage phenotypes as extremes (19, 20).
The interaction of chemokine receptors on circulating cells with their ligands enables the selective tissue-specific recruitment of subsets of circulating cells such as monocytes. Chemokines are a family of low molecular weight, secreted proteins with a prominent role in leukocyte activation and chemotaxis. Based on the NH2-terminal motif of two conserved cysteine residues, chemokines can be classified into 4 subfamilies: C, CC, CXC, and CX3C chemokines. Chemokines signal via G protein-coupled receptors (GPCRs), which are named XCR, CCR, CXCR, CX3CR according to the chemokine nomenclature (21). Additionally, chemokines can bind with high affinity to atypical chemokine receptors (ACKRs), a subgroup of seven-transmembrane receptors highly related to the classical GPCRs. Since these ACKRs lack or have a modified canonical DRYLAIV motif, activation of ACKRs does not lead to typical GPCR-mediated signaling and chemotactic functions (22).
The Effect of Chemokines on Macrophage Differentiation and Polarization in Physiological and Pathological Conditions
Neurological Diseases
Microglia, the resident, long-living macrophages in the central nervous system (CNS), act as the major inflammatory cell type in the brain and similar to peripheral macrophages they respond to pathogens and injury (23). Under physiological conditions, microglia are in a “quiescent” state or have a non-activated phenotype (24). Butofsky et al. demonstrated that this “resting” cell' phenotype is different from M1 or M2 microglia and expresses genes associated with neuronal development (25). This particular phenotype was found to be important for synaptic growth, maintenance, and neuronal growth. Furthermore, the “quiescent” state enables the intimate connection between neurons and microglial cells, which is tightly controlled by the CX3CL1-CX3CR1 axis (26). CX3CL1/fractalkine is the only member of the CX3C chemokine subfamily and differs from most other chemokines, as it can exist as a membrane-associated molecule with the chemokine motif being attached to a long mucin stalk. Alternatively, CX3CL1 is secreted as a soluble variant (27). CX3CL1 is expressed on healthy neurons, whereas the transmembrane protein receptor CX3CR1 is present on microglia (23, 28, 29).
The CX3CL1-CX3CR1 axis is an important neuroimmune interaction in the CNS and has been implicated in many neurophysiological and neuropathological conditions (Figure 1). For instance, in animal models of Parkinson's disease and amyotrophic lateral sclerosis (ALS), loss of CX3CR1 increased neuronal cell death (30). Using a murine model of diabetic retinopathy, Cardona et al. showed that in the absence of CX3CR1 the microglial response is dysregulated and associated with increased IL-1β cytokine release (Figure 1B) (31). Additionally, Mattison et al. found that CX3CL1 suppressed the release of pro-inflammatory and neurotoxic factors such as TNF-α and NO in activated microglia during neuroinflammation (Figure 1B) (32). Controlling neuroinflammation via CX3CR1 signaling was particularly beneficial in the pathogenesis of Alzheimer's disease (33). Furthermore, CX3CL1 promotes microglial phagocytosis of neuronal debris and increases the expression of heme oxygenase 1 (HMOX1), resulting in an anti-oxidant effect, which indirectly promotes neuronal survival (34). Conversely, some studies showed a neurotoxic role for CX3CL1 in CX3CR1−/− mice models for Alzheimer's disease (35) and stroke (36). Fuhrmann et al. also reported that neuronal loss neuronal loss in a model of Alzheimer's disease was prevented in CX3CR1 knock out mice (37).
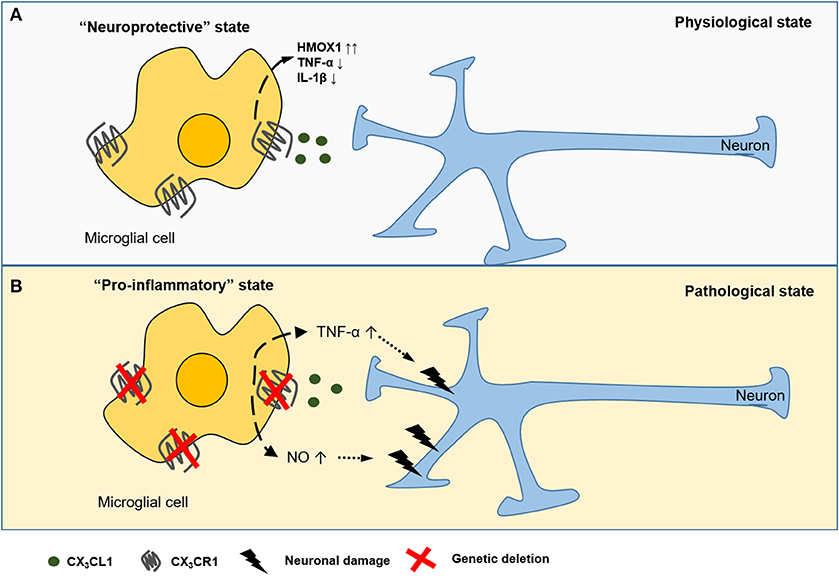
Figure 1. CX3CL1-CX3CR1 interaction between neurons and microglial cells in the CNS. CX3CL1 is released from the neurons and interacts with the CX3CR1 receptor expressed on CNS microglia. CX3CL1 signaling induces (dashed arrow) a neuroprotective state (A), characterized by the suppressed release of pro-inflammatory cytokines (TNF-α, IL-1β) and upregulation of heme oxygenese 1 (HMOX1). In several murine models of neurodegenerative diseases, genetic deficiency of CX3CR1 is associated with potentially detrimental secretion of pro-inflammatory cytokines and reactive nitrogen species (NO) causing (dotted arrow) neurotoxicity (B).
The atypical chemokine receptor CCRL2 was identified as an important regulator of microglial activation and polarization in experimental autoimmune encephalomyelitis (EAE) (38). Similar to ACKRs, CCRL2 lacks conventional GPCR signaling and chemotactic activity (39). More specifically, it was found that during the chronic disease phase microglia in CCRL2 KO mice develop a profound M1 phenotype compared to wild type (WT) mice after induction of EAE (38). These results highlight a potential role of CCRL2 in EAE-associated inflammatory responses and as such, provide a new potential target to control neuroinflammation.
Finally, using a neuron/microglia co-culture system, Yang et al. found that CCL2/MCP-1 (40) was able to activate microglia and stimulated production of pro-inflammatory cytokines such as TNF-α and IL-1β (41).
Fibrosis
Upon infection, activated macrophages use a set of innate immune defense strategies such as phagocytosis, release of proteases and production of antimicrobial mediators, such as reactive oxygen and nitrogen species. An important side effect of this efficient inflammatory response is partial tissue destruction, which is normally followed by a repair response to regenerate the tissue (42). However, when this repair phase is persistent, it leads to fibrosis or so-called scarring of the tissue, which is defined by the accumulation of excess extracellular matrix components. In the end, this causes progressive loss of function of the affected organ(s) (43, 44). Alternatively activated (M2) macrophages are known to play an important role in wound healing and acquire a pro-fibrotic phenotype (45, 46). Since this phenotype is observed during the peak of the fibrotic immune response, it is suggested that such M2 macrophages are important inducers and regulators of fibrosis (44). For instance, by producing transforming growth factor-β1 (TGF-β1), M2 macrophages directly stimulate collagen production in myofibroblasts (47, 48) and enhance the expression of tissue inhibitors of metalloproteinases (TIMPs) that block the degradation of extracellular matrix (ECM) (48). Additionally, M2-derived chemokines play a role in fibrosis. For instance, CCL18/PARC is pro-fibrotic by promoting collagen production in lung fibroblasts (49–51). Increased collagen deposition, in turn, can enhance CCL18 production in alveolar macrophages, thereby suggesting a positive feedback loop between alveolar macrophages and fibroblasts (50). In idiopathic pulmonary fibrosis (IPF), one of the most common types of interstitial lung disease, CCL18 levels correlated with severity of fibrosis (52). More recently, CCL18 was identified as a marker for early identification of progressive interstitial lung disease in systemic sclerosis (SS) (53). Pechkovsky et al. showed that the Th2 cytokines IL-4 and IL-10 induce M2 polarization of alveolar macrophages (54). Interestingly, IL-10 enhanced the IL-4-induced CCL18 expression (54).
Besides CCL18, also CCL2 directly mediates a pro-fibrotic effect on fibroblasts by affecting TGF-β signaling, which in turn stimulates collagen production (55). Mice lacking CCR2, the cognate receptor for CCL2, showed reduced infiltration of inflammatory macrophages in two models of hepatic fibrosis (56, 57). These CCR2−/− mice also developed less severe pulmonary fibrosis (58). Macrophages derived from CCR2 KO mice showed reduced production of matrix metalloproteinase (MMP)-2 and MMP-9 (59). Finally, the CCL2-CCR2 axis in macrophages has also been found to be important in renal fibrosis, where mononuclear cell infiltration and expression of chemokine receptors CCR1, CCR2, and CCR5 was enhanced in a spontaneous model of lupus nephritis (60).
Interestingly, in a commonly used model of bleomycin-induced lung fibrosis, CCR4−/− mice showed a decreased inflammatory and fibrotic response compared to WT mice. Further analysis revealed that CCR4 KO alveolar and bone marrow-derived macrophages exhibited a more pronounced M2 activation state, as evidenced by increased expression of the typical M2 markers ARG1 and “found in inflammatory zone 1” (FIZZ1). Further experiments showed that the CCR4 ligand CCL17/TARC (61) plays a role in CCR4-dependent M1 activation leading to iNOS induction and oxidative injury, thereby affecting the development of bleomycin-induced pulmonary fibrosis (62). Additionally, FIZZ1 activates fibroblasts and induces myofibroblast differentiation in bleomycin-induced pulmonary fibrosis (63, 64). Chvatchko et al. reported that CCR4−/− mice were more resistant to the effects of LPS compared to CCR4 WT mice (65). Further analysis revealed that peritoneal macrophages from CCR4 deficient mice possess an altered phenotype, more resembling M2 macrophages with elevated secretion of type 2 cytokines/chemokines and FIZZ1 protein (66). This study underscores the possible role of CCR4 in M1 activation.
In two different murine models of liver fibrosis, Heymann et al. demonstrated a protective role for the CCR8 receptor. Interestingly, hepatic macrophages from CCR8 KO mice showed an altered phenotype with more pronounced dendritic cell-like characteristics and enhanced CCL3 secretion (67).
Macrophage Polarization by Chemokines in Metabolic Disorders
Nowadays it is generally accepted that the immune system and metabolism are tightly connected and recent studies have demonstrated that macrophages, in particular, are critical effector cells in metabolic inflammation (68). Resident macrophages in the adipose tissue of lean mice constitute ~10–15% of the total cell population. These adipose tissue macrophages (ATMs) express predominantly M2 characteristics and were shown to be critical for maintaining insulin sensitivity in adipocytes (69, 70). Conversely, in obesity, a state of low-grade systemic inflammation (71), adipocytes secrete pro-inflammatory mediators, which recruit monocytes into the adipose tissue mainly via the CCL2-CCR2 and CCL5-CCR5 axis (72–74). During obesity the number of macrophages in white adipose tissue increases fourfold (69) and macrophages acquire an M1 phenotype that contributes to the pro-inflammatory environment (75). Via secretion of pro-inflammatory cytokines, M1 ATMs contribute to insulin resistance by counteracting the insulin sensitizing action of the adipokines adiponectin and leptin (69, 76, 77). More recently, it has been shown that macrophage polarization in obesity can also be modulated by chemokines and their receptors. Kitade et al. demonstrated that inactivation of CCR5 not only resulted in a reduced number of ATMs, but the recruited ATMs switched toward an M2 phenotype (73). Additionally, obesity-induced insulin resistance was attenuated in obese CCR5−/− mice (73). The question how CCR5 regulates M2 polarization is still unanswered. Obese mice with a genetic deficiency in CCR2 showed a reduced number of ATMs combined with a decreased expression of pro-inflammatory genes, compared to matched WT mice (72). Besides the CCR2 and CCR5 ligands, a recent study showed that during obesity CXCL12 recruits macrophages via CXCR4 to the adipose tissue (78). Moreover, CXCL12-CXCR4 signaling induced M1 macrophage accumulation and blocking this signaling diminished secretion of pro-inflammatory cytokines and improved insulin resistance (79).
The recruitment of macrophages, which stimulate the development of insulin resistance in obesity, is also critical in associated metabolic comorbidities such as nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH). NAFLD is characterized by excessive fat accumulation in the form of intrahepatic triglycerides in the liver. NAFLD exhibits as a spectrum ranging from steatosis of the liver to a more necro-inflammatory form, NASH, which may develop into hepatic fibrosis, cirrhosis, or hepatic carcinoma (80). In the liver, macrophages consist of distinct populations, namely the resident, self-renewing Kuppfer cells and the inflammatory monocyte-derived macrophages (81–83). Kuppfer cells line the liver sinusoids and are involved in cholesterol metabolism by taking up and clearing modified low-density lipoprotein (LDL) and bacterial endotoxins through their scavenger receptors (84).
In line with the improved insulin resistance in CCR2−/− obese mice, also hepatic steatosis was ameliorated (72). Besides CCR2, Karlmark et al. found that the CX3CL1-CX3CR1 axis is involved in the differentiation and survival of intrahepatic monocytes (85). The CX3CR1-mediated survival depends on the activation of the anti-apoptotic protein BCL2. Furthermore, in the absence of CX3CR1, hepatic macrophages showed a more pro-inflammatory phenotype characterized by increased TNF-α and iNOS production. These in vivo findings confirm earlier published data on elevated Tnfα expression and reduced ARG1 expression in CX3CR1-deficient macrophages in a carbon tetrachloride (CCl4)-induced NAFLD mouse model (86). The increased pro-inflammatory response of liver macrophages was associated with enhanced liver fibrosis (85). This latter observation suggests that activation of the CX3CL1-CX3CR1 axis can work as an antifibrotic liver therapy.
Macrophage Polarization in Cardio-Vascular Diseases
Cardiovascular disease (CVD) is the most common cause of mortality worldwide and accounts for 45% of all deaths in Europe (87). Atherosclerosis, an arterial narrowing due to plaque formation, is most often the underlying cause of myocardial infarction (88). The starting point of this pathology is the accumulation of lipoprotein particles in the intimal layer of the blood vessel. These lesions are mostly found at arterial branching points and bends, which are especially prone for local endothelial cell dysfunction. The stored lipoproteins are modified by several mechanisms such as oxidation, enzymatic processing, desialylation and aggregation, become pro-inflammatory and activate surrounding endothelial cells. Activated endothelial cells, in turn, release chemokines which recruit monocytes into the intimal and subintimal space of the artery where they differentiate into macrophages (89). These macrophages actively ingest cholesteryl ester-rich lipoproteins and eventually become “foam cells.” Although the uptake of lipoproteins by macrophages seems to be beneficial, these “foam cells” aggravate the disease through their secretion of pro-inflammatory mediators including cytokines and ROS and finally through their eventual death by necrosis or apoptosis. These latter processes result in the release of lipids and the formation of a pro-thrombotic core, which is a key-component of unstable plaques. Rupture of these plaques leads to the initiation of thrombosis, which limits or even blocks the flow of oxygen-rich blood to organs and other parts of the body (90, 91).
The first chemokine implicated in atherosclerosis was CCL2, which is normally not found in the blood vessel wall, but is induced in the early phase of atherosclerosis (92–94). Evidence for a prominent role of the CCL2-CCR2 axis came from a study by Boring et al. who reported that CCR2−/− mice exhibit severely reduced atherosclerotic lesions (95). Later on, CXCR2, CX3CR1 and CCR1 have been implicated in monocyte/macrophage accumulation in atherosclerotic plaques (96, 97).
Relatively large numbers of pro-inflammatory macrophages were found in plaques and M1 macrophages are associated with unstable plaques (98, 99). M2 macrophages have only been detected later on and are more common in asymptomatic lesions and the stable zones of plaques (100). In addition to M1 and M2 macrophages, atherosclerotic plaques also contain specific macrophage subtypes, which are different from the phenotypes suggested by the classical activation model. For instance, in mice, oxidized lipids induce a distinct proatherogenic phenotype, referred to as Mox macrophages (101). These are characterized by reduced phagocytic and chemotactic capacities compared to M1 and M2 macrophages (101). So far, this phenotype is only observed in mice, whether Mox macrophages are also present in human lesions remains to be investigated. Upon intraplaque hemorrhage, due to rupture of invaded microvessels in the plaque, red blood cells lyse quickly and release hemoglobin and free heme. These heme products can directly polarize macrophages toward the Mhem or M(Hb) phenotype. Functionally, these subtypes are resistant to lipid accumulation and foam cell formation (102). Macrophage polarization to the M(Hb) phenotype occurs via exposure to the hemoglobin-haptoglobin complex (102, 103). This M(Hb) subset expresses high levels of the scavenger receptors CD163 (the hemoglobin-haptoglobin complex receptor) and CD206 (the mannose receptor) and is resistant to cholesterol accumulation because of the increased expression of the cholesterol efflux receptors ABCA1 and ABCG1 (104). Heme induces atheroprotective Mhem macrophages, which have high levels of HMOX1 (105) and are able to engulf extravasated erythrocytes (erythrophagocytosis) (106).
Besides lipids and their derivatives, heme products and also chemokines and growth factors present in atherosclerotic lesions can contribute to macrophage phenotype determination. During the early atherogenic phase, platelets can adhere and act as a rich source of chemokines. The platelet-derived chemokine CXCL4/PF-4 (107), similar to M-CSF, has been shown to prevent monocyte apoptosis and to promote the differentiation into macrophages in vitro (8). Later on it was found that CXCL4-induced macrophages acquire a specific phenotype, with a mixture of M1 and M2 characteristics and distinct from their M-CSF-induced counterparts. These so-called M4 macrophages express the pro-inflammatory chemokines TNF-α and IL-6, MMP-7, and MMP-12 and the calcium binding protein S100A8 (108, 109). The complete loss of the hemoglobin-haptoglobin scavenger receptor CD163, which is required for effective hemoglobin clearance after plaque hemorrhage (108, 110) and low expression of the antigen-presenting molecule HLA-DR (8) are typical characteristics of these so-called M4 macrophages. When hemoglobin or the hemoglobin-haptoglobin complexes bind the CD163 receptor, the atheroprotective HMOX1 is induced. Consequently, HMOX1 activity is also completely abolished in CXCL4-stimulated monocytes (111). Interestingly, the marked downregulation of CD163 and the novel phenotype induced by CXCL4 was reported to be irreversible (108). The presence of M4 macrophages within human atherosclerotic lesions is associated with advanced plaque morphology (112). M4 macrophages can be considered pro-atherogenic, since these may promote destabilization of the plaque fibrous cap (113).
More recently, our group studied the effect of CXCL4L1/PF-4var (114), the non-allelic variant of CXCL4, on the differentiation of monocytes into macrophages (Figure 2) (115). Both variants are secreted by activated platelets and differ only in 3 amino acids near the carboxy-terminal end. The unique 3D structure of CXCL4L1 results in a decreased affinity for glycosaminoglycans (GAGs) and a more outspoken angiostatic potential compared to CXCL4 (116). Differently to M-CSF and CXCL4, CXCL4L1 is not a survival factor for monocytes. CXCL4L1-exposed monocytes display higher expression levels of the inflammatory chemokine receptors CCR2 and CCR5, suggesting that CXCL4L1 promotes a higher responsiveness to inflammatory chemokines, such as CCL2 and CCL3. Additionally, significantly higher amounts of CCL2 and CXCL8 (M1 marker) were measured in CXCL4L1-stimulated monocytes, whereas CXCL4 did modulate chemokine production in the same way as M-CSF. Finally, we found a lower expression of IL-1 receptor antagonist (IL-1RA) in CXCL4L1-treated monocytes, compared to CXCL4-treated monocytes, which is in line with the more inflammatory phenotype of macrophages generated in the presence of CXCL4L1 (115). Similar to CXCL4-treated monocytes, CXCL4L1-stimulated monocytes have a significantly lower expression of the CD163 receptor and the mannose receptor (MRC/CD206) compared to M-CSF treated monocytes (117). Interestingly, in contrast to M4 macrophages we found that HMOX1 expression was significantly increased in CXCL4L1-treated monocytes (Figure 2) (115). So far, the role of CXCL4L1 in atherosclerosis is not further investigated. However, we showed that patients with stable coronary artery disease have a worse prognosis when CXCL4L1 levels in the serum are low (118).
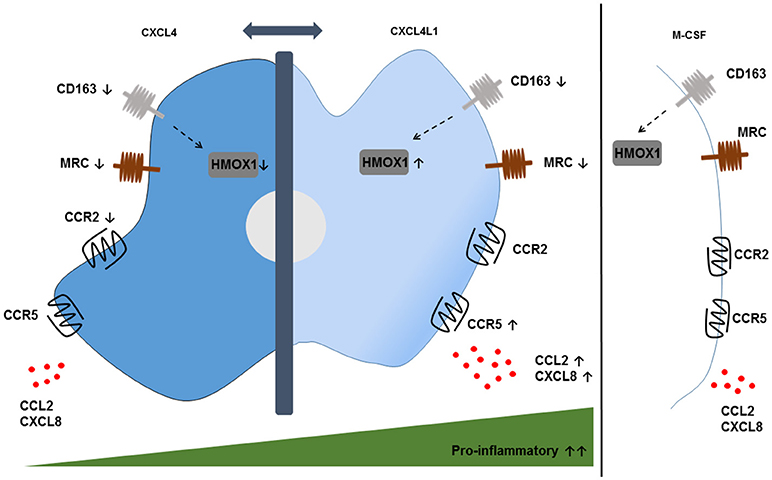
Figure 2. Phenotypic features of CXCL4- and CXCL4L1-induced macrophages. CXCL4-induced macrophages display a pro-atherogenic phenotype, characterized by the downregulation of the hemoglobin-haptoglobin scavenger receptor CD163 and the consequent downregulation of the HMOX1 enzyme compared to M-CSF-treated monocytes. Remarkably, the downregulation of HMOX1 is not observed in CXCL4L1-induced macrophages, which also show reduced expression of CD163. Both phenotypes show a downregulation of the mannose receptor (MRC) CD206. The expression of the chemokine receptors CCR2 and CCR5 and the secretion of pro-inflammatory chemokines CXCL8 and CCL2 are higher on CXCL4L1-treated monocytes compared to CXCL4-stimulated monocytes, thereby indicating more pro-inflammatory characteristics for CXCL4L1- than CXCL4-stimulated monocytes.
Role of TAMs in Cancer
It is generally accepted that macrophages are the most abundant component of the leukocyte infiltrate that is influencing tumor development. Macrophages that infiltrate the tumor microenvironment are usually referred to as tumor-associated macrophages (TAMs) (119). TAM infiltration is correlated with a poor prognosis in numerous cancers, suggesting that they promote tumor progression (1, 81, 120, 121). Indeed, TAMs can stimulate proliferation, invasion, metastasis of tumor cells, promote angiogenesis and suppress the anti-tumor response (122). Poor anti-tumoral activities are a consequence of the higher production of IL-10, TGF-β and prostaglandin E2 (PGE2) and reduced synthesis of inflammatory cytokines such as TNF-α and IL-6. Furthermore, TAMs display poor antigen-presenting capacities, leading to suppression rather than stimulation of T cell activation and proliferation (13). The decreased production of inflammatory mediators in TAMs is associated with a defective nuclear factor-kappa B (NF-κB) activation in response to LPS and proinflammatory cytokines (123). In addition to the production of the most potent angiogenic factor VEGF, TAMs were shown to produce platelet-derived growth factor (PDGF) (13) and VEGF-C (124), which was suggested to play a role in peri-tumoral lymphangiogenesis and subsequent lymphatic metastasis. As such, TAMs are generally characterized as M2-like macrophages (125).
However, extensive TAM density is associated with increased survival in some specific tumor types. These findings suggest that TAMs comprise multiple distinct pro- and anti-tumoral subpopulations with overlapping features depending on different micro-environmental stimuli. In an explant model of colorectal cancer liver metastasis, CCR5 blockade with Maraviroc, a highly specific CCR5 inhibitor originally developed to treat HIV patients (126), induced a repolarization from an M2 toward an anti-tumoral M1-like phenotype (127). This phenotypic switch was mediated via increased levels of the signal transducer and activator of transcription 3 (STAT3), which is commonly linked to an M1 activation state, due to abrogation of the suppressor of cytokine signaling 3 (SOCS3) activity (128). This so-called re-education of macrophages induced by CCR5 inhibition in human cancer patients could possibly contribute to the further development of chemokine-based anti-cancer therapy.
TAMs originate from circulating monocytes, which are recruited to the tumor by several growth factors and especially by chemokines, produced by stromal and tumor cells (120). Besides M-CSF, the CC chemokines CCL2, CCL3, CCL4, and CCL5 are well-recognized chemotactic factors for macrophage populations in the tumor (Figure 3) (129–133). CCL2 is dominantly expressed by many human carcinomas (134, 135) and detection of CCL2 in TAMs themselves even indicates the existence of an amplification loop for their recruitment (13, 136). Interestingly, once macrophages have entered the tumor microenvironment, the corresponding CCR2 is downregulated. It is suggested that receptor downregulation is a mechanism to trap recruited macrophages in the tumor micro-environment (137). Furthermore, in colon cancer models CCL20/LARC (138) chemoattracts monocytes that differentiate into TAMs. Additionally, in human breast cancer models CCL18 in collaboration with CSF-2 was involved in mobilization and recruitment of monocytes (139). Finally, VEGF-A was identified as a macrophage recruitment factor in an in vivo xenograft model, possibly acting indirectly through induction of chemoattractants (140).
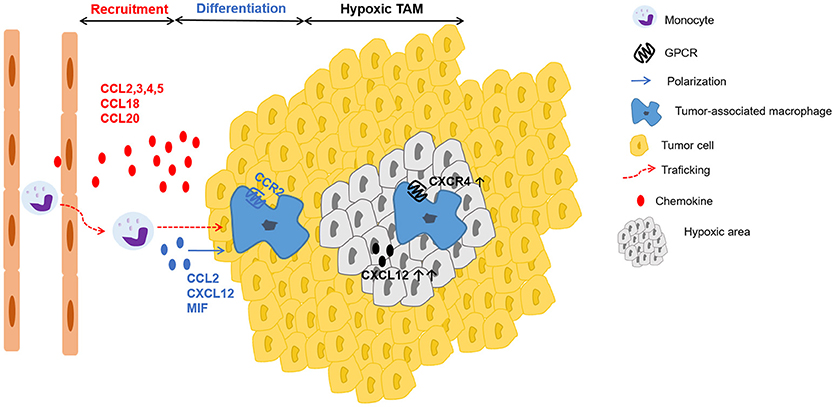
Figure 3. Schematic representation of chemokines involved in recruitment, differentiation and positioning of TAMs. Tumor-derived factors such as the chemokines CCL2, CCL3, CCL4, CCL5, CCL18, CCL20 actively recruit (red arrow) monocytes to the tumor, where they differentiate into tumor-associated macrophages (TAMs). In addition to several growth factors, a particular role in TAM polarization (blue arrow) has been described for the chemokines CCL2, CXCL12 and the chemokine-like protein MIF. In hypoxic areas, higher amounts of CXCL12 and increased expression of CXCR4 on macrophages enhance migration to and retention in these particular sites with low oxygen tension.
Once differentiated, TAMs preferentially accumulate in the hypoxic areas of the tumor (141). Casazza et al. found that the protein Neuropilin-1 (Nrp-1) is essential for TAM mobilization toward Semaphorin 3A (SEMA3A), which is upregulated in hypoxic regions of the tumor. When TAMs enter these hypoxic areas, Nrp-1 expression is downregulated and TAMs are trapped in the hypoxic environment (142). Further, these hypoxic TAMs upregulate hypoxia-regulated genes and alter the gene expression profile, acquiring an even more pronounced pro-angiogenic, immunosuppressive, and pro-metastatic phenotype (143). This hypoxia-induced response is partly mediated via the key transcription factor hypoxia-inducible factor (HIF)-1α (144). Interestingly, in endothelial cells HIF-1α induces CXCL12 expression, which is in direct proportion to the oxygen tension in hypoxic areas (145). Additionally, hypoxia induces the expression of CXCR4 on monocytes and macrophages, thereby highlighting a possible role of the CXCL12-CXCR4 axis for TAM trafficking to the hypoxic tumor areas (Figure 3) (146).
Besides functioning as chemoattractants, some chemokines can also affect TAM polarization. Sierra-Filardi et al. disclosed an important role for the CCL2-CCR2 axis in regulating macrophage polarization, since blocking CCL2 led to an upregulation of M1 polarization-associated genes and decreased expression of M2-associated markers in human macrophages (147). Additionally, in several animal models of non–small-cell lung cancer (NSCLC) CCL2 blockade significantly reduced tumor growth. Although the total number of recruited macrophages did not change, there was a clear change in the polarization state of TAMs toward a more anti-tumor phenotype after CCL2 blockade (148). These results are in line with the findings from Roca et al. who showed that CCL2 stimulation shifts human peripheral blood CD11b+ cells toward a CD206+ M2-polarized phenotype (149).
Furthermore, in multiple myeloma (MM) CCL2, CCL3, and CCL14/HCC-1 (150) stimulate macrophage polarization into MM-associated macrophages (139), which induce MM drug resistance in vitro and in MM mouse models in vivo (151, 152). Tripathi et al. showed that hypoxic cancer cell-derived oncostatin M and the chemokine CCL11/eotaxin skewed macrophages toward an M2 phenotype (153, 154).
Besides factors produced by tumor cells, some chemokines produced by the macrophages themselves can affect their polarization. As such, autocrine CXCL12 production modulated the differentiation of monocytes toward a proangiogenic and immunosuppressive phenotype (155).
Interestingly, migration inhibitory factor (MIF), a cytokine that is not a chemokine but considered to be a “chemokine-like” molecule, was found to be a regulator of TAM polarization in melanoma bearing mice. A small molecule MIF antagonist attenuated tumor-induced macrophage M2 polarization coinciding with a reduced angiogenic potential (156).
The final step of cancer progression is metastasis, i.e., the dissemination of cancer cells from the primary tumor to distant organs. This highly complex process involves cell detachment from the primary tumor site, local invasion, intravasation into adjacent circulatory blood and lymphatic vessels, extravasation at distant capillary beds and proliferation in/colonization of distant organs (157). Before metastatic tumor cells are able to colonize, primary tumor-derived products prepare a primed microenvironment at secondary sites, also known as the pre-metastatic niche (158). Soluble factors including VEGF and placental growth factor (PIGF) induce the recruitment of VEGF-receptor 1 (VEGFR1) positive myeloid cells, which form clusters in the lungs and liver, preparing a permissive niche for disseminating tumor cells. Depletion of these VEGFR1+ cells inhibited metastasis (158). Disseminated cancer cells, in turn, produce CCL2 that recruits inflammatory CCR2+ monocytes from the blood to the metastatic niche, where they differentiate into so-called metastasis-associated macrophages (MAMs) (159). By secreting VEGF-A, these MAMs cause vessel wall permeabilization, allowing subsequent tumor cell extravasation (159). Interestingly, activation of CCR2 on MAMs induces the expression of CCL3 (160). CCL3 signaling via CCR1, in turn, promotes the retention of MAMs in the lung through vascular cell adhesion molecule (VCAM1)-α4 integrin mediated signaling and promotes cancer cell extravasation and retention at the metastatic site (160). Furthermore, VCAM 1 – α4 signaling protects cancer cells from pro-apoptotic signals (161).
Thus, TAMs and MAMs are not only a target for chemokines but also considered as a source of chemotactic mediators. Among these CCL2, CCL3, CCL17, CCL18, and CCL22 have been found to be produced by TAMs/MAMs (61, 162). In ascitic fluid from ovarian cancer patients CCL18, an attractant for Th2 cells was identified, but this chemokine was not produced by ovarian carcinoma cell lines in vitro (163). Therefore, it was suggested that the inflammatory mononuclear cells infiltrating the tumor were the CCL18-producing cells (164). Furthermore, CCL17 and CCL22 induce migration of regulatory T (Treg) cells via interaction with the CCR4 receptor (165). Thus, attraction of immunosuppressive immune cells through chemokine production is one of the pro-tumoral characteristics of TAMs.
Concluding Remarks
Monocyte-derived macrophages respond to a variety of stimuli to modulate their phenotype, which underlines their phenotypic plasticity, one of the major features of macrophages. M1 and M2 macrophages represent the extremities of a continuum of macrophage polarization states with M1 and M2 representing a rather pro-inflammatory and anti-inflammatory phenotype, respectively. Besides their well-known role in monocyte migration, chemokines have also been found to play a role in long-term regulatory processes by inducing macrophage differentiation and polarization in physiological and pathological processes.
Author Contributions
PR wrote the review and designed the figures; JVD and PP corrected the manuscript; SS provided critical feedback, helped shaping, and corrected the manuscript.
Funding
This research was supported by the Fund for Scientific Research of Flanders (FWO-Vlaanderen Project G.0D25.17N) and C1 funding (grant C16/17/010) from KU Leuven.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. (2005) 5:953–64. doi: 10.1038/nri1733
2. Hume DA, Ross IL, Himes SR, Sasmono RT, Wells CA, Ravasi T. The mononuclear phagocyte system revisited. J Leukoc Biol. (2002) 72:621–7. doi: 10.1189/jlb.72.4.621
3. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. (2008) 8:533–44. doi: 10.1038/nri2356
4. Hanamura T, Motoyoshi K, Yoshida K, Saito M, Miura Y, Kawashima T, et al. Quantitation and identification of human monocytic colony-stimulating factor in human serum by enzyme-linked immunosorbent assay. Blood (1988) 72:886–92.
5. Tushinski RJ, Oliver IT, Guilbert LJ, Tynan PW, Warner JR, Stanley ER. Survival of mononuclear phagocytes depends on a lineage-specific growth factor that the differentiated cells selectively destroy. Cell (1982) 28:71–81.
6. Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood (1980) 56:947–58.
7. Gasson JC. Molecular physiology of granulocyte-macrophage colony-stimulating factor. Blood (1991) 77:1131–45.
8. Scheuerer B, Ernst M, Durrbaum-Landmann I, Fleischer J, Grage-Griebenow E, Brandt E, et al. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood (2000) 95:1158–66.
9. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496:445–55. doi: 10.1038/nature12034
10. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. (2013) 14:986–95. doi: 10.1038/ni.2705
11. Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. (2014) 262:36–55. doi: 10.1111/imr.12223
12. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. (2015) 33:643–75. doi: 10.1146/annurev-immunol-032414-112220
13. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
14. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature (2011) 472:476–80. doi: 10.1038/nature09973
15. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. (2008) 13:453–61.
16. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
17. Morris SM Jr. Arginine metabolism: boundaries of our knowledge. J Nutr. (2007) 137(6 Suppl. 2):1602s−9s. doi: 10.1093/jn/137.6.1602S
18. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
19. Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol. (2008) 180:2011–7. doi: 10.4049/jimmunol.180.4.2011
20. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
21. Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International Union of basic and clinical pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptorsPharmacol Rev. (2014) 66:1–79. doi: 10.1124/pr.113.007724
22. Bonecchi R, Graham GJ. Atypical chemokine receptors and their roles in the resolution of the inflammatory response. Front Immunol. (2016) 7:224. doi: 10.3389/fimmu.2016.00224
23. Perry VH, Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. (2013) 35:601–12. doi: 10.1007/s00281-013-0382-8
24. Olah M, Biber K, Vinet J, Boddeke HW. Microglia phenotype diversity. CNS Neurol Disord Drug Targets (2011) 10:108−18. doi: 10.2174/187152711794488575
25. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. (2014) 17:131–43. doi: 10.1038/nn.3599
26. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. (2014) 15:300–12. doi: 10.1038/nrn3722
27. Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature (1997) 385:640–4. doi: 10.1038/385640a0
28. Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, et al. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci. (2011) 31:9910–22. doi: 10.1523/jneurosci.2114-11.2011
29. Wolf Y, Yona S, Kim KW, Jung S. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci. (2013) 7:26. doi: 10.3389/fncel.2013.00026
30. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor Nat. Neurosci. (2006) 9:917–24. doi: 10.1038/nn1715
31. Cardona SM, Mendiola AS, Yang YC, Adkins SL, Torres V, Cardona AE. Disruption of fractalkine signaling leads to microglial activation and neuronal damage in the diabetic retina. ASN Neuro. (2015) 7:1759091415608204. doi: 10.1177/1759091415608204
32. Mattison HA, Nie H, Gao H, Zhou H, Hong JS, Zhang J. Suppressed pro-inflammatory response of microglia in CX3CR1 knockout mice. J Neuroimmunol. (2013) 257:110–5. doi: 10.1016/j.jneuroim.2013.02.008
33. Chen P, Zhao W, Guo Y, Xu J, Yin M. CX3CL1/CX3CR1 in alzheimer's disease: a target for neuroprotection. Biomed Res Int. (2016) 2016:8090918. doi: 10.1155/2016/8090918
34. Noda M, Doi Y, Liang J, Kawanokuchi J, Sonobe Y, Takeuchi H, et al. Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J Biol Chem. (2011) 286:2308–19. doi: 10.1074/jbc.M110.169839
35. Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, et al. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. (2010) 177:2549–62. doi: 10.2353/ajpath.2010.100265
36. Tang Z, Gan Y, Liu Q, Yin JX, Liu Q, Shi J, et al. CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J Neuroinflammation (2014) 11:26. doi: 10.1186/1742-2094-11-26
37. Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. (2010) 13:411–3. doi: 10.1038/nn.2511
38. Mazzon C, Zanotti L, Wang L, Del Prete A, Fontana E, Salvi V, et al. CCRL2 regulates M1/M2 polarization during EAE recovery phase. J Leukoc Biol. (2016) 99:1027–33. doi: 10.1189/jlb.3MA0915-444RR
39. Del Prete A, Bonecchi R, Vecchi A, Mantovani A, Sozzani S. CCRL2, a fringe member of the atypical chemoattractant receptor family. Eur J Immunol. (2013) 43:1418–22. doi: 10.1002/eji.201243179
40. Matsushima K, Larsen CG, DuBois GC, Oppenheim JJ. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J Exp Med. (1989) 169:1485–90.
41. Yang G, Meng Y, Li W, Yong Y, Fan Z, Ding H, et al. Neuronal MCP-1 mediates microglia recruitment and neurodegeneration induced by the mild impairment of oxidative metabolism. Brain Pathol. (2011) 21:279–97. doi: 10.1111/j.1750-3639.2010.00445.x
42. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. (2005) 115:56–65. doi: 10.1172/jci22675
43. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. (2008) 214:199–210. doi: 10.1002/path.2277
44. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. (2012) 18:1028–40. doi: 10.1038/nm.2807
45. Wynn TA, Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. (2004) 4:583–94. doi: 10.1038/nri1412
46. Xiao W, Hong H, Kawakami Y, Lowell CA, Kawakami T. Regulation of myeloproliferation and M2 macrophage programming in mice by Lyn/Hck, SHIP, and Stat5. J Clin Invest. (2008) 118:924–34. doi: 10.1172/jci34013
47. Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA. (1986) 83:4167–71.
48. Sunderkotter C, Steinbrink K, Goebeler M, Bhardwaj R, Sorg C. Macrophages and angiogenesis. J Leukoc Biol. (1994) 55:410–22.
49. Hieshima K, Imai T, Baba M, Shoudai K, Ishizuka K, Nakagawa T, et al. A novel human CC chemokine PARC that is most homologous to macrophage-inflammatory protein-1 alpha/LD78 alpha and chemotactic for T lymphocytes, but not for monocytes. J Immunol. (1997) 159:1140–9.
50. Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. (2006) 173:781–92. doi: 10.1164/rccm.200509-1518OC
51. Atamas SP, Luzina IG, Choi J, Tsymbalyuk N, Carbonetti NH, Singh IS, et al. Pulmonary and activation-regulated chemokine stimulates collagen production in lung fibroblasts. Am J Respir Cell Mol Biol. (2003) 29:743–9. doi: 10.1165/rcmb.2003-0078OC
52. Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR, et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2009) 179:717–23. doi: 10.1164/rccm.200808-1201OC
53. Hoffmann-Vold AM, Tennoe AH, Garen T, Midtvedt O, Abraityte A, Aalokken TM, et al. High level of chemokine ccl18 is associated with pulmonary function deterioration, lung fibrosis progression, and reduced survival in systemic sclerosis. Chest (2016) 150:299–306. doi: 10.1016/j.chest.2016.03.004
54. Pechkovsky DV, Prasse A, Kollert F, Engel KM, Dentler J, Luttmann W, et al. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin Immunol. (2010) 137:89–101. doi: 10.1016/j.clim.2010.06.017
55. Gharaee-Kermani M, Phan SH. Molecular mechanisms of and possible treatment strategies for idiopathic pulmonary fibrosis. Curr Pharm Des. (2005) 11:3943–71. doi: 10.2174/138161205774580561
56. Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology (2009) 50:185–97. doi: 10.1002/hep.22952
57. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology (2009) 50:261–74. doi: 10.1002/hep.22950
58. Moore BB, Paine R III, Christensen PJ, Moore TA, Sitterding S, Ngan R, et al. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J Immunol. (2001) 167:4368–77. doi: 10.4049/jimmunol.167.8.4368
59. Okuma T, Terasaki Y, Kaikita K, Kobayashi H, Kuziel WA, Kawasuji M, et al. C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases. J Pathol. (2004) 204:594–604. doi: 10.1002/path.1667
60. Perez de Lema G, Maier H, Nieto E, Vielhauer V, Luckow B, Mampaso F, et al. Chemokine expression precedes inflammatory cell infiltration and chemokine receptor and cytokine expression during the initiation of murine lupus nephritis. J Am Soc Nephrol. (2001) 12:1369–82.
61. Imai T, Yoshida T, Baba M, Nishimura M, Kakizaki M, Yoshie O. Molecular cloning of a novel T cell-directed CC chemokine expressed in thymus by signal sequence trap using Epstein-Barr virus vector. J Biol Chem. (1996) 271:21514–21.
62. Trujillo G, O'Connor EC, Kunkel SL, Hogaboam CM. A novel mechanism for CCR4 in the regulation of macrophage activation in bleomycin-induced pulmonary fibrosis. Am J Pathol. (2008) 172:1209–21. doi: 10.2353/ajpath.2008.070832
63. Liu T, Dhanasekaran SM, Jin H, Hu B, Tomlins SA, Chinnaiyan AM, et al. FIZZ1 stimulation of myofibroblast differentiation. Am J Pathol. (2004) 164:1315–26. doi: 10.1016/s0002-9440(10)63218-x
64. Liu T, Jin H, Ullenbruch M, Hu B, Hashimoto N, Moore B, et al. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. J Immunol. (2004) 173:3425–31. doi: 10.4049/jimmunol.173.5.3425
65. Chvatchko Y, Hoogewerf AJ, Meyer A, Alouani S, Juillard P, Buser R, et al. A key role for CC chemokine receptor 4 in lipopolysaccharide-induced endotoxic shock. J Exp Med. (2000) 191:1755−64. doi: 10.1084/jem.191.10.1755
66. Ness TL, Ewing JL, Hogaboam CM, Kunkel SL. CCR4 is a key modulator of innate immune responses. J Immunol. (2006) 177:7531–9. doi: 10.4049/jimmunol.177.11.7531
67. Heymann F, Hammerich L, Storch D, Bartneck M, Huss S, Russeler V, et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology (2012) 55:898–909. doi: 10.1002/hep.24764
68. McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity (2014) 41:36–48. doi: 10.1016/j.immuni.2014.05.010
69. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/jci19246
70. Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science (2013) 339:172–7. doi: 10.1126/science.1230721
71. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. (2011) 17:179–88. doi: 10.1038/nm.2279
72. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. (2006) 116:115–24. doi: 10.1172/jci24335
73. Kitade H, Sawamoto K, Nagashimada M, Inoue H, Yamamoto Y, Sai Y, et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes (2012) 61:1680–90. doi: 10.2337/db11-1506
74. Dahlman I, Kaaman M, Olsson T, Tan GD, Bickerton AS, Wahlen K, et al. A unique role of monocyte chemoattractant protein 1 among chemokines in adipose tissue of obese subjects. J Clin Endocrinol Metab. (2005) 90:5834–40. doi: 10.1210/jc.2005-0369
75. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84. doi: 10.1172/jci29881
76. Odegaard JI, Chawla A. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat Clin Pract Endocrinol Metab. (2008) 4:619–26. doi: 10.1038/ncpendmet0976
77. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. (2003) 112:1821–30. doi: 10.1172/jci19451
78. Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature (1996) 382:635–8. doi: 10.1038/382635a0
79. Kim D, Kim J, Yoon JH, Ghim J, Yea K, Song P, et al. CXCL12 secreted from adipose tissue recruits macrophages and induces insulin resistance in mice. Diabetologia (2014) 57:1456–65. doi: 10.1007/s00125-014-3237-5
80. Calzadilla Bertot L, Adams LA. The natural course of non-alcoholic fatty liver disease. Int J Mol Sci. (2016) 17:E774. doi: 10.3390/ijms17050774
81. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature (2015) 518:547–51. doi: 10.1038/nature13989
82. Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science (2016) 353:aaf4238. doi: 10.1126/science.aaf4238
83. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42:665–78. doi: 10.1016/j.immuni.2015.03.011
84. Haworth R, Platt N, Keshav S, Hughes D, Darley E, Suzuki H, et al. The macrophage scavenger receptor type A is expressed by activated macrophages and protects the host against lethal endotoxic shock. J Exp Med. (1997) 186:1431–9.
85. Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, et al. The fractalkine receptor CX(3)CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology (2010) 52:1769–82. doi: 10.1002/hep.23894
86. Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology (2010) 52:1390–400. doi: 10.1002/hep.23795
87. Townsend N, Wilson L, Bhatnagar P, Wickramasinghe K, Rayner M, Nichols M. Cardiovascular disease in Europe: epidemiological update 2016. Eur Heart J. (2016) 37:3232–3245. doi: 10.1093/eurheartj/ehw334
88. Davies MJ, Woolf N, Robertson WB. Pathology of acute myocardial infarction with particular reference to occlusive coronary thrombi. Br Heart J. (1976) 38:659–64.
89. Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol. (2004) 4:432–44. doi: 10.1038/nri1375
90. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. (2011) 17:1410–22. doi: 10.1038/nm.2538
91. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. (2013) 13:709–21. doi: 10.1038/nri3520
92. Rollins BJ. Chemokines and atherosclerosis: what Adam Smith has to say about vascular disease. J Clin Invest. (2001) 108:1269–71. doi: 10.1172/jci14273
93. Gu L, Rutledge B, Fiorillo J, Ernst C, Grewal I, Flavell R, et al. In vivo properties of monocyte chemoattractant protein-1. J Leukoc Biol. (1997) 62:577–80.
94. Rollins BJ. Monocyte chemoattractant protein 1: a potential regulator of monocyte recruitment in inflammatory disease. Mol Med Today (1996) 2:198–204.
95. Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature (1998) 394:894–7. doi: 10.1038/29788
96. Soehnlein O, Drechsler M, Doring Y, Lievens D, Hartwig H, Kemmerich K, et al. Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol Med. (2013) 5:471–81. doi: 10.1002/emmm.201201717
97. Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. (2007) 117:185–94. doi: 10.1172/jci28549
98. Chistiakov DA, Bobryshev YV, Nikiforov NG, Elizova NV, Sobenin IA, Orekhov AN. Macrophage phenotypic plasticity in atherosclerosis: the associated features and the peculiarities of the expression of inflammatory genes. Int J Cardiol. (2015) 184:436–45. doi: 10.1016/j.ijcard.2015.03.055
99. Cochain C, Zernecke A. Macrophages and immune cells in atherosclerosis: recent advances and novel concepts. Basic Res Cardiol. (2015) 110:34. doi: 10.1007/s00395-015-0491-8
100. Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. (2007) 6:137–43. doi: 10.1016/j.cmet.2007.06.010
101. Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. (2010) 107:737–46. doi: 10.1161/circresaha.109.215715
102. Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. (2012) 59:166–77. doi: 10.1016/j.jacc.2011.10.852
103. Boyle JJ, Harrington HA, Piper E, Elderfield K, Stark J, Landis RC, et al. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol. (2009) 174:1097–108. doi: 10.2353/ajpath.2009.080431
104. Habib A, Finn AV. The role of iron metabolism as a mediator of macrophage inflammation and lipid handling in atherosclerosis. Front Pharmacol. (2014) 5:195. doi: 10.3389/fphar.2014.00195
105. Boyle JJ, Johns M, Lo J, Chiodini A, Ambrose N, Evans PC, et al. Heme induces heme oxygenase 1 via Nrf2: role in the homeostatic macrophage response to intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. (2011) 31:2685–91. doi: 10.1161/atvbaha.111.225813
106. Boyle JJ Heme and haemoglobin direct macrophage Mhem phenotype and counter foam cell formation in areas of intraplaque haemorrhage. Curr Opin Lipidol. (2012) 23:453–61. doi: 10.1097/MOL.0b013e328356b145
107. Deuel TF, Keim PS, Farmer M, Heinrikson RL. Amino acid sequence of human platelet factor 4. Proc Natl Acad Sci USA. (1977) 74:2256–8.
108. Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol. (2010) 184:4810–8. doi: 10.4049/jimmunol.0901368
109. Erbel C, Tyka M, Helmes CM, Akhavanpoor M, Rupp G, Domschke G, et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun. (2015) 21:255–65. doi: 10.1177/1753425914526461
110. Gleissner CA. Macrophage Phenotype Modulation by CXCL4 in Atherosclerosis. Front Physiol. (2012) 3:1. doi: 10.3389/fphys.2012.00001
111. Schaer CA, Schoedon G, Imhof A, Kurrer MO, Schaer DJ. Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res. (2006) 99:943–50. doi: 10.1161/01.RES.0000247067.34173.1b
112. Erbel C, Wolf A, Lasitschka F, Linden F, Domschke G, Akhavanpoor M, et al. Prevalence of M4 macrophages within human coronary atherosclerotic plaques is associated with features of plaque instability. Int J Cardiol. (2015) 186:219–25. doi: 10.1016/j.ijcard.2015.03.151
113. Chistiakov DA, Bobryshev YV, Orekhov AN. Changes in transcriptome of macrophages in atherosclerosis. J Cell Mol Med. (2015) 19:1163–73. doi: 10.1111/jcmm.12591
114. Struyf S, Burdick MD, Proost P, Van Damme J, Strieter RM. Platelets release CXCL4L1, a nonallelic variant of the chemokine platelet factor-4/CXCL4 and potent inhibitor of angiogenesis. Circ Res. (2004) 95:855–7. doi: 10.1161/01.res.0000146674.38319.07
115. Gouwy M, Ruytinx P, Radice E, Claudi F, Van Raemdonck K, Bonecchi R, et al. CXCL4 and CXCL4L1 differentially affect monocyte survival and dendritic cell differentiation and phagocytosis. PLoS ONE (2016) 11:e0166006. doi: 10.1371/journal.pone.0166006
116. Kuo JH, Chen YP, Liu JS, Dubrac A, Quemener C, Prats H, et al. Alternative C-terminal helix orientation alters chemokine function: structure of the anti-angiogenic chemokine, CXCL4L1. J Biol Chem. (2013) 288:13522–33. doi: 10.1074/jbc.M113.455329
117. Struyf S, Salogni L, Burdick MD, Vandercappellen J, Gouwy M, Noppen S, et al. Angiostatic and chemotactic activities of the CXC chemokine CXCL4L1 (platelet factor-4 variant) are mediated by CXCR3. Blood (2011) 117:480–8. doi: 10.1182/blood-2009-11-253591
118. De Sutter J, Van de Veire NR, Struyf S, Philippe J, De Buyzere M, Van Damme J. PF-4var/CXCL4L1 predicts outcome in stable coronary artery disease patients with preserved left ventricular function. PLoS ONE (2012) 7:e31343. doi: 10.1371/journal.pone.0031343
119. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
120. Lahmar Q, Keirsse J, Laoui D, Movahedi K, Van Overmeire E, Van Ginderachter JA. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim Biophys Acta (2016) 1865:23–34. doi: 10.1016/j.bbcan.2015.06.009
121. Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol. (2013) 35:585–600. doi: 10.1007/s00281-013-0367-7
122. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
123. Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood (2006) 107:2112–22. doi: 10.1182/blood-2005-01-0428
124. Schoppmann SF, Birner P, Stockl J, Kalt R, Ullrich R, Caucig C, et al. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol. (2002) 161:947–56. doi: 10.1016/s0002-9440(10)64255-1
125. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. (2008) 267:204–15. doi: 10.1016/j.canlet.2008.03.028
126. Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. (2005) 49:4721–32. doi: 10.1128/aac.49.11.4721-4732.2005
127. Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-ccr5 therapy in cancer patients. Cancer Cell (2016) 29:587–601. doi: 10.1016/j.ccell.2016.03.005
128. Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol. (2012) 189:3439–48. doi: 10.4049/jimmunol.1201168
129. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. (2001) 193:727–40. doi: 10.1084/jem.193.6.727
130. De I, Steffen MD, Clark PA, Patros CJ, Sokn E, Bishop SM, et al. CSF1 overexpression promotes high-grade glioma formation without impacting the polarization status of glioma-associated microglia and macrophages. Cancer Res. (2016) 76:2552–60. doi: 10.1158/0008-5472.can-15-2386
131. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood (2004) 104:2224–34. doi: 10.1182/blood-2004-03-1109
132. Wu Y, Li YY, Matsushima K, Baba T, Mukaida N. CCL3-CCR5 axis regulates intratumoral accumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. J Immunol. (2008) 181:6384–93. doi: 10.4049/jimmunol.181.9.6384
133. Milliken D, Scotton C, Raju S, Balkwill F, Wilson J. Analysis of chemokines and chemokine receptor expression in ovarian cancer ascites. Clin Cancer Res. (2002) 8:1108–14.
134. Conti I, Rollins BJ. CCL2 (monocyte chemoattractant protein-1) and cancer. Semin Cancer Biol. (2004) 14:149–54. doi: 10.1016/j.semcancer.2003.10.009
135. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer (2004) 4:540–50. doi: 10.1038/nrc1388
136. Ueno T, Toi M, Saji H, Muta M, Bando H, Kuroi K, et al. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res. (2000) 6:3282−9.
137. Sica A, Saccani A, Bottazzi B, Bernasconi S, Allavena P, Gaetano B, et al. Defective expression of the monocyte chemotactic protein-1 receptor CCR2 in macrophages associated with human ovarian carcinoma. J Immunol. (2000) 164:733–8. doi: 10.4049/jimmunol.164.2.733
138. Hieshima K, Imai T, Opdenakker G, Van Damme J, Kusuda J, Tei H, et al. Molecular cloning of a novel human CC chemokine liver and activation-regulated chemokine (LARC) expressed in liver. Chemotactic activity for lymphocytes and gene localization on chromosome 2J Biol Chem. (1997) 272:5846–53. doi: 10.1074/jbc.272.9.5846
139. Li Y, Zheng Y, Li T, Wang Q, Qian J, Lu Y, et al. Chemokines CCL2, 3, 14 stimulate macrophage bone marrow homing, proliferation, and polarization in multiple myeloma. Oncotarget (2015) 6:24218–29. doi: 10.18632/oncotarget.4523
140. Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, Mueller MM. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol. (2012) 227:17–28. doi: 10.1002/path.3989
141. Lewis C, Murdoch C. Macrophage responses to hypoxia: implications for tumor progression and anti-cancer therapies. Am J Pathol. (2005) 167:627–35. doi: 10.1016/s0002-9440(10)62038-x
142. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell (2013) 24:695–709. doi: 10.1016/j.ccr.2013.11.007
143. Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. (2014) 74:24–30. doi: 10.1158/0008-5472.can-13-1196
144. Werno C, Menrad H, Weigert A, Dehne N, Goerdt S, Schledzewski K, et al. Knockout of HIF-1alpha in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis (2010) 31:1863–72. doi: 10.1093/carcin/bgq088
145. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. (2004) 10:858–64. doi: 10.1038/nm1075
146. Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. (2003) 198:1391–402. doi: 10.1084/jem.20030267
147. Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. (2014) 192:3858–67. doi: 10.4049/jimmunol.1302821
148. Fridlender ZG, Kapoor V, Buchlis G, Cheng G, Sun J, Wang LC, et al. Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am J Respir Cell Mol Biol. (2011) 44:230–7. doi: 10.1165/rcmb.2010-0080OC
149. Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem. (2009) 284:34342–54. doi: 10.1074/jbc.M109.042671
150. Schulz C, Massberg S. Platelets in atherosclerosis and thrombosis. Handb Exp Pharmacol. (2012). 210:111–33. doi: 10.1007/978-3-642-29423-5_5
151. Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, et al. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood (2009) 114:3625–8. doi: 10.1182/blood-2009-05-220285
152. Zheng Y, Yang J, Qian J, Qiu P, Hanabuchi S, Lu Y, et al. PSGL-1/selectin and ICAM-1/CD18 interactions are involved in macrophage-induced drug resistance in myeloma. Leukemia (2013) 27:702–10. doi: 10.1038/leu.2012.272
153. Jose PJ, Griffiths-Johnson DA, Collins PD, Walsh DT, Moqbel R, Totty NF, et al. Eotaxin: a potent eosinophil chemoattractant cytokine detected in a guinea pig model of allergic airways inflammation. J Exp Med. (1994) 179:881–7.
154. Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, et al. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget (2014) 5:5350–68. doi: 10.18632/oncotarget.2110
155. Sanchez-Martin L, Estecha A, Samaniego R, Sanchez-Ramon S, Vega MA, Sanchez-Mateos P. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood (2011) 117:88–97. doi: 10.1182/blood-2009-12-258186
156. Yaddanapudi K, Putty K, Rendon BE, Lamont GJ, Faughn JD, Satoskar A, et al. Control of tumor-associated macrophage alternative activation by macrophage migration inhibitory factor. J Immunol. (2013) 190:2984–93. doi: 10.4049/jimmunol.1201650
157. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
158. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature (2005) 438:820–7. doi: 10.1038/nature04186
159. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011). 475:222–5. doi: 10.1038/nature10138
160. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. (2015) 212:1043–59. doi: 10.1084/jem.20141836
161. Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell (2011) 20:538–49. doi: 10.1016/j.ccr.2011.08.025
162. Godiska R, Chantry D, Raport CJ, Sozzani S, Allavena P, Leviten D, et al. Human macrophage-derived chemokine (MDC), a novel chemoattractant for monocytes, monocyte-derived dendritic cells, and natural killer cells. J Exp Med. (1997) 185:1595–604.
163. Adema GJ, Hartgers F, Verstraten R, de Vries E, Marland G, Menon S, et al. A dendritic-cell-derived C-C chemokine that preferentially attracts naive T cells. Nature (1997) 387:713–7. doi: 10.1038/42716
164. Struyf S, Schutyser E, Gouwy M, Gijsbers K, Proost P, Benoit Y, et al. PARC/CCL18 is a plasma CC chemokine with increased levels in childhood acute lymphoblastic leukemia. Am J Pathol. (2003) 163:2065–75. doi: 10.1016/s0002-9440(10)63564-x
Keywords: macrophage polarization, chemokines, tumor-associated macrophage, leukocyte migration, inflammation and cancer
Citation: Ruytinx P, Proost P, Van Damme J and Struyf S (2018) Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front. Immunol. 9:1930. doi: 10.3389/fimmu.2018.01930
Received: 24 May 2018; Accepted: 06 August 2018;
Published: 07 September 2018.
Edited by:
Giovanni Bernardini, Sapienza Università di Roma, ItalyReviewed by:
Vanessa Pinho, Universidade Federal de Minas Gerais, BrazilNadia Lampiasi, Istituto di Biomedicina e di Immunologia Molecolare Alberto Monroy (IBIM), Italy
Copyright © 2018 Ruytinx, Proost, Van Damme and Struyf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Proost, paul.proost@kuleuven.be