 Karine Chemin
Karine Chemin Christina Gerstner
Christina Gerstner Vivianne Malmström
Vivianne Malmström- Division of Rheumatology, Department of Medicine, Karolinska Institute, Karolinska University Hospital Solna, Stockholm, Sweden
Infiltration of memory CD4+ T cells in synovial joints of Rheumatoid Arthritis (RA) patients has been reported since decades. Moreover, several genome wide association studies (GWAS) pinpointing a key genetic association between the HLA-DR locus and RA have led to the generally agreed hypothesis that CD4+ T cells are directly implicated in the disease. Still, RA is a heterogeneous disease and much effort has been made to understand its different facets. T cell differentiation is driven by mechanisms including antigen stimulation, co-stimulatory signals and cytokine milieu, all of which are abundant in the rheumatic joint, implying that any T cells migrating into the joint may be further affected locally. In parallel to the characterization and classification of T-cell subsets, the contribution of different effector T cells to RA has been investigated in numerous studies though sometimes with contradictory results. In particular, the frequency of Th1 and Th17 cells has been assessed in the synovial joints with various results that could, at least partly, be explained by the stage of the disease. For regulatory T cells, it is largely accepted that they accumulate in RA synovial fluid and that the equilibrium between regulatory T cells and effector cells is a key factor in controlling inflammation processes involved in RA. Recent phenotypic studies describe the possible implication of a novel subset of peripheral T helper cells (Tph) important for T-B cell cross talk and plasma cell differentiation in the RA joint of ACPA+ (autoantibodies against citrullinated proteins) RA patients. Finally, cytotoxic CD4+ T cells, historically described as increased in the peripheral blood of RA patients have attracted new attention in the last years. In view of the recently identified peripheral T-cell subsets, we will integrate immunological data as well as information on genetic variants and therapeutic strategy outcomes into our current understanding of the width of effector T cells. We will also integrate tissue-resident memory T cell aspects, and discuss similarities and differences with inflammatory conditions in skin (psoriasis) and mucosal organs (Crohn's disease).
Introduction
Rheumatoid Arthritis (RA) is a chronic inflammatory disease targeting peripheral joints leading to bone erosion, impairment of mobility, and decreased quality of life. It is affecting 0.5–1% of the population worldwide and is more prevalent in women than in men (1). The pathogenesis of RA is mainly localized in the synovial joint where immune cells composed of T cells, B cells, macrophages, and dendritic cells infiltrate the synovium. Moreover, fibroblast-like synoviocytes present in the sublining layer of the synovium proliferate and contribute to cartilage damage (2). Memory CD4+ T cells are enriched in affected joints of RA patients (3) and highly expanded CD4+ T cell clones are found in synovial tissue of early disease (4) suggesting that T cell expansion could be due to local antigen-induced proliferation. The efficiency of co-stimulation blockade targeting CD80/CD86-CD28 interaction further illustrates the importance of T cells in the pathogenesis of RA (5).
A central function of CD4+ T cells in RA has also been deducted from genetic studies. An early report by Stastny (6) identified an association between RA and HLA-DRB1 that was further confirmed by genome-wide association studies (GWAS) (7). This association led to the “shared epitope hypothesis” whereby a five-amino acid sequence found in certain HLA-DRB1 alleles was associated with increased susceptibility to RA (8). In about 2/3 of RA patients, serum antibodies to citrullinated protein antigens (ACPAs) are present and these are associated with the HLA-DRB1 risk alleles (1). Altogether, these findings have led to the hypothesis that citrullinated peptides might be preferentially presented by HLA-DRB1 risk alleles (9). Such peptide presentation has indeed been demonstrated both functionally (10) and by peptide-HLA crystal structure determination (11). Several citrullinated candidate peptides can be presented by HLA-DRB1*04:01 and other shared epitope alleles such as *04:04 and *10:01 (10, 12, 13) and the search for immunodominant T cell epitopes is still an important area of investigation in the field of RA. The relevance of antigen specificities has already been discussed elsewhere (13) and will not be detailed in this review but instead will be discussed in the context of effector T cell functions.
Infiltration of CD4+ T cells at the site of inflammation is a characteristic feature of several autoimmune syndromes. In the scope of this review, we present and discuss up-to-date understanding of effector functions of CD4+ T cells (Figure 1) present in the joint of RA patients. Examples of CD4+ T cell effector functions from other chronic inflammatory conditions (psoriasis and Crohn's disease) are selected to contrast and discuss our current knowledge in the field of RA. In particular, many common therapeutic strategies have been evaluated in RA, psoriasis and Crohn's disease with different outcomes that shed light on the different pathways implicated in the pathogenesis of these inflammatory disorders. Due to lack of space, this review will be mainly dedicated to findings in human inflammatory conditions.
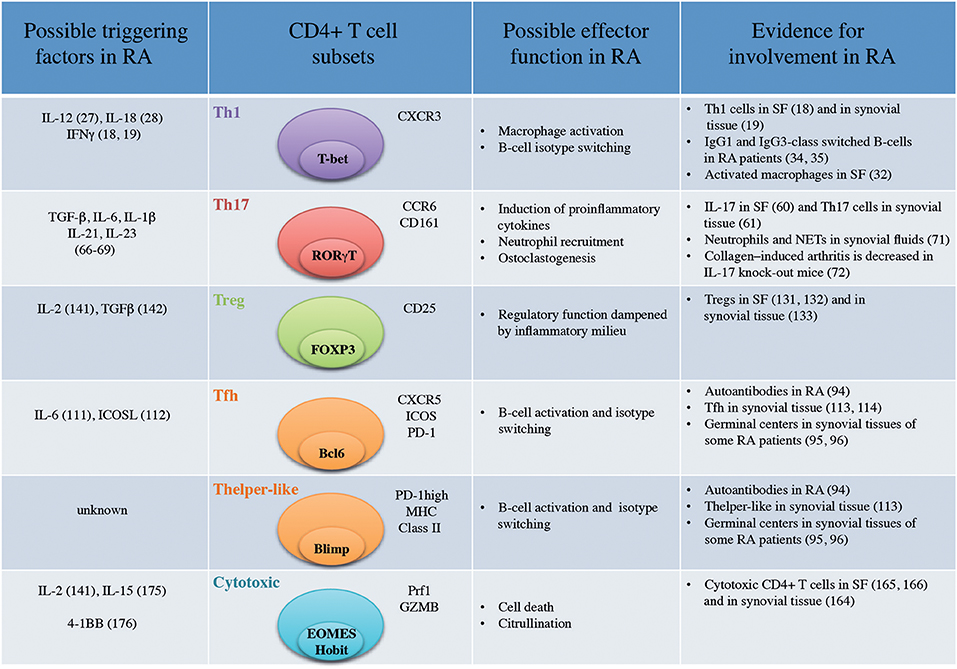
Figure 1. Important CD4+ T-cell subsets in Rheumatoid Arthritis (SF, Synovial Fluid; NETs, Neutrophil Extracellular Traps).
Th1 Cells and Associated Effector Functions
In 1986, Mosmann and Coffman proposed that mouse CD4+ helper T (Th) cells could be subdivided in Th1 or Th2 subsets based on their differential capacity to secrete IFNγ, IL-2, and TNF or IL-4, and IL-5, respectively (14). Subsequently, several reports identified human T cell clones separating into Th1 and Th2 categories (15). Th1 CD4+ T cells are crucial in the defense against intracellular pathogens such as mycobacteria (16) whereas Th2 CD4+ T cells mediate the immune defense against parasites such as helminths (17).
Th1 Cells in Circulation and at Site of Inflammation
CD4+ T cells prone to secrete IFNγ (18, 19) were identified in synovial fluids from RA patients while IL-4 production (18) and IL-4+ T cell clones (19) were not increased in synovial fluid compared to peripheral blood. RA was subsequently defined as a Th1-driven disease while Th2 immunity was proposed to have a therapeutic potential in RA (20). CXCR3 was identified as a surface marker for Th1 cells (21) and T-bet as a master transcription factor (22). CXCR3 binds the two IFNγ-induced chemokines CXCL9 and CXCL10 (23). CXCR3 expression on CD4+ T cells (24) as well as CXCL9 and CXCL10 are enriched in synovial fluids (25). Although the vast majority of CD4+ T cells present in synovial joints are of memory phenotype (CD45RO+) (3) and hence antigen-experienced, our insight into their antigen specificity is scarce. Non-specific CD4+ T cells infiltrating the inflamed joint are likely to bias the analysis of the phenotype of relevant CD4+ T cells.
In that context, ex vivo peptide-HLA-DR-tetramer analysis provides a more relevant picture of antigen-specific i.e., citrulline-reactive T cells. Hereby, around 40% of citrulline-reactive CD4+ T cells were found to be CXCR3+ in the blood of RA patients (26) pointing again toward a Th1 signature of autoreactive T cells in RA. Presence of IL-12, IL-18, IFNγ, drivers of Th1 differentiation has also been reported in the synovial tissues of RA patients but not in osteoarthritis patients (Figure 1) (27, 28). However, there is still a lack of information concerning the phenotype of antigen-specific CD4+ T cells at the site of inflammation. Finally, immunodominant T cells epitopes have yet to be discovered in RA that will facilitate the more common use of peptide-HLA-DR-tetramer.
Downstream Effects of Th1 Activity
Th1 cells classically induce macrophage activation (29) characterized in the context of the synovial joint by an increased capacity to produce pro-inflammatory cytokines such as TNF (30). Long-lived resident macrophages are present in synovial tissues from healthy donors (31) while inflammatory macrophages are mainly derived from blood monocytes in active RA (32). The interplay between Th1 cells and these two different subsets of macrophages in the context of the synovial joint is unknown. It will be particularly important to understand if Th1 cells can modify the properties of resident macrophages which could then contribute to perpetuation of the disease (33). Th1 cells have been proposed to influence class switching toward IgG1 and IgG3 in humans (20). In RA, polyclonal antibodies against type II collagen are predominantly of IgG1 and IgG3 subclasses (34) and autoantibodies against citrullinated fibrin are mainly IgG1 (35) suggesting previous interaction with IFNγ-producing cells. Nevertheless, Ig class switching is probably influenced by a multitude of other factors during the course of inflammation and should not be oversimplified by a link to a specific CD4+ T-cell subset. T helper cells also provide help to CD8+ T cells as demonstrated in the context of cancer immunology (36). Despite a reported presence of CD8+ T cells in synovial joints (37), the influence of CD4+ T cells on their activation is currently unknown.
Th1 Targeted Therapy
Evidences of pathogenic function of Th1 cells in RA were contradicted by the lack of efficiency of therapeutic strategy targeting IFNγ (Fontolizumab) initiated in a phase II clinical trial in active RA. This clinical trial was terminated because the first phase did not reach the goals of primary endpoint (38). In the same line, in IFNγ receptor knock-out mice, collagen-induced arthritis was accelerated (39). In this particular mouse model, it has been proposed that IFNγ suppresses inflammation through inhibition of Th17 responses (40). It is however currently unknown if this hypothesis holds true in a human setting. It should be mentioned that biologic therapies targeting TNF, a Th1 cytokine are successful treatments in RA (41). Hence, Th1 cells could act on at least two opposing levels by directly contributing to tissue damage through TNF production or by suppressing Th17 responses.
Since Th1 cells were one of the first T helper cell subsets described, their contribution to the pathogenesis of autoimmune diseases has been investigated in numerous studies. This is also the case both for psoriasis (42, 43) and Crohn's disease (44) that were both initially suggested to be Th1-driven diseases. IFNγ-producing cells were indeed identified at the site of inflammation in these two diseases (42, 45). However, in a phase II clinical trial, Fontolizumab did not induce a robust beneficial clinical effect in Crohn's disease (46). Similarly, in a small study, therapeutic targeting of IFNγ with a humanized anti-IFNγ (HuZAF) showed no significant efficacy in psoriasis patients (47). In these three diseases, despite the clear presence of Th1 cells at the site of inflammation, therapeutic targeting of IFNγ did not lead to beneficial results. IFNγ might be important in the very early phases of the disease through, for instance, the induction of TNF in macrophages (48). It has also been shown that IFNγ induces the expression of vascular cellular adhesion molecule-1 (VCAM-1) on endothelial cells (49) which facilitates lymphocyte migration to the tissue. Finally, through reciprocal regulation, Th1 cells might also suppress the generation of pathogenic T cells such as Th17 cells that contribute to tissue damage.
Th17 Cells and Associated Effector Functions
The Th1/Th2 hypothesis was revisited in 1995, when a third T-cell subset named Th17 cells based on the production of the newly identified cytokine IL-17 (50, 51) was discovered. The IL-17 family comprises 6 members with IL-17A (historically referred as IL-17) and IL-17F being the most closely related, in addition to IL-17B, IL-17C, IL-17D, and IL-17E (52). Th17 cells were initially described as co-expressing the chemokine receptors CCR6 and CCR4 (53) and expressing the master transcription factor RORγT (Figure 1) (54). In addition, CD161 was recently described as a marker of all IL-17 producing cells (55). In epithelial, endothelial and fibroblastic cells, IL-17A stimulates the production of pro-inflammatory cytokines such as IL-6, IL-8, and GM-CSF (56) and promotes neutrophil recruitment (57). Th17 cells are particularly important in protective immunity against fungal and extracellular bacterial infections (Staphylococcus aureus) (58).
Classic Th17 Responses and RA
Early on, production of IL-17 was demonstrated in synovial tissues (59) and synovial fluid (60) of RA patients but not in that of osteoarthritis patients. In addition, IL-17-producing CD4+ T cells from synovial tissues from RA patients could readily be identified (61). The reported frequency of Th17 cells in peripheral blood of RA patients varies according to different studies where either an increase (62, 63) or a status quo in their frequency (64, 65) has been observed. Moreover, only few citrulline-specific CD4+ T cells were CCR6+ positive in peripheral blood of RA when analyzed by ex vivo peptide-HLA-DR-tetramer analysis (26). Th17-inducing cytokines (IL-6 IL1-β, IL-21, TGF-β, and IL-23) (66–69) are present in the synovial joint (Figure 1). Further, synovial IL-17 from RA patients was shown to induce bone resorption (70). Finally, IL-17 contributes to neutrophil recruitment, a hallmark of RA synovial fluid (71). In IL-17-deficient mice, collagen-induced arthritis was decreased supporting the notion that Th17 cells play a pathogenic role in the development of the arthritis (72). It was therefore unexpected that therapeutic targeting of IL-17A (Secukinumab) or the IL-17 receptor (brodalumab) in phase II studies was less successful in RA than in other inflammatory conditions such as psoriasis (73, 74). It was recently proposed that Th17 cells might migrate to the synovium in CCP+ (anti-cyclic citrullinated peptide) early RA patients (75). Hence, in RA, IL-17-producing T cells might contribute during early stages of the disease or be more prominent in a subtype of RA patients.
The Different Facets of IL-17- Anti vs. Pro-inflammatory Features
Another level of complexity arises from evidence that Th17 cells are implicated in different immune responses depending on co-expressed cytokines (76). Indeed, T cells co-expressing IL-17 and IL-10 are thought to be important in mucosal defense but not pathogenic as T cells co-expressing IL-17, IFNγ, or GM-CSF are (66). After anti-TNF treatment, Th17 cells were shown to acquire IL-10 production in RA (77) implying that Th17 cells could also be protective and participate in dampening inflammation in RA. While GM-CSF appears to be a critical component of Th17 pathogenicity in the experimental autoimmune encephalomyelitis (EAE) mouse model (78), it is associated with the Th1 axis in multiple sclerosis (79). Likewise, in synovial joints of RA patients, GM-CSF production is enriched in Th1 cells and not in Th17 cells (80). Several clinical trials targeting GM-CSF are ongoing in RA (81) and will shed light on the pathogenic function of GM-CSF in the context of RA. It also implies that additional markers for Th17 pathogenic subsets are needed to predict which patients are more likely to respond to such therapies.
Different Responses to Anti-IL-17 Blockade in Inflammatory Diseases
While the importance of Th17 cells seems to vary according to the stage and subsets of RA, psoriasis vulgaris can currently be defined as an IL-17-mediated inflammatory skin disease (82). IL-17-secreting cells are found in psoriatic lesions and include CD4+ (83), CD8+ (84), and γδ T cells (85). Phase III trials with a human anti-IL-17A monoclonal antibody (Secukinumab) were successful in psoriasis with response rates of 72–82% at week 12 (86). Th17 cells are also present in the gut of patients with Crohn's disease (87) but IL-17 blockade with Secukinumab was not effective and adverse cases of fungal infections and worsening of the disease were observed (88). In this case, IL-10-producing Th17 cells having a regulatory function (89, 90) might have been targeted contributing to the exacerbation of the disease.
Although early studies suggested that Th17 cells are crucial in most of human inflammatory conditions, different responses to IL-17 blockade contradict this hypothesis. Th17 cells are present at the site of inflammation in RA, Crohn's disease and psoriasis but the difference in the response to anti-IL-17 therapies strongly suggests that their direct implications in the pathogenesis of these diseases differ and reflect different effector functions in tissues. In psoriasis, blocking of IL-17 will alleviate the direct effects of IL-17 on keratinocyte proliferation. In the gut, Th17 cells play an important role in mucosal host defense (58, 91), which is reflected by the secondary effects observed in Crohn's patients under anti-IL-17A therapies. Th17 cells also produce IL-22 which is involved in intestinal epithelial barrier integrity (92). In active RA, targeting IL-17 might not be sufficient to dampen the ongoing cytokine cascade and resorb migration of neutrophils already sequestered in the synovial joint. Targeting of IL-17 at earlier time points or in conjunction with other anti-cytokine blockade might be more effective. Indeed, the combination of IL-17 and TNF synergize to induce the expression of P and E-selectins on endothelial cells that induce an influx of neutrophils (93).
T Cells Providing B Cell Help–Tfh and Tph
Local CD4+ T-cell help to B cells is likely to be a prominent driver of humoral immunity in RA patients seropositive for ACPA (anti-citrullinated protein antibodies) and/or RF (Rheumatoid Factor). About 60–70% of RA patients present with ACPAs and 50-80% of patients are seropositive for RF (94). Already in 1992, synovial T cells from RA patients were shown to induce B cell Ig production in vitro (3). Moreover, ectopic germinal centers are observed in synovial tissues of some RA patients (95, 96). In 2000, a subset of follicular CD4+ T cells (Tfh) expressing CXCR5 and specialized in stimulating antibody responses was described in germinal centers in secondary lymphoid organs (97). They typically express the master transcription factor Bcl-6 as well as IL-21, PD-1, and ICOS. IL-6 and ICOS triggering regulate their differentiation (98) (Figure 1).
B Cell Helpers in Circulation
Circulating Tfh can be analyzed in peripheral blood where their characteristics slightly differ from the follicular ones with a lack of expression of Bcl-6 (98–100). Blood Tfh can be further subdivided into Th1, Th2, Th17 cell subsets with Tfh2 and Tfh17 being the only subsets capable of sustaining the B-cell Ig switch (101). Several studies have described an increased frequency of CXCR5+ICOS+CD4+ Tfh cells in peripheral blood of RA patients which correlates with serological anti-CCP titers and disease severity (102–105). This is accompanied by increased concentrations of IL-21 in the serum of RA patients when compared to healthy donors (102–106). Few reports have investigated the frequency and function of CXCR5+ follicular T cells in psoriasis and Crohn's disease probably due to the lower prevalence of humoral immunity in these patients as compared to RA patients (107, 108). One study reported an increase of Tfh17 CD4+ T cells in peripheral blood of psoriasis patients that correlated with disease activity (109). An increased frequency of Tfh1 and Tfh17 CD4+ T cells has been observed in peripheral blood of patients with Crohn's disease (110).
B Cell Helpers in Tissue
IL-6 (111) and ICOS expression on CD4+ T cells (112), inducers of Tfh differentiation, have been reported in the rheumatic joint. In synovial tissues, few CD4+ T cells express CXCR5 (113, 114) which is surprising given the reported expression of CXCL13, the ligand for CXCR5, in synovial fluids and tissues of RA patients (115, 116). It has recently been proposed that another subset of memory T cells, the peripheral T helper cell (Tph) subset expressing MHC class II and high levels of the co-inhibitory receptor PD-1 provide B-cell help in the synovial joint (Figure 1) (113). These cells lack Bcl-6 but express other typical markers for B-cell help such as IL-21, CXCL13, ICOS, and MAF. It is currently unknown if this subset shares the same ontogeny as Tfh. This study supports earlier reports showing the importance of PD-L1 (program death ligand-1)/PD-1 interaction in RA. Indeed, most of C57BL/6-Pdcd1-/- mice develop arthritis (117) and CD4+ T cells were shown to express PD-1 in synovial joints of RA patients (118). CD4+PD-1+ infiltrating T cells have also been described in the context of breast cancer (119) where they display comparable features with Tph such as ICOS and CXCL13 expression (120). Importantly, these cells also express IFNγ in both RA synovial fluid (113) and breast cancer (119) showing that these T cells have the capacity to convey multiple effector functions contradicting the original Th1/Th2 hypothesis. Recently, the occurrence of case reports of RA following PD-1 or PD-L1 blockade in the context of cancer therapies have also highlighted the role of this checkpoint regulation in the balance between cancer and autoimmunity (121). Cases of psoriasis have also been reported (122) suggesting the importance of PD-1 in the pathogenesis of this disease as well. Although this subset of pathogenic T cells has not been extensively studied in psoriasis, PD-1 expression on Th17 cells has been reported in psoriatic skin of patients (123). So far, the subset of pathogenic Tph cells driving B-cell responses has only been described in the synovial joint of RA patients but is probably relevant to other antibody-positive autoimmune diseases.
Regulatory T-Cell Subsets
The concept of regulatory T cells was revisited in 1995 when the group of Sakaguchi described a population of CD4+ CD25+ T cells capable of preventing the development of several autoimmune diseases in mice (124). Shortly after, the key function of the transcription factor FOXP3 (Forkhead box P3) in the development and function of regulatory T cells was demonstrated in mice (125) and humans (126). Regulatory T cells (Tregs) also express CTLA-4 (cytotoxic T lymphocyte-associated antigen-4) and other markers e.g., GITR (glucocorticoid-induced TNFR family related gene) and CD39 whose expression might vary depending on the context (127). In peripheral blood, Tregs can be divided into naïve and activated regulatory T cells based on the expression of CD45RA (128). Importantly, in humans, FOXP3 expression is not strictly restricted to regulatory T cells as it is transiently up-regulated also in effector T cells (129). Therefore, in humans, regulatory T cells cannot only be defined by the phenotypic expression of FOXP3 and CD25 but need to be supplemented by assessment of their in vitro suppressive capacity and/or the demethylation of the FOXP3 TSDR (Treg cell-specific demethylated region) (130).
Tregs at Site of Inflammation
In RA patients, FOXP3+CD25+CD4+ T cells accumulate in synovial fluid (131, 132) and in synovial tissue (Figure 1) (133). In vitro suppressive capacity and demethylation of the FOXP3 TSDR showed that these regulatory T cells are functional (133, 134). However, effector T-cell proliferation and pro-inflammatory cytokines have been demonstrated to reduce FOXP3 regulatory T-cell function in vitro (134), which is likely to happen also in situ. Indeed, adding TNF during co-culture experiments was shown to inhibit regulatory T-cell functions (135, 136). Whether this effect is mediated through a direct action on effector T cells or regulatory T cells is still a matter of debate (137) since TNF can also induce conventional T-cell proliferation (138). The ontogeny of FOXP3+CD4+ T cells in synovial tissues is still unknown. FOXP3+ Tregs can originate from the thymus (thymus Tregs or tTregs) or be induced in situ from conventional T cells during infections or other inflammatory processes (induced Tregs or iTregs) (139).
Regulatory T cells from peripheral blood and inflamed joints of juvenile arthritis patients were shown to harbor a different T cell Receptor Vβ usage than conventional T cells suggesting that Tregs would be generated independently of conventional T cells (140). Whether this is also the case in RA is currently unknown but IL-2 (141) and TGFβ (142), important for induced regulatory T-cell generation are present in synovial fluids of RA.
In plaques of psoriasis patients, the frequency of FOXP3+ CD4+ T cells is increased when compared to healthy skins (143) where only few Tregs are found in the dermis and epidermis (143). FOXP3+CD4+ T cells present in the psoriatic lesions also co-expressed IL-17 (143, 144). Hence, as for RA, it has been suggested that the inflammatory milieu through for instance IL-6 (145) may affect regulatory T-cell function.
Therapeutic Interventions Affecting Treg Function and/or Frequencies
Current therapies given in RA that alleviate inflammation are therefore likely to restore or increase regulatory T-cell function. In RA patients treated with anti-TNF therapy, an increased frequency of CD4+CD25 high T cells was observed in responders when compared to non-responders (146). Similarly, an expansion of CD4+CD25+FOXP3+ T cells has been observed after anti-IL-6R blockade and was accompanied by a decrease in Th17 frequency (147). Treg and Th17 cells have opposite functions but their differentiation both rely on the presence of TGFβ. In the absence of pro-inflammatory cytokines such as IL-6, Treg differentiation is enhanced whereas in the presence of IL-6, Th17 differentiation is promoted. Hence, targeting IL6R contributes to increase the ratio between Treg and Th17 cells in RA patients (147). Recently, a therapeutic strategy based on the use of low-dose of IL-2 has been developed to directly induce the expansion of Tregs in vivo in autoimmune patients (148). This concept relies on the fact that Treg development and expansion is dependent on low levels of IL2R signaling (149). Clinical trials investigating such treatments in RA are currently ongoing.
IL-10 Producing Tr1 Cells
Another subset of T regulatory cells named T regulatory type 1 cells (Tr1) is defined by their suppressive function combined with their capacity to produce IL-10 (150). So far, no unique cell surface marker specific for Tr1 cells has been identified but the expression of several markers such as ICOS, PD-1, CD49b, TIM-3, and LAG3 is increased on this subset (151). Tr1 cells have been extensively studied in the context of intestinal mucosal immunity and the prevention of colitis (152, 153). The importance of IL-10 in intestinal immunity is also illustrated by the identification of mutations in IL-10, IL-10RA, and IL-10RB genes in children suffering from inflammatory bowel disease (IBD) (153). The frequency of Tr1 cells (defined by production of IL-10 and low production of IL-2 and IL-4) was found to be decreased in peripheral blood and synovial fluid of RA patients when compared to osteoarthritis patients and healthy donors (154). However, IL-10 does not only have an anti-suppressive effect but is also involved in B-cell activation and antibody production (155) and is secreted by follicular helper T cells (156). Clearly, IL-10 alone is not sufficient to define Tr1 cells and additional markers are needed to understand their possible function in synovial tissues. As of today, Tr1 cells have been clearly implicated in intestinal mucosal immunity but their contribution to the synovial joint homeostasis is less clear.
Cytotoxic CD4+ T Cells
Although not part of the general text book, cytotoxic features of CD4+ T cells have been observed already more than 20 years ago but were initially described in T cell clones (157) raising the concern that their generation might be an artifact due to repeated in vitro stimulation. However, the presence of CD4+ T cells with cytotoxic activities (CD4+ CTLs) has been confirmed ex vivo in human diseases driven by a variety of viruses like CMV (158) or dengue (159) as reviewed in Juno et al. (160). In healthy individuals, the frequency of peripheral CD4+ CTLs is usually very low (161).
Cytotoxic T Cells in RA
In peripheral blood of a subset of RA patients, several groups have reported an increased frequency of a population of CD4+CD28null cells expressing perforin, granzymes, and other cytotoxic features (162–164) (Figure 1). Although CD4+ CD28null T cells are not enriched in synovial fluid, the presence of perforin+CD4+ T cells has been repeatedly reported in synovial fluids and tissues (164–166). No unique marker is associated with CD4+CD28null T cells but they express proteins related to their cytotoxic functions which are more commonly found in CD8+ CTLs and Natural Killer (NK) cells including granzyme B, granzyme A, and perforin-1. NK cell activating receptors such as NKG2D are also found on CD4+CD28null cells (167). Further investigation of CD4+CD28null cells or an updated approach of studying such cells in RA is warranted in the light of recent characterization of CD4+ CTLs at the single cell level (168, 169). In particular, the recently described transcription factor Hobit was identified in CD4+ CTLs where its precise function remains to be determined (170). We recently demonstrated that the transcription factor EOMES, implicated in terminal T-cell differentiation and the transcription of perforin-1 (171), is increased in CD4+ T cells from synovial fluids of RA patients (166). Using single cell transcriptomics, expanded T cell clones present in the synovium of RA patients were also shown to express EOMES and granzyme B when compared to circulating expanded clones (172). Antigen-specificity of CD4+ CTLs in RA is still debated although an increase in their frequency is more prominent in CMV-seropositive patients suggesting a link between CMV infections and the generation of this T-cell subset in RA patients (173). Nevertheless, repeated antigen stimulation, a classical feature of chronic inflammation, seems to be a constant feature in CD4+ CTL generation (160). Although CD4+ CTLs were initially suggested to derive from Th1 cells, it has also been proposed that they represent an independent lineage with CRTAM (class I-restricted T cell-associated molecule) as a possible marker for precursors of CD4+ CTLs (174). IL-2 and IL-15 as well as 4-1BB triggering are thought to favor their generation (160). Importantly, IL-2 (141) and IL-15 (175) are present in synovial fluids of RA patients while soluble forms of 4-1BB and 4-1BB ligand are increased in peripheral blood of RA patients (176). The functional implications of CD4+ CTLs interactions with HLA class-II expressing cells in synovial joints such as macrophages, dendritic cells, neutrophils (177), chondrocytes (178), or endothelial cells remains largely unknown. They might directly contribute to joint damage as it has been shown that CD4+ CTLs can directly lyse EBV-infected B cells (179). Another possibility is that they participate in the hypercitrullination of NETs (Neutrophil Extracellular Traps) through a perforin-dependent mechanism (180). We indeed observed that the level of ACPAs correlated with the frequency of perforin+CD4+ T cells in synovial joint of RA patients (166). It was recently shown that CIA was attenuated in granzyme A-/- mice (181) that presented reduced osteoclastogenesis. The source of granzyme A was not identified in this study but we have shown that CD4+ T cells producing granzyme A are present in synovial joints of RA patients (166). Granzyme A also stimulates monocytes to produce IL-6, IL-8, and TNF (182) which can contribute to increased inflammation in the RA joint. The presence of CD4+ CTL at the site of inflammation has been reported in several autoimmune diseases (183–185). Cytotoxic CD4+ T cells expressing NKG2D were identified in the lamina propria of patients with Crohn's disease (186). Likewise, perforin+ CD4+ T cells have been observed in skin lesions from patients with psoriasis (187).
Therapeutic Strategies Affecting Cytotoxic CD4+ T Cells
TNF has been shown to repress the expression of the CD28 gene (188). In an early study, the expression of CD28 was indeed increased on CD4+ T cells in RA patients undergoing anti-TNF therapy but markers of cytotoxicity were not investigated (189). Direct approaches to target cytotoxic CD4+ T cells can be achieved by targeting specific molecules expressed on these cells. For instance, an antibody targeting NKG2D induced a reduction in disease activity in some Crohn's disease patients in a phase II clinical trial (190). Cytotoxic CD4+ T cells also express CX3CR1, the receptor for fractalkine, a chemokine expressed on synoviocytes and endothelial cells of synovial tissues from RA patients (191). A phase II clinical trial is currently investigating the effect of fractalkine blockade in RA patients refractory to TNF inhibitors or methotrexate therapy. Clearly, the results of these new therapeutic blockades will bring new insights into the contribution of cytotoxic CD4+ T cells to RA.
Tissue-resident Memory T Cells
Tissue-resident memory T cells (Trm) are memory T cells that remain in a given tissue during a long period of time. They are well-described in mucosal tissues where they contribute to the first line of adaptive defense after re-exposure to a specific pathogen. For instance, influenza-specific resident memory CD8+ T cells have been described in the lung (192). The transcriptional signature of Trm cells differs from circulating T cells and includes genes important for their migration and retention in a given tissue (193). While the markers defining CD4+ Trm T cells are likely to slightly differ depending on the tissue, receptors such as CD69, CD49a, PD-1, and CXCR6 are commonly expressed (193). Persistence of memory T cells in tissues is beneficial in the rapid intervention against pathogenic infections infections but is also proposed to participate in the maintenance of pathogenicity in autoimmune inflammatory conditions.
Trm T Cells in Disease
Psoriasis is the best example of a clear implication of resident memory T cells in the pathogenesis and resurgence of the disease. Indeed, Th17 Trm cells are present in recurrent psoriatic skin lesions and persist in resolved skin even after effective treatment (194). These data highlight the implication of Trm T cells in the reappearance of psoriatic lesions in a site-specific manner (194). The presence of T cells in perivascular areas of healthy synovial joints has been reported but is largely inferior to the number of T cells observed in mucosal tissues at steady state (195). Persistence of inflammation in synovial joints is observed in RA patients even in clinical remission (196) and might be indicative of Trm involvement in RA as described in psoriasis. Recently, CD8+ T cells with features of Trm cells such as CD69, PD-1, and CD103 have been identified in synovial fluids of juvenile arthritis patients (197). A fraction of CD4+ T cells express PD-1 (118) and CD69 (198) in synovial fluid of RA patients but whether these cells are bonafide Trm cells is so far unknown. Importantly, the peripheral T helper cell subset recently described in synovial joints (113) also expresses PD-1 and CD69 suggesting at least some overlap with resident memory T-cell markers. In synovial fluids, T cell clones with identical TCR sequences persist over time indicative of retention mechanisms in the joint (199). However, clonal T-cell expansions have not been studied in the context of Trm markers. In particular, the maintenance of Trm T cells in synovial tissues during the course of the disease and during relapses has not been assessed in RA. This set of experiments would provide information about the nature of effector T-cell functions implicated in tissue damage as exemplified by the persistence of Th17 Trm cells in psoriasis.
Given the recent discovery of Trm T cells, no specific therapeutic strategy is currently targeting this population. The persistence of Trm T cells in the plaques of psoriasis patients show that they probably resist current therapies. Hence, future therapies targeting the maintenance of resident T cells in tissues represent an attractive perspective.
Genetic Risk Variants and T-Cell Subsets
The study of genetic risk variants can allow a better understanding of the pathogenesis of the disease and the cell subsets involved and also helps to validate therapeutic targets (200). The first genetic contribution to RA is located in the HLA-DRB1 locus (8). Genome-wide association studies have also identified 100 additional loci associated to RA (Table 1 and Supplementary Table 1) (201–203) that are predicted to target immune pathways. This set of gene loci does not correspond to a unique T-cell subset signature. However, epigenetic chromatin modifications (trimethylation of histone H3 at lysine 4) of RA-associated risk alleles are enriched in primary CD4+ regulatory T cells (201) suggesting that the function of this subset might be implicated in RA. Psoriasis represents a clear example where part of the 35 genetic loci can be assigned to the IL-23/Th17 pathway (204). Some of the genetic variants shared between psoriasis and Crohn's disease correspond to the IL-23 pathway and the T-helper 17 cell lineage (Table 1) based on gene ontology biological process analysis [Enrichr (205)]. Shared genetic variants between RA and Crohn's disease highlight a positive regulation of IFNγ secretion that might reflect part of the Th1 component of these diseases (Table 1). Many of the genetic variants associated with RA are not common to psoriasis or Crohn's disease emphasizing the importance of distinct mechanisms in the pathogenesis of the disease (Supplementary Table 1). For instance, genetic variants in the IL-10 and IL-10R loci are only found associated with Crohn's disease, which is striking given the importance of IL-10 regulatory function in the intestinal barrier as well as in inducing IgA class switch (206). Similarly, a risk locus encompassing PADI4 (peptidylarginine deiminase type 4) is found only in RA (Supplementary Table 1). PADI4 controls citrullination processes that are highly relevant in RA where anti-citrullinated peptides antibodies (ACPAs) are commonly found. Still, a clear correlation between genetic risk variants in RA and a specific T-cell subset is lacking. Nevertheless, this can be explained by several factors. First, although some of the locus variants directly have an effect on the expression of the assigned gene (201) (eQTL (expression quantitative trait loci effect)), in most cases the functional consequences of the genetic variants have not been elucidated. We recently demonstrated that the PTPN22 risk allele (rs2476601) favors the development of EOMES+ CD4+ T cells with cytotoxic features in RA (166). This finding and the fact that EOMES risk variants are associated with RA (Supplementary Table 1) suggest that cytotoxic T cells probably contribute to the disease. Second, RA is a complex disease that might encompass several sub-phenotypes with distinct stages and genetic signatures that are not uncovered in current GWAS. Finally, our understanding of genetic variants is evolving together with our knowledge on T-cell differentiation mechanisms and will be revisited in the light of emerging data on new T-cell subsets.
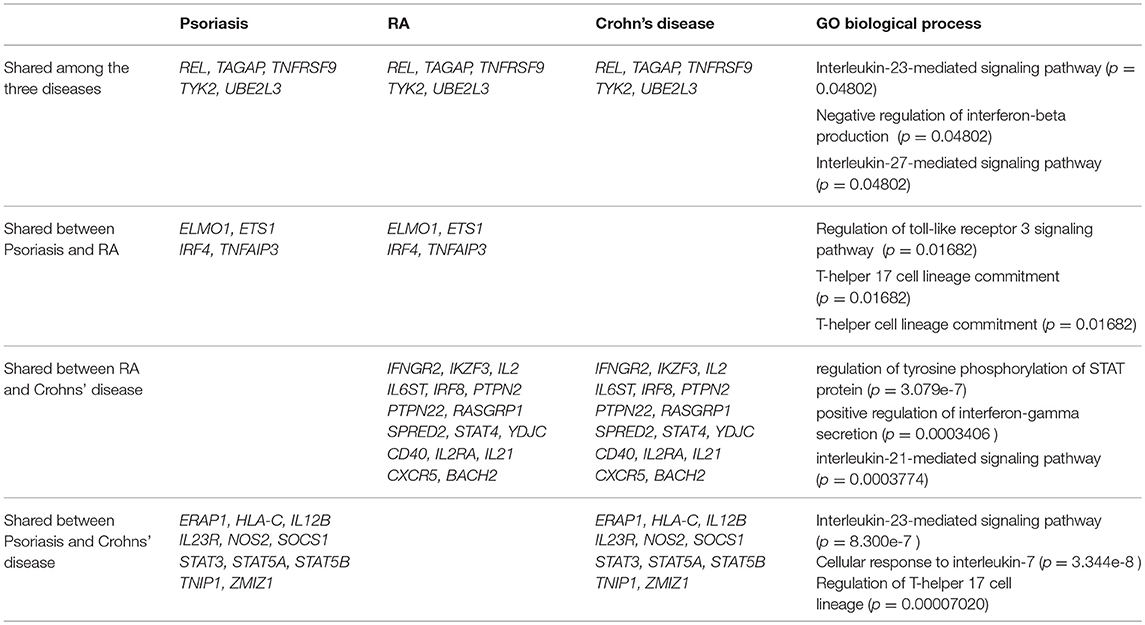
Table 1. Shared genetic variants associated to Rheumatoid Arthritis (RA), Psoriasis and Crohn's disease referenced at Immunobase corresponding to GO biological process (enrichr), p = adjusted p-value.
Conclusions and Future Directions
Rheumatoid Arthritis is a complex disease where several T-cell subsets have been proposed to be involved. During the last 10 years, new therapeutic trials as well as extended GWAS have provided new data to reinvestigate the contribution of T-cell subsets in RA. Based on the therapeutic intervention and the genetic data, RA cannot be classified as a Th17-driven disease such as for example psoriasis. Moreover, it has become clear that in human inflammatory contexts, CD4+ T cells harbor multiple effector function profiles that do not follow the classical dogma of T helper classification. IFNγ and IL-17 are present in synovial joints of RA patients but their blockade does not necessarily improve disease suggesting that their effector function is not rate-limiting for the downstream processes. These cytokines initiate a cascade of proinflammatory cytokines that may no longer be reversed by blocking IFNγ or IL-17 alone. Earlier targeting of these cytokines or combined therapeutic targeting of more downstream cytokines such as GM-CSF and TNF might be more effective. Th1 cells might also have already differentiated into cytotoxic CD4+ T cells capable of inducing cytotoxic damage and a cascade of proinflammatory cytokines. In that context, granzyme A represents a good candidate target since it induces osteoclastogenesis (181) as well as proinflammatory cytokines (182). In ACPA+ RA patients, recent identification of pathogenic Tph cells driving B-cell responses show that, in addition to IL-10, these cells also produce IFNγ and perforin-1 (113). How these multiple effector functions are integrated during T cell/APC interactions is currently not known. Still, the elevated expression of PD-1 on this subset confirms that this co-inhibitory signaling pathway is important in RA and represents a possible target. The emergence of the concept of resident memory T cells capable of perpetuating the disease represent a breakthrough in the understanding of the mechanisms behind disease chronicity and might also favor the development of new therapeutics. Based on the comparison between these three inflammatory conditions, it is clear that some pathogenic pathways are common to these diseases while some others are very distinct and are probably a reflection of different tissue-mediated immunity components. These data should also encourage us to stratify RA patients in subgroups who might be more likely to respond to certain therapies based on the stage of the disease as well as the genetic variants associated. Moreover, a more common use of single cell technologies will allow the dissection of functional properties of rare CD4+ T cells present in inflammatory tissues. However, caution should be taken when analyzing T-cell subsets present in inflammatory tissues since bystander T cells can bias our view of pathogenic T cells. The presence of specific T cell types in inflammatory tissues does not imply that they are necessarily involved in the pathogenesis of the disease. Hence, the analysis of antigen-specific T cells might give a more accurate picture of important effector T-cell functions in the aforementioned inflammatory conditions. Finally, instead of targeting a distinct T-cell subset or effector function, an alternative approach would be to perform antigen-specific targeting and hence to target pathogenic T cells irrespective of their phenotype. We hope that, with this review, we provide a better understanding of current knowledge of CD4+ T-cell functions in RA and highlight the possible ways to identify pathogenic T cells that could be therapeutically targeted.
Author Contributions
KC, CG, and VM discussed the content of the review. KC wrote the manuscript. CG and VM edited the manuscript.
Funding
This study is supported by grants from the Margaretha af Ugglas foundation, IMI RTCure (grant:777357), the Swedish research council, the Swedish association against rheumatism, the King Gustaf V 80-year Foundation and the Ulla and Gustaf af Ugglas foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr Bence Rethi (Division of Rheumatology, Department of Medicine, Karolinska Institute, Karolinska University Hospital Solna, Stockholm, Sweden) and Dr Sabrina Ruhrmann (Department of Clinical Neuroscience, Karolinska Institute, Sweden) for critical reading of the manuscript. The authors thank Annika van Vollenhoven (Division of Rheumatology, Department of Medicine, Karolinska Institute, Karolinska University Hospital Solna, Stockholm, Sweden) for proof reading of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00353/full#supplementary-material
References
1. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Primers. (2018) 4:18001. doi: 10.1038/nrdp.2018.1
2. Ospelt C. Synovial fibroblasts in 2017. RMD Open. (2017) 3:e000471. doi: 10.1136/rmdopen-2017-000471
3. Thomas R, Mcilraith M, Davis LS, Lipsky PE. Rheumatoid synovium is enriched in Cd45rbdim mature memory T-cells that are potent helpers for B-cell differentiation. Arthritis Rheumat. (1992) 35:1455–65. doi: 10.1002/art.1780351209
4. Klarenbeek PL, de Hair MJ, Doorenspleet ME, van Schaik BD, Esveldt RE, van de Sande MG, et al. Inflamed target tissue provides a specific niche for highly expanded T-cell clones in early human autoimmune disease. Ann Rheum Dis. (2012) 71:1088–93. doi: 10.1136/annrheumdis-2011-200612
5. Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. (2003) 349:1907–15. doi: 10.1056/NEJMoa035075
6. Stastny P. Association of the B-cell alloantigen DRw4 with rheumatoid arthritis. N Engl J Med. (1978) 298:869–71. doi: 10.1056/NEJM197804202981602
7. Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. (2007) 447:661–78. doi: 10.1038/nature05911
8. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis - an approach to understanding the molecular-genetics of susceptibility to rheumatoid-arthritis. Arthritis Rheumat. (1987) 30:1205–13. doi: 10.1002/art.1780301102
9. Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol. (2003) 171:538–41. doi: 10.4049/jimmunol.171.2.538
10. Gerstner C, Dubnovitsky A, Sandin C, Kozhukh G, Uchtenhagen H, James EA, et al. Functional and Structural characterization of a novel HLA-DRB1*04:01-restricted alpha-enolase T cell epitope in rheumatoid arthritis. Front Immunol. (2016) 7:494. doi: 10.3389/fimmu.2016.00494
11. Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. (2013) 210:2569–82. doi: 10.1084/jem.20131241
12. Chemin K, Pollastro S, James E, Ge C, Albrecht I, Herrath J, et al. A Novel HLA-DRB1*10:01-restricted T cell epitope from citrullinated type II Collagen relevant to rheumatoid arthritis. Arthritis Rheumatol. (2016) 68:1124–35. doi: 10.1002/art.39553
13. Chemin K, Klareskog L, Malmstrom V. Is rheumatoid arthritis an autoimmune disease? Curr Opin Rheumatol. (2016) 28:181–8. doi: 10.1097/Bor.0000000000000253
14. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. (1986) 136:2348–57.
15. Romagnani S. Human TH1 and TH2 subsets: doubt no more. Immunol Today. (1991) 12:256–7. doi: 10.1016/0167-5699(91)90120-I
16. Doffinger R, Jouanguy E, Altare F, Wood P, Shirakawa T, Novelli F, et al. Inheritable defects in interleukin-12- and interferon-gamma-mediated immunity and the TH1/TH2 paradigm in man. Allergy. (1999) 54:409–12.
17. Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol. (2018) 18:121–33. doi: 10.1038/nri.2017.118
18. Dolhain RJ, van der Heiden AN, ter Haar NT, Breedveld FC, Miltenburg AM. Shift toward T lymphocytes with a T helper 1 cytokine-secretion profile in the joints of patients with rheumatoid arthritis. Arthritis Rheum. (1996) 39:1961–9.
19. Miltenburg AM, van Laar JM, de Kuiper R, Daha MR, Breedveld FC. T cells cloned from human rheumatoid synovial membrane functionally represent the Th1 subset. Scand J Immunol. (1992) 35:603–10.
20. Schulze-Koops H, Kalden JR. The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best Pract Res Clin Rheumatol. (2001) 15:677–91. doi: 10.1053/berh.2001.0187
21. Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. (1998) 187:875–83.
22. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. (2000) 100:65–9. doi: 10.1016/s0092-8674(00)80702-3
23. Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, et al. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. (1996) 184:963–9.
24. Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. (1998) 101:746–54. doi: 10.1172/JCI1422
25. Patel DD, Zachariah JP, Whichard LP. CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin Immunol. (2001) 98:39–45. doi: 10.1006/clim.2000.4957
26. James EA, Rieck M, Pieper J, Gebe JA, Yue BB, Tatum M, et al. Citrulline-specific Th1 cells are increased in rheumatoid arthritis and their frequency is influenced by disease duration and therapy. Arthritis Rheumatol. (2014) 66:1712–22. doi: 10.1002/art.38637
27. Morita Y, Yamamura M, Nishida K, Harada S, Okamoto H, Inoue H, et al. Expression of interleukin-12 in synovial tissue from patients with rheumatoid arthritis. Arthritis Rheum. (1998) 41:306–14. doi: 10.1002/1529-0131(199802)41:2<306::AID-ART15>3.0.CO;2-4
28. Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest. (1999) 104:1393–401. doi: 10.1172/JCI7317
29. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
30. Maruotti N, Cantatore FP, Crivellato E, Vacca A, Ribatti D. Macrophages in rheumatoid arthritis. Histol Histopathol. (2007) 22:581–6. doi: 10.14670/HH-22.581
31. Alivernini S, Tolusso B, Petricca L, Bui L, Di Sante G, Peluso G, et al. Synovial features of patients with rheumatoid arthritis and psoriatic arthritis in clinical and ultrasound remission differ under anti-TNF therapy: a clue to interpret different chances of relapse after clinical remission? Ann Rheum Dis. (2017) 76:1228–36. doi: 10.1136/annrheumdis-2016-210424
33. Kurowska-Stolarska M, Alivernini S. Synovial tissue macrophages: friend or foe? RMD Open. (2017) 3:e000527. doi: 10.1136/rmdopen-2017-000527
34. Cook AD, Mackay IR, Cicuttini FM, Rowley MJ. IgG subclasses of antibodies to type II collagen in rheumatoid arthritis differ from those in systemic lupus erythematosus and other connective tissue diseases. J Rheumatol. (1997) 24:2090–6.
35. Chapuy-Regaud S, Nogueira L, Clavel C, Sebbag M, Vincent C, Serre G. IgG subclass distribution of the rheumatoid arthritis-specific autoantibodies to citrullinated fibrin. Clin Exp Immunol (2005) 139:542–50. doi: 10.1111/j.1365-2249.2004.02708.x
36. Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. (2018) 18:635–47. doi: 10.1038/s41577-018-0044-0
37. Cho BA, Sim JH, Park JA, Kim HW, Yoo WH, Lee SH, et al. Characterization of effector memory CD8+ T cells in the synovial fluid of rheumatoid arthritis. J Clin Immunol. (2012) 32:709–20. doi: 10.1007/s10875-012-9674-3
38. Pollard KM, Cauvi DM, Toomey CB, Morris KV, Kono DH. Interferon-gamma and systemic autoimmunity. Discov Med. (2013) 16:123–31. Available online at: http://www.discoverymedicine.com
39. Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated collagen-induced arthritis in IFN-gamma receptor-deficient mice. J Immunol. (1997) 158:5507–13.
40. Lee J, Lee J, Park MK, Lim MA, Park EM, Kim EK, et al. Interferon gamma suppresses collagen-induced arthritis by regulation of Th17 through the induction of indoleamine-2,3-deoxygenase. PLoS ONE. (2013) 8:e60900. doi: 10.1371/journal.pone.0060900
41. Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. (2002) 2:364–71. doi: 10.1038/nri802
42. Schlaak JF, Buslau M, Jochum W, Hermann E, Girndt M, Gallati H, et al. T cells involved in Psoriasis vulgaris belong to the Th1 subset. J Invest Dermatol. (1994) 102:145–9.
43. Diani M, Altomare G, Reali E. T helper cell subsets in clinical manifestations of Psoriasis. J Immunol Res. (2016) 2016:7692024. doi: 10.1155/2016/7692024
44. Brand S. Crohn's disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn's disease. Gut. (2009) 58:1152–67. doi: 10.1136/gut.2008.163667
45. Parronchi P, Romagnani P, Annunziato F, Sampognaro S, Becchio A, Giannarini L, et al. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn's disease. Am J Pathol. (1997) 150:823–32.
46. Reinisch W, de Villiers W, Bene L, Simon L, Racz I, Katz S, et al. Fontolizumab in moderate to severe Crohn's disease: a phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm Bowel Dis. (2010) 16:233–42. doi: 10.1002/ibd.21038
47. Harden JL, Johnson-Huang LM, Chamian MF, Lee E, Pearce T, Leonardi CL, et al. Humanized anti-IFN-gamma (HuZAF) in the treatment of psoriasis. J Allergy Clin Immunol. (2015) 135:553–6. doi: 10.1016/j.jaci.2014.05.046
48. Collart MA, Belin D, Vassalli JD, de Kossodo S, Vassalli P. Gamma interferon enhances macrophage transcription of the tumor necrosis factor/cachectin, interleukin 1, and urokinase genes, which are controlled by short-lived repressors. J Exp Med. (1986) 164:2113–8.
49. Wellicome SM, Thornhill MH, Pitzalis C, Thomas DS, Lanchbury JS, Panayi GS, et al. A monoclonal antibody that detects a novel antigen on endothelial cells that is induced by tumor necrosis factor, IL-1, or lipopolysaccharide. J Immunol. (1990) 144:2558–65.
50. Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. (1995) 3:811–21.
51. Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, et al. Human IL-17: a novel cytokine derived from T cells. J Immunol. (1995) 155:5483–6.
52. Jin W, Dong C. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect. (2013) 2:e60. doi: 10.1038/emi.2013.58
53. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. (2007) 8:639–46. doi: 10.1038/ni1467
54. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
55. Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol. (2010) 40:2174–81. doi: 10.1002/eji.200940257
56. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. (1996) 183:2593–603.
57. Borregaard N. Neutrophils, from marrow to microbes. Immunity. (2010) 33:657–70. doi: 10.1016/j.immuni.2010.11.011
58. Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. (2011) 332:65–8. doi: 10.1126/science.1200439
59. Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. (1998) 161:409–14.
60. Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, et al. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. (2000) 164:2832–8. doi: 10.4049/jimmunol.164.5.2832
61. Pene J, Chevalier S, Preisser L, Venereau E, Guilleux MH, Ghannam S, et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol. (2008) 180:7423–30. doi: 10.4049/jimmunol.180.11.7423
62. Leipe J, Grunke M, Dechant C, Reindl C, Kerzendorf U, Schulze-Koops H, et al. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum. (2010) 62:2876–85. doi: 10.1002/art.27622
63. van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM, et al. Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum. (2011) 63:73–83. doi: 10.1002/art.30093
64. Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, et al. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis. (2008) 67:1299–304. doi: 10.1136/ard.2007.080341
65. Shahrara S, Huang Q, Mandelin AM II, Pope RM. TH-17 cells in rheumatoid arthritis. Arthritis Res Ther. (2008) 10:R93. doi: 10.1186/ar2477
66. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
67. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. (2008) 9:641–9. doi: 10.1038/ni.1610
68. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. (2008) 9:650–7. doi: 10.1038/ni.1613
69. Cascao R, Moura RA, Perpetuo I, Canhao H, Vieira-Sousa E, Mourao AF, et al. Identification of a cytokine network sustaining neutrophil and Th17 activation in untreated early rheumatoid arthritis. Arthritis Res Ther. (2010) 12:R196. doi: 10.1186/ar3168
70. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with Rheumatoid Arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. (1999) 103:1345–52. doi: 10.1172/JCI5703
71. Kaplan MJ. Role of neutrophils in systemic autoimmune diseases. Arthritis Res Ther. (2013) 15:219. doi: 10.1186/ar4325
72. Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. (2003) 171:6173–7. doi: 10.4049/jimmunol.171.11.6173
73. Koenders MI, van den Berg WB. Secukinumab for rheumatology: development and its potential place in therapy. Drug Des Devel Ther. (2016) 10:2069–80. doi: 10.2147/DDDT.S105263
74. Baker KF, Isaacs JD. Novel therapies for immune-mediated inflammatory diseases: What can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic Lupus erythematosus, Psoriasis, Crohn's disease and Ulcerative colitis? Ann Rheum Dis. (2018) 77:175–87. doi: 10.1136/annrheumdis-2017-211555
75. Arroyo-Villa I, Bautista-Caro MB, Balsa A, Aguado-Acin P, Nuno L, Bonilla-Hernan MG, et al. Frequency of Th17 CD4+ T cells in early rheumatoid arthritis: a marker of anti-CCP seropositivity. PLoS ONE. (2012) 7:e42189. doi: 10.1371/journal.pone.0042189
76. Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature. (2012) 484:514–8. doi: 10.1038/nature10957
77. Evans HG, Roostalu U, Walter GJ, Gullick NJ, Frederiksen KS, Roberts CA, et al. TNF-alpha blockade induces IL-10 expression in human CD4+T cells. Nat Commun. (2014) 5:3199. doi: 10.1038/ncomms4199
78. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. (2011) 12:560–7. doi: 10.1038/ni.2027
79. Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med. (2014) 6:241ra280. doi: 10.1126/scitranslmed.3008706
80. Yamada H, Haraguchi A, Sakuraba K, Okazaki K, Fukushi JI, Mizu-Uchi H, et al. Th1 is the predominant helper T cell subset that produces GM-CSF in the joint of rheumatoid arthritis. RMD Open. (2017) 3:e000487. doi: 10.1136/rmdopen-2017-000487
81. Shiomi A, Usui T, Mimori T. GM-CSF as a therapeutic target in autoimmune diseases. Inflamm Regen. (2016) 36:8. doi: 10.1186/s41232-016-0014-5
82. Krueger JG, Fretzin S, Suarez-Farinas M, Haslett PA, Phipps KM, Cameron GS, et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. (2012) 130:145–54.e149. doi: 10.1016/j.jaci.2012.04.024
83. Matos TR, O'Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest. (2017) 127:4031–41. doi: 10.1172/JCI93396
84. Ortega C, Fernandez AS, Carrillo JM, Romero P, Molina IJ, Moreno JC, et al. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J Leukoc Biol. (2009) 86:435–43. doi: 10.1189/JLB.0109046
85. Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity. (2011) 35:596–610. doi: 10.1016/j.immuni.2011.08.001
86. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med. (2014) 371:326–38. doi: 10.1056/NEJMoa1314258
87. Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. (2007) 204:1849–61. doi: 10.1084/jem.20070663
88. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. (2012) 61:1693–700. doi: 10.1136/gutjnl-2011-301668
89. Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, et al. Control of TH17 cells occurs in the small intestine. Nature. (2011) 475:514–8. doi: 10.1038/nature10228
90. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. (2007) 8:1390–97. doi: 10.1038/ni1539
91. O'Connor W Jr. Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. (2009) 10:603–9. doi: 10.1038/ni.1736
92. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
93. Griffin GK, Newton G, Tarrio ML, Bu DX, Maganto-Garcia E, Azcutia V, et al. IL-17 and TNF-alpha sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol. (2012) 188:6287–99. doi: 10.4049/jimmunol.1200385
94. Conigliaro P, Chimenti MS, Triggianese P, Sunzini F, Novelli L, Perricone C, et al. Autoantibodies in inflammatory arthritis. Autoimmun Rev. (2016) 15:673–83. doi: 10.1016/j.autrev.2016.03.003
95. Schroder AE, Greiner A, Seyfert C, Berek C. Differentiation of B cells in the non-lymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci USA. (1996) 93:221–5.
96. Kim HJ, Krenn V, Steinhauser G, Berek C. Plasma cell development in synovial germinal centers in patients with rheumatoid and reactive arthritis. J Immunol. (1999) 162:3053–62.
97. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. (2000) 192:1553–62. doi: 10.1084/jem.192.11.1553
98. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
99. Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, et al. ICOS deficiency is associated with a severe reduction of CXCR5(+) CD4 germinal center Th cells. J Immunol. (2006) 177:4927–32. doi: 10.4049/jimmunol.177.7.4927
100. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. (2013) 39:770–81. doi: 10.1016/j.immuni.2013.09.007
101. Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. (2011) 34:108–21. doi: 10.1016/j.immuni.2010.12.012
102. Wang J, Shan Y, Jiang Z, Feng J, Li C, Ma L, et al. High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin Exp Immunol. (2013) 174:212–20. doi: 10.1111/cei.12162
103. Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C, et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol. (2012) 2012:827480. doi: 10.1155/2012/827480
104. Zhang Y, Li Y, Lv TT, Yin ZJ, Wang XB. Elevated circulating Th17 and follicular helper CD4(+) T cells in patients with rheumatoid arthritis. APMIS. (2015) 123:659–66. doi: 10.1111/apm.12399
105. Nakayamada S, Kubo S, Yoshikawa M, Miyazaki Y, Yunoue N, Iwata S, et al. Differential effects of biological DMARDs on peripheral immune cell phenotypes in patients with rheumatoid arthritis. Rheumatology(2018) 57:164–74. doi: 10.1093/rheumatology/kex012
106. Liu R, Wu Q, Su D, Che N, Chen H, Geng L, et al. A regulatory effect of IL-21 on T follicular helper-like cell and B cell in rheumatoid arthritis. Arthritis Res Ther. (2012) 14:R255. doi: 10.1186/ar4100
107. Singh S, Singh U, Singh S. Prevalence of autoantibodies in patients of psoriasis. J Clin Lab Anal. (2010) 24:44–8. doi: 10.1002/jcla.20365
108. Roggenbuck D, Reinhold D, Schierack P, Bogdanos DP, Conrad K, Laass MW. Crohn's disease specific pancreatic antibodies: clinical and pathophysiological challenges. Clin Chem Lab Med. (2014) 52:483–94. doi: 10.1515/cclm-2013-0801
109. Wang Y, Wang L, Shi Y, Wang F, Yang H, Han S, et al. Altered circulating T follicular helper cell subsets in patients with psoriasis vulgaris. Immunol Lett. (2017) 181:101–8. doi: 10.1016/j.imlet.2016.09.008
110. Wang Z, Wang Z, Diao Y, Qian X, Zhu N, Dong W. Circulating follicular helper T cells in Crohn's disease (CD) and CD-associated colorectal cancer. Tumour Biol. (2014) 35:9355–59. doi: 10.1007/s13277-014-2208-2
111. Waage A, Kaufmann C, Espevik T, Husby G. Interleukin-6 in synovial fluid from patients with arthritis. Clin Immunol Immunopathol. (1989) 50:394–8. doi: 10.1016/0090-1229(89)90146-3
112. Nanki T, Shimaoka T, Hayashida K, Taniguchi K, Yonehara S, Miyasaka N. Pathogenic role of the CXCL16-CXCR6 pathway in rheumatoid arthritis. Arthritis Rheum. (2005) 52:3004–14. doi: 10.1002/art.21301
113. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. (2017) 542:110–4. doi: 10.1038/nature20810
114. Chu Y, Wang F, Zhou M, Chen L, Lu Y. A preliminary study on the characterization of follicular helper T (Tfh) cells in rheumatoid arthritis synovium. Acta Histochem. (2014) 116:539–43. doi: 10.1016/j.acthis.2013.10.009
115. Rosengren S, Wei N, Kalunian KC, Kavanaugh A, Boyle DL. CXCL13: a novel biomarker of B-cell return following rituximab treatment and synovitis in patients with rheumatoid arthritis. Rheumatology. (2011) 50:603–10. doi: 10.1093/rheumatology/keq337
116. Bugatti S, Manzo A, Vitolo B, Benaglio F, Binda E, Scarabelli M, et al. High expression levels of the B cell chemoattractant CXCL13 in rheumatoid synovium are a marker of severe disease. Rheumatology. (2014) 53:1886–95. doi: 10.1093/rheumatology/keu163
117. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51.
118. Hatachi S, Iwai Y, Kawano S, Morinobu S, Kobayashi M, Koshiba M, et al. CD4+ PD-1+ T cells accumulate as unique anergic cells in rheumatoid arthritis synovial fluid. J Rheumatol. (2003) 30:1410–9. Available online at: http://www.jrheum.org/
119. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. (2013) 123:2873–92. doi: 10.1172/JCI67428
120. Gu-Trantien C, Willard-Gallo K. PD-1(hi)CXCR5(-)CD4(+) TFH cells play defense in cancer and offense in arthritis. Trends Immunol. (2017) 38:875–8. doi: 10.1016/j.it.2017.10.003
121. Belkhir R, Burel SL, Dunogeant L, Marabelle A, Hollebecque A, Besse B, et al. Rheumatoid arthritis and polymyalgia rheumatica occurring after immune checkpoint inhibitor treatment. Ann Rheum Dis. (2017) 76:1747–50. doi: 10.1136/annrheumdis-2017-211216
122. Bonigen J, Raynaud-Donzel C, Hureaux J, Kramkimel N, Blom A, Jeudy G, et al. Anti-PD1-induced psoriasis: a study of 21 patients. J Eur Acad Dermatol Venereol. (2017) 31:e254–7. doi: 10.1111/jdv.14011
123. Kim JH, Choi YJ, Lee BH, Song MY, Ban CY, Kim J, et al. Programmed cell death ligand 1 alleviates psoriatic inflammation by suppressing IL-17A production from programmed cell death 1-high T cells. J Allergy Clin Immunol. (2016) 137:1466–76.e1463. doi: 10.1016/j.jaci.2015.11.021
124. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995) 155:1151–64.
125. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. (2003) 4:330–6. doi: 10.1038/ni904
126. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–1. doi: 10.1038/83713
127. Chen X, Oppenheim JJ. Resolving the identity myth: key markers of functional CD4+FoxP3+ regulatory T cells. Int Immunopharmacol. (2011) 11:1489–96. doi: 10.1016/j.intimp.2011.05.018
128. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
129. Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated non-regulatory CD4+ T cells. Eur J Immunol. (2007) 37:129–38. doi: 10.1002/eji.200636435
130. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. (2008) 38:1654–63. doi: 10.1002/eji.200838105
131. Cao D, Malmstrom V, Baecher-Allan C, Hafler D, Klareskog L, Trollmo C. Isolation and functional characterization of regulatory CD25brightCD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur J Immunol. (2003) 33:215–23. doi: 10.1002/immu.200390024
132. van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. (2004) 50:2775–85. doi: 10.1002/art.20499
133. Herrath J, Chemin K, Albrecht I, Catrina AI, Malmstrom V. Surface expression of CD39 identifies an enriched Treg-cell subset in the rheumatic joint, which does not suppress IL-17A secretion. Eur J Immunol. (2014) 44:2979–89. doi: 10.1002/eji.201344140
134. Herrath J, Muller M, Amoudruz P, Janson P, Michaelsson J, Larsson PT, et al. The inflammatory milieu in the rheumatic joint reduces regulatory T-cell function. Eur J Immunol. (2011) 41:2279–90. doi: 10.1002/eji.201041004
135. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. (2006) 108:253–61. doi: 10.1182/blood-2005-11-4567
136. Zanin-Zhorov A, Ding Y, Kumari S, Attur M, Hippen KL, Brown M, et al. Protein kinase C-theta mediates negative feedback on regulatory T cell function. Science. (2010) 328:372–6. doi: 10.1126/science.1186068
137. Zaragoza B, Chen X, Oppenheim JJ, Baeyens A, Gregoire S, Chader D, et al. Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat Med. (2016) 22:16–7. doi: 10.1038/nm.4019
138. Tartaglia LA, Goeddel DV, Reynolds C, Figari IS, Weber RF, Fendly BM, et al. Stimulation of human T-cell proliferation by specific activation of the 75-kDa tumor necrosis factor receptor. J Immunol. (1993) 151:4637–41.
139. Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. (2009) 30:626–35. doi: 10.1016/j.immuni.2009.05.002
140. Bending D, Giannakopoulou E, Lom H, Wedderburn LR. Synovial regulatory T cells occupy a discrete TCR niche in human arthritis and require local signals to stabilize FOXP3 protein expression. J Immunol. (2015) 195:5616–24. doi: 10.4049/jimmunol.1500391
141. Ruschen S, Lemm G, Warnatz H. Interleukin-2 secretion by synovial fluid lymphocytes in rheumatoid arthritis. Br J Rheumatol. (1988) 27:350–6.
142. Fava R, Olsen N, Keski-Oja J, Moses H, Pincus T. Active and latent forms of transforming growth factor beta activity in synovial effusions. J Exp Med. (1989) 169:291–6.
143. Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, et al. Memory regulatory T cells reside in human skin. J Clin Invest. (2014) 124:1027–36. doi: 10.1172/JCI72932
144. Bovenschen HJ, van de Kerkhof PC, van Erp PE, Woestenenk R, Joosten I, Koenen HJ. Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J Invest Dermatol. (2011) 131:1853–60. doi: 10.1038/jid.2011.139
145. Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD. IL-6 signaling in psoriasis prevents immune suppression by regulatory T cells. J Immunol. (2009) 183:3170–76. doi: 10.4049/jimmunol.0803721
146. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. (2004) 200:277–85. doi: 10.1084/jem.20040165
147. Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, et al. Brief report: inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum. (2012) 64:2499–503. doi: 10.1002/art.34477
148. Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol. (2015) 15:283–94. doi: 10.1038/nri3823
149. Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity. (2009) 30:204–17. doi: 10.1016/j.immuni.2008.11.014
150. Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. (2006) 212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x
151. White AM, Wraith DC. Tr1-Like T Cells - an enigmatic regulatory T cell lineage. Front Immunol. (2016) 7:355. doi: 10.3389/fimmu.2016.00355
152. Groux H, OGarra A, Bigler M, Rouleau M, Antonenko S, deVries JE, et al. A CD4(+) T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. (1997) 389:737–42.
153. Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, et al. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol. (2014) 122:177–210. doi: 10.1016/B978-0-12-800267-4.00005-5
154. Yudoh K, Matsuno H, Nakazawa F, Yonezawa T, Kimura T. Reduced expression of the regulatory CD4+T cell subset is related to Th1/Th2 balance and disease severity in rheumatoid arthritis. Arthritis Rheum. (2000) 43:617–27. doi: 10.1002/1529-0131(200003)43:3<617::AID-ANR19>3.0.CO;2-B
155. AlJanadi M, AlDalaan A, AlBalla S, AlHumaidi M, Raziuddin S. Interleukin-10 (IL-10) secretion in systemic lupus erythematosus and rheumatoid arthritis: IL-10-dependent CD4+CD45RO+ T cell B cell antibody synthesis. J Clin Immunol. (1996) 16:198–207. doi: 10.1007/Bf01541225
156. Kim CH, Rott JS, Clark-Lewis I, Campbell DJ, Wu L, Butcher EC. Subspecialization of CXCR5(+) T cells: B helper activity is focused in a germinal center-localized subset of CXCR5(+) T cells. J Exp Med. (2001) 193:1373–81. doi: 10.1084/jem.193.12.1373
157. De Berardinis P, Londei M, James RF, Lake SP, Wise PH, Feldmann M. CD4+ T-cell damage to islet beta cells. Lancet. (1989) 1:329.
158. Pachnio A, Ciaurriz M, Begum J, Lal N, Zuo J, Beggs A, et al. Cytomegalovirus infection leads to development of high frequencies of cytotoxic virus-specific CD4+ T cells targeted to vascular endothelium. PLoS Pathog. (2016) 12:e1005832. doi: 10.1371/journal.ppat.1005832
159. Weiskopf D, Bangs DJ, Sidney J, Kolla RV, De Silva AD, de Silva AM, et al. Dengue virus infection elicits highly polarized CX3CR1+ cytotoxic CD4+ T cells associated with protective immunity. Proc Natl Acad Sci USA. (2015) 112:E4256–4263. doi: 10.1073/pnas.1505956112
160. Juno JA, van Bockel D, Kent SJ, Kelleher AD, Zaunders JJ, Munier CM. Cytotoxic CD4 T cells-friend or foe during viral infection? Front Immunol. (2017) 8:19. doi: 10.3389/fimmu.2017.00019
161. Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, et al. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell. (2014) 159:814–28. doi: 10.1016/j.cell.2014.10.026
162. Schmidt D, Goronzy JJ, Weyand CM. CD4+ CD7- CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest. (1996) 97:2027–37. doi: 10.1172/JCI118638
163. Fasth AE, Cao D, van Vollenhoven R, Trollmo C, Malmstrom V. CD28nullCD4+ T cells–characterization of an effector memory T-cell population in patients with rheumatoid arthritis. Scand J Immunol. (2004) 60:199–208. doi: 10.1111/j.0300-9475.2004.01464.x
164. Namekawa T, Wagner UG, Goronzy JJ, Weyand CM. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. (1998) 41:2108–16. doi: 10.1002/1529-0131(199812)41:12<2108::AID-ART5>3.0.CO;2-Q
165. Griffiths GM, Alpert S, Lambert E, McGuire J, Weissman IL. Perforin and granzyme A expression identifying cytolytic lymphocytes in rheumatoid arthritis. Proc Natl Acad Sci USA. (1992) 89:549–53.
166. Chemin K, Ramskold D, Diaz-Gallo LM, Herrath J, Houtman M, Tandre K, et al. EOMES-positive CD4(+) T cells are increased in PTPN22 (1858T) risk allele carriers. Eur J Immunol. (2018) 48:655–69. doi: 10.1002/eji.201747296
167. Namekawa T, Snyder MR, Yen JH, Goehring BE, Leibson PJ, Weyand CM, et al. Killer cell activating receptors function as costimulatory molecules on CD4+CD28null T cells clonally expanded in rheumatoid arthritis. J Immunol. (2000) 165:1138–45. doi: 10.4049/jimmunol.165.2.1138
168. Tian Y, Babor M, Lane J, Schulten V, Patil VS, Seumois G, et al. Unique phenotypes and clonal expansions of human CD4 effector memory T cells re-expressing CD45RA. Nat Commun. (2017) 8:1473. doi: 10.1038/s41467-017-01728-5
169. Patil VS, Madrigal A, Schmiedel BJ, Clarke J, O'Rourke P, de Silva AD, et al. Precursors of human CD4(+) cytotoxic T lymphocytes identified by single-cell transcriptome analysis. Sci Immunol. (2018) 3:eaan8664. doi: 10.1126/sciimmunol.aan8664
170. Oja AE, Vieira Braga FA, Remmerswaal EB, Kragten NA, Hertoghs KM, Zuo J, et al. The transcription factor hobit identifies human cytotoxic CD4(+) T cells. Front Immunol. (2017) 8:325. doi: 10.3389/fimmu.2017.00325
171. Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. (2009) 206:51–9. doi: 10.1084/jem.20081242
172. Ishigaki K, Shoda H, Kochi Y, Yasui T, Kadono Y, Tanaka S, et al. Quantitative and qualitative characterization of expanded CD4+ T cell clones in rheumatoid arthritis patients. Sci Rep. (2015) 5:12937. doi: 10.1038/srep12937
173. Broadley I, Pera A, Morrow G, Davies KA, Kern F. Expansions of cytotoxic CD4(+)CD28(-) T cells drive excess cardiovascular mortality in rheumatoid arthritis and other chronic inflammatory conditions and are triggered by CMV infection. Front Immunol. (2017) 8:195. doi: 10.3389/fimmu.2017.00195
174. Takeuchi A, Badr Mel S, Miyauchi K, Ishihara C, Onishi R, Guo Z, et al. CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J Exp Med. (2016) 213:123–38. doi: 10.1084/jem.20150519
175. Thurkow EW, van der Heijden IM, Breedveld FC, Smeets TJ, Daha MR, Kluin PM, et al. Increased expression of IL-15 in the synovium of patients with rheumatoid arthritis compared with patients with Yersinia-induced arthritis and osteoarthritis. J Pathol. (1997) 181:444–50. doi: 10.1002/(SICI)1096-9896(199704)181:4<444::AID-PATH778>3.0.CO;2-O
176. Jung HW, Choi SW, Choi JI, Kwon BS. Serum concentrations of soluble 4-1BB and 4-1BB ligand correlated with the disease severity in rheumatoid arthritis. Exp Mol Med. (2004) 36:13–22. doi: 10.1038/emm.2004.2
177. Cross A, Bucknall RC, Cassatella MA, Edwards SW, Moots RJ. Synovial fluid neutrophils transcribe and express class II major histocompatibility complex molecules in rheumatoid arthritis. Arthritis Rheum. (2003) 48:2796–806. doi: 10.1002/art.11253
178. Lance EM, Kimura LH, Manibog CN. The expression of major histocompatibility antigens on human articular chondrocytes. Clin Orthop Relat Res. (1993) 266–82.
179. Landais E, Saulquin X, Scotet E, Trautmann L, Peyrat MA, Yates JL, et al. Direct killing of Epstein-Barr virus (EBV)-infected B cells by CD4 T cells directed against the EBV lytic protein BHRF1. Blood. (2004) 103:1408–16. doi: 10.1182/blood-2003-03-0930
180. Romero V, Fert-Bober J, Nigrovic PA, Darrah E, Haque UJ, Lee DM, et al. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci Transl Med. (2013) 5:209ra150. doi: 10.1126/scitranslmed.3006869
181. Santiago L, Menaa C, Arias M, Martin P, Jaime-Sanchez P, Metkar S, et al. Granzyme A contributes to inflammatory arthritis in mice through stimulation of osteoclastogenesis. Arthritis Rheumatol. (2017) 69:320–34. doi: 10.1002/art.39857
182. Sower LE, Froelich CJ, Allegretto N, Rose PM, Hanna WD, Klimpel GR. Extracellular activities of human granzyme A. Monocyte activation by granzyme A versus alpha-thrombin. J Immunol. (1996) 156:2585–90.
183. Fasth AE, Dastmalchi M, Rahbar A, Salomonsson S, Pandya JM, Lindroos E, et al. T cell infiltrates in the muscles of patients with dermatomyositis and polymyositis are dominated by CD28null T cells. J Immunol. (2009) 183:4792–99. doi: 10.4049/jimmunol.0803688
184. Broux B, Pannemans K, Zhang X, Markovic-Plese S, Broekmans T, Eijnde BO, et al. CX(3)CR1 drives cytotoxic CD4(+)CD28(-) T cells into the brain of multiple sclerosis patients. J Autoimmun. (2012) 38:10–9. doi: 10.1016/j.jaut.2011.11.006
185. Raveney BJ, Oki S, Hohjoh H, Nakamura M, Sato W, Murata M, et al. Eomesodermin-expressing T-helper cells are essential for chronic neuroinflammation. Nat Commun. (2015) 6:8437. doi: 10.1038/ncomms9437
186. Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, et al. CD4+NKG2D+ T cells in Crohn's disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology. (2007) 132:2346–58. doi: 10.1053/j.gastro.2007.03.025
187. Lima XT, Cintra ML, Piaza AC, Mamoni RL, Oliveira RT, Magalhaes RF, et al. Frequency and characteristics of circulating CD4(+) CD28(null) T cells in patients with psoriasis. Br J Dermatol. (2015) 173:998–1005. doi: 10.1111/bjd.13993
188. Bryl E, Vallejo AN, Weyand CM, Goronzy JJ. Down-regulation of CD28 expression by TNF-alpha. J Immunol. (2001) 167:3231–8. doi: 10.4049/jimmunol.167.6.3231
189. Bryl E, Vallejo AN, Matteson EL, Witkowski JM, Weyand CM, Goronzy JJ. Modulation of CD28 expression with anti-tumor necrosis factor alpha therapy in rheumatoid arthritis. Arthritis Rheum. (2005) 52:2996–3003. doi: 10.1002/art.21353
190. Allez M, Skolnick BE, Wisniewska-Jarosinska M, Petryka R, Overgaard RV. Anti-NKG2D monoclonal antibody (NNC0142-0002) in active Crohn's disease: a randomised controlled trial. Gut. (2017) 66:1918–25. doi: 10.1136/gutjnl-2016-311824
191. Nanki T, Imai T, Nagasaka K, Urasaki Y, Nonomura Y, Taniguchi K, et al. Migration of CX3CR1-positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis Rheum. (2002) 46:2878–83. doi: 10.1002/art.10622
192. Turner DL, Bickham KL, Thome JJ, Kim CY, D'Ovidio F, Wherry EJ, et al. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. (2014) 7:501–10. doi: 10.1038/mi.2013.67
193. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
194. Cheuk S, Wiken M, Blomqvist L, Nylen S, Talme T, Stahle M, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol. (2014) 192:3111–20. doi: 10.4049/jimmunol.1302313
195. Singh JA, Arayssi T, Duray P, Schumacher HR. Immunohistochemistry of normal human knee synovium: a quantitative study. Ann Rheum Dis. (2004) 63:785–90. doi: 10.1136/ard.2003.013383
196. Brown AK, Quinn MA, Karim Z, Conaghan PG, Peterfy CG, Hensor E, et al. Presence of significant synovitis in rheumatoid arthritis patients with disease-modifying antirheumatic drug-induced clinical remission: evidence from an imaging study may explain structural progression. Arthritis Rheum. (2006) 54:3761–73. doi: 10.1002/art.22190
197. Petrelli A, Mijnheer G, van Konijnenburg DPH, van der Wal MM, Giovannone B, Mocholi E, et al. PD-1+CD8+ T cells are clonally expanding effectors in human chronic inflammation. J Clin Invest. (2018) 128:4669–81. doi: 10.1172/JCI96107
198. Afeltra A, Galeazzi M, Ferri GM, Amoroso A, De Pita O, Porzio F, et al. Expression of CD69 antigen on synovial fluid T cells in patients with rheumatoid arthritis and other chronic synovitis. Ann Rheum Dis. (1993) 52:457–60.
199. Kato T, Kurokawa M, Masuko-Hongo K, Sasakawa H, Sekine T, Ueda S, et al. T cell clonality in synovial fluid of a patient with rheumatoid arthritis: persistent but fluctuant oligoclonal T cell expansions. J Immunol. (1997) 159:5143–9.
200. Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. (2013) 12:581–94. doi: 10.1038/nrd4051
201. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. (2014) 506:376–81. doi: 10.1038/nature12873
202. Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet. (2012) 44:1336–40. doi: 10.1038/ng.2462
203. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. (2010) 42:508–14. doi: 10.1038/ng.582
204. Harden JL, Krueger JG, Bowcock AM. The immunogenetics of Psoriasis: a comprehensive review. J Autoimmun. (2015) 64:66–73. doi: 10.1016/j.jaut.2015.07.008
205. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. (2013) 14:128. doi: 10.1186/1471-2105-14-128
Keywords: rheumatoid arthritis, CD4+ T cells, effector function, T-cell subsets, helper, cytotoxic, tissue-resident
Citation: Chemin K, Gerstner C and Malmström V (2019) Effector Functions of CD4+ T Cells at the Site of Local Autoimmune Inflammation—Lessons From Rheumatoid Arthritis. Front. Immunol. 10:353. doi: 10.3389/fimmu.2019.00353
Received: 04 October 2018; Accepted: 11 February 2019;
Published: 12 March 2019.
Edited by:
Weiguo Cui, Bloodcenter of Wisconsin, United StatesReviewed by:
Shahram Salek-Ardakani, Pfizer, United StatesNicole L. La Gruta, Monash University, Australia
Copyright © 2019 Chemin, Gerstner and Malmström. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karine Chemin, karine.chemin@ki.se