Marine Sediment Sample Pre-processing for Macroinvertebrates Metabarcoding: Mechanical Enrichment and Homogenization
 Eva Aylagas
Eva Aylagas Iñaki Mendibil
Iñaki Mendibil  Ángel Borja
Ángel Borja Naiara Rodríguez-Ezpeleta
Naiara Rodríguez-Ezpeleta- AZTI, Marine Research Division, Sukarrieta, Spain
Metabarcoding is an accurate and cost-effective technique that allows for simultaneous taxonomic identification of multiple environmental samples. Application of this technique to marine benthic macroinvertebrate biodiversity assessment for biomonitoring purposes requires standardization of laboratory and data analysis procedures. In this context, protocols for creation and sequencing of amplicon libraries and their related bioinformatics analysis have been recently published. However, a standardized protocol describing all previous steps (i.e., processing and manipulation of environmental samples for macroinvertebrate community characterization) is lacking. Here, we provide detailed procedures for benthic environmental sample collection, processing, enrichment for macroinvertebrates, homogenization, and subsequent DNA extraction for metabarcoding analysis. Since this is the first protocol of this kind, it should be of use to any researcher in this field, having the potential for improvement.
Introduction
Biomonitoring has become essential to address changes in the quality of the environment as a response to the several pressures that are threatening marine ecosystems (Halpern et al., 2008). The rapid response of benthic organisms to a range of natural and anthropogenic pressures makes this community a suitable ecological component for marine biomonitoring (Johnston and Roberts, 2009). Above all, macroinvertebrates are widely used to assess environmental quality through the calculation of benthic indices (Diaz et al., 2004; Borja et al., 2015). Yet, the fast environmental degradation and the necessity of cost-effective methods for biodiversity assessment urge the need of new tools that allow species identification in a much faster way compared to morphological methodologies (Bourlat et al., 2013). The advent of high-throughput sequencing (HTS) technologies has favored the application of DNA-based biodiversity assessment methods (Creer et al., 2016) and in particular, DNA metabarcoding has become a promising technique for rapid, accurate, and cost-effective taxonomic identification of the benthic macroinvertebrate community in environmental samples (Elbrecht and Leese, 2015; Aylagas et al., 2016).
DNA metabarcoding involves the amplification of a particular DNA region (barcode) to resolve the total genomic DNA extracted from an environmental sample into distinct taxa, typically species, by using universal primers (Taberlet et al., 2012). Coupled with HTS, the technique enables the simultaneous identification of the taxonomic composition of several independent samples by matching the unknown amplified DNA barcode to a DNA reference database (ideally, every organism within a sample can be detected). Metabarcoding has been proven useful in the identification of metazoan community composition from a wide variety of aquatic environments (Chariton et al., 2010; Cowart et al., 2015; Dowle et al., 2015; Elbrecht and Leese, 2015; Lejzerowicz et al., 2015; Leray and Knowlton, 2015; Zaiko et al., 2015), and recent studies have proved that the ecological ecosystem condition addressed through the calculation of DNA-based biotic indices is comparable to that inferred using morphological identification (Dowle et al., 2015; Lejzerowicz et al., 2015; Aylagas et al., 2016). However, metabarcoding is not a fully established methodology for marine monitoring. Therefore, standardization of procedures is necessary, which requires of optimized protocols that allow the reliability and reproducibility of the approach. In this sense, significant efforts have been made to standardize different steps of the metabarcoding workflow by addressing the issues regarding to PCR amplification (Aylagas et al., 2016), barcode region (Carew et al., 2013), primer selection (Leray et al., 2013), library preparation (Bourlat et al., 2016), and bioinformatics analysis for data interpretation (Aylagas and Rodríguez-Ezpeleta, 2016).
A major limitation for environmental DNA metabarcoding studies of benthic macroinvertebrate communities that has not been properly addressed is the manipulation of the sample to be analyzed. Usually, sediment and organic matter carried over using marine benthic community sampling methods result in large sample volume, which needs to be correctly processed so that DNA representing the whole community can be extracted. However, the amount of collected material, the nature of the sample (e.g., mud sediments require different processing than coarse sands) and the size of the target organisms make, in some cases, DNA extraction of the entire sample unfeasible. The requisite of an adequate metabarcoding study is that the sample must be representative of the whole community. Thus, because each sample is different, the pre-processing strategy must be carefully considered in order to retrieve a reliable representation of the macroinvertebrate community. Additionally, routine application of metabarcoding for biomonitoring requires each step of sample collection, handling, pre-processing, DNA extraction, and DNA library preparation and sequencing be standardized so that results from different laboratories can be compared and combined (Deiner et al., 2015).
Different approaches can be used to recover DNA from sediment samples. Generally, the size range of the target organisms determines the amount of sediment to be processed and the protocol used (Creer et al., 2016). For studies targeting small size metazoans (e.g., meiofauna), the procedures can rely on extracting DNA from small sediment samples (i.e., 5 gr of sediment) without any pre-processing step (Lejzerowicz et al., 2015), targeting extracellular DNA (Guardiola et al., 2015; Pearman et al., 2016), or performing some separation via decantation/flotation (Creer et al., 2010). However, when the fraction to be investigated is larger (e.g., macroinvertebrates) samples need first be processed via decantation protocols so that the macroinvertebrate community is separated from the sediment. Recently, Aylagas et al. (2016) showed that following protocols to target the extracellular DNA from sediment samples, only a small proportion of the macroinvertebrate taxa is retrieved, whilst the isolation of organisms followed by homogenization and DNA extraction allows a reliable characterization of the macroinvertebrate community through DNA metabarcoding.
The objective of the present protocol is to extract good quality and integrity DNA from complex environmental samples which is representative of the whole macroinvertebrate community. For that purpose, we present guidelines for the processing of benthic sediment samples collected for metabarcoding-based biomonitoring. We detail the steps necessary to (i) preserve the benthic sample to ensure DNA integrity, (ii) isolate organic fraction from the sediment by decantation, (iii) homogenize the sample in order to achieve a good community representation, and (iv) extract DNA of good quality and integrity. The efficiency of sediment decantation and homogenization steps detailed in this protocol have previously shown to help providing accurate metabarcoding taxonomic inferences that are comparable to those inferred from morphology (Leray and Knowlton, 2015). Thus, followed by the well-established metabarcoding procedures for library preparation (Bourlat et al., 2016) and bioinformatics analysis (Aylagas and Rodríguez-Ezpeleta, 2016) this protocol represents the first steps of the procedure to gather the taxonomic list of several benthic samples simultaneously. This information can be ultimately used for a variety of applications that rely on the macroinvertebrate community characterization of the samples such as the calculation of benthic indices for ecological status assessment (Aylagas and Rodríguez-Ezpeleta, 2016), the detection of non-indigenous species (Zaiko et al., 2015), or large-scale spatio-temporal biodiversity assessments (Leray and Knowlton, 2015; Chain et al., 2016). Finally, a Notes section is dedicated to discuss various artifacts and pitfalls to consider throughout the description of the protocol.
Materials and Equipment
Sample Collection and Preservation
1. Gloves
2. 0.5 m2 sampling squares
3. Van Veen grab (0.07–0.1 m2)
4. 1 mm mesh size sieve (45 cm diameter)
5. Ethanol 96%
6. 1 L storing flasks
7. Spatula
Sample Processing
Decantation
8. Graduated cylinder with stopper (500 ml, 1 L, 2 L)
9. Deionized water
10. 1 mm mesh size sieve (20 cm diameter)
11. Tweezers
12. Stereomicroscope
13. Milli-Q water
14. Ethanol 96%
Homogenization and DNA Extraction
15. Blender (PHILIPS hr2095 700 W 2 L glass jar) for large volume samples or porcelain mortar (Thermo Scientific) for small volume samples
16. 50 ml falcon tubes
17. Ethanol 96%
18. 20 μm mesh size filter
19. Spatula
20. Mo Bio PowerMax® Soil DNA Isolation Kit (for large volume samples) or Mo Bio PowerSoil® DNA Isolation Kit (for small volume samples)
21. Proteinase K (20 mg/ml)
22. Shaking incubator
23. Water bath
DNA Overall Quality Assessment, Purification and Normalization
24. Agarose
25. SYBR® Safe DNA Gel Stain (Thermo Scientific)
26. HyperLadderTM 1 kbp (BIOLINE)
27. Electrophoresis equipment
28. Nanodrop® ND-1000 (Thermo Scientific)
29. Qubit dsDNA HS Assay Kit (Thermo Scientific)
30. 1.5 ml Eppendorf tubes
31. Mo Bio PowerClean Pro DNA Clean-Up Kit
32. MilliQ water
Procedures
Sample Collection and Preservation
DNA-free materials thoroughly cleaned between locations must be used to avoid cross-contamination (see Note 1), and samples should be preserved under appropriate conditions to guarantee DNA integrity.
1. Collect soft benthic samples using 0.5 m2 sampling squares in intertidal locations concurring with the low tide or using a van Veen grab from a boat on sublittoral stations.
2. Pass through a 1 mm mesh size sieve.
3. Preserve the retained material in 96% ethanol (Note 2) in a 5:1 volumetric ratio using 1 L flask and store at 4 °C until further analysis (Note 3a: Safe stopping point).
Sample Processing
Decantation (0.5 h)
Humic substances, co-extracted with DNA, inhibit enzymes such as the Taq Polymerase used in PCR reactions to amplify DNA, representing the primary inhibitory compound associated with sediment samples (Matheson et al., 2010). This inhibition represents a potential bias for DNA metabarcoding studies performed on sediment samples and, if not properly addressed, can lead to generation of false negative results (Thomsen and Willerslev, 2015). At the same time, the heterogenic composition of the benthic macroinvertebrate community would require extracting all DNA within a sample in order to detect all species present. As this step is logistically unfeasible, the homogenization of the sample is required, so that a subsample is representative of the whole community. The volume of sediment processed may significantly vary among samples, which could imply a great impact on the sample representativeness. In this sense, low amounts of sediment in the sample allow for more representative homogenized subsamples. For these reasons, it is recommended to separate the organic fraction from the sediment before proceeding with DNA extraction. Depending on sediment type (Figure 1), this separation can be totally or partially performed through a decantation process. Medium to coarse grain sediments can often be completely removed through decantation but muddy or fine sediments may decant with the organic matter and impede the complete sediment removal. The sample processing workflow is shown in Figure 2.

Figure 1. Different types of sediment samples collected from intertidal and sublittoral benthic environments. (A) Coarse Sands, (B) Medium Sands, (C) Fine Sands and (D) Mud.

Figure 2. Illustration of workflow for bulk sample processing.
1. Transfer each sample into a graduated cylinder up to ¼. For 50–200 ml volume samples use the 500 ml cylinder; for 200–500 ml, the 1 L; and for 500–2 L the 2 L graduate cylinder.
2. Fill up with deionized water, cover the cylinder, and shake vigorously to resuspend animals and other organic matter.
3. After 5 s or when the sediment has been deposited on the bottom of the flask, gently pour the water with the suspended matter onto a 1 mm mesh size sieve so that resuspended organic material decants onto the sieve and the sediment is retained in the cylinder.
4. Repeat steps 2 and 3 five times or until no organic particles can be observed after shaking.
5. Collect the organic material into the corner of the sieve and pour into a blender-jar containing ethanol 96% or into a mortar (Figure 2). Large amounts of recovered material (i.e., organisms together with a fraction of organic matter) require sampling homogenization using a blender unit that allows big volume sample processing. In contrast, samples from sediments with low amount of organic matter allow the successful isolation of organisms which can be easily homogenized using a mortar.
6. Check sieve under a stereomicroscope for attached animals and examine sediment for remaining shelled organisms that are not separated through decantation (e.g., bivalves, gastropods); recover with the help of tweezers and add to the previously decanted material (Note 3b: Safe stopping point).
Homogenization and DNA Extraction (2 h, Overnight and 3 h)
The biomass of the decanted organic material may greatly differ among samples, which predetermines subsequent sample pre-processing and DNA extraction procedures. Large amounts of organic material recovered (i.e., the recovered material contains macroinvertebrates and lots of organic matter or big-sized organisms) are followed by Blender homogenization and DNA extraction using the PowerMax Soil DNA Isolation Kit; conversely, samples with a range of recovered biomass from 10 to 200 mg (i.e., the recovered material contains animals for the most part) are processed using Mortar homogenization followed by DNA extraction using the PowerSoil DNA Isolation Kit (see Figure 2 for schematic representation of the workflow).
Blender Homogenization
1. Homogenize the sample until no fragments of animals and other organic material can be observed in the final homogenate.
2. Pour the material through a 20 μm sieve to remove the ethanol and mix the blended material using a spatula. Rinse using ethanol until no material remains in the blender jar.
3. Take two subsamples of 10 gr from the homogenized sample and preserve the remaining material in a flask with ethanol 96% in a 5:1 volumetric ratio using 50 ml falcon tube and store at −20 °C (Note 3c: Safe stopping point).
4. Extract DNA from each of the two subsamples (Note 4) using the PowerMax Soil DNA Isolation Kit following manufacturer's instructions but replacing the initial bead-beating step by adding proteinase K (0.4 mg/ml) to the power bead solution and incubating samples in a shaking incubator overnight at 56 °C (Leray and Knowlton, 2015).
Mortar Homogenization
1. Pour isolated organisms through a 20 μm sieve to remove the ethanol if sample has been stored before homogenization and place in a mortar.
2. Homogenize animals for 5 min or until a mixture has been formed and collect homogenized material in 2 ml Eppendorf tubes (Note 3c: Safe stopping point).
3. Extract DNA from whole homogenate or from a subsample of up to 25 mg using the PowerSoil DNA Isolation Kit following manufacturer's instructions but replacing the initial bead-beating step, by adding proteinase K (0.4 mg/ml) to the power bead solution and incubating samples in a shaking incubator overnight at 56 °C (Leray and Knowlton, 2015).
DNA Overall Quality Assessment, Purification and Normalization (3 h)
1. Assess DNA integrity migrating about 100 ng of DNA on an agarose 1.0% gel stained with SYBR® Safe (Figure 3), purity using the Nanodrop® ND-100 system, and quantity using a Qubit® 2.0 Fluorometer with the Qubit® dsDNA HS Assay Kit.
2. Pool the same amount of DNA derived from each extraction replicate in a single tube.
3. Purify DNA using PowerClean Pro DNA Clean-Up Kit following manufacturer's instructions (Note 5).
4. Normalize DNA at 5 ng/μl using milliQ water (Note 3d: Safe stopping point)
5. Use DNA as a template for downstream analysis.
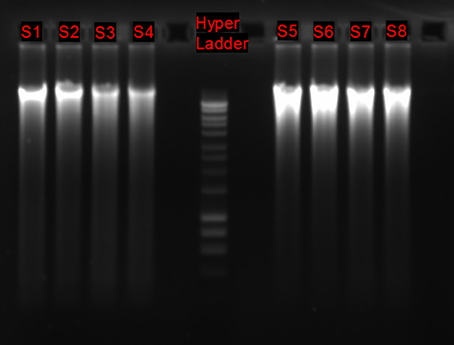
Figure 3. DNA integrity of 8 environmental samples processed as described in the present protocol. DNA extraction was performed using the PowerMax Soil DNA Isolation Kit. HyperLadder™1 kbp.
Anticipated Results
The protocol described here provides guidelines to resolve the first steps needed for metabarcoding-based benthic macroinvertebrate community assessment: sample collection and preservation, processing, and extraction of representative. DNA of good quality and integrity. The standardization of these three steps is crucial to further obtaining accurate taxonomic inferences from metabarcoding data.
Macroinvertebrate samples used for benthic monitoring can occur in different types of sediment (coarse, medium and fine sands, and muds), and contain organisms of heterogeneous size (from 1 mm to several cm) and nature (soft or containing hard, shell, or spiny calcium carbonate exoskeleton, gelatinous, etc.), which implies that DNA extraction may not be equally efficient for all types of sediment or organismal types. Our protocol is based on large sediment volumes (>100 ml) to ensure that all organisms are present, preserved in appropriate conditions to prevent DNA degradation, that are mortar or blender beaten to ensure breaking of hard exoskeletons.
DNA extracted from complex environmental samples need to be representative and of good quality and integrity. The steps presented here ensure both (i) macroinvertebrate community representation by homogenizing samples from which subsamples are taken before DNA extraction and (ii) good quality and integrity DNA by utilizing kits-based extraction protocols specifically designed for isolating high-quality environmental DNA from soil or sediment. The procedures described in the present protocol for decantation, homogenization, and DNA extraction have been recently applied to sediment samples from estuarine and coastal locations with different level of anthropogenic pressures. The DNA extracted from each environmental sample was amplified following the protocol for amplicon library preparation and sequencing (Bourlat et al., 2016) and the resulting reads analyzed using the pipeline for bioinformatics analysis of metabarcoding data (Aylagas and Rodríguez-Ezpeleta, 2016). Using the retrieved macroinvertebrate taxonomic list from each sample, the marine biotic index AMBI (Borja et al., 2000) was calculated, showing comparable results to that inferred using morphological species identification from samples of the same locations (Aylagas et al., in preparation). Thus, the promising results obtained using the present protocol for environmental biomonitoring contributes to accelerating the implementation of metabarcoding for environmental status assessment.
Finally, in response to the necessity of more cost-effective approaches than the traditional morphological species identification, the present protocol followed by DNA amplification coupled with HTS proves to be a suitable cheaper alternative for biodiversity assessment. Although several procedures involving less sample manipulation prior DNA extraction are well-established for small metazoans metabarcoding studies (Guardiola et al., 2015; Lejzerowicz et al., 2015; Pearman et al., 2016), these approaches cannot be accommodated for macroinvertebrates. In this context, the standardization of the sample pre-processing through mechanical enrichment and homogenization before DNA extraction will ensure the reproducibility of the results and may help to the establishment of macroinvertebrates metabarcoding for environmental biomonitoring.
Notes
Note 1. Recommendations to Prevent Cross-Contamination
DNA-based approach to characterize metazoan communities is very sensitive to contamination. Avoiding cross-contamination is essential to ensure the success of DNA metabarcoding-based biodiversity studies. During sample collection, decantation and homogenization steps, material (sieves, graduated cylinders, blender jar, mortar, and tweezers) must be cleaned between samples by soaking in 10% bleach for a minimum of 5 min and gently rinsing with deionized water. Finally, these recommendations must be followed:
– The working area must be cleared and previously cleaned using 10% bleach
– Gloves and lab coat must be worn during manipulation of samples
– Pre and post-amplification laboratory areas should be differentiated
– Sterile filter pipette tips must be used and changed between samples
Note 2. Environmental Sample Preservation for DNA-Based Studies
DNA degradation is critical for metabarcoding marine benthic community assessment. In this sense, the detection of some of the species present in an environmental sample may be reduced if DNA integrity has been altered. The process of DNA degradation starts at the moment an organism dies, when cell membranes break and allow entrance of bacteria and other threats with the subsequent release of DNAses that degrade DNA. Thus, avoiding DNA degradation requires storing the sample as soon as collected in appropriate preserving agents (ethanol or other reagents such as RNA later) that prevent DNAse activity (Rodriguez-Ezpeleta et al., 2013). Although formalin has traditionally been used to store marine benthic organism samples, as it preserves morphological structure and allows visual identification, it is toxic and degrades DNA (Serth et al., 2000); thus, ethanol 96% is recommended to preserve samples for molecular studies (Stein et al., 2013).
Note 3. Safe Stopping Points
a. If sample processing is not immediately performed, bulk benthic sample must be preserved in ethanol at 4 °C until further use (Stein et al., 2013).
b. If homogenization is not immediately performed, pour decanted material into a 2 ml Eppendorf tube, a 50 ml falcon tube or a 1 L flask (depending on the amount of recovered material) containing ethanol 96% and store at −20 °C until homogenization.
c. If DNA extraction is not immediately performed, store homogenized sample in a falcon tube containing ethanol 96% at −20 °C until DNA extraction.
d. Preserve DNA at −20 °C for downstream analysis.
Note 4. Subsample Representativeness
Homogenization is performed in order to solve the problem of representativeness issues in large volume samples from which the whole macroinvertebrate community is aimed to be characterized. The best community characterization using DNA-based approaches would require the DNA extraction of the total sample; yet, this cannot be achieved in a reasonable time and commercial kits are not designed for samples up to 10 g. Therefore, a good homogenization step is crucial to ensure the representativeness of the whole community in a subsample. However, we recommend performing two DNA extractions on two subsamples from the homogenized sample to further guarantee a reliable representation of the whole community. In order to ease following steps of the protocol, the DNA replicates are pooled and purified prior amplicon library preparation. Finally, one of the issues related with metabarcoding of different size organisms (from 1 mm to several cm) is the homogenization of exceptionally large specimens with the remaining sample. The DNA of large organisms may mask the presence of other biota in the sample, which may lead to false negative results. In this case, body parts from large specimens can be subsampled or set aside for standard DNA barcoding.
Note 5. Recommendation to Avoid Inhibition Issues Related to Humic Substances
Even though DNA extraction kits used in this protocol are appropriate to remove humic substances, applying cleaning columns further removes other potential PCR inhibitors such as calcium carbonates, silicates, proteins, and algal polysaccharides.
Author Contributions
Conceived and designed the protocol: EA and NR. Developed and performed the protocol: EA and IM. Wrote the first draft of the protocol: EA and NR. All authors contributed equally in writing the last version of the protocol.
Funding
This work was funded by the European Union (7th Framework Program “The Ocean of Tomorrow” Theme, grant agreement no. 308392) through the DEVOTES (DEVelopment Of innovative Tools for understanding marine biodiversity and assessing good Environmental Status—http://www.devotes-project.eu) project and by the Basque Water Agency (URA) through a Convention with AZTI. EA is supported by a doctoral grant from Fundación Centros Tecnológicos—IG.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a collaboration with the authors and states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
The authors would like to thank Dr. Maria C. Uyarra for kindly advising us on some details on the manuscript. This paper is contribution number 776 from AZTI (Marine Research Division).
References
Aylagas, E., Borja, A., Irigoien, X., and Rodriguez-Ezpeleta, N. (2016). Benchmarking DNA metabarcoding for biodiversity-based monitoring and assessment. Front. Mar. Sci. 3:96. doi: 10.3389/fmars.2016.00096
Aylagas, E., and Rodríguez-Ezpeleta, N. (2016). “Analysis of Illumina MiSeq amplicon reads: application to benthic indices for environmental monitoring,” in Marine Genomics Methods and Protocols, Methods in Molecular Biology, ed S. J. Bourlat (New York, NY: Springer), 1452.
Borja, A., Franco, J., and Perez, V. (2000). A marine biotic index to establish the ecological quality of soft-bottom benthos within european estuarine and coastal environments. Mar. Pollut. Bull. 40, 12. doi: 10.1016/S0025-326X(00)00061-8
Borja, Á., Marín, S. L., Muxika, I., Pino, L., and Rodríguez, J. G. (2015). Is there a possibility of ranking benthic quality assessment indices to select the most responsive to different human pressures? Mar. Pollut. Bull. 97, 85–94. doi: 10.1016/j.marpolbul.2015.06.030
Bourlat, S. J., Borja, A., Gilbert, J., Taylor, M. I., Davies, N., Weisberg, S. B., et al. (2013). Genomics in marine monitoring: new opportunities for assessing marine health status. Mar. Pollut. Bull. 74, 19–31. doi: 10.1016/j.marpolbul.2013.05.042
Bourlat, S. J., Haenel, Q., Finnman, J., and Leray, M. (2016). “Preparation of amplicon libraries for metabarcoding of marine eukaryotes using Illumina MiSeq: the dual-PCR method,” in Marine Genomics Methods and Protocols, Methods in Molecular Biology, ed S. J. Bourlat (New York, NY: Springer), 1452.
Carew, M. E., Pettigrove, V. J., Metzeling, L., and Hoffmann, A. A. (2013). Environmental monitoring using next generation sequencing: rapid identification of macroinvertebrate bioindicator species. Front. Zool. 10:45. doi: 10.1186/1742-9994-10-45
Chain, F. J. J., Brown, E. A., MacIsaac, H. J., Cristescu, M. E., and Cowie, R. (2016). Metabarcoding reveals strong spatial structure and temporal turnover of zooplankton communities among marine and freshwater ports. Divers. Distrib. 22, 493–504. doi: 10.1111/ddi.12427
Chariton, A. A., Court, L. N., Hartley, D. M., Colloff, M. J., and Hardy, C. M. (2010). Ecological assessment of estuarine sediments by pyrosequencing eukaryotic ribosomal DNA. Front. Ecol. Environ. 8:233–238. doi: 10.1890/090115
Cowart, D. A., Pinheiro, M., Mouchel, O., Maguer, M., Grall, J., Miné, J., et al. (2015). Metabarcoding is powerful yet still blind: a comparative analysis of morphological and molecular surveys of seagrass communities. PLoS ONE 10:e0117562. doi: 10.1371/journal.pone.0117562
Creer, S., Deiner, K., Frey, S., Porazinska, D., Taberlet, P., Thomas, W. K., et al. (2016). The ecologist's field guide to sequence-based identification of biodiversity. Methods Ecol. Evol. 7, 1008–1018. doi: 10.1111/2041-210X.12574
Creer, S., Fonseca, V. G., Porazinska, D. L., Giblin-Davis, R. M., Sung, W., Power, D. M., et al. (2010). Ultrasequencing of the meiofaunal biosphere: practice, pitfalls and promises. Mol. Ecol. 19(Suppl.1), 4–20. doi: 10.1111/j.1365-294X.2009.04473.x
Deiner, K., Walser, J.-C., Mächler, E., and Altermatt, F. (2015). Choice of capture and extraction methods affect detection of freshwater biodiversity from environmental DNA. Biol. Conserv. 183, 53–63. doi: 10.1016/j.biocon.2014.11.018
Diaz, R. J., Solan, M., and Valente, R. M. (2004). A review of approaches for classifying benthic habitats and evaluating habitat quality. J. Environ. Manage. 73, 165–181. doi: 10.1016/j.jenvman.2004.06.004
Dowle, E. J., Pochon, X., Banks, J., Shearer, K., and Wood, S. A. (2015). Targeted gene enrichment and high throughput sequencing for environmental biomonitoring: a case study using freshwater macroinvertebrates. Mol. Ecol. Resour. 16, 1240–1254. doi: 10.1111/1755-0998.12488
Elbrecht, V., and Leese, F. (2015). Can DNA-Based ecosystem assessments quantify species abundance? testing primer bias and biomass–sequence relationships with an innovative metabarcoding protocol. PLoS ONE 10:e0130324. doi: 10.1371/journal.pone.0130324
Guardiola, M., Uriz, M. J., Taberlet, P., Coissac, E., Wangensteen, O. S., and Turon, X. (2015). Deep-Sea, deep-sequencing: metabarcoding extracellular DNA from sediments of marine canyons. PLoS ONE 10:e0139633. doi: 10.1371/journal.pone.0139633
Halpern, B. S., Walbridge, S., Selkoe, K. A., Kappel, C. V., Micheli, F., D'Agrosa, C., et al. (2008). A global map of human impact on marine ecosystems. Science 319, 948–952. doi: 10.1126/science.1149345
Johnston, E. L., and Roberts, D. A. (2009). Contaminants reduce the richness and evenness of marine communities: a review and meta-analysis. Environ. Pollut. 157, 1745–1752. doi: 10.1016/j.envpol.2009.02.017
Lejzerowicz, F., Esling, P., Pillet, L., Wilding, T. A., Black, K. D., and Pawlowski, J. (2015). High-throughput sequencing and morphology perform equally well for benthic monitoring of marine ecosystems. Sci. Rep. 5:13932. doi: 10.1038/srep13932
Leray, M., and Knowlton, N. (2015). DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proc. Natl. Aacd. Sci. U.S.A. 112, 2076–2081. doi: 10.1073/pnas.1424997112
Leray, M., Yang, Y. J., Meyer, P. C., Mills, C. S., Agudelo, N., Ranwez, V., et al. (2013). New versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Front. Zool. 10:34. doi: 10.1186/1742-9994-10-34
Matheson, C. D., Gurney, C., Esau, N., and Lehto, R. (2010). Assessing PCR inhibition from humic substances. Open Enzym. Inhib. J. 3, 38–45. doi: 10.2174/1874940201003010038
Pearman, J. K., Irigoien, X., and Carvalho, S. (2016). Extracellular DNA amplicon sequencing reveals high levels of benthic eukaryotic diversity in the central Red Sea. Mar. Genomics 26, 29–39. doi: 10.1016/j.margen.2015.10.008
Rodriguez-Ezpeleta, N., Mendibil, I., Álvarez, P., and Cotano, U. (2013). Effect of fish sampling and tissue storage conditions in DNA quality: considerations for genomic studies. Rev. Invest. Mar. 20, 77–87.
Serth, J., Kuczyk, M. A., Paeslack, U., Lichtinghagen, R., and Jonas, U. (2000). Quantitation of DNA extracted after micropreparation of cells from frozen and formalin-fixed tissue sections. Am. J. Pathol. 156, 1189–1196. doi: 10.1016/S0002-9440(10)64989-9
Stein, E. D., White, B. P., Mazor, R. D., Miller, E. P., and Pilgrim, E. M. (2013). Evaluating ethanol-based sample preservation to facilitate use of DNA barcoding in routine freshwater biomonitoring programs using benthic macroinvertebrates. PLoS ONE 8:e51273. doi: 10.1371/journal.pone.0051273
Taberlet, P., Coissac, E., Pompanon, F., Brochmann, C.-., and Willerslev, E. (2012). Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 21, 2045–2050. doi: 10.1111/j.1365-294X.2012.05470.x
Thomsen, P. F., and Willerslev, E. (2015). Environmental DNA – An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 183, 4–18. doi: 10.1016/j.biocon.2014.11.019
Keywords: environmental samples, laboratory procedures, sample manipulation, DNA, biomonitoring
Citation: Aylagas E, Mendibil I, Borja Á and Rodríguez-Ezpeleta N (2016) Marine Sediment Sample Pre-processing for Macroinvertebrates Metabarcoding: Mechanical Enrichment and Homogenization. Front. Mar. Sci. 3:203. doi: 10.3389/fmars.2016.00203
Received: 15 June 2016; Accepted: 30 September 2016;
Published: 18 October 2016.
Edited by:
Marianna Mea, Ecoreach s.r.l, Italy; Jacobs University of Bremen, GermanyReviewed by:
Franck Lejzerowicz, University of Geneva, SwitzerlandErik Michael Pilgrim, U.S. Environmental Protection Agency, USA
Copyright © 2016 Aylagas, Mendibil, Borja and Rodríguez-Ezpeleta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eva Aylagas, eaylagas@azti.es