 Jackie K. Zorz
Jackie K. Zorz Jessica A. Kozlowski
Jessica A. Kozlowski Lisa Y. Stein
Lisa Y. Stein Marc Strous
Marc Strous Manuel Kleiner
Manuel Kleiner- 1Department of Geoscience, University of Calgary, Calgary, AB, Canada
- 2Department of Ecogenomics and Systems Biology, Division Archaea Biology and Ecogenomics, University of Vienna, Vienna, Austria
- 3Department of Biological Sciences, University of Alberta, Edmonton, AB, Canada
- 4Department of Plant and Microbial Biology, North Carolina State University, Raleigh, NC, United States
Ammonia-oxidizing bacteria (AOB) are important members of terrestrial, marine, and industrial microbial communities and play a fundamental role in the Nitrogen cycle within these systems. They are responsible for the first step of nitrification, ammonia oxidation to nitrite. Although AOB are widespread and essential to environmental and industrial systems, where they regularly experience fluctuations in ammonia availability, no comparative studies of the physiological response of diverse AOB species at the protein level exist. In the present study, we used 1D-LC-MS/MS proteomics to compare the metabolism and physiology of three species of ammonia AOB, Nitrosomonas europaea, Nitrosospira multiformis, and Nitrosomonas ureae, under ammonia replete and ammonia starved conditions. Additionally, we compared the expression of orthologous genes to determine the major differences in the proteome composition of the three species. We found that approximately one-third of the predicted proteome was expressed in each species and that proteins for the key metabolic processes, ammonia oxidation and carbon fixation, were among the most abundant. The red copper protein, nitrosocyanin was highly abundant in all three species hinting toward its possible role as a central metabolic enzyme in AOB. The proteomic data also allowed us to identify pyrophosphate-dependent 6-phosphofructokinase as the potential enzyme replacing the Calvin-Benson-Bassham cycle enzyme Fructose-1,6-bisphosphatase missing in N. multiformis and N. ureae. Additionally, between species, there were statistically significant differences in the expression of many abundant proteins, including those related to nitrogen metabolism (nitrite reductase), motility (flagellin), cell growth and division (FtsH), and stress response (rubrerythrin). The three species did not exhibit a starvation response at the proteome level after 24 h of ammonia starvation, however, the levels of the RuBisCO enzyme were consistently reduced after the starvation period, suggesting a decrease in capacity for biomass accumulation. This study presents the first published proteomes of N. ureae and N. multiformis, and the first comparative proteomics study of ammonia-oxidizing bacteria, which gives new insights into consistent metabolic features and differences between members of this environmentally and industrially important group.
Introduction
Nitrification is an essential process of the nitrogen cycle in terrestrial, aquatic, and wastewater systems that links reduced and oxidized pools of inorganic nitrogen (Gruber and Galloway, 2008). Nitrification is classically considered a two-step process with the first and rate limiting step, ammonia (NH3) oxidation to nitrite (NO2-), performed by ammonia-oxidizing bacteria (AOB) and archaea, and NO2- oxidation to nitrate (NO3-) performed by nitrite-oxidizing microorganisms (Kowalchuk and Stephen, 2001; Klotz and Stein, 2011). Recently organisms have been discovered that can perform the complete process of NH3 oxidation to NO3- (Daims et al., 2015; Van Kessel et al., 2015). This study focuses solely on the AOB. Ammonia-oxidation by AOB is catalyzed aerobically via two characterized enzymes, ammonia monooxygenase (AMO) which oxidizes NH3 to hydroxylamine (NH2OH), and hydroxylamine dehydrogenase (HAO) which oxidizes NH2OH most likely to nitric oxide (NO) (Caranto and Lancaster, 2017). A third enzyme that oxidizes NO to nitrite (NO2-) has not yet been characterized and was only recently proposed as a third requisite enzyme in the ammonia-oxidation pathway.
The AOB are generally obligate chemolithotrophs living solely off the energy from oxidizing NH3 with oxygen as the terminal electron acceptor. They are autotrophic and obtain carbon through fixation of carbon dioxide using the Calvin-Benson-Bassham (CBB) cycle (Schramm et al., 1998; Utåker et al., 2002), though the capacity for mixotrophy has been observed in Nitrosomonas europaea (Hommes et al., 2003). AOB are also capable of reducing NO2- to N2O via NO through a process termed nitrifier denitrification, which enables maintenance of intracellular redox balance under conditions of oxygen limitation (Stein, 2011).
Niche differentiation among strains of AOB and AOA has been attributed to differences in affinities for NH3, temperature, and pH, among other factors (Bollmann and Laanbroek, 2002; Nicol et al., 2008; Fierer et al., 2009; Martens-Habbena et al., 2009). Differences in NH3 affinities in particular dictate oligotrophic versus eutrophic environmental preferences within distinct groups of AOB (Klotz and Stein, 2011). The three species investigated in this study, N. europaea ATCC 19718, Nitrosospira multiformis ATCC 25196, and Nitrosomonas ureae Nm10 are all ecologically relevant strains of AOB and have complete genome sequences available (Chain et al., 2003; Norton et al., 2008; Kozlowski et al., 2016b). They are phylogenetically diverse representing different clusters and ecotypes within betaproteobacterial AOB. N. europaea is a representative of cluster 7, N. ureae is a representative of cluster 6a, and N. multiformis is a representative of Nitrosospira cluster 3 (Purkhold et al., 2000; Kozlowski et al., 2016a). These strains are generally widespread, however, N. europaea is more often recovered from nitrogen-rich environments like wastewater treatment plants, whereas Nitrosospira strains are more often found in terrestrial or agricultural soil systems (Prosser et al., 2014). Organisms from Nitrosomonas cluster 6a (N. ureae) are often isolated from freshwater or marine environments (Speksnijder et al., 1998; Bollmann and Laanbroek, 2001) and are thus considered comparatively oligotrophic (Prosser et al., 2014).
The AOB in both environmental and industrial settings experience fluctuations in nutrient and substrate availability (Geets et al., 2006). Previous work has aimed to identify the starvation response of specific species of AOB by observing growth and enzyme activity (Sayavedra-Soto et al., 1996; Bollmann and Laanbroek, 2002), mRNA levels (Wei et al., 2004, 2006; Stein et al., 2013; Pérez et al., 2014; Mellbye et al., 2016), and more recently proteomes (Pellitteri-Hahn et al., 2011; Jiang et al., 2015; Sedlacek et al., 2016; Yu et al., 2018). However, past work has mainly been conducted on single species (primarily N. europaea) and in most cases, has focused on the response of genes related to nitrogen cycling, rather than whole-genome expression at the proteome level. This study uses proteomics to compare the diversity in gene expression across phylogenetically coherent and functionally similar bacteria under the same growth conditions: ammonia replete and ammonia starved. Because each of the three AOB species is adapted to its own range of physicochemical parameters, the variance in protein expression will clearly show the range of genomic variability and flexibility. The results observed in this paper provide evidence about how genomic and proteomic expression governs niche preferences by individual species.
Materials and Methods
Culture Growth and Starvation
The three species of AOB, N. europaea ATCC 19718, N. multiformis ATCC 25196, and N. ureae Nm10, were grown at 22°C, in triplicate 300 ml cultures with 100 rpm of shaking, in the dark. We used 5 mM (NH4)2SO4 HEPES-buffered HK medium (0.2 mM MgSO4.7H2O, 1.0 mM CaCl2.2H2O, 1.0 mM KCl, 0.02% Phenol red, 15 mM HEPES buffer, trace solution, pH 7.8) (Krummel and Harms, 1982). Each 300 ml culture was inoculated with 6 ml of a 4 × 107 to 5 × 107 cells ml-1 culture, resulting in an initial density of approximately 1 × 106 cells ml-1. We determined cell density daily using OD600 measurements (Supplementary Figure 1), and we converted OD600 values to cell density (cells ml-1) using standard curves of cell counts, which we generated by comparing cell counts with a Neubauer counting chamber to OD600 values. The pH of the cultures was maintained using sterile 10% sodium bicarbonate. Once cell density had reached late exponential phase and at least 1 × 107 cells ml-1 (Supplementary Figure 1), we spun down the cultures at 15,000 × g for 15 min. Cells were resuspended and washed in 200 ml of ammonia-free HK media and spun down again (15,000 × g for 15 min). The pellet was then resuspended in 10 ml of ammonia-free HK media. We split each replicate, adding 5 ml of the cell suspension to 100 ml HK media with ammonia to act as a control and the remaining 5–100 ml HK media without ammonia for the starved treatment. This resulted in a total of six cultures per species. One of the starved cultures of N. ureae was lost during downstream sample processing. Both control and starved cultures were grown for 24 h at 100 rpm in the dark. After 24 h, we centrifuged the cultures (4,816 × g for 20 min) in a Sorvall ST40R centrifuge with a swinging bucket rotor (75003608) (Thermo Fisher Scientific), followed by centrifugation at 21,000 × g for 5 min in a microcentrifuge. Pellets were immediately stored at -80°C. Previous studies (e.g., Wei et al., 2006), demonstrated that less than 24 h of substrate starvation resulted in a significant starvation response in the transcriptome of N. europaea, so 24 h was chosen as the starvation period for the present study.
Ammonia and nitrite concentrations of media in the pre-culture prior to starvation, in starved cultures, and in control cultures were measured using standard colorimetric assays (Supplementary Table 1). Ammonia was measured using the assay from Sims et al. (1995), and nitrite was measured using a Griess test (Griess-Romijn van Eck, 1966).
Protein Extraction
Protein was extracted using the filter-aided sample preparation (FASP) protocol as previously described (Wisniewski et al., 2009). Briefly, for lysis, we added SDT-lysis buffer [4% (w/v) SDS, 100 mM Tris–HCl, 0.1 M DTT] in a 1:10 pellet:buffer ratio to sample pellets. We heated samples at 95°C for 10 min with periodic vortexing. This was followed by 5 min of sonication in a Branson 8800 sonication water bath (Branson). Once lysed, we centrifuged samples for 5 min at 21,000 × g to clear debris. After this we followed the FASP protocol for the remaining steps. Approximate peptide concentrations were determined using a Micro BCA Protein Assay Kit (Thermo Fisher Scientific).
Proteomics
Samples were analyzed by one-dimensional Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS) on a Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific). The LC-MS/MS analysis was done as previously described (Kleiner et al., 2017). For each run, ∼1200 ng of peptide was loaded onto a 5 mm, 300 μm ID C18 Acclaim PepMap100 pre-column (Thermo Fisher Scientific) using an UltiMateTM 3000 RSLCnano Liquid Chromatograph (Thermo Fisher Scientific). Peptides were then separated on a 50 cm × 75 μm analytical EASY-Spray column packed with PepMap RSLC C18, 2-μm material (Thermo Fisher Scientific) using a 260-min gradient as described in Kleiner et al. (2017). The column was heated to 45°C via an integrated heating module. The analytical column was connected via an EASY-Spray source to the Orbitrap Mass Spectrometer. In between each sample, two washes with acetonitrile and one blank were run to reduce and assess carry over. Eluting peptides were analyzed in the Orbitrap Mass Spectrometer as described by Petersen et al. (2016). Roughly 140,000 MS/MS spectra were acquired per sample run (Supplementary Table 1).
Proteomics Analysis
A protein sequence database for each species was made using the reference proteomes from Uniprot of N. europaea (AL954747) (Chain et al., 2003), N. multiformis (CP000103) (Norton et al., 2008), and N. ureae (FNLN01000000) (Kozlowski et al., 2016b). The sequences of common contaminating proteins were also added to each database1. We used the program Proteome Discoverer version 2.0.0.802 (Thermo Fisher Scientific) to analyze the proteome data for each species separately. The MS/MS spectra for each species were searched against the protein sequence database using the Sequest HT node in Proteome Discoverer as described by Petersen et al. (2016). False discovery rates (FDRs) for peptide spectral matches (PSMs) were calculated and filtered using the Percolator Node in Proteome Discoverer (Spivak et al., 2009), using an FDR of 5% as the cut-off. Protein level FDRs were determined using FidoCT and filtered at 5% as well.
Search results for all samples were combined into a multiconsensus report with Proteome Discoverer. Relative protein abundances were calculated based on spectral counts that were normalized for protein length and the total number of spectra using the normalized spectral abundance factor (NSAF) approach (Zybailov et al., 2006). Perseus version 1.5.6.0 (Tyanova et al., 2016) and R (R Development Core Team, 2015) version 3.2.4 were used for the statistical analysis and visualization of the data. Prior to any statistical testing, we added a constant of 10-10 to all NSAF values and then applied a centered log-ratio (clr) transform (Aitchison, 1982; Fernandes et al., 2014) using the clr function from the chemometrics package in R (Kumar et al., 2014). For statistical tests, we also removed very low abundance proteins that did not have at least 20 spectral counts in total across all samples. We used t-tests to determine proteins that were statistically different between control and starved groups for each of the strains (p <0.05). In Perseus, to correct for multiple testing, we used permutation based calculation of FDR (Tusher et al., 2001). Average values reported in this study were calculated based on the triplicate control cultures.
Analysis of Orthologs
We identified orthologs shared between the three strains of AOB using the program OrthoVenn (Wang et al., 2015) with the protein sequence files obtained from Uniprot as input. OrthoVenn provided tab-delimited tables of protein sequence accession numbers that indicated which protein sequences represented orthologs. We merged the ortholog tables with the protein quantification tables using the VLOOKUP function in Excel. We used pairwise t-tests (p < 0.05) to identify statistically significant differences in the expression of orthologous proteins between strains. We corrected for multiple testing using permutation-based FDR implemented in the program Perseus version 1.5.6.0 (Tyanova et al., 2016). The NSAF values were transformed using the clr transformation prior to testing as described above.
Results and Discussion
Major Protein Expression Patterns in AOB Proteomes Under Ammonia Replete Conditions
A total of 891, 1064, and 814 proteins were identified from N. ureae, N. multiformis, and N. europaea, representing approximately 30, 37, and 32% of the predicted proteome for each species, respectively. Of these, 181 (20% of expressed proteins), 111 (10% of expressed proteins), and 107 (13% of expressed proteins) were listed as uncharacterized proteins for N. ureae, N. multiformis, and N. europaea, respectively.
A previous proteomics study on N. europaea found a total of 876 expressed proteins (34% of predicted proteome) (Pellitteri-Hahn et al., 2011), which is remarkably similar to the 814 expressed proteins (32% of predicted proteome) identified in this present study. In comparison, proteome analysis of Nitrosomonas eutropha, an eutrophic species, found 24% of the predicted proteome expressed (Wessels et al., 2011). In oligotrophic AOA species, separate proteome analyses revealed that Nitrososphaera viennensis expressed 1503 proteins, representing 48% of its genome (Kerou et al., 2016), while Nitrosopelagicus brevis which has a very streamlined genome, expressed 70% of its predicted proteome (Santoro et al., 2015).
There were some major differences in the most highly expressed proteins across the species (Table 1). In all three strains, AMO subunit B was either the most highly expressed (N. ureae, N. multiformis), or the second most highly expressed (N. europaea) protein. The most abundant protein in N. europaea was a general diffusion Gram-negative porin (Q82S02), which accounted for 4.36% of the strain’s normalized proteome. Orthologous porin proteins in N. ureae (A0A0S3AG80) and N. multiformis (Q2Y5X1) were also relatively highly expressed (Table 1). In the previous proteomics study conducted on N. europaea, the same general diffusion Gram-negative porin was identified as the most abundant protein (Pellitteri-Hahn et al., 2011). This confirms that there is some continuity between quantitative proteomics studies, even when using varying proteomics techniques and instruments. The second most abundant protein in N. ureae was nitrite reductase, but this enzyme was not highly expressed in the other two AOB (see following sections). Other highly expressed proteins included chaperonin proteins, which assist protein folding, elongation factor Tu, involved in protein translation, nitrosocyanin (NcyA), and rubrerythrin. In general, these highly expressed proteins play a role in key metabolic pathways, as well as basic cell function and maintenance.
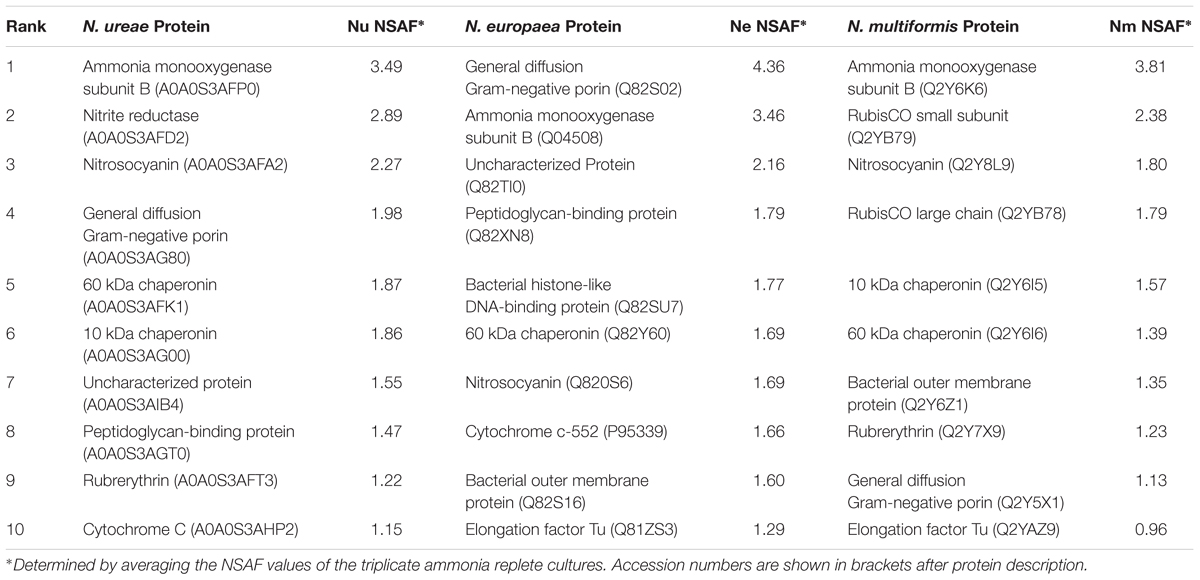
TABLE 1. Most highly expressed proteins from N. ureae, N. europaea, and N. multiformis under ammonia replete conditions.
Significant Differences in Expression of Proteins Involved in Ammonia Oxidation
The AMO was highly expressed in all three strains, which was unsurprising given the central role of this enzyme in the ammonia-oxidation pathway. Specifically, the beta subunit (AmoB) was the most highly expressed of the three subunits (N. europaea: 3.46%; N. ureae: 3.49%; N. multiformis: 3.81%) (Figure 1). The beta subunit is thought to form part of a complex with subunit A (AmoA), although its functional role is still not entirely clear (McTavish et al., 1993; Guo et al., 2013). AmoA contains the active site of the enzyme (Hyman and Wood, 1985), as further evidenced by the resolved structure of the related particulate methane monooxygenase (Lieberman and Rosenzweig, 2005), and was the next most highly expressed subunit (N. europaea: 1.28%, N. multiformis: 0.88%, and N. ureae: 0.61%). Subunit C (AmoC) was also highly expressed, and is thought to function as a chaperone as well as stabilizing the AMO subunits under stress-response (Klotz et al., 1997; Berube and Stahl, 2012).
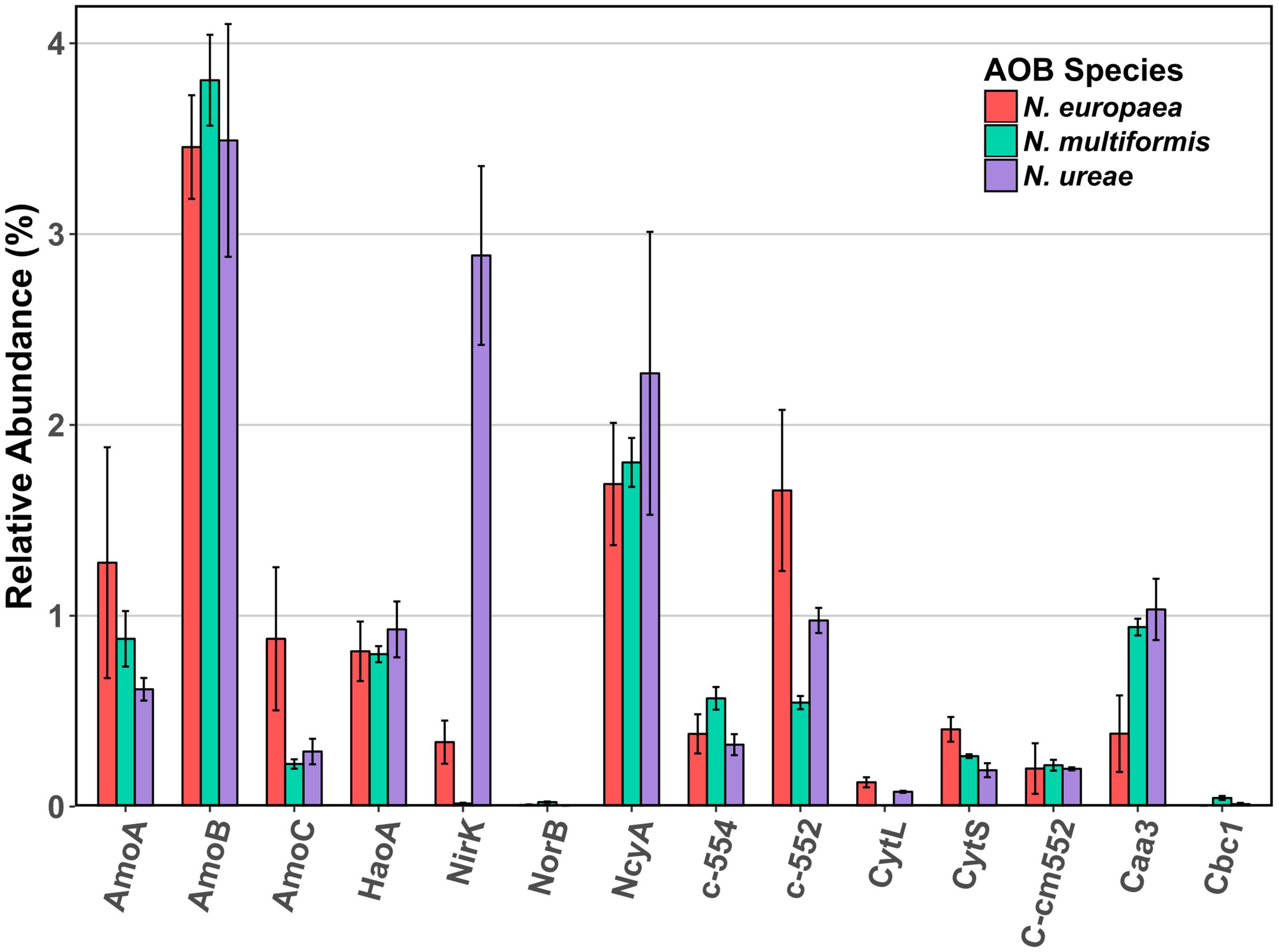
FIGURE 1. Mean relative abundance (%) of select enzymes involved in ammonia oxidation and electron transport in the proteomes of three AOB species under ammonia replete conditions. Error bars indicate SD of triplicate experiments. When multiple copies of a gene were expressed, their abundances were summed. Accession numbers for each of the enzymes for each species are listed in Supplementary Table 2. AmoA: ammonia monooxygenase subunit A, AmoB: ammonia monooxygenase subunit B, AmoC: ammonia monooxygenase subunit C, HaoA: hydroxylamine dehydrogenase, NirK: nitrite reductase, NorB: nitric oxide reductase, NcyA: nitrosocyanin, c-552: cytochrome c-552, c-554: cytochrome c-554, CytL: cytochrome P460, CytS: cytochrome c′ beta, cm552: cytochrome cm-552, caa3: cytochrome aa3, cbc1: cytochrome bc1.
The HAO was another highly expressed metabolic enzyme, but to a lesser extent than AMO in all three strains (N. europaea: 0.81%; N. ureae: 0.93%; N. multiformis: 0.80%). Recent evidence suggests that, contrary to previous literature, HAO catalyzes a three electron oxidation of NH2OH to NO, instead of a four electron oxidation to NO2- (Caranto and Lancaster, 2017). Thus, there is likely a third enzyme involved in the ammonia oxidation process that completes the oxidation of NO to NO2-. Such an enzyme would likely be highly expressed and present in all AOB species.
From our results, another highly expressed metabolic protein apart from AMO and HAO was the red copper protein, NcyA (Figure 2), which was expressed to between 1.7 and 2.3% of the proteomes (Table 1). Previous studies of AOB in aerobic growth conditions also found NcyA in high protein concentrations and transcript levels, similar to the concentrations of other key components of the ammonia oxidizing system (Whittaker et al., 2000; Klotz and Stein, 2011). Due to its abundance, it has been previously proposed that NcyA is part of the central ammonia oxidation pathway either with an enzymatic function, or through mediating electron transfer (Arciero et al., 2002; Basumallick et al., 2005; Klotz and Stein, 2011). Specifically, it has been suggested that NcyA participates in recycling electrons from the quinone pool to AMO or functions as a relay for electrons from hydroxylamine to oxygen (Arp et al., 2007). Other specific hypotheses of roles include a functional link to nitrite reductase (Arciero et al., 2002), or a role in NO binding and reduction to N2O (Basumallick et al., 2005). Increased expression was previously found when N. europaea cultures were exposed to NO (Schmidt et al., 2004), and the protein has also been linked to the ammonia starvation response (Klotz and Stein, 2011). However, the latter role was not supported by the results of this study as there was no significant difference in expression levels of NcyA between control and starved conditions in any species (NSAF values starved cultures: replete cultures; 2.27:1.69% N. europaea (Q820S6), 2.49:2.27% N. ureae (A0A0S3AFA2), 1.99:1.80% N. multiformis (Q2Y8L9), p > 0.05, t-test). Given its expression at the protein level and structural potential for NO binding and enzymatic activity, it seems a likely candidate for the missing nitric oxide oxidase in AOB, although this hypothesis awaits further testing. One case against this hypothesis is that the genome of Nitrosomonas sp. Is79 lacks the ncyA gene, putting into question its universality as a central metabolic enzyme in AOB (Bollmann et al., 2013). Regardless, more experiments are needed to elucidate the function of this highly expressed protein.
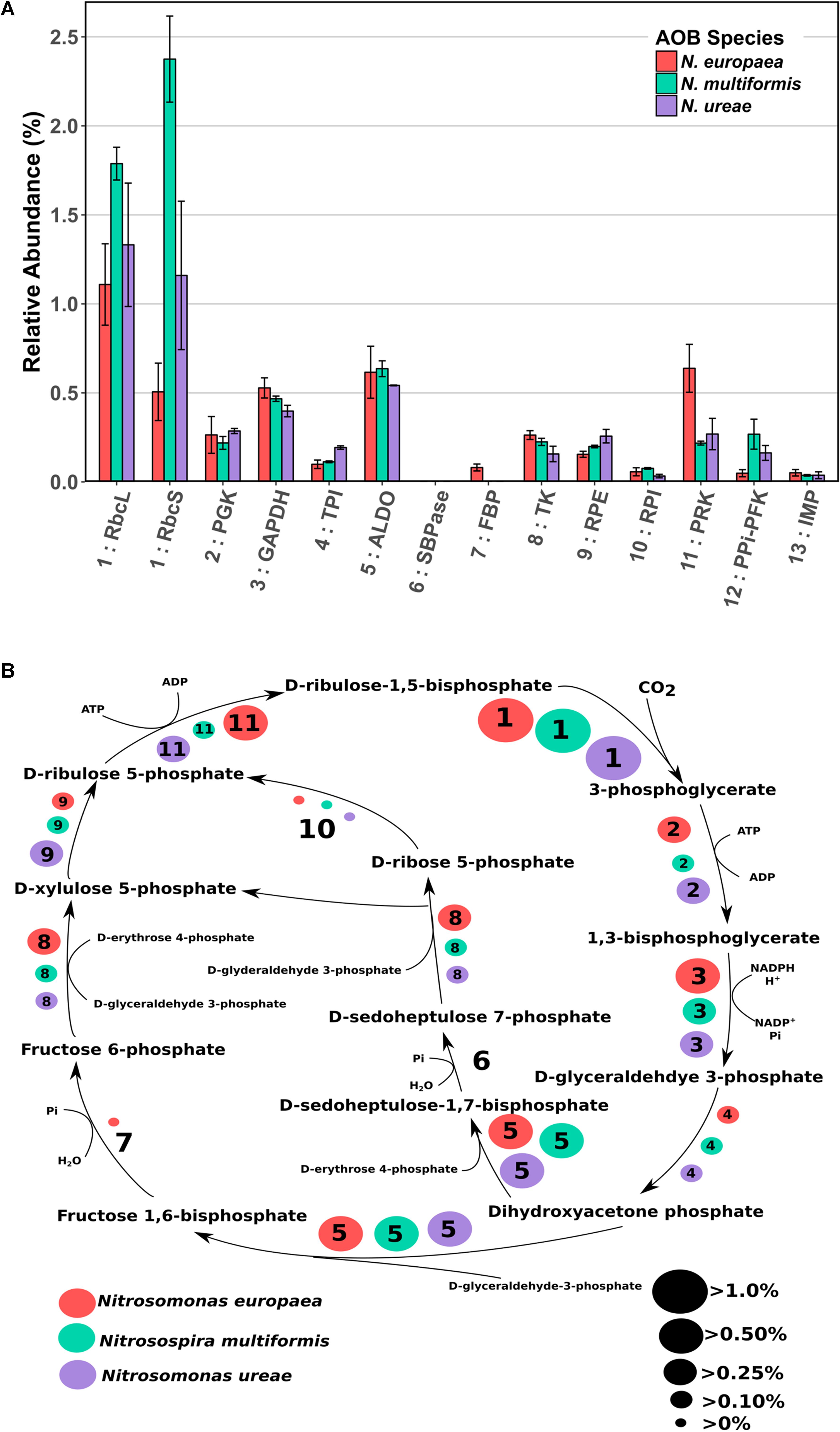
FIGURE 2. (A) Mean relative abundance (%) of CBB cycle enzymes in the proteomes of three AOB species under ammonia replete conditions. Error bars indicate SD of triplicate experiments. If a species had two expressed copies of a particular enzyme (see Supplementary Table 3) expression values were summed. Numbers preceding enzyme name are matched to their respective reaction in B. RbcL: RuBisCO large chain, RbcS: RuBisCO small chain, PGK: Phosphoglycerate kinase, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, TPI: Triosephosphate isomerase, ALDO: Fructose-bisphosphate aldolase, SBPase: Sedoheptulose bisphosphatase, FBP: Fructose-1,6-bisphosphatase, TK: Transketolase, RPE: Ribulose-phosphate-3-epimerase, RPI: Ribose-5-phosphate isomerase, PRK: Phosphoribulokinase, PPi-PFK: Pyrophosphate phosphofructokinase, IMP: Inositol monophosphatase/type IV F1P6Pase. PPi-PFK and IMP are suggestions from the literature for enzymes that could potentially complete the CBB cycle in AOB. (B) CBB cycle with relative abundances (%) of enzymes depicted by size of corresponding circle. Numbered circles are linked to the corresponding enzyme number depicted in A. Red circles indicate the abundance in N. europaea, teal circles indicate the abundance in N. multiformis, and purple circles indicate the abundance in N. ureae. Enzyme 6: SBPase is missing from all species, and enzyme 7: FBP is either missing, or expressed to a low amount (N. europaea).
Another potential candidate NO oxidase is a reversely operating nitrite reductase, NirK, however, our results showed minimal expression of NirK in N. multiformis (0.02%) and several AOB, like Nitrosomonas communis, lack the nirK gene (Cantera and Stein, 2007; Kozlowski et al., 2016c), again calling into question its universality as a NO oxidase. Furthermore, only one study has shown the potential for NirK to operate as a NO oxidase rather than as a nitrite reductase (Wijma et al., 2004), making this enzyme an unlikely candidate as a central participant in the ammonia oxidation pathway of AOB.
Of interest, the highest NirK expression from the three species was found in N. ureae where the enzyme was the second most highly expressed protein (2.89%; A0A0S3AFD2). Its non-orthologous equivalents in the other two strains were expressed to a much lower level (N. europaea: 0.34% Q82TG8; N. multiformis: 0.02% Q2Y7H8), and this disparity in expression patterns is one of the most pronounced differences between the nitrogen metabolism enzymes of these three AOB (Figure 1). NirK is the enzyme responsible for reducing nitrite (NO2-) to NO (Wrage et al., 2001; Stein, 2011; Kozlowski et al., 2016a), and in AOB, NirK was shown to play a role in aiding in the efficient oxidation of NH3 to NO2-, as opposed to functioning in nitrifier denitrification (Kozlowski et al., 2014). AMO can have a higher turnover rate than HAO, and thus when excess NH3 is oxidized, the intermediate NH2OH can accumulate. NirK in AOB can aid HAO in oxidizing NH2OH by alleviating the electron flow bottle-neck through transferring electrons from the cytochrome pool onto NO2-. A possible explanation for the increased amounts of NirK in N. ureae is that the strain is generally considered oligotrophic, and although it is able to grow at the ammonia concentrations used in this study (Kozlowski et al., 2016a), N. ureae may not be naturally adapted to these conditions. N. ureae grown at NH3-concentrations higher than it is adapted (i.e., > 5 mM) likely causes an imbalance between the rates of NH3 and NH2OH-oxidation, which AOB adapted to higher NH3 concentrations are not likely to experience at the NH3 concentration (10 mM) used in this study. Thus, N. ureae could potentially be using NirK to speed the oxidation of excess NH2OH.
Nitric oxide reductase is a key enzyme in the AOB nitrifier denitrification pathway, converting NO to nitrous oxide (N2O), a potent greenhouse gas much stronger than carbon dioxide. Homologs of the nitric oxide reductases NorB and NorY are present in all AOB except for N. ureae, and Nitrosomonas sp. Is79 (Bollmann et al., 2013; Kozlowski et al., 2016b). The NorB enzyme was expressed at very low levels in N. multiformis and N. europaea (Figure 1). The low to negligible expression of NorB in these AOB may be due to a decrease in nitrifier denitrification under the growth conditions in this study.
Cytochrome P460 (CytL) has been implicated in the oxidation of NH2OH and NO to NO2- (Elmore et al., 2007; Stein, 2011), and more recently in the direct oxidation of NH2OH to N2O, producing NO2- only as a side product when NO dissociates from the active site and reacts abiotically with oxygen (Caranto et al., 2016). In the former hypothesis, CytL was thought to be important for alleviating nitrosative stress in AOB that lack nitric oxide reductase, such as N. ureae (Kozlowski et al., 2016a). CytL was relatively highly expressed in N. europaea (H2VFU9; 0.13%), moderately expressed in N. ureae (A0A0S3AFC9; 0.08%), and is absent in N. multiformis (Norton et al., 2008). Our finding of a higher expression of CytL in N. europaea, which has a nitric oxide reductase, as compared to N. ureae, suggests that the primary role of CytL is unrelated to the absence of nitric oxide reductase under these conditions.
Other cytochromes implicated in the energy metabolism of AOB were also expressed (Figure 1). Cytochrome aa3 was expressed relatively highly in all three species (N. europaea: 0.38%, N. multiformis: 0.94%, and N. ureae: 1.03%). Cytochrome bc1 was not expressed in N. europaea, and was expressed at 0.05% and 0.01% in N. multiformis, and N. ureae, respectively. Cytochrome cm-552 and cytochrome c-552 were also relatively highly expressed in all three species (>0.2%). Cytochrome c-554 was very highly expressed in N. europaea (1.7%, Q57142), compared to N. ureae (0.98%, A0A0S3AN35), and N. multiformis (0.54%, Q2YA34).
Proposal of Novel Enzyme Substitution for Missing Calvin-Benson-Bassham Cycle Enzymes of AOB
As autotrophs, AOB almost exclusively use the CBB cycle to acquire carbon. However, due to a few missing enzymes in AOB species, the CBB cycle in these organisms appears to deviate from the classical version (Chain et al., 2003; Norton et al., 2008; Kozlowski et al., 2016b). N. europaea lacks one enzyme necessary for the classical version of the CBB cycle, sedoheptulose-1,7-bisphosphatase (SBPase), whereas N. multiformis and N. ureae lack two enzymes; fructose-1,6-bisphosphatase (FBP) and SBPase (Figure 2 and Supplementary Table 3). Potential alternative enzymes that could fulfill these missing roles in AOB have been suggested (Norton et al., 2008; Kleiner et al., 2012). For the missing SBPase, Chain et al. (2003) suggested that in N. europaea it could be replaced by the FBP, which is known to frequently operate as a dual function FBP/SBPase enzyme in bacteria (Yoo and Bowien, 1995; Shively et al., 1998; Miyagawa et al., 2001). For the missing FBP in N. multiformis, Norton et al. (2008) suggested that it could be replaced by genes that have some similarity to archaeal inositol monophosphatase/type IV FBP (IMP/FBP). They specifically identified the gene Nmul_A2147 (Q2Y731) as the most likely candidate for FBP replacement, because of similarities in domain structure and active site residues. Lastly, Kleiner et al. (2012) suggested that the missing enzymatic reactions could be performed by pyrophosphate-dependent 6-phosphofructokinase (PPi-PFK). This hypothesis is based on the fact that the PPi-PFK of Methylococcus capsulatus, which is highly similar to the one in AOB (75% nucleotide sequence similarity in N. europaea and N. multiformis, and 77% in N. ureae) was experimentally shown to catalyze reactions analogous to the FBP and SBPase reactions in a pyrophosphate-dependent manner (Reshetnikov et al., 2008). Thus PPi-PFK could enable the completion of the CBB cycle in the AOB missing two CBB cycle enzymes.
To gain a better understanding of how the CBB cycle functions in AOB we did a careful analysis of CBB cycle enzyme expression. Enzymes of the CBB cycle were highly expressed in all three species, and each enzyme accounted for between 0.03 and 2.3% of each species’ proteome, with the total expression of all CBB cycle enzymes accounting for 4.3, 6.3, and 4.6% percent of the proteomes of N. europaea, N. multiformis, and N. ureae, respectively. The key enzyme of the CBB cycle, RuBisCO, including both small and large subunits, was the most abundant CBB cycle enzyme (Figure 2A). One copy of each RuBisCO large and small subunit was expressed in N. europaea and N. multiformis, and two copies of each were expressed in N. ureae. Of the two copies in N. ureae, one pair (A0A0S3AHX5, A0A0S3AHZ4) was orthologous to the subunits from N. multiformis, corresponding to RuBisCO Form IC. The other RuBisCO pair (A0A0S3AFL1, A0A0S3AFG4) was orthologous to the subunits from N. europaea RuBisCO form IAq. The RuBisCO Form IC, which was more highly expressed by N. ureae in this study, is thought to have a slightly lower affinity for CO2 compared to RuBisCO Form IAq (Badger and Bek, 2008). The RuBisCO sequences in N. ureae resemble the RuBisCO copies found in the closely related Nitrosomonas sp. Is79, and suggests that these Nitrosomonas species have higher flexibility with respect to CO2 availability in their environments compared to other AOB species (Bollmann et al., 2013). The enzyme that was consistently the lowest expressed from the CBB cycle (<0.08%) was ribose-5-phosphate isomerase (RPI), which converts D-ribose-5 phosphate to D-ribulose-5 phosphate (Figure 2). In general, specific enzymes were expressed to similar levels across the species, with exceptions being phosphoribulokinase (PRK) which was expressed approximately twofold higher in N. europaea, and triosephosphate isomerase (TPI) which was expressed roughly twofold higher in N. ureae. Both sets of comparisons were significant (p < 0.05) with a pairwise t-test (Supplementary Tables 7–9).
It would be expected that the alternate enzymes performing the missing CBB cycle reactions in the three AOB species would be expressed to levels comparable to the traditional CBB cycle enzymes. The first proposed enzyme replacement was FBP, which was suggested to perform both its own function as well as that of SBPase. This functional overlap may be possible in N. europaea (although FBP was not expressed highly at 0.08%, in comparison with most other CBB cycle enzymes), however, this replacement enzyme is not present in N. multiformis or N. ureae. For the specific IMP/FBP (Q2Y731) proposed by Norton et al. (2008) as a SBPase and FBP substitute, expression was undetectable in N. multiformis in this study. Furthermore, other inositol monophosphatases present in N. ureae (A0A0S3AHX4, A0A0S3AIS2) and N. multiformis (Q2YB92), which bear some similarity to the IMP/FBP, accounted for on average, less than 0.035% of their proteomes, making them much less abundant than all other CBB cycle enzymes. N. europaea also has a copy of inositol monophosphatase (Q82TE3), and it was expressed at 0.05% of the proteome.
The PPi-PFK is a likely candidate for a FBP/SBPase substitute, based on the proteome expression data obtained in this study, and the fact that the PPi-PFK enzyme is able to perform the two missing reactions. PPi-PFK was expressed (0.05% in N. europaea, 0.16% in N. ureae, and 0.27% in N. multiformis) at levels comparable to the other CBB cycle enzymes (Figure 2A). In addition, N. europaea, which does contain FBP, showed lower expression of PPi-PFK than the other two species.
It has further been suggested that the use of PPi-PFK in the CBB cycle has the potential to save 10% of the energy in comparison with the classical version of the CBB cycle, when PPi-PFK is used in conjunction with a proton-translocating pyrophosphatase (HPPase) (Kleiner et al., 2012). The HPPase and the PPi-PFK form an operon in the genomes of all three analyzed AOB species, which indicates a tight metabolic coupling between the two enzymes. Hydrolysis of PPi, which is produced by PPi-PFK when cleaving the phosphate of sedoheptulose-1,7-bisphosphate and fructose-1,6-bisphosphate, by the HPPase would lead to energy conservation in form of a proton motive force. All three species expressed HPPase [0.17% in N. europaea (Q82TF3), 0.13% in N. ureae (A0A0S3AI31), and 0.10% in N. multiformis (Q2YB25)], indicating that a PPi-dependent energy economy plays a role in AOB. In summary, our proteomic data supports the hypothesis that PPi-PFK substitutes for FBP and SBPase in the CBB cycle of AOB and that the CBB cycle of AOB may be more energy efficient due to PPi-dependent energy conservation.
Major Differences in Expression of Orthologous Genes Across the AOB
Using the program Orthovenn (Wang et al., 2015), we identified orthologous protein encoding genes shared between either two or all three AOB species, as well as genes that are unique to individual species. By comparing the expression of orthologous proteins, we could directly determine differences in the allocation of cellular resources toward cellular functions. Of all the protein encoding genes, 1253 were identified as having orthologs in all three species (1207 of which only occurred in one copy per genome), 648 were considered orthologs common to two species, and 162 were found in multiple copies but only in one species. Of these unique genes, 41 were found in N. europaea, 57 in N. ureae, and 64 in N. multiformis (Supplementary Figure 2). The remaining genes were found in only one copy in one species and resulted in 524 singletons in N. europaea, 873 in N. ureae, and 825 in N. multiformis.
To identify orthologous proteins that differed in expression between species, we applied pairwise t-tests (p < 0.05) corrected for multiple testing with permutation based FDR to all culture replicates, and detected hundreds of statistically significant differences. A comprehensive list of all orthologous proteins and the corresponding statistical information is presented in Supplementary Tables 7–9 and Supplementary Table 4 provides a description of each of the tables’ contents. A total of 798 proteins out of the 1207 single gene orthologs were detectable to some degree in one of the three species at the protein level. Out of these 798 proteins, 585 proteins were statistically differentially expressed between N. europaea and N. multiformis (73% of three species orthologs), 626 proteins were statistically differentially expressed between N. multiformis and N. ureae (78% of three species orthologs), and 508 proteins were statistically differentially expressed between N. ureae and N. europaea (64% of three species orthologs). N. europaea and N. multiformis shared 90 two-species orthologs, and out of these 80 were differentially expressed. N. multiformis and N. ureae shared 94 two-species orthologs, and out of these 87 were expressed differentially. N. europaea and N. ureae shared 58 two-species orthologs, and out of these 46 were expressed differentially.
The betaproteobacteria AOB as a tight phylogenetic group offer a compelling system to examine how functionally similar microbes make use of differential gene expression, enabling competitiveness within a range of physicochemical factors including pH, temperature, substrate concentration, oxygen tension, trace metals, and salinity among others (Bernhard et al., 2005; Fierer et al., 2009; Martens-Habbena et al., 2009; Wells et al., 2009). Prior studies comparing genome content, regulation of genes and proteins, and relative gene abundance at ecosystem levels suggest that this genomic flexibility enables each species to occupy a preferred niche (Norton, 2011). Hence, it is not surprising that many orthologous genes showed differential expression at the protein level in this study as each of the three compared species is found occupying different habitats, and thus, were expected to show a wide range of responses to identical growth conditions (Prosser et al., 2014). The top 50 most abundant proteins from each species and the abundance of orthologous proteins in the other species (if present) are shown in Figure 3 and listed in Supplementary Table 5. Due to the large number of differences in orthologous protein expression we only highlight a few interesting examples from cellular subsystems below.
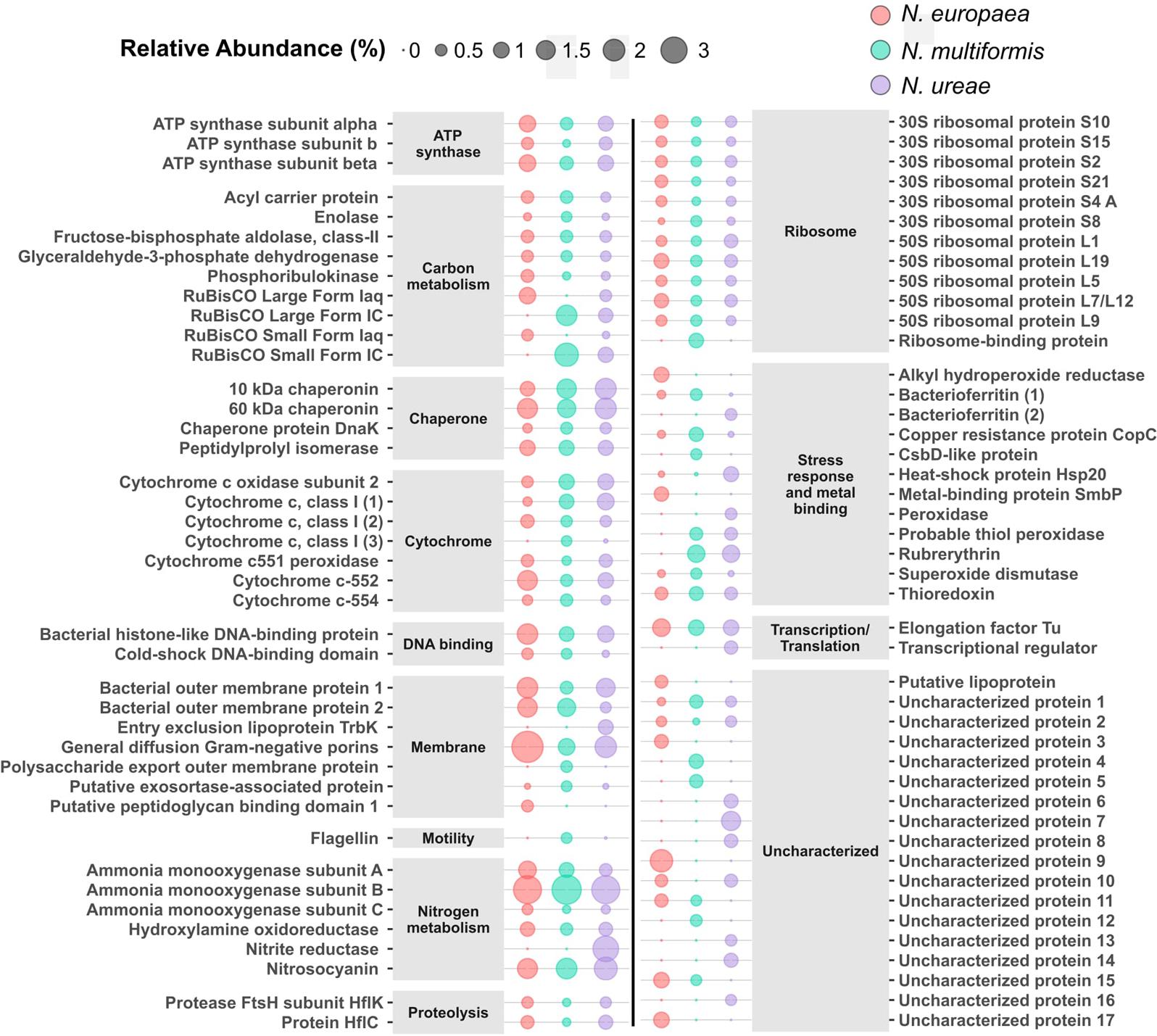
FIGURE 3. Top 50 most abundant proteins in each species with orthologs from the other species. Supplementary Table 5 contains accession numbers for each protein. Size relates to relative abundance (%) in the proteome of each species.
Motility
The three AOB species investigated have the capacity for motility with orthologous proteins encoding flagellin and flagellin regulation present in each genome. Under the present growth conditions, however, the flagellin protein was not expressed at all in N. europaea, and only slightly in N. ureae (0.006%, A0A0S3AJU9). In contrast, N. multiformis expressed its flagellin protein (Q2Y9D1) to a significantly higher level than the other two strains, and it accounted for 0.43% of its proteome. Additionally, the sigma factor controlling for the expression of flagella-related genes (FliA) was only expressed in N. multiformis. Thus, N. multiformis allocates significant resources to motility compared to the other two species.
Cell Division
The MraZ transcription regulator, involved in inhibition of cell division among other functions (Eraso et al., 2014), was more highly expressed in N. ureae (0.07%, A0A0S3AHI1) than the other two species (N. europaea: 0.005% Q82VT2, N. multiformis: 0.02% Q2Y629). N. ureae had both higher expression of the MraZ transcriptional regulator and slower growth rates than the other two species agreeing with its functional description (Supplementary Figure 1). Furthermore, there was a statistically significant relationship between generation time and MraZ expression across these AOB species (Spearman’s correlation = 0.85, p < 0.005, n = 9). In contrast to MraZ expression, the ATP-dependent zinc metalloprotease FtsH, was most highly expressed in N. europaea (0.13%, Q82VZ3), followed by N. multiformis (0.04%, Q2Y826), and at the lowest level in N. ureae (0.01%, A0A0S3AFK9). FtsH regulates the expression of genes required for the synthesis of cell wall lipopolysaccharide components, an important process in actively dividing cells. Correlation of FtsH expression with the generation time of the AOB cultures was highly significant (Spearman’s correlation = -0.85, p < 0.005, n = 9). Additionally, there was a negative correlation between the expression levels of MraZ and FtsH across AOB replicates (Spearman’s correlation = -0.87, n = 9, p = 0.003, Supplementary Figure 3). FtsZ, the ubiquitous key protein involved in cell division, was significantly more highly expressed in N. europaea (Q820N2, 0.12%) and N. multiformis (Q2Y644, 0.14%) than in N. ureae (A0A0S3AHF2, 0.03%). The relationship between FtsZ and generation time across the species, however, was not significant (p > 0.05). Generation time was highly correlated with the expression of 40 additional proteins (see Supplementary Table 6 for more details). Of these proteins, the majority that were correlated with faster growing cells were related to protein synthesis or cell division (Supplementary Table 6).
Stress Response
Several proteins involved in AOB stress response, including nitrosative and oxidative stresses, showed strong differences in expression between the three species. Nitrosative and oxidative stress from exposure to reactive nitrogen species (RNS) and reactive oxygen species (ROS), respectively, are common in environmental bacteria like AOB. The origin of these RNS/ROS can be from interactions with other organisms, by-products of aerobic cellular metabolism, or they can be photochemically produced from organic solutes in soil and other environments (Wood and Sørensen, 2001). Rubrerythrin belongs to the ferritin-like superfamily and is involved in oxidative stress relief mainly by acting as a peroxide scavenger (Cardenas et al., 2016). Originally evolving to protect anaerobic species in increasingly aerobic conditions, this protein has since evolved to be used by aerobic organisms. Among the AOB species included in this study, N. ureae (A0A0S3AFT3) and N. multiformis (Q2Y7X9) had orthologous copies of rubrerythrin that were expressed at high levels (1.22 and 1.23% of proteomes, respectively). Interestingly, although highly expressed in the other two species, N. europaea does not contain an ortholog for this gene. Without a rubrerythrin copy in N. europaea, alternate enzymes are likely responsible for this particular oxidative stress function. One candidate is alkyl hydroxide reductase (Q820H3), a highly expressed enzyme in N. europaea (0.91%), which also scavenges peroxide radicals (Seaver and Imlay, 2001). The gene has one ortholog in N. multiformis, which was not expressed, but no orthologs in N. ureae.
Superoxide dismutase catalyzes the partitioning of superoxide radicals, which can form during oxygen metabolism, into either molecular oxygen or hydrogen peroxide. The enzyme was expressed to high levels in all three species, but was significantly more highly expressed in N. multiformis (0.39%, Q2YBR2), than N. europaea (0.19%, Q82W28), and N. ureae (0.10%, A0A0S3ALT7). Catalase, the enzyme that converts hydrogen peroxide (the potential product of superoxide dismutase) to water and oxygen was expressed to low levels (< 0.05%) in N. europaea (Q82TK1) and N. ureae (A0A0S3AIK6), while the ortholog in N. multiformis (Q2Y8V3) was not detected.
Heat shock protein 20 (Hsp20) was much more highly expressed in N. ureae (0.82%, A0A0S3AHM5) than N. multiformis (0.02%, Q2Y862), and N. europaea (0.06%, Q82VT2). Heat shock proteins are normally expressed when an organism is exposed to environmental stress, and in these conditions, they act as protein chaperones, preventing misfolding of other proteins. In the present study, the three species were exposed to the exact same growth conditions, suggesting that the high levels in N. ureae are either due to constitutive expression of the gene, or suboptimal growth conditions (e.g., high NH3 concentrations) for the strain.
The copper resistance protein CopC is important for protecting against copper toxicity, as well as for copper utilization. The copCD genes are located within the amoCAB gene cluster, indicating functional relatedness to the AMO enzyme. CopC was more highly expressed in N. multiformis (0.70%, Q2Y5C3) than N. europaea (0.18%, Q82T65) or N. ureae (0.093%, A0A0S3AIQ1). Copper is in the active site of AMO and NirK and as CopC is important in copper binding and uptake, it is also essential for the functioning of these two enzymes. The other subunit to the enzyme CopD was expressed to much lower levels in all species (N. europaea: 0.02% Q820J3, N. multiformis: 0.03% Q2Y5C4, and not expressed in N. ureae).
Urea Utilization
Under certain conditions (i.e., low pH environments), urea can be used as both a source of nitrogen and carbon in some AOB (Marsh et al., 2005). N. ureae, as its name suggests, is one of the AOB species capable of using urea (Koops et al., 1991). The three subunits of the enzyme urease, which converts urea and water to carbon dioxide and ammonia, were moderately expressed in N. ureae (0.06–0.11%). Urease was barely expressed in N. multiformis (less than 0.02%) and N. europaea lacks a homologous gene (Chain et al., 2003). Urea carboxylase, which catalyzes the reaction of urea, ATP, and bicarbonate to urea-1-carboxylate (allophanate), was represented by three copies (A0A0S3AHN5, A0A0S3AI24, and A0A0S3AHN2) in the N. ureae proteome, but none of the three were expressed to greater than 0.04%. Orthologs of urea carboxylase were present in the other two genomes but were undetectable at the protein level. Low expression of urea related proteins is to be expected as no urea was added to the media, however, the observed consistent expression of urease in N. ureae suggests it could be constitutively expressed in this species.
Uncharacterized Proteins Without Orthologs
Many uncharacterized proteins without orthologs in the other two species (singletons) were highly expressed at the protein level. For instance, Q2Y9X6, Q2Y9X8 and Q2Y844 (DUF2795 domain) were expressed to 0.81, 0.69, and 0.37% of the proteome, respectively, from N. multiformis. From N. ureae, uncharacterized singleton proteins A0A0S3AIB4, A0A0S3AFI8, and A0A0S3AKF1 were expressed to 1.5, 0.78, and 0.71%, respectively. N. europaea also had uncharacterized singleton proteins Q82VY9 (PepSY domain), Q82V70 (DUF533 domain), and Q82TW7 (LTXXQ motif) highly expressed to 0.75, 0.38, and 0.23% of the proteome. High expression of uncharacterized proteins without orthologs in these AOB suggests that more work is needed in elucidating the function of hypothetical proteins with potentially important roles central to AOB metabolism.
Response to Ammonia Starved Conditions
Overall, surprisingly few proteins exhibited a statistically significant change in expression in response to 24 h of ammonia starvation. Assays confirmed that the control cultures contained ammonia and that the starved cultures did not. Furthermore, after 24 h of incubation, the production of nitrite had started in the control cultures, suggesting active ammonia oxidation, but no nitrite was measured in the starved cultures (Supplementary Table 1). The control cultures had accumulated 0.25–1 mM of nitrite, and these values are comparable to the daily nitrite production observed in pre-cultures. We also performed correlation analysis between substrate concentrations after 24 h (nitrification activity) and relative protein abundances and found no significant relationships. The lack of response to a full day of ammonia starvation in the AOB species tested shows that these AOB regulate their proteomes similarly despite ecological differences. Important to point out, however, is that in the present study we aimed to identify changes in the relative abundance of proteins, but we cannot rule out the possibility of alternate regulation of protein function in the form of post-translational modifications, or substrate level activation or inhibition.
Nitrosospira multiformis was the only strain in which proteins showed statistically significant differences between control and starved cultures. These differences were minor, however, as from the 1064 proteins expressed by this strain only three were identified as statistically significant using t-tests (p < 0.05) after correcting for multiple tests using permutation based FDR. The differentially expressed proteins were heat shock protein Hsp20 (Q2Y862, 0.1% average in starved compared to 0.02% average in control), RND efflux system outer membrane lipoprotein NodT (Q2Y897, 0.006% average in starved compared to 0.02% average in control), and RuBisCO large chain (Q2YB78, 0.96% average in starved compared to 1.8% average in control).
Steady expression levels of AMO throughout the experiment were a strong indication of a slow functional response to ammonia starvation by these organisms. In support of this, it has been suggested based on similar observations in prior studies that N. europaea cells maintain a basal level of AMO enzyme activity that is largely insensitive to changes in ammonia concentration (Stein et al., 1997; Geets et al., 2006). This basal level of AMO expression in AOB could present an immediate advantage to the cells when ammonia is available again and could be a strategy indicative of cells adapted to substrate fluctuations.
A proteomics study by Pellitteri-Hahn et al. (2011) identified only 27 proteins that were differentially expressed after a 2-week period of ammonia starvation in N. europaea, significantly longer than the present study. The proteins with greater abundance in growing cells from that study were geared toward biosynthesis, while energy starved cells had more proteins related to survival functions. In contrast, a study that looked at the starvation response of N. europaea, through transcriptomics and with respect to both ammonia and carbonate, saw 90% of transcripts present at twofold greater levels in growing cells compared to cells that had been deprived of ammonia for only 16 h (Wei et al., 2006). This suggests that at least in some AOB there is a relatively quick regulatory response to ammonia depletion on the level of transcription likely leading to a decrease in production of new protein. Consequently, the energy that was invested in initial protein synthesis is conserved in AOB. Keeping protein machinery intact would allow these species the opportunity to quickly respond to increases in nutrients. This hypothesis is also supported by previous studies that observed extremely quick renewal of NH3-oxidation activity of AOB following starvations that varied in length from weeks to many months (Wilhelm et al., 1998; Tappe et al., 1999; Bollmann et al., 2005). Furthermore, a recent study monitoring the response of N. europaea to daily anoxic-oxic cycles found that short-term responses (a few hours) to stress were regulated at the level of mRNA, whereas long-term responses (after 12 days) to the same stress were regulated at the level of proteins (Yu et al., 2018). Thus, our finding of a minimal response at the protein level to ammonia starvation after 24 h is in agreement with the recent study by Yu et al. (2018).
Despite the absence of a coordinated starvation response, the RuBisCO enzyme consistently decreased in abundance in starved cells as compared to control cells in all three species (Figure 4), and this decrease was statistically significant for the large subunit of N. multiformis. In N. multiformis the ratio of starved to control for the large subunit was 0.53, in N. europaea it was 0.68, and in N. ureae it was 0.32 and 0.56 for the two expressed copies. The small subunit of RuBisCO also decreased between starved and control conditions. In N. multiformis the ratio was 0.69, in N. europaea the ratio was 0.63, and in N. ureae the ratio was 0.35 and 0.56. In a previous starvation study using transcript abundance, RuBisCO transcripts were found to decrease significantly in ammonia starved conditions, and they decreased to a greater extent than amoCAB and haoAB transcript levels (Wei et al., 2004). Additionally, in a study observing the transcriptome of Nitrosococcus oceani, an AOB from the class of Gammaproteobacteria, transcripts involved in carbon fixation (i.e., RuBisCO) strongly decreased in response to a change in energy status due to ammonia starvation (Stein et al., 2013). RuBisCO catalyzes the rate limiting step of the CBB cycle, which often behaves as the ultimate electron sink in chemolithoautotrophic bacteria (Bar-Even et al., 2012). Thus, the reduction of RuBisCO in the proteome could act to immediately reduce the requirement for electrons and energy from ammonia oxidation in times of scarcity, without having to break down other CBB cycle enzymes. In this manner, the AOB are limiting anabolic metabolism by specifically targeting the enzyme that best controls these processes. Tappe et al. (1999) suggested that in non-spore forming bacteria, maintenance energy demand during periods of starvation should be as low as possible, but still sufficient to ensure a fast response when nutrients become available again (Tappe et al., 1999). Maintenance energy is defined as the energy consumed during the activities that allow the cell to survive without biomass production. As RuBisCO and the CBB cycle are responsible for the build-up of biomass in AOB, the reduction of RuBisCO in the proteome is in line with this hypothesis.
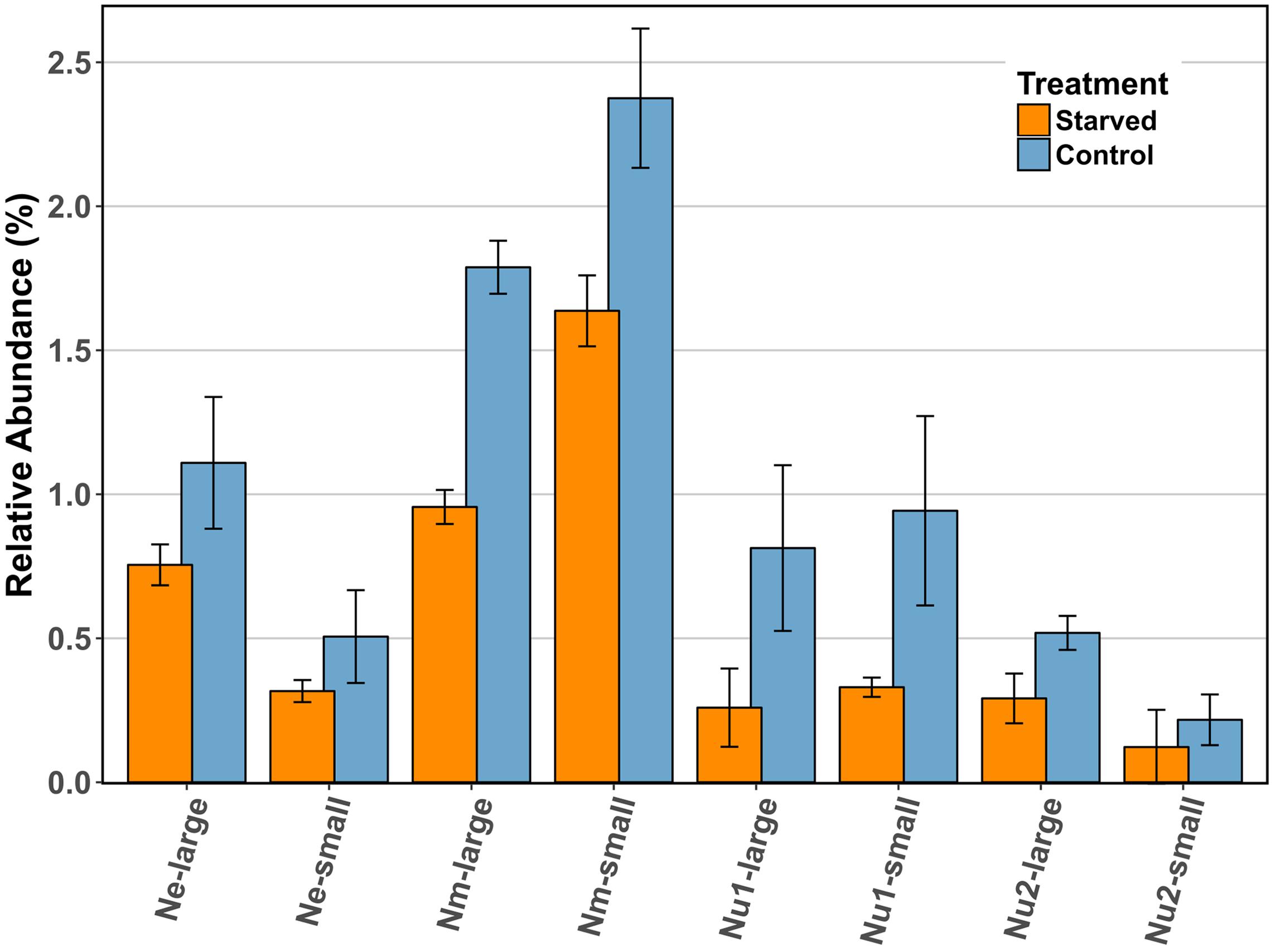
FIGURE 4. Comparison of the relative abundance (%) of RuBisCO large and small subunits across species in control and starved conditions. Ne: N. europaea, Nm: N. multiformis, and Nu: N. ureae. Nu1 refers to the copy of N. ureae RuBisCO subunits that are orthologous to N. multiformis RuBisCO subunits (A0A0S3AHX5, A0A0S3AHZ4), while Nu2 refers to the other copy, orthologous to N. europaea RuBisCO subunits (A0A0S3AFL1, A0A0S3AFG4). Only N. multiformis RuBisCO large subunit (Nm-large) was identified as statistically significant with a t-test corrected for FDR.
Conclusion
In summary, a 24-h period of ammonia starvation did not trigger a robust response in the proteomes of the three AOB species analyzed in this study. This suggests that even with the ecological differences between the species, they respond to fluctuations in substrate availability in this time-period in a similar manner at the protein level. We would like to suggest, however, that longer starvation times and the corresponding shift in the AOB proteome should be explored in the future as the AOB response to these changes may be slow (Yu et al., 2018). Although all AOB perform the same core function of ammonia oxidation to nitrite, they inhabit a variety of environments, from the oligotrophic ocean to eutrophic wastewater treatment plants, sometimes with multiple AOB species coexisting in the same environment (Gao et al., 2014). There were major differences in the presence and expression of a large number of orthologous genes between the three species, likely representing adaptations to these different environments and partitioning of the AOB species into their ecological niches (Norton, 2011). From the present study, various other insights were gained regarding how AOB allocate resources at the protein level. For instance, enzymes of the central energy-generating NH3-oxidation pathway, such as AMO, are highly expressed in all three species, whereas other enzymes in this pathway, like NirK, are expressed at significantly different levels, suggesting differing responses among the AOB to similar conditions and the potential for alternate flux of intermediates in the pathway.
Data Deposition
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al., 2016) partner repository with the dataset identifier PXD008954.
Author Contributions
JZ conceived study, planned, and performed the starvation experiments, prepared the samples for mass spectrometry, analyzed the mass spectrometric data, and wrote the paper with input from all co-authors. JK revised the manuscript, provided the expertise in the field of AOB, and aided in the analysis of mass spectrometric data. LS revised the manuscript, provided the expertise in the field of AOB, and aided in the analysis of mass spectrometric data. MS conceived the study, revised the manuscript, and aided in the analysis of mass spectrometric data. MK conceived the study, helped plan the experiments, helped in the generation of mass spectrometric data, aided in the analysis of mass spectrometric data, and revised the manuscript.
Funding
This study was supported by the Campus Alberta Innovation Chair Program (MS), the Canadian Foundation for Innovation (MS), the NC State Chancellor’s Faculty Excellence Program Cluster on Microbiomes and Complex Microbial Communities (MK), and the Natural Sciences and Engineering Research Council (NSERC) of Canada through a Banting fellowship (MK), an NSERC Postgraduate Doctoral Scholarship (JZ), and NSERC Discovery Grants to MS and LS. JZ was also supported by an Izaak Walton Killam Pre-Doctoral Scholarship, an Alberta Innovates Graduate Student Scholarship, and a University of Calgary Eyes High Doctoral Recruitment Scholarship. We acknowledge the support of the International Microbiome Center as well as funding support from the Government of Canada through Genome Canada, the Government of Alberta through Genome Alberta, Genome Prairie, Research Manitoba, and Genome Quebec. This research was undertaken thanks in part to funding from the Canada First Research Excellence Fund.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Maryam Ataeian, Tjorven Hinzke, and Angela Kouris for their help with proteomics sample preparation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00938/full#supplementary-material
Footnotes
References
Aitchison, J. (1982). The statistical analysis of compositional data. J. R. Stat. Soc. Ser. B 44, 139–177. doi: 10.2307/2345821
Arciero, D. M., Pierce, B. S., Hendrich, M. P., and Hooper, A. B. (2002). Nitrosocyanin, a red cupredoxin-like protein from Nitrosomonas europaea. Biochemistry 41, 1703–1709. doi: 10.1021/bi015908w
Arp, D. J., Chain, P. S. G., and Klotz, M. G. (2007). The impact of genome analyses on our understanding of ammonia-oxidizing bacteria. Annu. Rev. Microbiol. 61, 503–528. doi: 10.1146/annurev.micro.61.080706.093449
Badger, M. R., and Bek, E. J. (2008). Multiple Rubisco forms in proteobacteria: their functional significance in relation to CO2 acquisition by the CBB cycle. J. Exp. Bot. 59, 1525–1541. doi: 10.1093/jxb/erm297
Bar-Even, A., Noor, E., and Milo, R. (2012). A survey of carbon fixation pathways through a quantitative lens. J. Exp. Bot. 63, 2325–2342. doi: 10.1093/jxb/err417
Basumallick, L., Sarangi, R., DeBeer George, S., Elmore, B., Hooper, A. B., Hedman, B., et al. (2005). Spectroscopic and density functional studies of the red copper site in nitrosocyanin: role of the protein in determining active site geometric and electronic structure. J. Am. Chem. Soc. 127, 3531–3544. doi: 10.1021/ja044412+
Bernhard, A. E., Donn, T., Giblin, A. E., and Stahl, D. A. (2005). Loss of diversity of ammonia-oxidizing bacteria correlates with increasing salinity in an estuary system. Environ. Microbiol. 7, 1289–1297. doi: 10.1111/j.1462-2920.2005.00808.x
Berube, P. M., and Stahl, D. A. (2012). The divergent AmoC3 subunit of ammonia monooxygenase functions as part of a stress response system in Nitrosomonas europaea. J. Bacteriol. 194, 3448–3456. doi: 10.1128/JB.00133-12
Bollmann, A., and Laanbroek, H. (2001). Continuous culture enrichments of ammonia-oxidizing bacteria at low ammonium concentrations. FEMS Microbiol. Ecol. 37, 211–221. doi: 10.1016/S0168-6496(01)00163-5
Bollmann, A., and Laanbroek, J. (2002). Growth at low ammonium concentrations and starvation response as potential factors involved in niche differentiation among ammonia-oxidizing bacteria. Appl. Environ. Microbiol. 68, 4751–4757. doi: 10.1128/AEM.68.10.4751
Bollmann, A., Schmidt, I., Saunders, A. M., and Nicolaisen, M. H. (2005). Influence of starvation on potential ammonia-oxidizing activity and amoA mRNA levels in Nitrosospira briensis. Appl. Environ. Microbiol. 71, 1276–1282. doi: 10.1128/AEM.71.3.1276-1282.2005
Bollmann, A., Sedlacek, C. J., Norton, J., Laanbroek, H. J., Suwa, Y., Stein, L. Y., et al. (2013). Complete genome sequence of Nitrosomonas sp. Is79, an ammonia oxidizing bacterium adapted to low ammonium concentrations. Stand. Genomic Sci. 7, 469–482. doi: 10.4056/sigs.3517166
Cantera, J. J. L., and Stein, L. Y. (2007). Molecular diversity of nitrite reductase genes (nirK) in nitrifying bacteria. Environ. Microbiol. 9, 765–776. doi: 10.1111/j.1462-2920.2006.01198.x
Caranto, J. D., and Lancaster, K. M. (2017). Nitric oxide is an obligate bacterial nitrification intermediate produced by hydroxylamine oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 114, 8217–8222. doi: 10.1073/pnas.1704504114
Caranto, J. D., Vilbert, A. C., and Lancaster, K. M. (2016). Nitrosomonas europaea cytochrome P460 is a direct link between nitrification and nitrous oxide emission. Proc. Natl. Acad. Sci. U.S.A. 113, 14704–14709. doi: 10.1073/pnas.1611051113
Cardenas, J. P., Quatrini, R., and Holmes, D. S. (2016). Aerobic lineage of the oxidative stress response protein rubrerythrin emerged in an ancient microaerobic, (hyper)thermophilic environment. Front. Microbiol. 7:1822. doi: 10.3389/fmicb.2016.01822
Chain, P., Lamerdin, J., Larimer, F., Regala, W., Lao, V., Land, M., et al. (2003). Complete genome sequence of the ammonia-oxidizing bacterium and obligate chemolithoautotroph Nitrosomonas europaea. J. Bacteriol. 185, 2759–2773. doi: 10.1128/JB.185.9.2759
Daims, H., Lebedeva, E. V., Pjevac, P., Han, P., Herbold, C., Albertsen, M., et al. (2015). Complete nitrification by Nitrospira bacteria. Nature 528, 504–509. doi: 10.1038/nature16461
Elmore, B. O., Bergmann, D. J., Klotz, M. G., and Hooper, A. B. (2007). Cytochromes P460 and c’-beta; a new family of high-spin cytochromes c. FEBS Lett. 581, 911–916. doi: 10.1016/j.febslet.2007.01.068
Eraso, J. M., Markillie, L. M., Mitchell, H. D., Taylor, R. C., Orr, G., and Margolin, W. (2014). The highly conserved MraZ protein is a transcriptional regulator in Escherichia coli. J. Bacteriol. 196, 2053–2066. doi: 10.1128/JB.01370-13
Fernandes, A. D., Reid, J. N., Macklaim, J. M., McMurrough, T. A., Edgell, D. R., and Gloor, G. B. (2014). Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2:15. doi: 10.1186/2049-2618-2-15
Fierer, N., Carney, K. M., Horner-Devine, M. C., and Megonigal, J. P. (2009). The biogeography of ammonia-oxidizing bacterial communities in soil. Microb. Ecol. 58, 435–445. doi: 10.1007/s00248-009-9517-9
Gao, J., Luo, X., Wu, G., Li, T., and Peng, Y. (2014). Abundance and diversity based on amoA genes of ammonia-oxidizing archaea and bacteria in ten wastewater treatment systems. Appl. Microbiol. Biotechnol. 98, 3339–3354. doi: 10.1007/s00253-013-5428-2
Geets, J., Boon, N., and Verstraete, W. (2006). Strategies of aerobic ammonia-oxidizing bacteria for coping with nutrient and oxygen fluctuations. FEMS Microbiol. Ecol. 58, 1–13. doi: 10.1111/j.1574-6941.2006.00170.x
Griess-Romijn van Eck, E. (1966). Physiological and Chemical Tests for Drinking Water. NEN 1056 IV-2. Rijswijk: Nederlands Normalisatie Instituut.
Gruber, N., and Galloway, J. N. (2008). An Earth-system perspective of the global nitrogen cycle. Nature 451, 293–296. doi: 10.1038/nature06592
Guo, J., Peng, Y., Wang, S., Ma, B., Ge, S., Wang, Z., et al. (2013). Pathways and organisms involved in ammonia oxidation and nitrous oxide emission. Crit. Rev. Environ. Sci. Technol. 43, 2213–2296. doi: 10.1080/10643389.2012.672072
Hommes, N. G., Sayavedra-Soto, L. A., and Arp, D. J. (2003). Chemolithoorganotrophic growth of Nitrosomonas europaea on fructose. J. Bacteriol. 185, 6809–6814. doi: 10.1128/JB.185.23.6809-6814.2003
Hyman, M. R., and Wood, P. M. (1985). Suicidal inactivation and labelling of ammonia mono-oxygenase by acetylene. Biochem. J. 227, 719–725. doi: 10.1042/BJ2270719
Jiang, D., Khunjar, W. O., Wett, B., Murthy, S. N., and Chandran, K. (2015). Characterizing the metabolic trade-off in Nitrosomonas europaea in response to changes in inorganic carbon supply. Environ. Sci. Technol. 49, 2523–2531. doi: 10.1021/es5043222
Kerou, M., Offre, P., Valledor, L., Abby, S. S., Melcher, M., Nagler, M., et al. (2016). Proteomics and comparative genomics of Nitrososphaera viennensis reveal the core genome and adaptations of archaeal ammonia oxidizers. Proc. Natl. Acad. Sci. U.S.A. 113, E7937–E7946. doi: 10.1073/pnas.1601212113
Kleiner, M., Thorson, E., Sharp, C. E., Dong, X., Liu, D., Li, C., et al. (2017). Assessing species biomass contributions in microbial communities via metaproteomics. Nat. Commun. 8:1558. doi: 10.1038/s41467-017-01544-x
Kleiner, M., Wentrup, C., Lott, C., Teeling, H., Wetzel, S., Young, J., et al. (2012). PNAS Plus: metaproteomics of a gutless marine worm and its symbiotic microbial community reveal unusual pathways for carbon and energy use. Proc. Natl. Acad. Sci. U.S.A. 109, E1173–E1182. doi: 10.1073/pnas.1121198109
Klotz, M. G., Alzerreca, J., and Norton, J. M. (1997). A gene encoding a membrane protein exists upstream of the amoA/amoB genes in ammonia oxidizing bacteria: a third member of the amo operon? FEMS Microbiol. Lett. 150, 65–73. doi: 10.1111/j.1574-6968.1997.tb10351.x
Klotz, M. G., and Stein, L. Y. (2011). “Genomics of ammonia-oxidizing bacteria and insights into their evolution,” in Nitrification, eds B. B. Ward, D. J. Arp, and M. G. Klotz (Washington, DC: ASM Press), 57–94.
Koops, H. P., Boettcher, B., Moller, U. C., Pommerening-Roeser, A., and Stehr, G. (1991). Classification of eight new species of ammonia-oxidizing bacteria?. J. Gen. Microbiol. 137, 1689–1699. doi: 10.1099/00221287-137-7-1689
Kowalchuk, G. A., and Stephen, J. R. (2001). Ammonia-oxidizing bacteria: a model for molecular microbial ecology. Annu. Rev. Microbiol. 55, 485–529. doi: 10.1146/annurev.micro.55.1.485
Kozlowski, J. A., Kits, K. D., and Stein, L. Y. (2016a). Comparison of nitrogen oxide metabolism among diverse ammonia-oxidizing bacteria. Front. Microbiol. 7:1090. doi: 10.3389/fmicb.2016.01090
Kozlowski, J. A., Kits, K. D., and Stein, L. Y. (2016b). Complete genome sequence of Nitrosomonas ureae strain Nm10, an oligotrophic group 6a nitrosomonad. Am. Soc. Microbiol. 4, 4–5. doi: 10.1128/genomeA.00094-16.Copyright
Kozlowski, J. A., Kits, K. D., and Stein, L. Y. (2016c). Genome sequence of Nitrosomonas communis strain Nm2, a mesophilic ammonia-oxidizing bacterium isolated from Mediterranean soil. Genome Announc. 4:e01541-15. doi: 10.1128/genomeA.01541-15
Kozlowski, J. A., Price, J., and Stein, L. Y. (2014). Revision of N2O-producing pathways in the ammonia-oxidizing bacterium Nitrosomonas europaea ATCC 19718. Appl. Environ. Microbiol. 80, 4930–4935. doi: 10.1128/AEM.01061-14
Krummel, A., and Harms, H. (1982). Effect of organic matter on growth and cell yield of ammonia-oxidizing bacteria. Arch. Microbiol. 133, 50–54. doi: 10.1007/BF00943769
Kumar, N., Bansal, A., Sarma, G. S., and Rawal, R. K. (2014). Chemometrics tools used in analytical chemistry: an overview. Talanta 123, 186–199. doi: 10.1016/j.talanta.2014.02.003
Lieberman, R. L., and Rosenzweig, A. C. (2005). Crystal structure of a membrane-bound metalloenzyme that catalyses the biological oxidation of methane. Nature 434, 177–182. doi: 10.1038/nature03311
Marsh, K. L., Sims, G. K., and Mulvaney, R. L. (2005). Availability of urea to autotrophic ammonia-oxidizing bacteria as related to the fate of 14C- and 15N-labeled urea added to soil. Biol. Fertil. Soils 42, 137–145. doi: 10.1007/s00374-005-0004-2
Martens-Habbena, W., Berube, P. M., Urakawa, H., de la Torre, J. R., Stahl, D. A., Torre, J., et al. (2009). Ammonia oxidation kinetics determine niche separation of nitrifying Archaea and Bacteria. Nature 461, 976–979. doi: 10.1038/nature08465
McTavish, H., Fuchs, J. A., and Hooper, A. B. (1993). Sequence of the gene coding for ammonia monooxygenase in Nitrosomonas europaea. J. Bacteriol. 175, 2436–2444. doi: 10.1128/jb.175.8.2436-2444.1993
Mellbye, B. L., Giguere, A., Chaplen, F., Bottomley, P. J., and Sayavedra-Soto, L. A. (2016). Steady state growth under inorganic carbon limitation increases energy consumption for maintenance and enhances nitrous oxide production in Nitrosomonas europaea. Appl. Environ. Microbiol. 82:AEM.00294-16. doi: 10.1128/AEM.00294-16
Miyagawa, Y., Tamoi, M., and Shigeoka, S. (2001). Overexpression of a cyanobacterial fructose-1,6-/sedoheptulose-1,7-bisphosphatase in tobacco enhances photosynthesis and growth. Nat. Biotechnol. 19, 965–969. doi: 10.1038/nbt1001-965
Nicol, G. W., Leininger, S., Schleper, C., and Prosser, J. I. (2008). The influence of soil pH on the diversity, abundance and transcriptional activity of ammonia oxidizing archaea and bacteria. Environ. Microbiol. 10, 2966–2978. doi: 10.1111/j.1462-2920.2008.01701.x
Norton, J. M. (2011). “Diversity and environmental distribution of ammonia-oxidizing bacteria,” in Nitrification, eds B. B. Ward, M. G. Klotz, and D. J. Arp (Washington, DC: ASM Press), 39–55. doi: 10.1128/9781555817145.ch3
Norton, J. M., Klotz, M. G., Stein, L. Y., Arp, D. J., Bottomley, P. J., Chain, P. S. G., et al. (2008). Complete genome sequence of Nitrosospira multiformis, an ammonia-oxidizing bacterium from the soil environment. Appl. Environ. Microbiol. 74, 3559–3572. doi: 10.1128/AEM.02722-07
Pellitteri-Hahn, M. C., Halligan, B. D., Scalf, M., Smith, L., and Hickey, W. J. (2011). Quantitative proteomic analysis of the chemolithoautotrophic bacterium Nitrosomonas europaea: comparison of growing- and energy-starved cells. J. Proteomics 74, 411–419. doi: 10.1016/j.jprot.2010.12.003
Pérez, J., Buchanan, A., Mellbye, B., Ferrell, R., Chang, J. H., Chaplen, F., et al. (2014). Interactions of Nitrosomonas europaea and Nitrobacter winogradskyi grown in co-culture. Arch. Microbiol. 197, 79–89. doi: 10.1007/s00203-014-1056-1
Petersen, J. M., Kemper, A., Gruber-Vodicka, H., Cardini, U., van der Geest, M., Kleiner, M., et al. (2016). Chemosynthetic symbionts of marine invertebrate animals are capable of nitrogen fixation. Nat. Microbiol. 2:16195. doi: 10.1038/nmicrobiol.2016.195
Prosser, J., Head, I., and Stein, L. (2014). “The family nitrosomonadaceae,” in The Prokaryotes, eds M. Dworkinm, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (Berlin: Springer).
Purkhold, U., Pommerening-röser, A., Schmid, M. C., Koops, H.-P., Juretschko, S., and Wagner, M. (2000). Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis?: implications for molecular diversity surveys. Appl. Environ. Microbiol. 66, 5368–5382. doi: 10.1128/AEM.66.12.5368-5382.2000
R Development Core Team (2015). R: A Language and Environment for Statistical Computing. Available at: http://www.r-project.org
Reshetnikov, A. S., Rozova, O. N., Khmelenina, V. N., Mustakhimov, I. I., Beschastny, A. P., Murrell, J. C., et al. (2008). Characterization of the pyrophosphate-dependent 6-phosphofructokinase from Methylococcus capsulatus Bath. FEMS Microbiol. Lett. 288, 202–210. doi: 10.1111/j.1574-6968.2008.01366.x
Santoro, A. E., Dupont, C. L., Richter, R. A., Craig, M. T., Carini, P., McIlvin, M. R., et al. (2015). Genomic and proteomic characterization of “Candidatus Nitrosopelagicus brevis: an ammonia-oxidizing archaeon from the open ocean. Proc. Natl. Acad. Sci. U.S.A. 112, 1173–1178. doi: 10.1073/pnas.1416223112
Sayavedra-Soto, L. A., Hommes, N. G., Russell, S. A., and Arp, D. J. (1996). Induction of ammonia monooxygenase and hydroxylamine oxidoreductase mRNAs by ammonium in Nitrosomonas europaea. Mol. Microbiol. 20, 541–548. doi: 10.1046/j.1365-2958.1996.5391062.x
Schmidt, I., Steenbakkers, P. J. M., op den Camp, H. J. M., Schmidt, K., and Jetten, M. S. M. (2004). Physiologic and proteomic evidence for a role of nitric oxide in biofilm formation by Nitrosomonas europaea and other ammonia oxidizers. J. Bacteriol. 186, 2781–2788. doi: 10.1128/JB.186.9.2781-2788.2004
Schramm, A., De Beer, D., Wagner, M., and Amann, R. (1998). Identification and activities in situ of Nitrosospira and Nitrospira spp. as dominant populations in a nitrifying fluidized bed reactor. Appl. Environ. Microbiol. 64, 3480–3485.
Seaver, L. C., and Imlay, J. A. (2001). Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J. Bacteriol. 183, 7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001
Sedlacek, C. J., Nielsen, S., Greis, K. D., Haffey, W. D., Revsbech, P., Ticak, T., et al. (2016). Effects of bacterial community members on the proteome of the ammonia-oxidizing bacterium Nitrosomonas sp. Strain Is79. Appl. Environ. Microbiol. 82, 4776–4788. doi: 10.1128/AEM.01171-16
Shively, J. M., van Keulen, G., and Meijer, W. G. (1998). Something from almost nothing: carbon dioxide fixation in chemoautotrophs. Annu. Rev. Microbiol. 52, 191–230. doi: 10.1146/annurev.micro.52.1.191
Sims, G. K., Ellsworth, T. R., and Mulvaney, R. L. (1995). Microscale determination of inorganic nitrogen in water and soil extracts. Commun. Soil Sci. Plant Anal. 26, 303–316. doi: 10.1080/00103629509369298
Speksnijder, A. G. C. L., Kowalchuk, G. A., Roest, K., and Laanbroek, H. J. (1998). Recovery of a Nitrosomonas-like 16S rDNA sequence group from freshwater habitats. Syst. Appl. Microbiol. 21, 321–330. doi: 10.1016/S0723-2020(98)80040-4
Spivak, M., Weston, J., Bottou, L., Käll, L., and Stafford, W. (2009). Improvements to the percolator algorithm for peptide identification from shotgun proteomics data sets. J. Proteome Res. 8, 3737–3745. doi: 10.1021/pr801109k
Stein, L. Y. (2011). “Heterotrophic nitrification and nitrifier denitrification,” in Nitrification, eds B. Ward, D. Arp, and M. Klotz (Washington, DC: ASM Press), 95–114. doi: 10.1128/9781555817145.ch5
Stein, L. Y., Arp, D. J., and Hyman, M. R. (1997). Regulation of the synthesis and activity of ammonia monooxygenase in Nitrosomonas europaea by altering pH to affect NH3 availability. Appl. Environ. Microbiol. 63, 4588–4592.
Stein, L. Y., Campbell, M. A., and Klotz, M. G. (2013). Energy-mediated vs. ammonium-regulated gene expression in the obligate ammonia-oxidizing bacterium, Nitrosococcus oceani. Front. Microbiol. 4:277. doi: 10.3389/fmicb.2013.00277
Tappe, W., Laverman, A., Bohland, M., Braster, M., Rittershaus, S., Groeneweg, J., et al. (1999). Maintenance energy demand and starvation recovery dynamics of Nitrosomonas europaea and Nitrobacter winogradskyi cultivated in a retentostat with complete biomass retention. Appl. Environ. Microbiol. 65, 2471–2477.
Tusher, V. G., Tibshirani, R., and Chu, G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116–5121. doi: 10.1073/pnas.091062498
Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M. Y., Geiger, T., et al. (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. doi: 10.1038/nmeth.3901
Utåker, J. B., Andersen, K., Aakra, A., Moen, B., and Nes, I. F. (2002). Phylogeny and functional expression of ribulose 1,5-bisphosphate carboxylase/oxygenase from the autotrophic ammonia-oxidizing bacterium Nitrosospira sp. isolate 40KI. J. Bacteriol. 184, 468–478. doi: 10.1128/JB.184.2.468-478.2002
Van Kessel, M. A. H. J., Speth, D. R., Albertsen, M., Nielsen, P. H., Op Den Camp, H. J. M., Kartal, B., et al. (2015). Complete nitrification by a single microorganism. Nature 528, 555–559. doi: 10.1038/nature16459
Vizcaíno, J. A., Csordas, A., del-Toro, N., Dianes, J. A., Griss, J., Lavidas, I., et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456. doi: 10.1093/nar/gkv1145
Wang, Y., Coleman-Derr, D., Chen, G., and Gu, Y. Q. (2015). OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 43, W78–W84. doi: 10.1093/nar/gkv487
Wei, X., Sayavedra-Soto, L. A., and Arp, D. J. (2004). The transcription of the cbb operon in Nitrosomonas europaea. Microbiology 150, 1869–1879. doi: 10.1099/mic.0.26785-0
Wei, X., Yan, T., Hommes, N. G., Liu, X., Wu, L., McAlvin, C., et al. (2006). Transcript profiles of Nitrosomonas europaea during growth and upon deprivation of ammonia and carbonate. FEMS Microbiol. Lett. 257, 76–83. doi: 10.1111/j.1574-6968.2006.00152.x
Wells, G. F., Park, H. D., Yeung, C. H., Eggleston, B., Francis, C. A., and Criddle, C. S. (2009). Ammonia-oxidizing communities in a highly aerated full-scale activated sludge bioreactor: betaproteobacterial dynamics and low relative abundance of Crenarchaea. Environ. Microbiol. 11, 2310–2328. doi: 10.1111/j.1462-2920.2009.01958.x
Wessels, H. J., Gloerich, J., van der Biezen, E., Jetten, M. S., and Kartal, B. (2011). Chapter Twenty-One - Liquid Chromatography—Mass Spectrometry-Based Proteomics of Nitrosomonas, 1st Edn. New York City, NY: Elsevier Inc. doi: 10.1016/B978-0-12-381294-0.00021-3
Whittaker, M., Bergmann, D., Arciero, D., and Hooper, A. B. (2000). Electron transfer during the oxidation of ammonia by the chemolithotrophic bacterium Nitrosomonas europaea. Biochim. Biophys. Acta 1459, 346–355. doi: 10.1016/S0005-2728(00)00171-7
Wijma, H. J., Canters, G. W., de Vries, S., and Verbeet, M. P. (2004). Bidirectional catalysis by copper-containing nitrite reductase. Biochemistry 43, 10467–10474. doi: 10.1021/BI0496687
Wilhelm, R., Abeliovich, A., and Nejidat, A. (1998). Effect of long-term ammonia starvation on the oxidation of ammonia and hydroxylamine by Nitrosomonas europaea. J. Biochem. 124, 811–815. doi: 10.1093/oxfordjournals.jbchem.a022184
Wisniewski, J. R., Zougman, A., Nagaraj, N., and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. doi: 10.1038/nmeth.1322
Wood, N. J., and Sørensen, J. (2001). Catalase and superoxide dismutase activity in ammonia-oxidising bacteria. FEMS Microbiol. Ecol. 38, 53–58. doi: 10.1016/S0168-6496(01)00173-8
Wrage, N., Velthof, G. L., Van Beusichem, M. L., and Oenema, O. (2001). Role of nitrifier denitrification in the production of nitrous oxide. Soil Biol. Biochem. 33, 1723–1732. doi: 10.1016/S0038-0717(01)00096-7
Yoo, J.-G., and Bowien, B. (1995). Analysis of the cbbF genes from Alcaligenes eutrophus that encode fructose-1,6-/sedoheptulose-1,7-bisphosphatase. Curr. Microbiol. 31, 55–61. doi: 10.1007/BF00294635
Yu, R., Perez-Garcia, O., Lu, H., and Chandran, K. (2018). Nitrosomonas europaea adaptation to anoxic-oxic cycling: insights from transcription analysis, proteomics and metabolic network modeling. Sci. Total Environ. 615, 1566–1573. doi: 10.1016/j.scitotenv.2017.09.142
Keywords: nitrification, Nitrosomonas europaea, Nitrosomonas ureae, Nitrosospira multiformis, Calvin-Benson-Bassham cycle, Q Exactive Plus, proteomics, ammonia-oxidizing bacteria (AOB)
Citation: Zorz JK, Kozlowski JA, Stein LY, Strous M and Kleiner M (2018) Comparative Proteomics of Three Species of Ammonia-Oxidizing Bacteria. Front. Microbiol. 9:938. doi: 10.3389/fmicb.2018.00938
Received: 22 February 2018; Accepted: 23 April 2018;
Published: 14 May 2018.
Edited by:
Satoshi Tsuneda, Waseda University, JapanReviewed by:
Brett Mellbye, Oregon State University, United StatesOctavio Perez-Garcia, The University of Auckland, New Zealand
Copyright © 2018 Zorz, Kozlowski, Stein, Strous and Kleiner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jackie K. Zorz, jacqueline.zorz@ucalgary.ca Manuel Kleiner, manuel_kleiner@ncsu.edu