Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities
 Anne-Laure Hemonnot
Anne-Laure Hemonnot Jennifer Hua†
Jennifer Hua†  Lauriane Ulmann
Lauriane Ulmann Hélène Hirbec
Hélène Hirbec- Institute for Functional Genomics (IGF), University of Montpellier, Centre National de la Recherche Scientififique, Institut National de la Santé et de la Recherche Médicale, Montpellier, France
Microglia are the resident macrophages of the central nervous system. They play key roles in brain development, and physiology during life and aging. Equipped with a variety of molecular sensors and through the various functions they can fulfill, they are critically involved in maintaining the brain’s homeostasis. In Alzheimer disease (AD), microglia reaction was initially thought to be incidental and triggered by amyloid deposits and dystrophic neurites. However, recent genome-wide association studies have established that the majority of AD risk loci are found in or near genes that are highly and sometimes uniquely expressed in microglia. This leads to the concept of microglia being critically involved in the early steps of the disease and identified them as important potential therapeutic targets. Whether microglia reaction is beneficial, detrimental or both to AD progression is still unclear and the subject of intense debate. In this review, we are presenting a state-of-knowledge report intended to highlight the variety of microglial functions and pathways shown to be critically involved in AD progression. We first address both the acquisition of new functions and the alteration of their homeostatic roles by reactive microglia. Second, we propose a summary of new important parameters currently emerging in the field that need to be considered to identify relevant microglial targets. Finally, we discuss the many obstacles in designing efficient therapeutic strategies for AD and present innovative technologies that may foster our understanding of microglia roles in the pathology. Ultimately, this work aims to fly over various microglial functions to make a general and reliable report of the current knowledge regarding microglia’s involvement in AD and of the new research opportunities in the field.
Introduction
Microglia cells are the main immunocompetent cells in the brain. They colonize the brain in the early prenatal period (Ginhoux et al., 2010), but contrary to other tissue resident macrophages, they remain secluded within the CNS throughout life and self-renew at slow pace (Ajami et al., 2007). Importantly, the CNS microenvironment significantly shapes the microglia’s phenotype, endowing them with specific important homeostatic and supportive brain functions (Kierdorf and Prinz, 2017). Should the brain homeostasis be compromised, microglia change their phenotype and initiate a defense program. Thus, under pathological conditions, they adopt reactive states characterized by multiple morphological and functional changes including but not limited to increased phagocytosis and increased expression of receptors, cytokines, chemokines and additional inflammation related molecules (Wolf et al., 2017).
Alzheimer’s disease (AD) classical hallmarks include brain atrophy, extracellular amyloid-beta (Aβ) deposits, intracellular aggregated phosphorylated tau, dystrophic neurites, synapses and neurons loss (Bedner et al., 2015). The presence of reactive glial cells within the neuritic plaques was described by Alois Alzheimer himself (Alzheimer, 1907; Graeber et al., 1997) and further studies identified both reactive astrocytes and microglia in the vicinity of the Aβ deposits (Verkhratsky et al., 2016). Long considered as a consequence of the pathology, reactive glia and associated neuroinflammation are now regarded as playing key roles in both disease initiation and progression. Indeed, Human genetic studies identified over 25 genetic loci that robustly associate with AD risk (Hansen et al., 2018; Verheijen and Sleegers, 2018). Among them, most of the common (ApoE, Sp1l) or rare (Trem2, Cd33) genetic variants code for proteins that are preferentially or exclusively expressed in microglia. These findings strongly support a causal involvement of microglial cells in AD pathogenesis and generated a strong interest for studying these cells in AD. Yet, the roles of microglia in AD initiation and progression are unclear and heavily debated, with conflicting reports regarding their detrimental or protective contribution to the disease.
In the present review, we have summarized the main findings regarding the role of microglia in AD. Microglia reaction is known to be associated with the acquisition of many immune functions which are triggered by the activation of receptors designed to recognize danger or pathogen associated molecular patterns (DAMPs/PAMPs). Its role is to restore homeostasis and is also associated with the loss or the alteration of homeostatic functions which are important for brain physiological functioning. In the two first parts, we thus provide an overview of the microglial functions and pathways that are known to be altered during AD. We then highlight factors such as mouse models, sex, age whose influence may have been under-examined in assessing the contribution of microglial cells to the disease progression. Finally, we identified new research topics that are likely to foster our understanding of the roles of microglia to AD initiation and progression and may help design more targeted therapeutic strategies.
New Functions for Reactive Microglia in AD
Neuroinflammation is a common feature of neurodegenerative diseases and inflammatory processes are thus among the most studied microglial functions in AD. Microglia, which represent the main immune cells of the brain, have been shown to play key roles in orchestrating this brain inflammation. In the following section, we are reporting the main microglial processes involved in neuroinflammation (Figure 1, top part). However, more detailed description of these processes can be found in recent reviews that are focusing on these specific points (Labzin et al., 2017; Nizami et al., 2019).
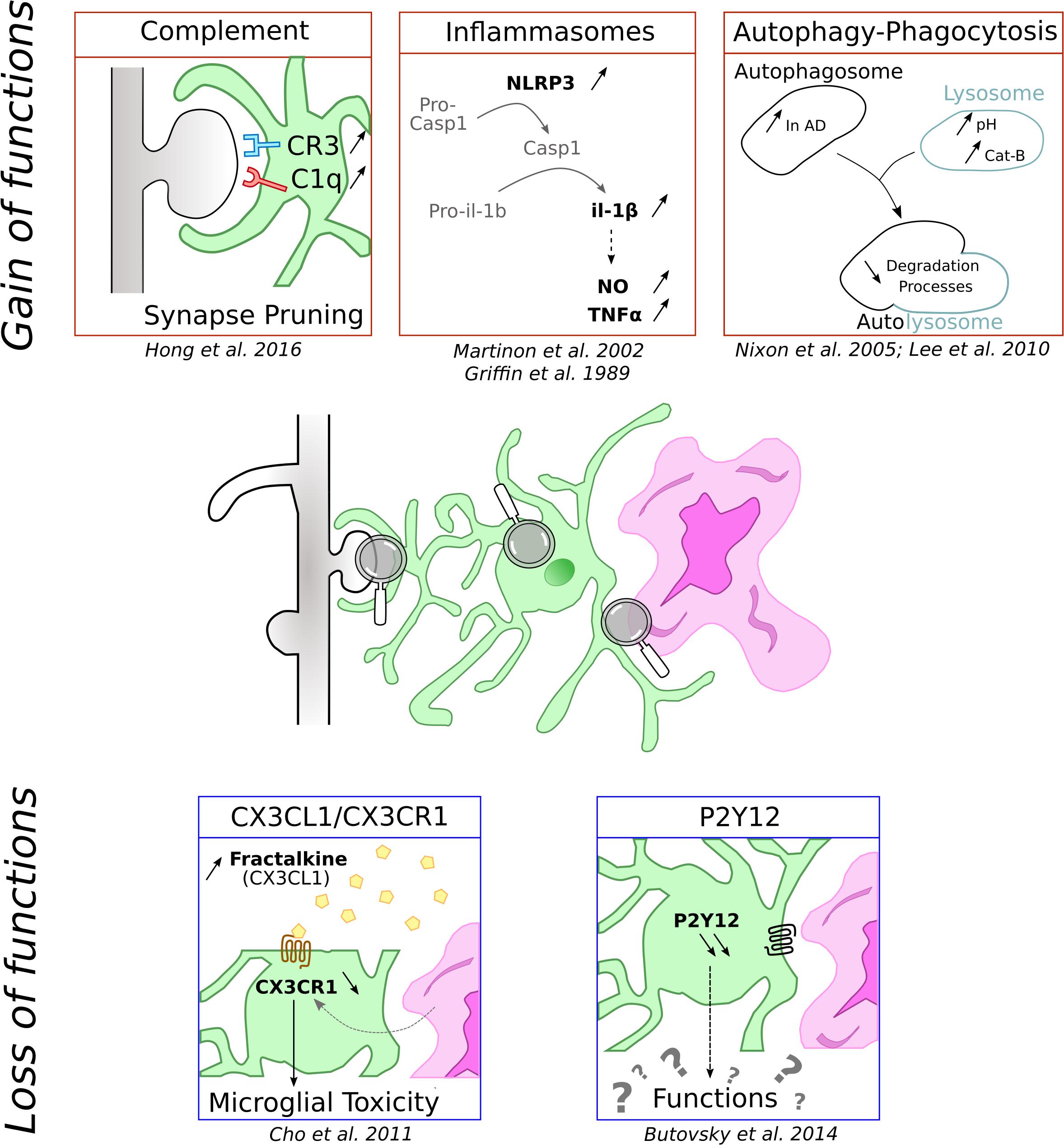
Figure 1. Schematic representation of functions microglial cells can loose and gain in context of AD. Microglia is represented in green associated to amyloid-β deposit in purple and dendritic spines in gray.
Inflammasomes Are Central Hubs for Cytokines Production
One of inflammation hallmarks is the release of cytokines. This secretion requires the activation of inducible multiproteic complexes called inflammasomes. Several inflammasomes have been characterized but the most important microglial contributor in pathologies is certainly the NLRP3. It is composed of the sensor protein NLRP3 and the adaptor protein apoptosis associated Speck-like protein (ASC), which contains a caspase recruitment domain. ASC can recruit and activate pro-caspase-1. When stimulated, the complex induces the cleavage of pro-caspase-1 into active caspase-1, which in turn cleaves pro-IL-1β and IL-18 triggering their release in the extracellular space (Martinon et al., 2002). NLRP3 activation pathway is not fully characterized, but the current view is that NLRP3 activation requires the occurrence of two independent but co-concomitant priming and activation signals (Próchnicki et al., 2016).
In the context of AD, IL-1β is known to elicit the secretion of NO and TNFα, promoting the formation of deleterious amyloid plaques and neuronal degeneration (Griffin et al., 1989). In accordance with these data, frontal cortex from AD patients exhibit an increase of caspase-1 which is correlated with an attenuation of Aβ peptide phagocytosis (Burguillos et al., 2011; Heneka et al., 2013). Likewise, genetic deletion of NLRP3 in mice with familiar AD associated mutation reduces the level of IL-1β and Aβ deposits, and correlates with positive impacts on synaptic dysfunction and cognitive performances (Heneka et al., 2013).
Other members of the caspase family are also involved in AD pathology. Interestingly, the activity of caspase-3 prematurely increases in hippocampal neurons in an AD mouse model (D’Amelio et al., 2011). Similarly, in samples from AD patients, caspase-8 and -3 are upregulated in cortical microglia (Burguillos et al., 2011). Microglial caspase-3 around amyloid plaques also present a cytosolic location, suggesting a non-apoptotic role of caspases in AD (Burguillos et al., 2011). Such evidence has also been described in others degenerative models such as brain ischemia, in which reactive astrocytes and microglia express cytoplasmic non-apoptotic caspase-3 (Wagner et al., 2011). This non-nuclear localization could be involved in cytoplasmic rearrangement and modifications of cell populations surrounding lesions. Although it is undeniable that caspase signaling is crucial in the development of AD, the delay of activation and the pathway downstream of each of them need to be clarified.
The Complement System, the Microglial Way of Eating Synapses?
The complement pathway is part of the innate immune system and mediates the recognition and elimination of pathogens and cellular debris. It is involved in many physiological and pathological functions throughout life, including activity-dependent synapse elimination during development (Schafer et al., 2012). In AD, reactivation of this pathway by Aβ deposits has been associated with synapse loss.
The complement cascade is composed of a large panel of mediators including C1q and C3 complex proteins which can be activated by three different pathways, all of which are capable of triggering phagocytosis (Stephan et al., 2012). In the CNS, complement proteins are expressed in neurons and glial cells, although microglia and astrocytes are the major sources of complement. Particularly, microglia expressed high level of C1q and CR3 (Veerhuis et al., 2011) and microglial CR3 have been shown to be crucial in synapse pruning during development (Schafer et al., 2012).
In AD context, patients showed elevated CSF concentrations of C3 and CR1, pointing out an alteration of complement system in the pathology (Daborg et al., 2012). The complement has been associated with Aβ but it is not clear whether it is protective or detrimental. Indeed, some studies reported that complement inhibition or deficiency results in accelerated amyloid pathology (Wyss-Coray et al., 2002; Maier et al., 2008) and that C3 along with CR3 contribute to Aβ phagocytosis (Fu et al., 2012). On the other hand, other studies pointed out that elimination and modulation of microglial CR3 decrease Aβ level (Czirr et al., 2017) and C3 antagonist ameliorates plaque load (Lian et al., 2016). Further studies are thus required to better understand the role of complement in Aβ pathology.
As AD is marked by an important synapse loss, questions have been raised on whether the complement could mediate such synapse elimination. Fonseca et al. (2004) demonstrated that C1q deficiency in an AD mouse model partly restores synapse integrity pointing out a role of the complement system in AD. More recently, works from Hong et al. demonstrated that microglial C3/CR3 mediates synapse elimination when challenged with oligomeric Aβ (Hong et al., 2016). More specifically, they found that C1q is upregulated into synapse early in J20 mice and that Aβ oligomers increase C1q and microglia phagocytic activity. This resulted in synapse elimination by microglia, a process which is lost in CR3 KO mice. This study proposes a model in which C1q and oligomer Aβ would activate the complement cascade to drive synapse elimination through microglial CR3.
Eat Me and Eat Myself
Autophagy and phagocytosis are cellular degradation processes, necessary to degrade additional or damaged particles in lysosomes. These processes, ensured by a large enzymatic degradation system, are dysregulated during aging and are of particular importance during AD, as shown with the autophagy failure and the increase of autophagosomes in AD patients (Nixon et al., 2005). Moreover, lysosomal acidification and autophagy are disrupted by Alzheimer-related PS1 mutation (Lee et al., 2010). Numerous studies demonstrate that microglial Aβ phagocytosis contribute to degeneration by triggering NLRP3 and lysosomal cathepsin-B that subsequently results in maturation and release of IL-1β (Halle et al., 2008). Cellular degradation processes could thus, by differently modulating the inflammasome, present opposite effects. It could be protective in normal physiological states and during the premature state of the pathology, and detrimental during chronic and late phases of diseases.
Loss of Homeostatic Functions in Reactive Microglia
Although the majority of the studies concentrate on the microglial reactivity-acquired functions and assess their contribution to neurodegeneration, loss of key homeostatic functions may also be detrimental to neuronal functions and may contribute to the detrimental effects of microglia reaction. In the following part, we are reviewing few key microglial functions that are compromised in AD (Figure 1, bottom part).
Consequences of CX3CL1/CX3CR1 Signaling Loss in AD
In brain, the CX3CR1 receptor is predominantly expressed in microglia. Its ligand is the secreted soluble form of fractalkine, also named CX3CL1, and is constitutively expressed by neurons. CX3CL1 exerts an inhibitory signal, maintaining microglia in a resting state. CX3CL1-CX3CR1 is a critical signaling pathway during development as shown by the delay of glutamatergic synapse maturation (Paolicelli et al., 2011; Hoshiko et al., 2012) and functional consequences in adult synapses (Basilico et al., 2019) in CX3CR1–/– mice. Age is also a decisive factor in the regulation of CX3CR1 expression as LPS challenge is responsible for a more pronounced impairment of CX3CR1 expression in aged compared to young rats (Lyons et al., 2009).
The roles of CX3CL1/CX3CR1 communication during neuroinflammation are still subject to debate since CX3CR1 deletion effects differ depending on the challenge. In CX3CR1–/– mice and in both PD and ALS models, Cardona et al. (2006) demonstrated an extensive neuronal loss due to an alteration of cytokines production. CX3CR1 decrease is also observed in AD models. In neurodegenerative conditions, this disruption is associated with a strong microglial toxicity and an aggravation of the pathology (Keren-Shaul et al., 2017). The involvement of the CX3CL1/CX3CR1 signaling pathway in AD is confirmed by an increase in the plasmatic concentration of CX3CL1 in AD and MCI patients compared to healthy control subjects (Kim et al., 2008). However, the role of this pathway might be more complex as CX3CR1 deletion was shown to prevent neuronal loss in 3 × Tg AD mice (Fuhrmann et al., 2010) but worsen cellular and behavioral deficits in hAPP-J20 mice (Cho et al., 2011).
All these data point out to critical roles for CX3CL1–CX3CR1 signaling during neurodegenerative diseases, including AD, but also demonstrate that its complex spectrum of responses may depend on the genetic model of the disease.
The Yet Unresolved Role of P2Y12 Down-Regulation in AD
In physiological conditions, P2Y12 receptor is involved in chemotaxis and mice lacking this receptor showed altered microglia migration and polarization (Haynes et al., 2006). P2ry12 was identified as a unique microglia gene in the CNS (Butovsky et al., 2014). It is one of the most highly expressed genes in microglia and is downregulated in reactive microglia (Haynes et al., 2006). Consequently, P2ry12 gene expression levels have been proposed to be a marker of the homeostatic microglial signature (Butovsky et al., 2014). In agreement, in AD transgenic mouse model, microglia located at proximity of amyloid plaques do not express P2Y12R whereas the receptor is observed in plaque-distant ones (Butovsky et al., 2014; Jay et al., 2015). Similar findings have also been reported in human AD patients (Sanchez-Mejias et al., 2016; Mildner et al., 2017). However, so far, the consequences of this down-regulation for microglia functions are unknown and merit further attention.
Most Studied Microglial Molecular Targets in AD
In the past years, genome-wide association studies (GWAS) identified over 25 genetic loci that robustly associate with risk of late onset Alzheimer disease (LOAD); many of these, relate to neuroinflammation and are preferentially or exclusively expressed in microglial cells, implicating microglia reaction as not only a consequence of Alzheimer’s but likely also a cause. In this section, we are reviewing the current knowledge regarding the roles of the most studied genes in this context.
APOE: Beyond Aβ Modulation, a Microglia-Function Modifier
The ε4 isoform of the Apolipoprotein E (APOE) represents a common genetic variant associated with AD and is the most significant known risk factor. APOE is an apolipoprotein implicated in cholesterol and lipid transfer between cells. In the brain, it is produced mainly by astrocytes, but also by microglia and to a lesser extent by neurons. In humans, APOE is found in three main isoforms: ε2, ε3, and ε4. The APOE-ε4 isoform represents the most significant risk factor for LOAD: the presence of one APOE-ε4 copy increases the risk of developing LOAD by three-fold whereas two APOE-ε4 copies lead to a nine-fold increase (Corder et al., 1993). On the opposite, individuals carrying the rare ε2 variant are less likely to develop AD and the most common APOE-ε3 isoform is thought to be neutral (Serrano-Pozo et al., 2015). How APOE isoforms affect the onset and development of AD remains unclear. Based on the early-described interaction between APOE and Aβ, studies mainly focused on the effects of APOE on amyloid load and oligomerization, which was shown to be isoforms-dependent (Strittmatter et al., 1993; Naslund et al., 1995; Hashimoto et al., 2012). Thus, APOE-ε4 patients have more Aβ plaques and oligomers than ε3 carriers (Schmechel et al., 1993; Tiraboschi et al., 2004; Hashimoto et al., 2012; Koffie et al., 2012), and mouse models expressing human APOE isoforms mimics the isoform-dependent modifications on Aβ deposits (Fagan et al., 2002; Hudry et al., 2013; Zhao et al., 2014). It was also reported that APOE can also influence Aβ clearance (DeMattos et al., 2004; Castellano et al., 2011), however, APOE-knockdown mice develop less Aβ deposits (Holtzman et al., 1999; Fagan et al., 2002).
Little is known regarding the microglial role of APOE. Early post-mortem studies in humans found a higher number of reactive microglia in APOE-ε4 carriers compared to APOE-ε3 (Egensperger et al., 1998), and recently Minett et al. (2016) found that APOE-ε4 allele was strongly associated with reactive microglia. Studies using mouse models expressing human isoforms also showed increased microgliosis in APOE-ε4 expressing mice compared to APOE-ε3 (Belinson and Michaelson, 2009; Rodriguez et al., 2014). Overall, these studies highlight a relation between APOE isoforms and reactive microglia. Evidences have also been pointing out a role of APOE in inflammatory processes (Lynch et al., 2001; Thangavel et al., 2017). Thus, APOE is up-regulated in disease-associated microglia (DAMs) and modulates transcription of homeostatic and inflammatory factors (Krasemann et al., 2017). Moreover, APOE modifies inflammatory response in an isoform manner; APOE-ε4 expressing mice releasing more pro-inflammatory cytokines than APOE-ε3 (Guo et al., 2004; Vitek et al., 2009; Zhu et al., 2012). However, the concept of APOE triggering more pro-inflammatory response has been challenged since APOE in the presence of Aβ can induce a decrease in pro-inflammatory cytokine release (Guo et al., 2004), indicating a more complex link between APOE, Aβ and glial cells. Overall, APOE is undoubtedly implicated in LOAD through Aβ aggregation and clearance but also by modulating microglia activation and cytokine release. However, additional studies are needed to explore these processes.
TREM2, the Well-Known Risk Factor With Unclarified Roles
Aside from APOE, the other major well-studied gene associated with LOAD is the Triggering receptor expressed on myeloid cells 2 (TREM2). Indeed, GWAS studies identified several rare Trem2 gene variants associated with LOAD (Guerreiro et al., 2013; Jin et al., 2014). Among them, the rs75932628 variant, which causes the loss-of-function R47H mutation, showed significant association with AD and suggests a protective role for TREM2 activation pathway in AD (Guerreiro et al., 2013; Jonsson et al., 2013). TREM2 is a cell surface receptor expressed in myeloid cells, including microglia (Schmid et al., 2002; Kiialainen et al., 2005) and was shown to modulate inflammation (Piccio et al., 2007), phagocytosis (Takahashi et al., 2005) and chemokine secretion (Bouchon et al., 2002; Sieber et al., 2013). How TREM2 affect AD is currently not well understood but TREM2 is increased in APP/PS1 hippocampus and cortices (Jiang et al., 2014) and its expression increases with age (Jay et al., 2015). Moreover, TREM2 expression is increased in plaque-associated microglia (Frank et al., 2008; Guerreiro et al., 2013), and modulating TREM2 expression can reprogram microglia response (Krasemann et al., 2017; Lee C.Y.D. et al., 2018). In particular, Keren-Shaul et al. demonstrated that TREM2 is required for a complete activation of DAMs (Keren-Shaul et al., 2017), pointing out a major role of TREM2 in those cells.
Most of the studies on TREM2 in AD relate to microglia-mediated Aβ phagocytosis, however, they do not all agree on whether it has beneficial or detrimental effects. Indeed, Ulrich et al. (2014) found no change in amyloid burden in 3-month-old TREM2-heterozygous APP-21 mice whereas Jay et al. (2015) showed reduced 6E10 staining in 4-month-old TREM2 deficient APP/PS1 mice. On the opposite, Wang et al. (2015) suggested using 8-month-old 5xFAD mice, that TREM2 deficiency would be detrimental as it increases hippocampal Aβ peptide. Similarly, Jiang et al. (2014) showed, in vitro, that TREM2 facilitates Aβ 1-42 phagocytosis and, in vivo, that TREM2 overexpression reduces plaque density in the cortex and hippocampus of APP/PS1 mice. Altogether, these different studies suggest a complex role of TREM2 on Aβ and suggest an age- or stage-dependent effect.
Contradictory results have also been found in TREM2-dependent inflammatory response. Jay et al. (2015) showed that TREM2 deficiency in APP/PS1 mice reduces the pro-inflammatory response. On the contrary, in in vitro studies, TREM2 overexpression was shown to reduce - while TREM2 deficiency increases - pro-inflammatory cytokine production (Takahashi et al., 2005; Jiang et al., 2014). Therefore, further studies are required to clarify how TREM2 influences inflammatory response.
Despite discrepancies found in TREM2 implication in inflammatory response and Aβ deposition, it is of agreement that TREM2 deficiency causes a decrease in Aβ-associated microglia (Jay et al., 2015; Wang et al., 2015; Zhao et al., 2018). Altogether, those studies show TREM2 implication in AD by modulating inflammatory processes and Aβ deposition. Moreover, several studies found that Aβ binds to TREM2 and that this interaction can modulate microglia functions such as proliferation, Aβ degradation and inflammatory response (Zhong et al., 2018). Interestingly, a link between TREM2 and APOE has recently been highlighted suggesting that TREM2 modulates APOE expression and signaling (Krasemann et al., 2017; Parhizkar et al., 2019).
Many studies aim to decipher the role of TREM2 in AD. However, current studies mainly used TREM2 overexpression or knockout although it might be relevant to also study TREM2 variants.
Other AD Risk Factors
Alzheimer disease-related GWAS have identified many other microglia genes as potential risk factors in AD. This includes genes such as Cd33, Cr1, or Abca7, which have been implicated in phagocytosis (Villegas-Llerena et al., 2016). Additionally, CD33 was shown to inhibit Aβ clearance whereas CR1 is part of the complement pathway that helps eliminating Aβ plaques. Various SNPs of CD33 have been found: the rs3826656 and rs3865444 variants are associated with LOAD whereas the rs3865444 variant confers protection (McQuade and Blurton-Jones, 2019). Overall, more than 25 microglia genes have been highlighted by GWAS studies (Verheijen and Sleegers, 2018). Their contribution to AD pathogenesis is yet unknown and merit scientists’ attention as they tend to be more common genetic variants.
Emerging Microglial Targets in AD
Emerging microglial targets are highlighted in Figure 2.
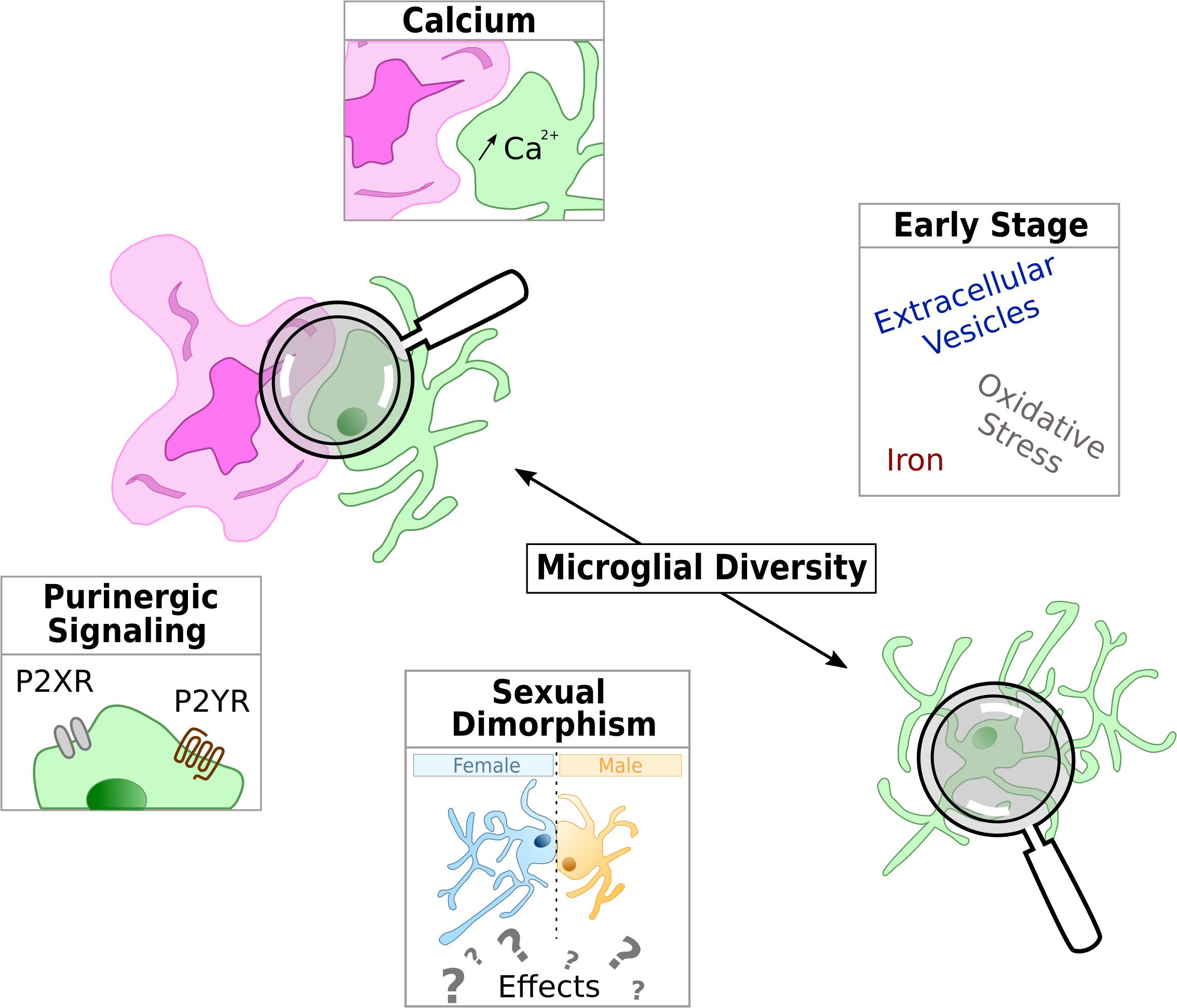
Figure 2. Schematic representation of the current hot topics and emerging microglial targets in AD physiopathology studied in this review. Microglia is represented in green and either with a reactive shape associated to amyloid-β deposit in purple or in the parenchyma with an homeostatic shape, away from Aβ.
Purinergic Signaling
Of the many mechanisms implicated in microglial functions, purinergic signaling is one worth mentioning. Indeed, microglial purinergic receptors are known to modulate several processes including phagocytosis, chemotaxis and cytokine release (Abbracchio and Ceruti, 2007; Calovi et al., 2019). Purinergic receptors are divided into two super families: P1 receptors respond to adenosine whereas P2 respond to ADP and ATP. Purinergic receptors are widely expressed in several CNS cell types including microglia (Butovsky et al., 2014), and both ionotropic P2X and metabotropic P2Y receptors have been implicated in neurological diseases including AD (Burnstock, 2016).
Of the G protein-coupled P2Y receptors, P2Y6R, 12, and 13 are the three most investigated subtypes expressed in microglia (Calovi et al., 2019). In AD, the most studied is P2Y12 receptor which plays important role in homeostatic microglia and whose role is described above (see section “Loss of Homeostatic Functions in Reactive Microglia”). As for the other P2YRs, P2Y6R might be implicated in amyloid phagocytosis as Wendt et al. (2017) demonstrate an impaired P2Y6R-dependent phagocytosis mechanism in 9-month-old 5xFAD mice. Although P2Y2R expression in microglia seems very low (Calovi et al., 2019), studies have also investigate its implication in AD. In vitro, Aβ treatment of primary microglia induces an increase of P2Y2R expression and Aβ-treated P2Y2R–/– microglia showed loss of motility and altered ATP/UDP-triggered Aβ uptake (Kim et al., 2012) suggesting that P2Y2R up-regulation enhances microglia-dependent Aβ degradation. Interestingly, P2Y2R expression is up-regulated in 10 weeks old TgCRND8 AD mouse models and P2Y2R heterozygous mice showed accelerated pathology compared to wild-type mice (Ajit et al., 2014). In contradiction with data from the TgCRND8 mice but supporting the idea of P2Y2R displaying protective effect, data demonstrate a downregulation of P2Y2R in parietal cortex of AD patients (Lai et al., 2008). Discrepancies between mice and human P2Y2R expression regulation may be explained by the stage at which the studies were conducted. Altogether, these results suggest a protective role of P2Y2R signaling in the context of AD.
The other major P2 receptors are the ATP-gated ion channels P2X receptors. There are seven P2XR subunits whose assembly in trimers forms cation-permeable channels that are widely expressed in the CNS. To date, P2X4R and P2X7R are the only P2XRs pertinently characterized in microglia. P2X7 is the most studied in AD context because of its well-established pro-inflammatory role, being a key step in the NLRP3 inflammasome activation leading to IL-1β release (Bhattacharya and Biber, 2016; Adinolfi et al., 2018). P2X7R is up-regulated in AD patient brains (McLarnon et al., 2006; Martin et al., 2018) and administration of Aβ peptide in rat hippocampus increases P2X7R expression (McLarnon et al., 2006). Moreover, downregulating or blocking P2X7R decreases pro-inflammatory cytokines release in microglia cell cultures stimulated by ATP and Aβ (Ni et al., 2013). On the other hand, Martin et al. (2018) demonstrate that P2X7R deficiency ameliorates cognitive functions and reduces amyloid load without altering IL-1β level. This set of data suggest a pro-inflammatory role of P2X7R in AD. Lastly, the C489T polymorphism of P2X7R, which causes a loss of function, was found to be less frequent in AD patients, supporting a potential contribution of P2X7R in AD pathogenesis (Sanz et al., 2014).
Another highly expressed P2X receptor in reactive microglia is P2X4R. P2X4R are highly up-regulated upon microglia activation and have been implicated in neurological disorders and inflammatory processes (Suurväli et al., 2017). Although P2X4R is decreased in AD brain patients and neuronal P2X4R seems to modulate Aβ-induced neuronal death (Varma et al., 2009), no work has yet been performed on microglial P2X4R in AD.
The third main purinergic receptor family is that of adenosine P1Rs, which comprises A1, A2a, A2B, and A3 receptors. P1R are G protein-coupled receptors abundantly expressed in the CNS and all P1 subtypes are expressed in microglia. A1 and A2a receptor expressions have been found to be dysregulated in post-mortem tissues from AD patients (Erb et al., 2018), thus suggesting that P1 receptors might be implicated in AD. However, to date, not much is known regarding the contribution of microglial P1 signaling in the context of AD.
Overall, those few studies suggest the implication of microglia purinergic receptors in AD through different processes such as inflammation or phagocytosis. However, since purinergic receptors are expressed in all neural cell types incriminated in AD, more specific tools such as cell-specific KO models are required to decipher the roles of microglial purinergic signaling.
Microglial Calcium Signaling, a Poorly Studied Messenger in AD
Calcium signaling is a key second messenger in almost all cell type and is essential for the normal CNS functioning. Many microglia functions are mediated by Ca2+ (McLarnon, 2005; Färber and Kettenmann, 2006). In particular, microglia reaction is accompanied with intracellular calcium increase, a process that is required to induce cytokines and chemokines release (Hoffmann et al., 2003).
Calcium signaling dysregulation in AD has been widely studied in neurons (Tong et al., 2018), but very little is known about calcium signaling in microglia in the context of AD. In an early study, Combs et al. (1999) demonstrated that stimulation of cultured microglia with Aβ25–35 peptide results in a transient increase in intracellular [Ca2+]. This calcium is released from intracellular store and leads to microglia activation and neurotoxic factors production. More recently, McLarnon et al. showed that cultured microglia from AD patients have higher basal Ca2+ level but lower amplitude and longer-lasting ATP-induced calcium response with respect to that measured in microglia from non-demented individuals, thus suggesting that calcium signaling is impaired in microglia from AD patients (McLarnon et al., 2005).
Altered P2YR-dependent calcium signaling has been observed in plaque-associated microglia in AD mouse models whereas plaque-distant microglia showed similar Ca2+ activity than matched controls (Brawek et al., 2014). As calcium mediates many microglial functions, it might be interesting to further investigate microglia calcium signaling in the context of AD. Particularly since some purinergic receptors are likely implicated in AD, it might be worth studying whether calcium signaling dysregulation involves purinergic signaling impairment.
Microglia in Alzheimer Disease: Current Hot Topics
The involvement of microglia in AD is a relatively new area of research, but it is growing at a fast pace. In addition to the pathways described above, new areas of investigation are emerging or revisited based on our current knowledge of microglial functions. In the following part, we highlight few current hot topics (Figure 2). These new areas of research will help to increase our understanding of AD pathogenesis, and may, on a longer term, help to better stratify the patients and to design tailored therapeutic strategies.
Sexual Dimorphism, a Key Factor to Design Efficient Therapeutic Strategies
The impact of sexual dimorphism in biology is a hot topic, and AD research makes no exception. Indeed, AD prevalence is two-fold higher in women (Hebert et al., 2013), and women suffering from AD display specific cognitive alterations, biomarker patterns or risk factors susceptibility (Ferretti et al., 2018). This sex effect was, at first, thought to be due to higher longevity but recent reports show that it is clearly not the only explanation (Viña and Lloret, 2010). The involvement of sex is still a subject of intense debate and biological mechanisms involved in human pathology are still controversial. Because most AD pre-clinical studies use either males or females, but rarely both, the cellular and molecular mechanisms involved in AD sexual dimorphism are still poorly understood. Yet, the few studies comparing the influence of sexual dimorphism in AD mouse models, all agree on a sooner precocious and stronger AD phenotype in females (Table 1). Sex-based effects are observed in different AD mouse models, both at behavioral and histological levels. Globally, cognitive alterations in females appear at younger ages and remain stronger than in males (Clinton et al., 2007; Carroll et al., 2010; Gallagher et al., 2013; Yang et al., 2018). Whatever brain areas, models and ages, females display more amyloid plaques and higher amount of soluble Aβ (Wang et al., 2003; Carroll et al., 2010; Gallagher et al., 2013; Ben Haim et al., 2015; Janus et al., 2015; Jiao et al., 2016; Yang et al., 2018). Furthermore, in late stages, Tau phosphorylation levels and number of Phospho-Tau positive cells are higher in females (Clinton et al., 2007). Synaptic and neuronal degeneration processes seem also to be stronger in aged females (Jiao et al., 2016). Relative to neuroinflammation, glial cells from old female AD mice present stronger reactive states compared to males and are associated with higher levels of pro-inflammatory factors (Jiao et al., 2016; Yang et al., 2018). While sex effects are clearly established in both AD mice and patients, it remains unknown whether these modifications are due to systemic/hormonal effects or whether it also exists at the cellular level.
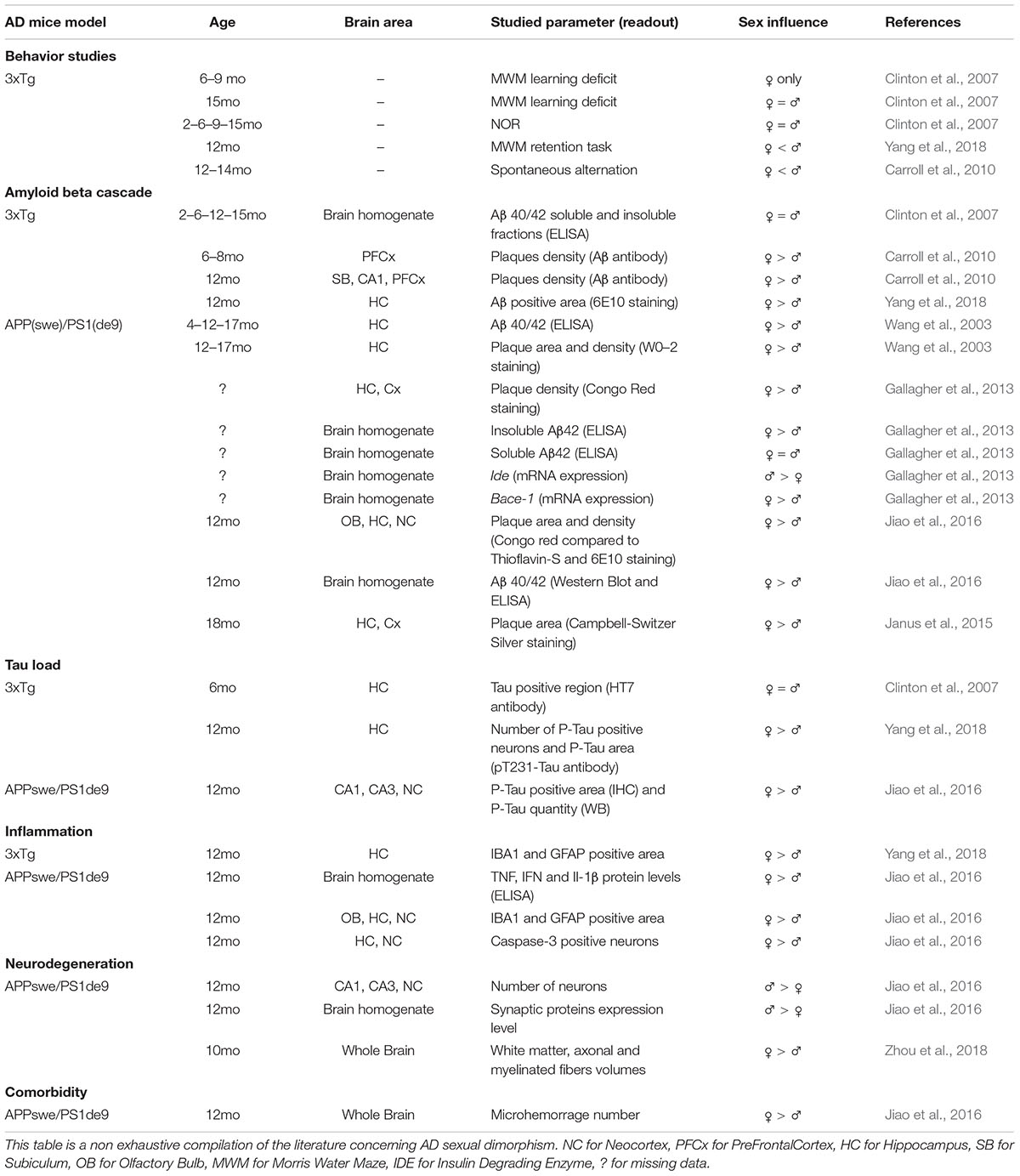
Table 1. Impact of sexual dimorphism on AD pathophyiology in various mice models.
A growing number of studies highlights the impact of sexual dimorphism on microglia shape and functions (Table 2). Many sex mediated effects on microglia have been demonstrated. They seem to be highly dependent on age and brain region: (1) in both mice and rats, adult males show higher microglia densities (Guneykaya et al., 2018; Perkins et al., 2018); (2) adult females microglia also display higher proportion of cells with long and thick processes while male microglia have larger soma (Schwarz et al., 2012; Guneykaya et al., 2018; Weinhard et al., 2018). These differences may indicate that adult female microglia are in a more homeostatic state while males display a more reactive state. Functional differences are also observed, with microglia from male brain displaying higher migration rates while female’s present stronger phagocytic activity related to higher expression of phagocytosis associated genes (Nelson et al., 2017; Yanguas-Casás et al., 2018). RNA sequencing also revealed a more protective transcriptomic profile for female microglia while male microglia displayed a more inflammatory phenotype (Villa et al., 2018). A recent study also reports a sex impact on microglial electrical properties, suggesting that sex may affect microglia secretory profile, a property that can directly influence inflammatory response abilities (Guneykaya et al., 2018). It was initially thought that microglia sex differences depend from circulating sexual hormones (Nissen, 2017), but this view was recently challenged as it was shown that female microglia retain their protective properties when transplanted in males brain (Villa et al., 2018).
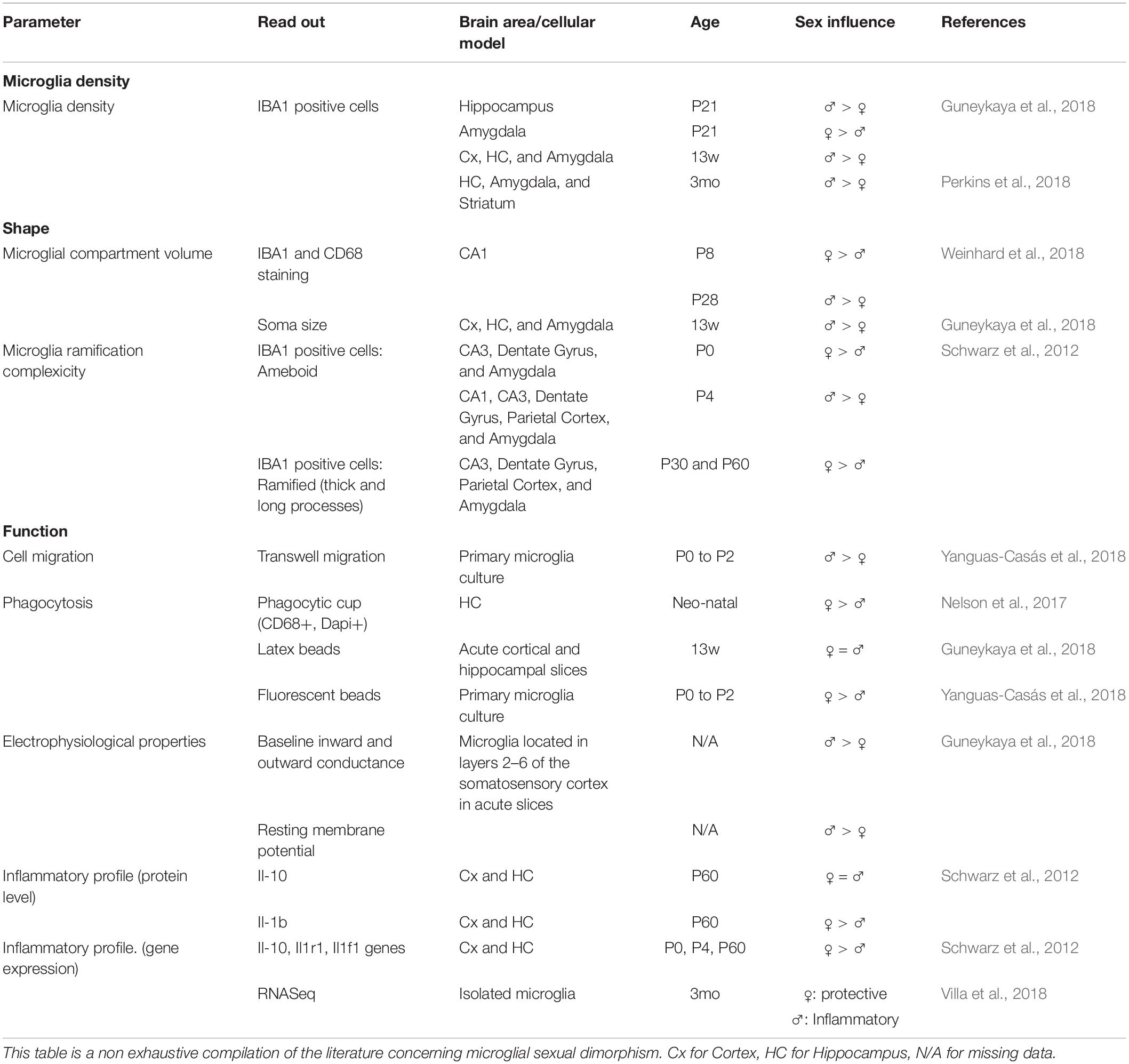
Table 2. Impact of sexual dimorphism on microglial shape and function.
Thus, although the molecular pathways involved in this microglial sexualization remain largely unknown, the current data suggest that, at least in adulthood, female microglia are in a more homeostatic and protective state. In the context of AD, it could be speculated that female microglia committed to homeostatic functions would need longer time exposure or stronger stimuli to polarize their shape and functions to answer correctly to harmful stimuli. Additionally, when they finally react to Aβ, they will change their local environment from a low- to a high-inflammatory environment. This drastic change could lead to more harmful effects on microglia themselves but also on all other surrounding cells and may explain why female are more likely to develop the pathology.
Although the effect of sex on microglial functions has been implicated in various CNS pathologies (Sorge et al., 2015; Charriaut-Marlangue et al., 2018; Jullienne et al., 2018; Mapplebeck et al., 2018; Thion et al., 2018), further studies are needed to understand the impact of microglial sexual dimorphism in AD initiation and progression and to which extend it relies to sex disbalance in AD. Whether sexual dimorphism mechanisms are similar in human and animal models also need to be clarified.
Microglial Diversity Is Both a Challenge and an Opportunity
Microglia cell diversity has been recognized since their first description by Pio del Rio-Hortega in 1919 (Sierra et al., 2016), who reported the existence of different morphological microglia phenotypes in the human brain. He also established that microglia morphology is considerably altered in disease conditions, introducing the concept that microglia tightly adapt to their local environment. More recent functional, morphological, immunohistochemical, and medium throughput analyses of microglia in pathological conditions also pointed out to the existence of a diversity of reactive phenotypes (Hanisch and Ketternmann, 2007; Ransohoff and Perry, 2009; Kierdorf and Prinz, 2017). In this respect, several semi-automated tools are now available to enable the identification of phenotypically distinct microglial cell subpopulations (Wagner et al., 2013; Verdonk et al., 2016; Salamanca et al., 2019).
In the last decade, the development of high throughput transcriptomic approaches combined with improved cell purification techniques helped refine our understanding of the molecular diversity of microglia both in physiological (Grabert et al., 2016) and pathophysiological conditions (Hirbec et al., 2017; Holtman et al., 2017; Sousa et al., 2017; Dubbelaar et al., 2018). They established that microglia cannot be categorized in a discrete number of physiological and pathological states. However, the breakthrough in the appreciation of the microglia diversity arose from the emergence of single-cell high throughput approaches. In particular single-cell RNAseq (scRNA-seq) enables investigating the diversity at cellular resolution and allows a detailed examination of cell states diversity and changes that are reflective of those in vivo (Macosko et al., 2015).
Two very recent scRNA-seq studies established the spatial and temporal diversity of microglia during the mouse development, and in either neurodegenerative or inflammatory conditions (Hammond et al., 2019; Masuda et al., 2019). These studies, together with more focused previous ones, (Keren-Shaul et al., 2017; Friedman et al., 2018) established that different microglia subpopulations co-exist at a given physiological or pathological state. Such heterogeneity within the microglia cell population represents both a challenge and an opportunity: the existence of distinct subpopulations supports the design of specific treatments targeting specific subpopulations with the aims of either promoting the beneficial subpopulations and/or hampering the deleterious ones.
What about microglia diversity in AD? It is known for years that AD brains exhibit at least two very distinct morphological microglia phenotypes: cells associated to the amyloid plaques display a “reactive”/amoeboid-like phenotype whereas those present in the rest of the parenchyma have a homeostatic-like morphology (Krabbe et al., 2013). As a mark of the importance of this diversity, it was shown that transcripts up-regulated in microglia isolated from AD mice were more highly expressed in tissues isolated in the vicinity of amyloid plaques compared to those distant from plaques (Orre et al., 2014). This suggested that microglia associated to plaques activate specific signaling pathways and functions. Using scRNA-seq of sorted 5xFAD mouse brain immune cells, Keren-Shaul et al. (2017) identified three distinct subtypes of microglia, including DAMs whose relative abundance increases with disease progression and which are preferentially located around amyloid plaques. The translational relevance of the DAMs was confirmed in human postmortem brains in which specific DAM markers are expressed in a subset of microglia in AD patients, but not in control subjects. More recently, in the knock-in APPNLFG AD mouse model (Saito et al., 2014; Sala Frigerio et al., 2019) identified activated- and interferon response microglia (ARM and IRM, respectively). ARMs partly overlap with DAMs, but by applying cell trajectory inference methods, these last authors demonstrated that ARMs were not solely present in disease conditions and are part of the normal brain aging process. However, many unresolved questions remain regarding the functional significance of this diversity. In particular, when it arises during AD progression and whether it is already present at the prodromal stages of the disease. With progress of both technology and analytic methods, it is now feasible to get a deeper understanding of the microglia diversity across AD progression. Identification of specific, potentially small in size, microglia subpopulations may have important implications for translational applications. Identification of factors released by these subpopulations and that can diffuse to the CSF may lead to the identification of new biomarkers. Additionally, identification of specific subpopulations will help discover new pathways and functions that contribute to the disease progression and may lead to the development of more specific functional positron emission tomography (PET) tracers. Deciphering, whether these subpopulations have beneficial, neutral or detrimental effects on brain cells, including neurons, will open the way for the design innovative disease-modifying therapeutic strategies.
Studying AD at Early Stages to Identify Early Biomarkers
After years of unsuccessful clinical trials, scientists are now realizing that AD is a pathology we may want to prevent (or stop) rather than to cure. This is calling for the identification of early diagnostic biomarkers, which are sorely lacking to help prodromal diagnostic, or design innovative disease-modifying therapeutic strategies. Although AD early stages are still less studied, increasing number of studies are now focusing on the prodromal phases to unravel early dysregulations.
Because one of microglia’s main role is to sense their environment and to react to danger, they are likely to play key roles in initial brain response to AD-driven changes. In agreement, a recent PET and magnetic resonance imaging (MRI) based study showed that AD patients have a first microglial activation peak before they display any other hallmarks of the pathology, suggesting that microglial dysfunction is critically involved in AD initiation (Fan et al., 2017). The involvement of microglia in AD initiation stages raises the question of which microglial targets could be modulated at an early stage to slow down the progression of the disease.
Extracellular vesicles (EVs) are a family of small membrane vesicles, which includes exosomes and microvesicles (MVs), transporting various types of molecules. In the recent years, EVs have emerged as a new mean of communication between cells. Microglia abilities to secrete EVs and MVs are part of their essential inflammatory functions. In the Aβ phagocytosis process, overloaded microglia can release Aβ-containing small secretory vesicles (Joshi et al., 2014), as well as Tau or P-Tau through exosomes (Saman et al., 2012; Asai et al., 2015). Accordingly, the levels of myeloid MVs detected in the CSF of MCI or early stage AD patients, is correlated with the extent of their white matter damage (Agosta et al., 2014). Microglial EVs may thus be regarded as a possible mean to spread the pathology during early stages. Interestingly, because they are secreted and can access first the CSF compartment and then the blood, microglial EVs may represent valuable diagnosis tools (Trotta et al., 2018).
Oxidative stress is known as a necessary but potentially harmful process. In response to fibrillary Aβ, reactive microglia produce free radicals, notably superoxide via the NADPH oxidase (NOX) (Harrigan et al., 2008). Moreover, NOX activity is increased in MCI patients compared to controls (Bruce-Keller et al., 2010). These findings open the way for using NOX activity as the marker of early microglia reaction in AD. Interfering with NOX activity through specific compounds may also provide a mean to slow down the pathology (Dumont and Beal, 2011).
Iron is an important metal implicated in vital biological processes. However, intracellular iron overload can lead to neuronal degeneration (Zhang et al., 2014). Microglia are able to interact, transport and metabolize iron. When they secrete pro-inflammatory factors, microglia can stimulate neuronal iron uptake leading to an increase in neurodegeneration (Zhang et al., 2014). Interestingly, patients with specific iron overload disorder are subject to earlier onset of AD, suggesting that iron may be involved in the initiation steps of the pathology. Moreover, iron chelation seems to be beneficial to AD patients. Further work is needed to understand the early implication of microglial iron regulations in AD (Nnah and Wessling-Resnick, 2018).
Barriers to Discoveries
Although fundamental knowledge is of importance, the ultimate goal of characterizing cells functions and dysfunctions in physiological and pathological conditions is to design efficient therapeutic strategies with clinical benefit. Understanding the complexity of Alzheimer’s disease is one of the XXIth century challenges. To do so we mainly rely on animal models that mimic the symptoms of the pathology (amyloid deposits, hyperphosphorylated tau, cognitive alterations, etc.). However, so far, the results generated from these models have generally failed to translate to clinic, and only symptomatic and poorly efficient treatments exist (Ransohoff, 2018). In the two following paragraphs, we explore two important factors that may have been under-estimated in AD pre-clinical research: the relevance of current AD mouse models and the immunological differences between mice and humans. These remarks do not only apply to the study of microglia’ roles in AD, but are important issues to consider for the design of any preclinical studies.
Are the Current Mouse Models Tailored to Understand AD and Identify Drug Targets?
Most of the animal models are genetically engineered to overexpress human protein mutations also leading to an overexpression of their respective by-products, thus generating potential confounding factors (Sasaguri et al., 2017). In addition, most AD models are based on familial mutations whereas early-onset familial AD is estimated to account for only 3.5% of total AD cases (Harvey et al., 2003). In addition, some of these models are based on a combination of mutations that were never found in patients. Because it is the first hallmark of AD, most of the models focused on the amyloidogenic pathway (Sasaguri et al., 2017). However, it is clear that, in humans, there is a concomitant effect of Aβ and P-Tau. To solve this issue, the 3x-Tg model is combining Aβ and Tau associated features. However, it only poorly represents the pathology in term of kinetic of appearance of the symptoms as cognitive deficits appear way too early and before any amyloid accumulation or Tau dysregulation (AlzForum1). To solve these issues, new mouse-based or human-based models are created (see section “New opportunities in AD research”).
To study microglia impact on AD, new models have been created by combining microglial dedicated tools with AD models (Zhou et al., 2018). One of the most commonly used models is the CX3CR1+/eGFP x APP/PS1 strain, in which an allele of the fractalkine receptor CX3CR1 is replaced by a GFP allowing to track microglial cells. However, to our knowledge, the impact of the CX3CR1 haplo-deficiency on AD development has not yet been thoroughly investigated. Finally, AD studies are generally conducted in old mice, when the pathology symptoms and hallmarks are well established. This stage of the pathology is probably too advanced for the identification of efficient disease modifying drugs.
Human and Mice Are Not Immunologically Similar
In the field of inflammation, the comprehensive study by Seok et al. (2013) revealed that gene dysregulation in mouse models of severe human inflammatory conditions (endotoxemia, burns, and trauma) do not correlate with human genomic changes. This raises the question of the relevance of mouse models to study the role of immune cells in diseases, including brain disease. Several factors known to be of importance for immune functions differ between rodents and human studies. Most often, rodent models and studies use inbred strains whereas human genetic backgrounds are much more diverse. Additionally, humans are exposed to multiple diseases whereas research mouse models are raised in tightly controlled environment (Davis, 2012).
Functional studies investigated the response of human microglia to either endogenous (i.e., M-CSF) or exogenous (i.e., LPS) stimuli (Melief et al., 2012; Smith et al., 2013). For the most part, they show comparable results with mouse analyses, thus agreeing with two comprehensive transcriptomic studies which compared gene expression profiles in human and mouse microglia and concluded that, overall, gene expression is very similar in the two species (Galatro et al., 2017; Gosselin et al., 2017). Of interest to brain diseases, a good correlation between human and mouse microglia in response to neurodegeneration was also observed by other authors (Holtman et al., 2015; Keren-Shaul et al., 2017; Krasemann et al., 2017).
However, notwithstanding their global resemblance, significant number of genes are differentially expressed in mouse and human microglia. In particular, specific immune genes are only expressed in human samples (Gosselin et al., 2017). In addition, differences in the relative expression of lineage- and signal-dependent transcription factors between mice and humans were observed (Holtman et al., 2017). In line, previous studies identified several molecular pathways and signaling functions that significantly differ between mouse and human microglia. This includes proliferation, response to TGFβ1, Siglecs signaling, nitric oxide (NO) production, and response to certain drugs such as valproic acid (VPA) (Smith and Dragunow, 2014).
Of relevance to AD, Siglec-3 (CD33) that has been identified as a risk factor for AD (Bertram et al., 2008) displays substantial species differences in expression patterns and ligand recognition (Lajaunias et al., 2005). Additionally, age-related changes of immune and cognitive functions may not be correctly modeled in rodents, whose life expectancy is far less compared to humans. Accordingly, gene expression changes in aged human brain are significantly different from those in the mouse brain (Loerch et al., 2008; Bishop et al., 2010). In agreement, Galatro et al. (2017) revealed that there is a limited overlap in age-related changes in human and mouse microglia, highlighting that data obtained in aged mice should be extrapolated to the human situation with caution. Translation of results from mice to humans is also hampered by the lack of tools to precisely characterize microglial reactivity in patients: TSPO binding is so far the only way to study microglia reaction in a clinical context.
Although methods to purify, culture and experiment on human microglia have been established, access to human samples that meet the requirements of high-quality studies is limited. AD mouse models are thus undoubtedly useful to decipher the roles of specific microglial functions and signaling pathways. However, the species differences highlighted above should prompt scientists to confirm the results they obtained in mouse models on human samples.
New Opportunities in AD Research
While current models to study AD have multiple limitations, scientists are trying to generate new models with limited pitfalls. In this last part, we are reviewing new approaches currently developed. These approaches are expected to improve our knowledge on microglial functions and help bridge the gap between in vitro studies, rodent models and the human disease.
Promising Animal Models to Overcome Identified Pitfalls
Although mouse models present significant pitfalls, they are attractive models in preclinical studies. Indeed, they are small, easy to raise, mammals with reasonable life expectancy. They reproduce well in captivity and can be engineered and/or humanized in a relatively easy way. Furthermore, with the exponential development of a wide repertoire of mouse lines (Model Organism Development and Evaluation for Late-Onset Alzheimer’s Disease2), it is possible for scientists to explore new hypotheses by combining different mouse lines. In this context and to overcome the problems associated with APP overexpression, Drs Saito and Saido developed new knock-in AD mouse models, namely the APPNLF and APPNLFG mice (Saito et al., 2014). In these models, mutated APP is expressed from its endogenous promoter, leading to a kinetic of appearance of the symptoms which is more comparable with human pathology (Sakakibara et al., 2018). These models are not thoroughly characterized yet and still excludes Tau-associated dysfunctions. However, in the APPNLF model, the delay before Aβ deposits appearance supports that it may represent an interesting model to study the prodromal phase of the disease.
Other models, not based on genetic modifications, have also emerged in the last decades. Compared to humans, dogs share an almost identical enzymatic machinery to process APP. Moreover, they may naturally develop an age-related cognitive dysfunction that reproduces several aspects of AD (Schmidt et al., 2015). With its high life span, the canine model can be useful specifically to study pathways involved in Aβ deposition and clearance in the early phases of the disease (Sarasa and Pesini, 2009). To get closer to human pathology, several primate models are currently used to study AD. Depending on their phylogenetic distance to humans, they neither display the same kinetic nor kind of neurological alterations (Heuer et al., 2012). Among primates, Microcebus Murinus also known as the gray mouse lemur, is particularly interesting (Bons et al., 2006). Indeed, they are small animals, just over the size of a mouse. They can be raised in cohorts of several individuals, and can reproduce in captivity. Along aging some individuals naturally develop an AD like pathology with Aβ plaques (Mestre-Francés et al., 2000), associated with specific transcriptomic remodeling (Abdel Rassoul et al., 2010). Histological features also seem to be associated with cognitive alterations (Trouche et al., 2010). Although we know that, at least in the spinal cord, the gray mouse lemur microglial shape and distribution is closer to human compared to mouse (Le Corre et al., 2018), microglia are still poorly studied in this model. Further studies are needed to investigate the involvement of microglia in this age-related AD like pathology.
Although, both dog and primate models represent interesting models, they are not so easy to use. For example, there are only few breeding centers for Microcebus Murinus worldwide. Those animals also need more housing space and their use is subjected to drastic ethic rules. Moreover, many of the research tools are designed on mouse genome/proteins limiting the use of these peculiar models.
hiPSCs-Derived Microglia: An Emerging Tool to Decipher the Role of Microglia in AD
To overcome species issues, microglial cell lines of human origin can be useful. They allow to study specific biological functions or functional pathways; however, they cannot reproduce the complexities of cells. This particularly true for cells which, like microglia, are highly adapted to their environment. Bone marrow-derived or blood monocytes-derived macrophages represent another source of human microglia-like cells. However, because these cells are of fundamentally different embryonic origin (Hoeffel and Ginhoux, 2015), they are likely poor models of adult microglia. Adult primary microglia can be harvested from dissociated brain area, but this process generally lead to low yields and can only sparsely be obtained for human samples. Moreover, isolation processes and culturing methods impact on gene expression pattern thus calling for caution in interpreting results based on these approaches (Gosselin et al., 2017).
An alternative to those issues is the development of cell reprograming methods, which offer the possibility to generate human pluripotent cell lines (hiPSCs) from healthy individuals but also from patients with specific diseases (Sullivan and Young-Pearse, 2017). Human iPSCs-derived neural cells can be produced in consistent yields. In the context of AD, iPSC-derived neurons issued from either familial AD (FAD) patients carrying mutations found in AD or sporadic AD (SAD) patients recapitulate some of the main hallmarks of the disease (Poon et al., 2017). Similarly, astrocytes derived from AD-patient iPSC show significant morphological and functional alterations, including up-regulation of inflammatory cytokines (Chandrasekaran et al., 2016; Auboyer et al., 2019).
Human pluripotent cell lines have recently been successfully differentiated into microglia (Muffat et al., 2016; Abud et al., 2017; Douvaras et al., 2017; Haenseler et al., 2017; Takata et al., 2017; Pocock and Piers, 2018). Although there is yet no consensus protocol to obtain hiPSCs-derived microglia, all protocols follow similar steps based on this cell type ontogeny. Based on such protocol, Garcia-Reitboeck et al. (2018) recently described microglial dysfunctions in hiPSCs-derived microglia from TREM2 T66M and W50C carriers, thus outlining the interest of such iPSCs approaches to decipher the role of microglia functions and dysfunctions in AD. Deciphering to which extend hiPSCs microglia derived from different patients (i.e., different SADs and/or different FAD mutations) share the same molecular and/or functional characteristics will help better understand the heterogeneity of AD, and may open the way toward the use of hiPSCs to implement personalized treatment in AD.
An important emerging concept is that, because microglia are CNS resident macrophages highly adapted to the CNS environment, a two-steps protocol is required to obtain hiPSCs-derived microglia with genuine microglial functions. In this scheme, the first step is to obtain iPSCs-derived macrophages progenitors and second to grow them in a conditioned environment, i.e., in the presence of other neural cells (Lee C.Z.W. et al., 2018). Under such experimental settings, hiPSCs-derived microglia exhibit phenotype, gene expression profile and functional properties close to brain-isolated microglia (Pandya et al., 2017; Takata et al., 2017).
Maintenance of tissue architecture is an important aspect of in vivo studies. Interestingly, hiPSCs derived cells can be grown in 3D cultures to model the cytoarchitecture and the connectivity of the brain while allowing in vitro manipulation and experimentation. Such 3D or organoids models open the way for studying human microglia under conditions that are close to physiological and physio-pathological context. Although specific pitfalls exist with such approaches, including the need to develop isogenic controls to alleviate the risk of cofounding factors due to difference in genetic backgrounds between patients and controls, there is little doubt that these approaches will be instrumental to decipher the role of microglia in the progression of AD.
Concluding Remarks
Glial cells, including microglia, have long been suspected to play a role in Alzheimer’s disease but only because of their ability to react to neuronal dysfunctions (e.g., Amyloid and Tau aggregates). This neurocentric view, which considered glial cells as secondary, has been challenged recently by the results of genetic association studies identifying genetic loci associated with risk of Alzheimer’s that are associated to genes preferentially or exclusively expressed in glial cells (Verheijen and Sleegers, 2018). This has refined our view of how Alzheimer disease initiates and progresses, and introduced new concepts and ideas for Alzheimer’s pathophysiological mechanisms, both at the molecular and cellular levels.
Because of their abilities to sense and react to their environment, reactive microglia are likely playing key early roles in the disease progression and may lead to the identification of early biomarkers. Because they can drive functional changes in astrocytes (Liddelow and Barres, 2017) and crosstalk with non-neuronal immune cells (Dionisio-Santos et al., 2019), they also represent attractive drug targets to stop or limit the disease progression.
As reported here, the exact contribution of the different reactive microglia subtypes to AD is currently unclear and the subject of intense researches. Over the recent years, several technological breakthroughs have been achieved, allowing scientists to address new challenging questions. These technical developments now allow studying microglia roles with medium or high throughput flows, and perform fine analysis of their functions in preserved environments. A better understanding of the contribution of microglia cells to AD initiation and progression is expected to renew the interest of big pharma to re-invest in the field and will pave the way toward better designed strategies.
Many factors need to be considered, including sex, age, species, molecular diversity, health status, communication with the periphery, etc., to fully decipher the role of microglia in AD. These are undoubtedly challenging but also very exciting fields of research, which hold the promise of defining innovative therapeutic strategies and reduce the socio-economic burden of this devastating disease.
Author Contributions
A-LH, JH, LU, and HH designed and drafted the sections of the manuscript. A-LH prepared the tables and the figures.
Funding
This study was supported by the France Alzheimer, Fondation Alzheimer, LABEX ICST and the Fondation NRJ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Abbracchio, M. P., and Ceruti, S. (2007). P1 receptors and cytokine secretion. Purinergic Signal. 3, 13–25. doi: 10.1007/s11302-006-9033-z
Abdel Rassoul, R., Alves, S., Pantesco, V., De Vos, J., Michel, B., Perret, M., et al. (2010). Distinct transcriptome expression of the temporal cortex of the primate Microcebus murinus during brain aging versus Alzheimer’s disease-like pathology. PLoS One 5:e12770. doi: 10.1371/journal.pone.0012770
Abud, E. M., Ramirez, R. N., Martinez, E. S., Healy, L. M., Nguyen, C. H. H., Newman, S. A., et al. (2017). iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94, 278–293.e9. doi: 10.1016/j.neuron.2017.03.042
Adinolfi, E., Giuliani, A. L., De Marchi, E., Pegoraro, A., Orioli, E., and Di Virgilio, F. (2018). The P2X7 receptor: a main player in inflammation. Biochem. Pharmacol. 151, 234–244. doi: 10.1016/j.bcp.2017.12.021
Agosta, F., Dalla Libera, D., Spinelli, E. G., Finardi, A., Canu, E., Bergami, A., et al. (2014). Myeloid microvesicles in cerebrospinal fluid are associated with myelin damage and neuronal loss in mild cognitive impairment and Alzheimer disease. Ann. Neurol. 76, 813–825. doi: 10.1002/ana.24235
Ajami, B., Bennett, J. L., Krieger, C., Tetzlaff, W., and Rossi, F. M. (2007). Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10, 1538–1543. doi: 10.1038/nn2014
Ajit, D., Woods, L. T., Camden, J. M., Thebeau, C. N., El-Sayed, F. G., Greeson, G. W., et al. (2014). Loss of P2Y2 nucleotide receptors enhances early pathology in the TgCRND8 mouse model of Alzheimer’s disease. Mol. Neurobiol. 49, 1031–1042. doi: 10.1007/s12035-013-8577-5
Alzheimer, A. (1907). über eine eigenartige Erkrankung der Hirnrinde. Allg. Zeitschrift für Psychiatr. und phychish-Gerichtliche Medizin 64, 146–148.
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Auboyer, L., Monzo, C., Wallon, D., Rovelet-Lecrux, A., Gabelle, A., Gazagne, I., et al. (2019). Generation of induced pluripotent stem cells (IRMBi001-A) from an Alzheimer’s disease patient carrying a G217D mutation in the PSEN1 gene. Stem Cell Res. 34:1013183. doi: 10.1016/j.scr.2018.101381
Basilico, B., Pagani, F., Grimaldi, A., Cortese, B., Di Angelantonio, S., Weinhard, L., et al. (2019). Microglia shape presynaptic properties at developing glutamatergic synapses. Glia 67, 53–67. doi: 10.1002/glia.23508
Bedner, P., Dupper, A., Huttmann, K., Muller, J., Herde, M. K., Dublin, P., et al. (2015). Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138, 1208–1222. doi: 10.1093/brain/awv067
Belinson, H., and Michaelson, D. M. (2009). ApoE4-dependent Aβ-mediated neurodegeneration is associated with inflammatory activation in the hippocampus but not the septum. J. Neural Transm. 116, 1427–1434. doi: 10.1007/s00702-009-0218-9
Ben Haim, L., Carrillo-de Sauvage, M.-A., Ceyzériat, K., and Escartin, C. (2015). Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell. Neurosci. 9:278. doi: 10.3389/fncel.2015.00278
Bertram, L., Lange, C., Mullin, K., Parkinson, M., Hsiao, M., Hogan, M. F., et al. (2008). Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83, 623–632. doi: 10.1016/j.ajhg.2008.10.008
Bhattacharya, A., and Biber, K. (2016). The microglial ATP-gated ion channel P2X7 as a CNS drug target. Glia 64, 1772–1787. doi: 10.1002/glia.23001
Bishop, N. A., Lu, T., and Yankner, B. A. (2010). Neural mechanisms of ageing and cognitive decline. Nature 464, 529–535. doi: 10.1038/nature08983
Bons, N., Rieger, F., Prudhomme, D., Fisher, A., and Krause, K.-H. (2006). Microcebus murinus: a useful primate model for human cerebral aging and Alzheimer’s disease? Genes Brain Behav. 5, 120–130. doi: 10.1111/j.1601-183X.2005.00149.x
Bouchon, A., Hernández-Munain, C., Cella, M., and Colonna, M. (2002). A Dap12-mediated pathway regulates expression of Cc chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 194, 1111–1122. doi: 10.1084/jem.194.8.1111
Brawek, B., Schwendele, B., Riester, K., Kohsaka, S., Lerdkrai, C., Liang, Y., et al. (2014). Impairment of in vivo calcium signaling in amyloid plaque-associated microglia. Acta Neuropathol. 127, 495–505. doi: 10.1007/s00401-013-1242-2
Bruce-Keller, A. J., Gupta, S., Parrino, T. E., Knight, A. G., Ebenezer, P. J., Weidner, A. M., et al. (2010). NOX activity is increased in mild cognitive impairment. Antioxid. Redox Signal. 12, 1371–1382. doi: 10.1089/ars.2009.2823
Burguillos, M. A., Deierborg, T., Kavanagh, E., Persson, A., Hajji, N., Garcia-Quintanilla, A., et al. (2011). Caspase signalling controls microglia activation and neurotoxicity. Nature 472, 319–324. doi: 10.1038/nature09788
Burnstock, G. (2016). An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology 104, 4–17. doi: 10.1016/j.neuropharm.2015.05.031
Butovsky, O., Jedrychowski, M. P., Moore, C. S., Cialic, R., Lanser, A. J., Gabriely, G., et al. (2014). Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. doi: 10.1038/nn.3599
Calovi, S., Mut-Arbona, P., and Sperlágh, B. (2019). Microglia and the Purinergic Signaling System. Neuroscience 405, 137–147. doi: 10.1016/j.neuroscience.2018.12.021
Cardona, A. E., Pioro, E. P., Sasse, M. E., Kostenko, V., Cardona, S. M., Dijkstra, I. M., et al. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 9, 917–924. doi: 10.1038/nn1715
Carroll, J. C., Rosario, E. R., Kreimer, S., Villamagna, A., Gentzschein, E., Stanczyk, F. Z., et al. (2010). Sex differences in β-amyloid accumulation in 3xTg-AD mice: role of neonatal sex steroid hormone exposure. Brain Res. 1366, 233–245. doi: 10.1016/j.brainres.2010.10.009
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., DeMattos, R. B., Patterson, B. W., et al. (2011). Human apoE isoforms differentially regulate brain amyloid- peptide clearance. Sci. Transl. Med. 3:89ra57. doi: 10.1126/scitranslmed.3002156
Chandrasekaran, A., Avci, H. X., Leist, M., Kobolak, J., and Dinnyes, A. (2016). Astrocyte differentiation of human pluripotent stem cells: new tools for neurological disorder research. Front. Cell. Neurosci. 10:215. doi: 10.3389/fncel.2016.00215
Charriaut-Marlangue, C., Leconte, C., Csaba, Z., Chafa, L., Pansiot, J., Talatizi, M., et al. (2018). Sex differences in the effects of PARP inhibition on microglial phenotypes following neonatal stroke. Brain. Behav. Immun. 73, 375–389. doi: 10.1016/j.bbi.2018.05.022
Cho, S. H., Sun, B., Zhou, Y., Kauppinen, T. M., Halabisky, B., Wes, P., et al. (2011). CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 286, 32713–32722. doi: 10.1074/jbc.M111.254268
Clinton, L. K., Billings, L. M., Green, K. N., Caccamo, A., Ngo, J., Oddo, S., et al. (2007). Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol. Dis. 28, 76–82. doi: 10.1016/j.nbd.2007.06.013
Combs, C. K., Johnson, D. E., Cannady, S. B., Lehman, T. M., and Landreth, G. E. (1999). Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of β-amyloid and prion proteins. J. Neurosci. 19, 928–939. doi: 10.1523/JNEUROSCI.19-03-00928.1999
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923.
Czirr, E., Castello, N. A., Mosher, K. I., Castellano, J. M., Hinkson, I. V., Lucin, K. M., et al. (2017). Microglial complement receptor 3 regulates brain Aβ levels through secreted proteolytic activity. J. Exp. Med. 214, 1081–1092. doi: 10.1084/jem.20162011
Daborg, J., Andreasson, U., Pekna, M., Lautner, R., Hanse, E., Minthon, L., et al. (2012). Cerebrospinal fluid levels of complement proteins C3, C4 and CR1 in Alzheimer’s disease. J. Neural Transm. 119, 789–797. doi: 10.1007/s00702-012-0797-8
D’Amelio, M., Cavallucci, V., Middei, S., Marchetti, C., Pacioni, S., Ferri, A., et al. (2011). Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 14, 69–79. doi: 10.1038/nn.2709
Davis, M. M. (2012). Immunology taught by humans. Sci. Transl. Med. 4:117fs2. doi: 10.1126/scitranslmed.3003385
DeMattos, R. B., Cirrito, J. R., Parsadanian, M., May, P. C., O’Dell, M. A., Taylor, J. W., et al. (2004). ApoE and clusterin cooperatively suppress Aβ levels and deposition: evidence that ApoE regulates extracellular Aβ metabolism in vivo. Neuron 41, 193–202. doi: 10.1016/S0896-6273(03)00850-X
Dionisio-Santos, D. A., Olschowka, J. A., and O’Banion, M. K. (2019). Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflammation 16:74. doi: 10.1186/s12974-019-1453-0
Douvaras, P., Sun, B., Wang, M., Kruglikov, I., Lallos, G., Zimmer, M., et al. (2017). Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Rep. 8, 1516–1524. doi: 10.1016/j.stemcr.2017.04.023
Dubbelaar, M. L., Kracht, L., Eggen, B. J. L., and Boddeke, E. W. G. M. (2018). The Kaleidoscope of microglial phenotypes. Front. Immunol. 9:1753. doi: 10.3389/fimmu.2018.01753
Dumont, M., and Beal, M. F. (2011). Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic. Biol. Med. 51, 1014–1026. doi: 10.1016/j.freeradbiomed.2010.11.026
Egensperger, R., Kösel, S., Eitzen, U., and Graeber, M. B. (1998). Microglial activation in Alzheimer disease: association with APOE genotype. Brain Pathol. 8, 439–447. doi: 10.1111/j.1750-3639.1998.tb00166.x
Erb, L., Woods, L. T., Khalafalla, M. G., and Weisman, G. A. (2018). Purinergic signaling in Alzheimer’s disease. Brain Res. Bull. doi: 10.1016/j.brainresbull.2018.10.014 [Epub ahead of print].
Fagan, A. M., Watson, M., Parsadanian, M., Bales, K. R., Paul, S. M., and Holtzman, D. M. (2002). Human and murine ApoE markedly alters Aβ metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 9, 305–318. doi: 10.1006/nbdi.2002.0483
Fan, Z., Brooks, D. J., Okello, A., and Edison, P. (2017). An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 140, 792–803. doi: 10.1093/brain/aww349
Färber, K., and Kettenmann, H. (2006). Functional role of calcium signals for microglial function. Glia 54, 656–665. doi: 10.1002/glia.20412
Ferretti, M. T., Iulita, M. F., Cavedo, E., Chiesa, P. A., Schumacher Dimech, A., Santuccione Chadha, A., et al. (2018). Sex differences in Alzheimer disease - the gateway to precision medicine. Nat. Rev. Neurol. 14, 457–469. doi: 10.1038/s41582-018-0032-9
Fonseca, M. I., Zhou, J., Botto, M., and Tenner, A. J. (2004). Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 24, 6457–6465. doi: 10.1523/jneurosci.0901-04.2004
Frank, S., Burbach, G. J., Bonin, M., Walter, M., Streit, W., Bechmann, I., et al. (2008). TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56, 1438–1447. doi: 10.1002/glia.20710
Friedman, B. A., Srinivasan, K., Ayalon, G., Meilandt, W. J., Lin, H., Huntley, M. A., et al. (2018). Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 22, 832–847. doi: 10.1016/j.celrep.2017.12.066
Fu, H., Liu, B., Frost, J. L., Hong, S., Jin, M., Ostaszewski, B., et al. (2012). Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia 60, 993–1003. doi: 10.1002/glia.22331
Fuhrmann, M., Bittner, T., Jung, C. K. E., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Galatro, T. F., Holtman, I. R., Lerario, A. M., Vainchtein, I. D., Brouwer, N., Sola, P. R., et al. (2017). Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci. 20, 1162–1171. doi: 10.1038/nn.4597
Gallagher, J. J., Minogue, A. M., and Lynch, M. A. (2013). Impaired performance of female APP/PS1 mice in the morris water maze is coupled with increased Aβ accumulation and microglial activation. Neurodegener. Dis. 11, 33–41. doi: 10.1159/000337458
Garcia-Reitboeck, P., Phillips, A., Piers, T. M., Villegas-Llerena, C., Butler, M., Mallach, A., et al. (2018). Human induced pluripotent stem cell-derived microglia-like cells harboring TREM2 missense mutations show specific deficits in phagocytosis. Cell Rep. 24, 2300–2311. doi: 10.1016/j.celrep.2018.07.094
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Gosselin, D., Skola, D., Coufal, N. G., Holtman, I. R., Schlachetzki, J. C. M., Sajti, E., et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356:eaal3222. doi: 10.1126/science.aal3222
Grabert, K., Michoel, T., Karavolos, M. H., Clohisey, S., Baillie, J. K., Stevens, M. P., et al. (2016). Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 19, 504–516. doi: 10.1038/nn.4222
Graeber, M. B., Kösel, S., Egensperger, R., Banati, R. B., Müller, U., Bise, K., et al. (1997). Rediscovery of the case described by Alois Alzheimer in 1911: historical, histological and molecular genetic analysis. Neurogenetics 1, 73–80. doi: 10.1007/s100480050011
Griffin, W. S., Stanley, L. C., Ling, C., White, L., MacLeod, V., Perrot, L. J., et al. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci.U.S.A. 86, 7611–7615. doi: 10.1073/pnas.86.19.7611
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guneykaya, D., Ivanov, A., Hernandez, D. P., Haage, V., Wojtas, B., Meyer, N., et al. (2018). Transcriptional and translational differences of microglia from male and female brains. Cell Rep. 24, 2773–2783.e6. doi: 10.1016/J.CELREP.2018.08.001
Guo, L., Ladu, M. J., and Van Eldik, L. J. (2004). A dual role for apolipoprotein E in neuroinflammation anti-and pro-inflammatory activity. J. Mol. Neurosci. 23, 205–212.
Haenseler, W., Sansom, S. N., Buchrieser, J., Newey, S. E., Moore, C. S., Nicholls, F. J., et al. (2017). A highly efficient human pluripotent stem cell microglia model displays a neuronal-co-culture-specific expression profile and inflammatory response. Stem Cell Rep. 8, 1727–1742. doi: 10.1016/j.stemcr.2017.05.017
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
Hammond, T. R., Dufort, C., Dissing-Olesen, L., Giera, S., Young, A., Wysoker, A., et al. (2019). Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50, 253–271.e6. doi: 10.1016/j.immuni.2018.11.004
Hanisch, U. K., and Ketternmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472. doi: 10.1083/jcb.201709069
Harrigan, T. J., Abdullaev, I. F., Jourd’heuil, D., and Mongin, A. A. (2008). Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J. Neurochem. 106, 2449–2462. doi: 10.1111/j.1471-4159.2008.05553.x
Harvey, R. J., Skelton-Robinson, M., and Rossor, M. N. (2003). The prevalence and causes of dementia in people under the age of 65 years. J. Neurol. Neurosurg. Psychiatry 74, 1206–1209. doi: 10.1136/JNNP.74.9.1206
Hashimoto, T., Serrano-Pozo, A., Hori, Y., Adams, K. W., Takeda, S., Banerji, A. O., et al. (2012). Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid peptide. J. Neurosci. 32, 15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012
Haynes, S. E., Hollopeter, G., Yang, G., Kurpius, D., Dailey, M. E., Gan, W.-B., et al. (2006). The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 9, 1512–1519. doi: 10.1038/nn1805
Hebert, L. E., Weuve, J., Scherr, P. A., and Evans, D. A. (2013). Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80, 1778–1783. doi: 10.1212/WNL.0b013e31828726f5
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Heuer, E., Rosen, R. F., Cintron, A., and Walker, L. C. (2012). Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr. Pharm. Des. 18, 1159–1169.
Hirbec, H. E., Noristani, H. N., and Perrin, F. E. (2017). Microglia responses in acute and chronic neurological diseases: what microglia-specific transcriptomic studies taught (and did Not Teach) Us. Front. Aging Neurosci. 9:227. doi: 10.3389/fnagi.2017.00227
Hoeffel, G., and Ginhoux, F. (2015). Ontogeny of tissue-resident macrophages. Front. Immunol. 6:486. doi: 10.3389/fimmu.2015.00486
Hoffmann, A., Kann, O., Ohlemeyer, C., Hanisch, U.-K., and Kettenmann, H. (2003). Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J. Neurosci. 23, 4410–4419. doi: 10.1523/JNEUROSCI.23-11-04410.2003
Holtman, I. R., Raj, D. D., Miller, J. A., Schaafsma, W., Yin, Z., Brouwer, N., et al. (2015). Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol. Commun. 3:31. doi: 10.1186/s40478-015-0203-5
Holtman, I. R., Skola, D., and Glass, C. K. (2017). Transcriptional control of microglia phenotypes in health and disease. J. Clin. Invest. 127, 3220–3229. doi: 10.1172/JCI90604
Holtzman, D. M., Bales, K. R., Wu, S., Bhat, P., Parsadanian, M., Fagan, A. M., et al. (1999). Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer’s disease. J. Clin. Invest. 103, R15–R21. doi: 10.1172/JCI6179
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hoshiko, M., Arnoux, I., Avignone, E., Yamamoto, N., and Audinat, E. (2012). Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci. 32, 15106–15111. doi: 10.1523/JNEUROSCI.1167-12.2012
Hudry, E., Dashkoff, J., Roe, A. D., Takeda, S., Koffie, R. M., Hashimoto, T., et al. (2013). Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med. 5:212ra161. doi: 10.1126/scitranslmed.3007000
Janus, C., Flores, A. Y., Xu, G., and Borchelt, D. R. (2015). Behavioral abnormalities in APPSwe/PS1dE9 mouse model of AD-like pathology: comparative analysis across multiple behavioral domains. Neurobiol. Aging 36, 2519–2532. doi: 10.1016/j.neurobiolaging.2015.05.010
Jay, T. R., Miller, C. M., Cheng, P. J., Graham, L. C., Bemiller, S., Broihier, M. L., et al. (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 212, 287–295. doi: 10.1084/jem.20142322
Jiang, T., Tan, L., Zhu, X.-C., Zhang, Q.-Q., Cao, L., Tan, M.-S., et al. (2014). Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 39, 2949–2962. doi: 10.1038/npp.2014.164
Jiao, S.-S., Bu, X.-L., Liu, Y.-H., Zhu, C., Wang, Q.-H., Shen, L.-L., et al. (2016). Sex dimorphism profile of Alzheimer’s disease-type pathologies in an APP/PS1 mouse model. Neurotox. Res. 29, 256–266. doi: 10.1007/s12640-015-9589-x
Jin, S. C., Benitez, B. A., Karch, C. M., Cooper, B., Skorupa, T., Carrell, D., et al. (2014). Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum. Mol. Genet. 23, 5838–5846. doi: 10.1093/hmg/ddu277
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Joshi, P., Turola, E., Ruiz, A., Bergami, A., Libera, D. D., Benussi, L., et al. (2014). Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 21, 582–593. doi: 10.1038/cdd.2013.180
Jullienne, A., Salehi, A., Affeldt, B., Baghchechi, M., Haddad, E., Avitua, A., et al. (2018). Male and female mice exhibit divergent responses of the cortical vasculature to traumatic brain injury. J. Neurotrauma 35, 1646–1658. doi: 10.1089/neu.2017.5547
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17. doi: 10.1016/j.cell.2017.05.018
Kierdorf, K., and Prinz, M. (2017). Microglia in steady state. J. Clin. Invest. 127, 3201–3209. doi: 10.1172/JCI90602
Kiialainen, A., Hovanes, K., Paloneva, J., Kopra, O., and Peltonen, L. (2005). Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiol. Dis. 18, 314–322. doi: 10.1016/j.nbd.2004.09.007
Kim, H. J., Ajit, D., Peterson, T. S., Wang, Y., Camden, J. M., Gibson Wood, W., et al. (2012). Nucleotides released from Aβ1-42-treated microglial cells increase cell migration and Aβ1-42 uptake through P2Y2 receptor activation. J. Neurochem. 121, 228–238. doi: 10.1111/j.1471-4159.2012.07700.x
Kim, T. S., Lim, H. K., Lee, J. Y., Kim, D. J., Park, S., Lee, C., et al. (2008). Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 436, 196–200. doi: 10.1016/j.neulet.2008.03.019
Koffie, R. M., Hashimoto, T., Tai, H.-C., Kay, K. R., Serrano-Pozo, A., Joyner, D., et al. (2012). Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-β. Brain 135, 2155–2168. doi: 10.1093/brain/aws127
Krabbe, G., Halle, A., Matyash, V., Rinnenthal, J. L., Eom, G. D., Bernhardt, U., et al. (2013). Functional impairment of microglia coincides with beta-amyloid deposition in mice with alzheimer-like pathology. PLoS One 8:e60921. doi: 10.1371/journal.pone.0060921
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9. doi: 10.1016/j.immuni.2017.08.008
Labzin, L. I., Heneka, M. T., and Latz, E. (2017). Innate Immunity and Neurodegeneration. Annu. Rev. Med. 69, 437–449. doi: 10.1146/annurev-med-050715-104343
Lai, M. K. P., Tan, M. G. K., Kirvell, S., Hobbs, C., Lee, J., Esiri, M. M., et al. (2008). Selective loss of P2Y2 nucleotide receptor immunoreactivity is associated with Alzheimer’s disease neuropathology. J. Neural Transm. 115, 1165–1172. doi: 10.1007/s00702-008-0067-y
Lajaunias, F., Dayer, J. M., and Chizzolini, C. (2005). Constitutive repressor activity of CD33 on human monocytes requires sialic acid recognition and phosphoinositide 3-kinase-mediated intracellular signaling. Eur. J. Immunol. 35, 243–251. doi: 10.1002/eji.200425273
Le Corre, M., Noristani, H. N., Mestre-Frances, N., Saint-Martin, G. P., Coillot, C., Goze-Bac, C., et al. (2018). A Novel Translational Model of Spinal Cord Injury in Nonhuman Primate. Neurotherapeutics 15, 751–769. doi: 10.1007/s13311-017-0589-9
Lee, C. Y. D., Daggett, A., Gu, X., Jiang, L.-L., Langfelder, P., Li, X., et al. (2018). Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 97, 1032–1048.e5. doi: 10.1016/j.neuron.2018.02.002
Lee, C. Z. W., Kozaki, T., and Ginhoux, F. (2018). Studying tissue macrophages in vitro: are iPSC-derived cells the answer? Nat. Rev. Immunol. 18, 716–725. doi: 10.1038/s41577-018-0054-y
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Lian, H., Litvinchuk, A., Chiang, A. C.-A., Aithmitti, N., Jankowsky, J. L., and Zheng, H. (2016). Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J. Neurosci. 36, 577–589. doi: 10.1523/jneurosci.2117-15.2016
Liddelow, S. A., and Barres, B. A. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006
Loerch, P. M., Lu, T., Dakin, K. A., Vann, J. M., Isaacs, A., Geula, C., et al. (2008). Evolution of the aging brain transcriptome and synaptic regulation. PLoS One 3:e3329. doi: 10.1371/journal.pone.0003329
Lynch, J. R., Morgan, D., Mance, J., Matthew, W. D., and Laskowitz, D. T. (2001). Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J. Neuroimmunol. 114, 107–113. doi: 10.1016/S0165-5728(00)00459-8
Lyons, A., Lynch, A. M., Downer, E. J., Hanley, R., O’Sullivan, J. B., Smith, A., et al. (2009). Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J. Neurochem. 110, 1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x
Macosko, E. Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman, M., et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. doi: 10.1016/j.cell.2015.05.002
Maier, M., Peng, Y., Jiang, L., Seabrook, T. J., Carroll, M. C., and Lemere, C. A. (2008). Complement C3 deficiency leads to accelerated amyloid plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J. Neurosci. 28, 6333–6341. doi: 10.1523/jneurosci.0829-08.2008
Mapplebeck, J. C. S., Dalgarno, R., Tu, Y., Moriarty, O., Beggs, S., Kwok, C. H. T., et al. (2018). Microglial P2X4R-evoked pain hypersensitivity is sexually dimorphic in rats. Pain 159, 1752–1763. doi: 10.1097/j.pain.0000000000001265
Martin, E., Amar, M., Dalle, C., Youssef, I., Boucher, C., Le Duigou, C., et al. (2018). New role of P2X7 receptor in an Alzheimer’s disease mouse model. Mol. Psychiatry 24, 108–125. doi: 10.1038/s41380-018-0108-3
Martinon, F., Burns, K., and Tschopp, J. (2002). The Inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426. doi: 10.1016/S1097-2765(02)00599-3
Masuda, T., Sankowski, R., Staszewski, O., Bottcher, C., Amann, L., Scheiwe, C., et al. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392. doi: 10.1038/s41586-019-0924-x
McLarnon, J. G. (2005). Purinergic mediated changes in Ca2+ mobilization and functional responses in microglia: effects of low levels of ATP. J. Neurosci. Res. 81, 349–356. doi: 10.1002/jnr.20475
McLarnon, J. G., Choi, H. B., Lue, L.-F., Walker, D. G., and Kim, S. U. (2005). Perturbations in calcium-mediated signal transduction in microglia from Alzheimer’s disease patients. J. Neurosci. Res. 81, 426–435. doi: 10.1002/jnr.20487
McLarnon, J. G., Ryu, J. K., Walker, D. G., and Choi, H. B. (2006). Upregulated expression of purinergic P2X 7 receptor in Alzheimer disease and amyloid-β peptide-treated microglia and in peptide-injected rat hippocampus. J. Neuropathol. Exp. Neurol. 65, 1090–1097. doi: 10.1097/01.jnen.0000240470.97295.d3
McQuade, A., and Blurton-Jones, M. (2019). Microglia in Alzheimer’s disease: exploring how genetics and phenotype influence risk. J. Mol. Biol. 431, 1805–1817. doi: 10.1016/J.JMB.2019.01.045
Melief, J., Koning, N., Schuurman, K. G., Van De Garde, M. D. B., Smolders, J., Hoek, R. M., et al. (2012). Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia 60, 1506–1517. doi: 10.1002/glia.22370
Mestre-Francés, N., Keller, E., Calenda, A., Barelli, H., Checler, F., and Bons, N. (2000). Immunohistochemical analysis of cerebral cortical and vascular lesions in the primate Microcebus murinus reveal distinct amyloid beta1-42 and beta1-40 immunoreactivity profiles. Neurobiol. Dis. 7, 1–8. doi: 10.1006/nbdi.1999.0270
Mildner, A., Huang, H., Radke, J., Stenzel, W., and Priller, J. (2017). P2Y 12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 65, 375–387. doi: 10.1002/glia.23097
Minett, T., Classey, J., Matthews, F. E., Fahrenhold, M., Taga, M., Brayne, C., et al. (2016). Microglial immunophenotype in dementia with Alzheimer’s pathology. J. Neuroinflammation 13:135. doi: 10.1186/s12974-016-0601-z
Muffat, J., Li, Y., Yuan, B., Mitalipova, M., Omer, A., Corcoran, S., et al. (2016). Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 22, 1358–1367. doi: 10.1038/nm.4189
Naslund, J., Thyberg, J., Wernstedt, C., Ft Karlstrom, A., Bogdanovic, N., Samuel Gandy, I. E., et al. (1995). Characterization of stable complexes involving apolipoprotein E and the amyloid B peptide in Alzheimer’s disease brain. Neuron 15, 219–228.
Nelson, L. H., Warden, S., and Lenz, K. M. (2017). Sex differences in microglial phagocytosis in the neonatal hippocampus. Brain. Behav. Immun. 64, 11–22. doi: 10.1016/j.bbi.2017.03.010
Ni, J., Wang, P., Zhang, J., Chen, W., and Gu, L. (2013). Silencing of the P2X7 receptor enhances amyloid-β phagocytosis by microglia. Biochem. Biophys. Res. Commun. 434, 363–369. doi: 10.1016/j.bbrc.2013.03.079
Nissen, J. C. (2017). Microglial function across the spectrum of age and gender. Int. J. Mol. Sci. 18:E561. doi: 10.3390/ijms18030561
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
Nizami, S., Hall-Roberts, H., Warrier, S., Cowley, S. A., and Di Daniel, E. (2019). Microglial inflammation and phagocytosis in Alzheimer’s disease: potential therapeutic targets. Br. J. Pharmacol. doi: 10.1111/bph.14618 [Epub ahead of print].
Nnah, I. C., and Wessling-Resnick, M. (2018). Brain iron homeostasis: a focus on microglial iron. Pharmaceuticals 11:E129. doi: 10.3390/ph11040129
Orre, M., Kamphuis, W., Osborn, L. M., Jansen, A. H., Kooijman, L., Bossers, K., et al. (2014). Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 35, 2746–2760. doi: 10.1016/j.neurobiolaging.2014.06.004
Pandya, H., Shen, M. J., Ichikawa, D. M., Sedlock, A. B., Choi, Y., Johnson, K. R., et al. (2017). Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 20, 753–759. doi: 10.1038/nn.4534
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Parhizkar, S., Arzberger, T., Brendel, M., Kleinberger, G., Deussing, M., Focke, C., et al. (2019). Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 22, 191–204. doi: 10.1038/s41593-018-0296-9
Perkins, A. E., Piazza, M. K., and Deak, T. (2018). Stereological analysis of microglia in aged male and female fischer 344 rats in socially relevant brain regions. Neuroscience 377, 40–52. doi: 10.1016/j.neuroscience.2018.02.028
Piccio, L., Buonsanti, C., Mariani, M., Cella, M., Gilfillan, S., Cross, A. H., et al. (2007). Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur. J. Immunol. 37, 1290–1301. doi: 10.1002/eji.200636837
Pocock, J. M., and Piers, T. M. (2018). Modelling microglial function with induced pluripotent stem cells: an update. Nat. Rev. Neurosci. 19, 445–452. doi: 10.1038/s41583-018-0030-3
Poon, A., Zhang, Y., Chandrasekaran, A., Phanthong, P., Schmid, B., Nielsen, T. T., et al. (2017). Modeling neurodegenerative diseases with patient-derived induced pluripotent cells: possibilities and challenges. N. Biotechnol. 39, 190–198. doi: 10.1016/j.nbt.2017.05.009
Próchnicki, T., Mangan, M. S., and Latz, E. (2016). Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Research 5:1469. doi: 10.12688/f1000research.8614.1
Ransohoff, R. M. (2018). All (animal) models (of neurodegeneration) are wrong. Are they also useful? J. Exp. Med. 215, 2955–2958. doi: 10.1084/jem.20182042
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145. doi: 10.1146/annurev.immunol.021908.132528
Rodriguez, G. A., Tai, L. M., LaDu, M. J., and Rebeck, G. W. (2014). Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflammation 11:111. doi: 10.1186/1742-2094-11-111
Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., et al. (2014). Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 17, 661–663. doi: 10.1038/nn.3697
Sakakibara, Y., Sekiya, M., Saito, T., Saido, T. C., and Iijima, K. M. (2018). Cognitive and emotional alterations in App knock-in mouse models of Aβ amyloidosis. BMC Neurosci. 19:46. doi: 10.1186/s12868-018-0446-8
Sala Frigerio, C., Wolfs, L., Fattorelli, N., Thrupp, N., Voytyuk, I., Schmidt, I., et al. (2019). The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 27, 1293–1306.e6. doi: 10.1016/j.celrep.2019.03.099
Salamanca, L., Mechawar, N., Murai, K. K., Balling, R., Bouvier, D. S., and Skupin, A. (2019). MIC-MAC: An automated pipeline for high-throughput characterization and classification of three-dimensional microglia morphologies in mouse and human postmortem brain samples. Glia 67, 1496–1509. doi: 10.1002/glia.23623
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Saman, S., et al. (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.M111.277061
Sanchez-Mejias, E., Navarro, V., Jimenez, S., Sanchez-Mico, M., Sanchez-Varo, R., Nuñez-Diaz, C., et al. (2016). Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol. 132, 897–916. doi: 10.1007/s00401-016-1630-5
Sanz, J. M., Falzoni, S., Rizzo, R., Cipollone, F., Zuliani, G., and Di Virgilio, F. (2014). Possible protective role of the 489C>T P2X7R polymorphism in Alzheimer’s disease. Exp. Gerontol. 60, 117–119. doi: 10.1016/j.exger.2014.10.009
Sarasa, M., and Pesini, P. (2009). Natural non-trasgenic animal models for research in Alzheimer’s disease. Curr. Alzheimer Res. 6, 171–178. doi: 10.2174/156720509787602834
Sasaguri, H., Nilsson, P., Hashimoto, S., Nagata, K., Saito, T., De Strooper, B., et al. (2017). APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 36, 2473–2487. doi: 10.15252/embj.201797397
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schmechel, D. E., Saunders, A. M., Strittmatter, W. J., Crain, B. J., Hulette, C. M., Joo, S. H., et al. (1993). Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 9649–9653.
Schmid, C. D., Sautkulis, L. N., Danielson, P. E., Cooper, J., Hasel, K. W., Hilbush, B. S., et al. (2002). Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 83, 1309–1320. doi: 10.1046/j.1471-4159.2002.01243.x
Schmidt, F., Boltze, J., Jäger, C., Hofmann, S., Willems, N., Seeger, J., et al. (2015). Detection and quantification of β-amyloid, pyroglutamyl Aβ, and tau in aged canines. J. Neuropathol. Exp. Neurol. 74, 912–923. doi: 10.1097/NEN.0000000000000230
Schwarz, J. M., Sholar, P. W., and Bilbo, S. D. (2012). Sex differences in microglial colonization of the developing rat brain. J. Neurochem. 120, 948–963. doi: 10.1111/j.1471-4159.2011.07630.x
Seok, J., Cuenca, H. S. W. A. G., Mindrinos, M. N., Bakerc, H. V., Xua, W., Richardsd, D. R., et al. (2013). Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. U.S.A. 110, 3507–3512. doi: 10.1073/pnas.1222878110
Serrano-Pozo, A., Qian, J., Monsell, S. E., Betensky, R. A., Hyman, B. T., and Massachusetts Alzheimer, P. (2015). APOEε2 is associated with milder clinical and pathological Alzheimer’s disease. Ann. Neurol. 77, 917–929. doi: 10.1002/ana.24369
Sieber, M. W., Jaenisch, N., Brehm, M., Guenther, M., Linnartz-Gerlach, B., Neumann, H., et al. (2013). Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One 8:e52982. doi: 10.1371/journal.pone.0052982
Sierra, A., de Castro, F., del Río-Hortega, J., Rafael Iglesias-Rozas, J., Garrosa, M., and Kettenmann, H. (2016). The “Big-Bang” for modern glial biology: translation and comments on Pío del Río-Hortega 1919 series of papers on microglia. Glia 64, 1801–1840. doi: 10.1002/glia.23046
Smith, A. M., and Dragunow, M. (2014). The human side of microglia. Trends Neurosci. 37, 125–135. doi: 10.1016/j.tins.2013.12.001
Smith, A. M., Gibbons, H. M., Oldfield, R. L., Bergin, P. M., Mee, E. W., Curtis, M. A., et al. (2013). M-CSF increases proliferation and phagocytosis while modulating receptor and transcription factor expression in adult human microglia. J. Neuroinflammation 10:85. doi: 10.1186/1742-2094-10-85
Sorge, R. E., Mapplebeck, J. C. S., Rosen, S., Beggs, S., Taves, S., Alexander, J. K., et al. (2015). Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 18, 1081–1085. doi: 10.1038/nn.4053
Sousa, C., Biber, K., and Michelucci, A. (2017). Cellular and molecular characterization of microglia: a unique immune cell population. Front. Immunol. 8:198. doi: 10.3389/fimmu.2017.00198
Stephan, A. H., Barres, B. A., and Stevens, B. (2012). The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369–389. doi: 10.1146/annurev-neuro-061010-113810
Strittmatter, W. J., Weisgraber, K. H., Huang, D. Y., Dong, L.-M., Salvesen, G. S., Pericak-Vance, M., et al. (1993). Binding of human apolipoprotein E to synthetic amyloid, B peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 8098–8102.
Sullivan, S. E., and Young-Pearse, T. L. (2017). Induced pluripotent stem cells as a discovery tool for Alzheimer×s disease. Brain Res. 1656, 98–106. doi: 10.1016/j.brainres.2015.10.005
Suurväli, J., Boudinot, P., Kanellopoulos, J., and Rüütel Boudinot, S. (2017). P2X4: a fast and sensitive purinergic receptor. Biomed. J. 40, 245–256. doi: 10.1016/j.bj.2017.06.010
Takahashi, K., Rochford, C. D. P., and Neumann, H. (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657. doi: 10.1084/jem.20041611
Takata, K., Kozaki, T., Lee, C. Z. W., Thion, M. S., Otsuka, M., Lim, S., et al. (2017). Induced-pluripotent-stem-cell-derived primitive macrophages provide a platform for modeling tissue-resident macrophage differentiation and function. Immunity 47, 183–198.e6. doi: 10.1016/j.immuni.2017.06.017
Thangavel, R., Bhagavan, S. M., Ramaswamy, S. B., Surpur, S., Govindarajan, R., Kempuraj, D., et al. (2017). Co-expression of glia maturation factor and apolipoprotein E4 in Alzheimer’s disease brain. J. Alzheimer’s Dis. 61, 553–560. doi: 10.3233/JAD-170777
Thion, M. S., Low, D., Silvin, A., Chen, J., Grisel, P., Schulte-Schrepping, J., et al. (2018). Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell 172, 500–516.e16. doi: 10.1016/j.cell.2017.11.042
Tiraboschi, P., Hansen, L. A., Masliah, E., Alford, M., Thal, L. J., and Corey-Bloom, J. (2004). Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 62, 1977–1983. doi: 10.1212/01.WNL.0000128091.92139.0F
Tong, B. C.-K., Wu, A. J., Li, M., and Cheung, K.-H. (2018). Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta Mol. Cell Res. 1865, 1745–1760. doi: 10.1016/j.bbamcr.2018.07.018
Trotta, T., Panaro, M. A., Cianciulli, A., Mori, G., Di Benedetto, A., and Porro, C. (2018). Microglia-derived extracellular vesicles in Alzheimer’s disease: a double-edged sword. Biochem. Pharmacol. 148, 184–192. doi: 10.1016/j.bcp.2017.12.020
Trouche, S. G., Maurice, T., Rouland, S., Verdier, J.-M., and Mestre-Francés, N. (2010). The three-panel runway maze adapted to Microcebus murinus reveals age-related differences in memory and perseverance performances. Neurobiol. Learn. Mem. 94, 100–106. doi: 10.1016/j.nlm.2010.04.006
Ulrich, J. D., Finn, M., Wang, Y., Shen, A., Mahan, T. E., Jiang, H., et al. (2014). Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener 9:20. doi: 10.1186/1750-1326-9-20
Varma, R., Chai, Y., Troncoso, J., Gu, J., Xing, H., Stojilkovic, S. S., et al. (2009). Amyloid-β induces a caspase-mediated cleavage of P2X4 to promote purinotoxicity. Neuromolecular Med. 11, 63–75. doi: 10.1007/s12017-009-8073-2
Veerhuis, R., Nielsen, H. M., and Tenner, A. J. (2011). Complement in the brain. Mol. Immunol. 48, 1592–1603. doi: 10.1016/j.molimm.2011.04.003
Verdonk, F., Roux, P., Flamant, P., Fiette, L., Bozza, F. A., Simard, S., et al. (2016). Phenotypic clustering: a novel method for microglial morphology analysis. J. Neuroinflammation 13:153. doi: 10.1186/S12974-016-0614-7
Verheijen, J., and Sleegers, K. (2018). Understanding Alzheimer disease at the interface between genetics and transcriptomics. Trends Genet. 34, 434–447. doi: 10.1016/j.tig.2018.02.007
Verkhratsky, A., Zorec, R., Rodríguez, J. J., and Parpura, V. (2016). Astroglia dynamics in ageing and Alzheimer’s disease. Curr. Opin. Pharmacol. 26, 74–79. doi: 10.1016/j.coph.2015.09.011
Villa, A., Gelosa, P., Castiglioni, L., Cimino, M., Rizzi, N., Pepe, G., et al. (2018). Sex-specific features of microglia from adult mice. Cell Rep. 23, 3501–3511. doi: 10.1016/j.celrep.2018.05.048
Villegas-Llerena, C., Phillips, A., Reitboeck, P. G., Hardy, J., and Pocock, J. M. (2016). Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 36, 74–81. doi: 10.1016/j.conb.2015.10.004
Viña, J., and Lloret, A. (2010). Why women have more Alzheimer’s disease than men: gender and mitochondrial toxicity of amyloid-β peptide. J. Alzheimer’s Dis. 20, S527–S533. doi: 10.3233/JAD-2010-100501
Vitek, M. P., Brown, C. M., and Colton, C. A. (2009). APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 30, 1350–1360. doi: 10.1016/j.neurobiolaging.2007.11.014
Wagner, D.-C., Riegelsberger, U. M., Michalk, S., Härtig, W., Kranz, A., and Boltze, J. (2011). Cleaved caspase-3 expression after experimental stroke exhibits different phenotypes and is predominantly non-apoptotic. Brain Res. 1381, 237–242. doi: 10.1016/j.brainres.2011.01.041
Wagner, D.-C., Scheibe, J., Glocke, I., Weise, G., Deten, A., Boltze, J., et al. (2013). Object-based analysis of astroglial reaction and astrocyte subtype morphology after ischemic brain injury. Acta Neurobiol. Exp. 73, 79–87.
Wang, J., Tanila, H., Puoliväli, J., Kadish, I., and van Groen, T. (2003). Gender differences in the amount and deposition of amyloidβ in APPswe and PS1 double transgenic mice. Neurobiol. Dis. 14, 318–327. doi: 10.1016/J.NBD.2003.08.009
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., and Robinette, M. L. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Weinhard, L., Neniskyte, U., Vadisiute, A., di Bartolomei, G., Aygün, N., Riviere, L., et al. (2018). Sexual dimorphism of microglia and synapses during mouse postnatal development. Dev. Neurobiol. 78, 618–626. doi: 10.1002/dneu.22568
Wendt, S., Maricos, M., Vana, N., Meyer, N., Guneykaya, D., Semtner, M., et al. (2017). Changes in phagocytosis and potassium channel activity in microglia of 5xFAD mice indicate alterations in purinergic signaling in a mouse model of Alzheimer’s disease. Neurobiol. Aging 58, 41–53. doi: 10.1016/j.neurobiolaging.2017.05.027
Wolf, S. A., Boddeke, H. W. G. M., and Kettenmann, H. (2017). Microglia in physiology and disease. Annu. Rev. Physiol. 79, 619–643. doi: 10.1146/annurev-physiol-022516-034406
Wyss-Coray, T., Yan, F., Lin, A. H.-T., Lambris, J. D., Alexander, J. J., Quigg, R. J., et al. (2002). Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc. Natl. Acad. Sci. U.S.A. 99, 10837–10842. doi: 10.1073/pnas.162350199
Yang, J.-T., Wang, Z.-J., Cai, H.-Y., Yuan, L., Hu, M.-M., Wu, M.-N., et al. (2018). Sex differences in neuropathology and cognitive behavior in APP/PS1/tau triple-transgenic mouse model of Alzheimer’s disease. Neurosci. Bull. 34, 736–746. doi: 10.1007/s12264-018-0268-9
Yanguas-Casás, N., Crespo-Castrillo, A., de Ceballos, M. L., Chowen, J. A., Azcoitia, I., Arevalo, M. A., et al. (2018). Sex differences in the phagocytic and migratory activity of microglia and their impairment by palmitic acid. Glia 66, 522–537. doi: 10.1002/glia.23263
Zhang, W., Yan, Z., Gao, J., Sun, L., Huang, X., Liu, Z., et al. (2014). Role and mechanism of microglial activation in iron-induced selective and progressive dopaminergic neurodegeneration. Mol. Neurobiol. 49, 1153–1165. doi: 10.1007/s12035-013-8586-4
Zhao, W., Dumanis, S. B., Tamboli, I. Y., Rodriguez, G. A., Jo LaDu, M., Moussa, C. E. H., et al. (2014). Human APOE genotype affects intraneuronal A 1-42 accumulation in a lentiviral gene transfer model. Hum. Mol. Genet. 23, 1365–1375. doi: 10.1093/hmg/ddt525
Zhao, Y., Wu, X., An, Z., Huang, T. Y., Xu, H., Li, X., et al. (2018). TREM2 Is a receptor for β-amyloid that mediates microglial function. Neuron 97, 1023–1031.e7. doi: 10.1016/j.neuron.2018.01.031
Zhong, L., Wang, Z., Wang, D., Wang, Z., Martens, Y. A., Wu, L., et al. (2018). Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 13:15. doi: 10.1016/j.neuron.2016.05.003
Zhou, C.-N., Chao, F.-L., Zhang, Y., Jiang, L., Zhang, L., Luo, Y.-M., et al. (2018). Sex differences in the white matter and myelinated fibers of APP/PS1 mice and the effects of running exercise on the sex differences of AD mice. Front. Aging Neurosci. 10:243. doi: 10.3389/fnagi.2018.00243
Keywords: Alzheimer disease, microglia, neuroinflammation, microglia diversity, purinergic signaling, sexual dimorphism, early stage, hiPSCs
Citation: Hemonnot A-L, Hua J, Ulmann L and Hirbec H (2019) Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 11:233. doi: 10.3389/fnagi.2019.00233
Received: 27 May 2019; Accepted: 14 August 2019;
Published: 30 August 2019.
Edited by:
Patrizia Giannoni, University of Nîmes, FranceReviewed by:
Anna Maria Colangelo, University of Milano-Bicocca, ItalyJohannes Boltze, University of Warwick, United Kingdom
Copyright © 2019 Hemonnot, Hua, Ulmann and Hirbec. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hélène Hirbec, helene.hirbec@igf.cnrs.fr
†These authors have contributed equally to this work as co-first authors
‡These authors have contributed equally to this work as co-last authors