Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity
 James Gilbert1
James Gilbert1  Heng-Ye Man1,2*
Heng-Ye Man1,2*- 1Department of Biology, Boston University, Boston, MA, United States
- 2Department of Pharmacology & Experimental Therapeutics, Boston University School of Medicine, Boston, MA, United States
Autism spectrum disorder (ASD) is a set of neurodevelopmental disorders with a high prevalence and impact on society. ASDs are characterized by deficits in both social behavior and cognitive function. There is a strong genetic basis underlying ASDs that is highly heterogeneous; however, multiple studies have highlighted the involvement of key processes, including neurogenesis, neurite growth, synaptogenesis and synaptic plasticity in the pathophysiology of neurodevelopmental disorders. In this review article, we focus on the major genes and signaling pathways implicated in ASD and discuss the cellular, molecular and functional studies that have shed light on common dysregulated pathways using in vitro, in vivo and human evidence.
Highlights
• Autism spectrum disorder (ASD) has a prevalence of 1 in 68 children in the United States.
• ASDs are highly heterogeneous in their genetic basis.
• ASDs share common features at the cellular and molecular levels in the brain.
• Most ASD genes are implicated in neurogenesis, structural maturation, synaptogenesis and function.
Introduction
Autism spectrum disorder (ASD) comprises a heterogeneous class of neurodevelopmental disorders characterized by impaired social interactions, restrictive interests and repetitive behaviors (Landa, 2008). ASD typically presents with other mental and physical disabilities such as anxiety, attention-deficit/hyperactivity disorder (ADHD), intellectual disability (ID), epilepsy and impairments in motor coordination. In up to 25% of individuals diagnosed with ASD, an identifiable or genetic variant can be identified, providing valuable insights into the mechanisms involved in proper neurodevelopment (Huguet et al., 2013). A large number of ASD-linked genes are also associated with broad processes such as metabolism, chromatin remodeling, mRNA regulation, protein synthesis and synaptic function.
The human brain contains about 86 billion neurons making trillions of connections (Azevedo et al., 2009). Most neurons are produced in the ventricular zone (VZ) and migrate radially out into the developing neocortex, and it is estimated that about 75% of rodent, and up to 90% of human neurons use glial-guided migration (Letinic et al., 2002). The movement of neurons along migratory routes is guided by a number of guidance molecules that direct their movement into the cortex and the formation of an organized 6-layered structure (Marín and Rubenstein, 2001; Huang, 2009; Valiente and Marin, 2010).
After neuronal migration, neurons must undergo extensive morphological changes. Long axonal processes are extended and are required to connect to target neurons with precision, while complex dendritic arbors must grow and branch to occupy specified dendritic field volumes. These processes take place during prenatal and early postnatal periods and lay the foundations for neuronal connectivity within and across brain regions. Ultimately, with activity-dependent structural remodeling, neurons form synaptic connections and incorporate into functional neuronal networks for proper brain function. It’s of no surprise that disruptions in any of these intricate processes would cause abnormalities in brain development and function, leading to neurodevelopmental disorders including ASD.
While ASD shares characteristic features at the behavioral level, its underlying causes are highly heterogeneous. Developmental dysregulation in ASD may affect processes ranging from progenitor cell proliferation and neuronal differentiation to neuron migration, axon guidance, dendrite outgrowth, synaptogenesis, synaptic function and neural circuitry.
Studies of ASD-related brain pathologies indicate that abnormal acceleration of brain growth in early childhood (Wegiel et al., 2010) accompanied with impaired neuron morphological development and brain cytoarchitecture are common features in ASDs (Bauman and Kemper, 1985; Bailey et al., 1998; van Kooten et al., 2008). Additionally, impairments in synapse formation and synaptic plasticity (Bourgeron, 2007, 2009), which ultimately lead to functional and cognitive impairments, are fundamental causative factors underlying ASD pathology.
Analysis of the Simmons Foundation Autism Research Initiative (SFARI) gene database shows that ASD causative genes display vast diversity involving up to one thousand genes (Banerjee-Basu and Packer, 2010; Abrahams et al., 2013). Additionally, a large number of rare genetic variants in protein-coding genes are causative for ASD, none of which individually account for more than 1% of the total number of ASD diagnoses. Therefore, the complexity and heterogeneity of autism genetics is a major challenge when investigating the underlying neurobiological pathways that are shared within ASD (Happe et al., 2006).
The ability to classify ASD patients according to genetics has been enhanced by the substantial amount of work done to understand ASD-linked genes, and the role of their encoded genes, in the underlying neuropathologies. For example, well known ASD-linked genes, such as neurexin, neuroligin and Shank have been well characterized for their roles in synaptic formation and function (Toro et al., 2010). Additionally, co-expression network analyses of ASD-linked genes have identified that early developmental periods when neurogenesis and synaptogenesis occur are commonly disrupted processes in ASD (Parikshak et al., 2013). Genes involved with regulating developmental processes such as Chromodomain Helicase DNA-binding protein 8 (CHD8), T-Box Brain Protein 1 (TBR1), and Fragile X Mental Retardation 1 (FMR1), have also been linked to ASD (Parikshak et al., 2013; Willsey et al., 2013). These findings suggest that multiple processes during prenatal and early postnatal brain growth are linked to ASD, underscoring the vast heterogeneity of causative factors leading to ASD pathogenesis. ASD-risk genes, their associated disorders and the neurodevelopmental processes they affect are shown in Table 1.
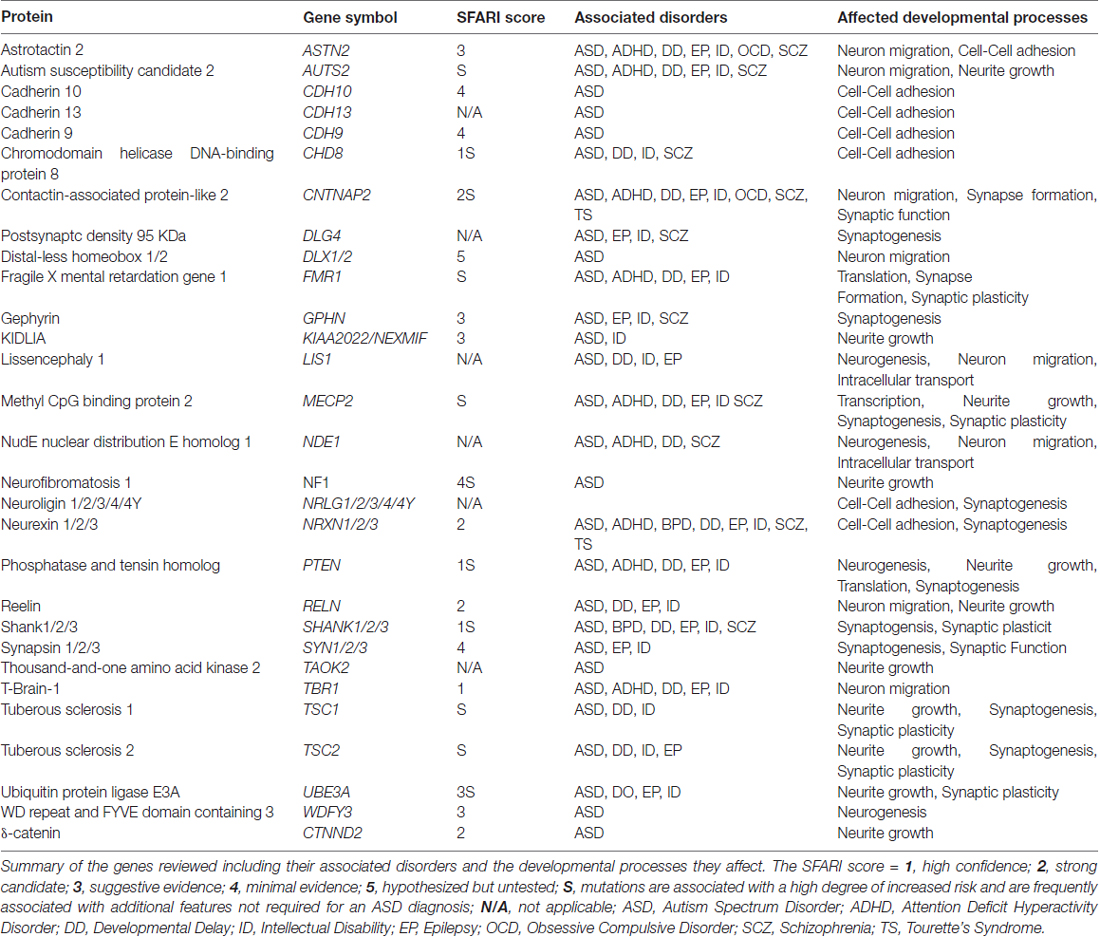
Table 1. Autism spectrum disorder (ASD) genes and associated disorders.
Alterations in Neurogenesis and Neuron Migration in ASD
Increased Neuronal Proliferation and Macrocephaly in ASD
A growing body of literature has provided strong evidence that subsets of individuals with ASD show aberrant brain growth patterns. In patients, cerebral size may be normal at birth but display a period of increased overgrowth and a subsequent period of decline compared to unaffected control patients. An increase in neuronal numbers in prefrontal cortex has recently been observed, indicating that excess neurogenesis may be the underlying cause for an increase in cerebral size in ASD (Courchesne et al., 2011). Additionally, children diagnosed with ASD sometimes show regions of abnormal laminar positioning of cortical projection neurons (Wegiel et al., 2010). Both the development of proper lamination and the number of cortical neurons rely on temporally controlled proliferation of neural progenitors, therefore defects in neural progenitors may underlie ASD subtypes associated with overgrowth in the developing brain.
Projection neurons comprise roughly 80% of neurons in the cortex and when taking into account their dendrites, axons, myelin and synapses, they indeed could contribute to an increase in both the gray and white matter volumes in adolescents with ASD. Indeed, support for this theory has recently been shown with the comparison of neuron numbers in postmortem tissue from ASD and normal adolescents, with a surprising 67% more neurons detected in the prefrontal cortex of ASD patients (Courchesne et al., 2011). An aberrant increase in neuron numbers during prenatal neurodevelopment supports a role for a disruption of neurogenesis, which occurs mostly during gestational weeks 7–20 in humans, which coincides with the end of the embryonic period and early fetal period. Because new cell production is estimated to outnumber cell elimination by at least 100-fold in the developing brain, the role of cell death may play less of a role in macrocephaly (Rakic and Zecevic, 2000). It is important to note that programmed cell death can eliminate up to 50% of particular neuron subtypes (De Zio et al., 2005; Yamaguchi and Miura, 2015), suggesting that a shift in the balance between neuron generation and elimination could play a role in macrocephaly.
Radial glia cells must undergo a regulated series of symmetric divisions to generate more radial glia, and asymmetric divisions, to produce intermediate progenitors and post-mitotic neurons. Recently born projection neurons, principally produced via symmetric divisions of intermediate progenitor cells in the sub-ventricular zone (SVZ), will migrate in an “inside-out” fashion to establish the six layered neocortex. This mode of migration forms deeper layer neurons first, followed by later born neurons that migrate to form the outer cortical layers (Gupta et al., 2002; Nadarajah and Parnavelas, 2002; Kriegstein and Noctor, 2004). Thus, alterations in neurogenesis could lead to changes in total number neuron numbers yielded from the progenitor populations, and/or in the overall laminar structure. For example, a study identified abnormalities in lamination of the neocortex and excess neuron numbers in seven out of eight ASD cases that they examined, via magnetic resonance imaging (MRI) and postmortem histology (Hutsler et al., 2007). Cerebral dysplasia in multiple regions has been reported in ASD cases, indicative of dysregulated neurogenesis, neuronal migration and/or maturation (Wegiel et al., 2010). Additionally, Stoner et al. (2014) identified regions of disorganized cortical lamination in ten out of the eleven ASD cases they examined. These studies have provided strong evidence to support the theory of dysregulated proliferation from neural progenitors as an underlying pathology associated with macrocephaly and cortical lamination defects in ASD. Although astrocytes are generated from the same progenitor pool as neurons, the underlying cause of macrocephaly may be largely due to increased numbers of cortical projection neurons, as an associated increase in glia cells is not observed (Courchesne et al., 2011; Morgan et al., 2014). Additionally, data suggests that the cerebral white matter, which contains glia and the myelinated projections from cortical neurons, is not increased in ASD patients with macrocephaly; however, reports do indicate delayed maturation and a compromised integrity of the white matter (Hazlett et al., 2005; Friedman et al., 2006; Bakhtiari et al., 2012). These findings suggest that a preferential up-regulation in neuronal differentiation and proliferation play a major role in ASD.
The phosphatase and tensin homolog (PTEN) gene was the first gene to be clearly associated with macrocephaly in ASD (Goffin et al., 2001; Butler et al., 2005; Buxbaum et al., 2007; Varga et al., 2009). Pten was originally identified as a tumor suppressor and key negative regulator of phosphatidylinositol 3-kinase (PI3K) signaling, with Pten mutations found in multiple cancers (Zhao et al., 2004). In Pten-deficient mouse models, enhanced levels of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) lead to activation of protein kinase B (AKT) and downstream mammalian target of rapamycin (mTOR). The mTOR pathway is well known for its ability to regulate cell growth and proliferation and consistently Pten-deficient animals show neuronal over-growth, brain enlargement, seizures and premature death (Kwon et al., 2006; Ogawa et al., 2007; Garcia-Junco-Clemente and Golshani, 2014).
CHD8 has emerged as a key ASD-linked gene. Strikingly, 80% of individuals with mutations in CHD8 alleles display macrocephaly, which composes a much higher percentage of total macrocephaly cases in ASD patients without CHD8 mutations (Bernier et al., 2014). Using a mouse model, Katayama et al. (2016) have shown increased brain weight, mirroring the macrocephaly observed in humans. Using transcriptome analysis of the entire brain, the authors concluded that major targets of Chd8 were genes regulated by the RE-1 silencing transcription factor (REST), which is a neuronal transcriptional repressor (Katayama et al., 2016).
WD repeat and FYVE domain containing 3 (WDFY3) was identified through surveys of de novo variants linked to ASD (Iossifov et al., 2012, 2014), which is implicated in macrocephaly and altered neural progenitor proliferation. Wdfy3 is a scaffold protein, involved in macroautophagy of large aggregation-prone proteins (Filimonenko et al., 2010). Decreased Wdfy3 expression in mice produces pronounced effects on neuronal proliferation and migration. Wdfy3 mutant mice also display macrocephaly resulting from a shift of radial glia divisions from asymmetric to symmetric (Orosco et al., 2014). This change in division ultimately produces greater numbers of neuronal progenitors and therefore brain size. Wdfy3’s function in regulating cellular division is unclear, however expression studies have shown that it is up-regulated during cellular division and Wdfy3 plays a role in autophagy and regulation of proteins that control the cell cycle, ultimately resulting in a shortened cell cycle in Wdfy3 mutant mice (Orosco et al., 2014), and that additionally, because progenitor expansion and neurogenesis initiates anterolaterally and concludes posteromedially (Caviness et al., 2009), Wdfy3 mutant mice show a more pronounced affect in the anterolateral areas. This finding is in line with MRI performed in ASD adolescents, wherein temporal and frontal cortical areas showed the largest size increases (Carper et al., 2002; Hazlett et al., 2005; Schumann et al., 2010). Notably, these region-specific changes of cerebral growth could be associated with key behavioral symptoms observed in ASD. In humans, areas such as the superior temporal sulcus and parts of the prefrontal and temporal cortex, which are key regions involved in reward and reinforcement pathways as well as social and emotional pathways are susceptible in ASD patients (Pelphrey and Carter, 2008; Redcay, 2008; Gotts et al., 2012; Gasquoine, 2014).
The association of macrocephaly with clinical phenotypes in autism has been characterized in an inconsistent manner and previous studies have indicated higher levels of cognitive function in patients with macrocephaly and ASD compared to normal controls (Courchesne and Pierce, 2005; Sacco et al., 2007). An increase in head circumference has been shown in ASD patients with special capabilities, compared to those without them (Ben-Itzchak et al., 2013a). However, additional studies have not discovered similar correlations with increased head circumferences and special abilities (Gillberg and de Souza, 2002; Ben-Itzchak and Zachor, 2007).
Genes Associated with Mitotic Dysregulation of Neural Progenitors and Microcephaly in ASD
Microcephaly has not been studied as thoroughly as macrocephaly in ASD patients. The reports have indicated an increased prevalence of microcephaly in ASD, with up to 20% of cases, in comparison to 3% reported in the general population. Additionally, microcephaly is more frequent in individuals with ID and higher ASD severity (Fombonne et al., 1999; Cody et al., 2002; Miles et al., 2005; Ben-Itzchak et al., 2013b).
Autosomal recessive primary microcephaly (MCPH) is a condition that displays with significantly reduced head circumference that develops during the prenatal period (Tunca et al., 2006). The development of the forebrain is prominently affected in this form of microcephaly ultimately results in ID (Roberts et al., 2002; Bond et al., 2003). Disruption in genes encoding proteins that localize to the centrosome are known to result in MCPH (Kaindl et al., 2010), including Microcephalin 1 (MCPH1). MCPH1 is a gene expressed during fetal development and mutations in MCPH1 produce microcephaly (Jackson et al., 1998, 2002). Studies have found rare variants in the MCPH1 gene that are linked to ASD (Ozgen et al., 2009; Neale et al., 2012), and play a role in DNA-damage repair, chromosome condensation, and the regulation of DNA damage genes (Thornton and Woods, 2009; Richards et al., 2010). Mechanistically, it has been shown that Mcph1 regulates neuroprogenitor division by coupling centrosomal maturation and mitotic spindle orientation with mitotic entry (Gruber et al., 2011).
Abnormal Spindle Primary Microcephaly (ASPM, MCPH5) is involved in orientation of the mitotic spindle, regulation of mitosis and cytokinesis (Fish et al., 2006; Passemard et al., 2009, 2016; Higgins et al., 2010). Mutant Aspm mice have mild microcephaly without aberrant increase in cell death, suggesting that disruptions in the proliferation of embryonic neuronal progenitor cells underlie MCPH (Pulvers et al., 2010). Additionally, Aspm positively regulates Wnt signaling (Major et al., 2008), and over-expression of β-catenin, a positive transducer of the Wnt pathway, can rescue neurogenesis deficits in mice (Buchman et al., 2011).
Mutations in WD Repeat Domain 62 (WDR62) are associated with microcephaly and other cortical abnormalities in humans (Bilgüvar et al., 2010; Yu et al., 2010). Wdr62 deficient mice have reduced brain size due to decreased neural progenitor cell population cells show mitotic spindle instability, mitotic arrest and cell death after loss of Wdr62 expression (Chen et al., 2014). Mutations or loss of Wdr62 expression therefore leads to mitotic delay and death of neural progenitor cells, thereby resulting in microcephaly.
Alterations in Neuron Migration in ASD
Excitatory Projection Neuron Migration
The time of birth of neocortical neurons, as well as their proper movement from the proliferative zone, dictates their final position within the layers of the cerebral cortex (Angevine and Sidman, 1961). Neurons born later during neurogenesis will ultimately reach the superficial layers above earlier born neurons, due to an “inside-out” mode of neuronal migration. Initially, multipolar neurons will migrate via cellular locomotion. As neurons continue into the neocortex, they adhere to and migrate along radial glia, which provide aid in the direction of their migration (Rakic, 1972; Hatten, 1999; Kriegstein and Noctor, 2004; Ayala et al., 2007). Neurons adopt a bipolar morphology as they move along radial glia and detach upon reaching their proper laminar position (Figure 1). Additionally, daughter neurons in different cortical layers that were generated from the same mother progenitor cell will align radially into highly connected mini-columnar structures, most likely constituting a functional unit in the brain (Gao et al., 2013).
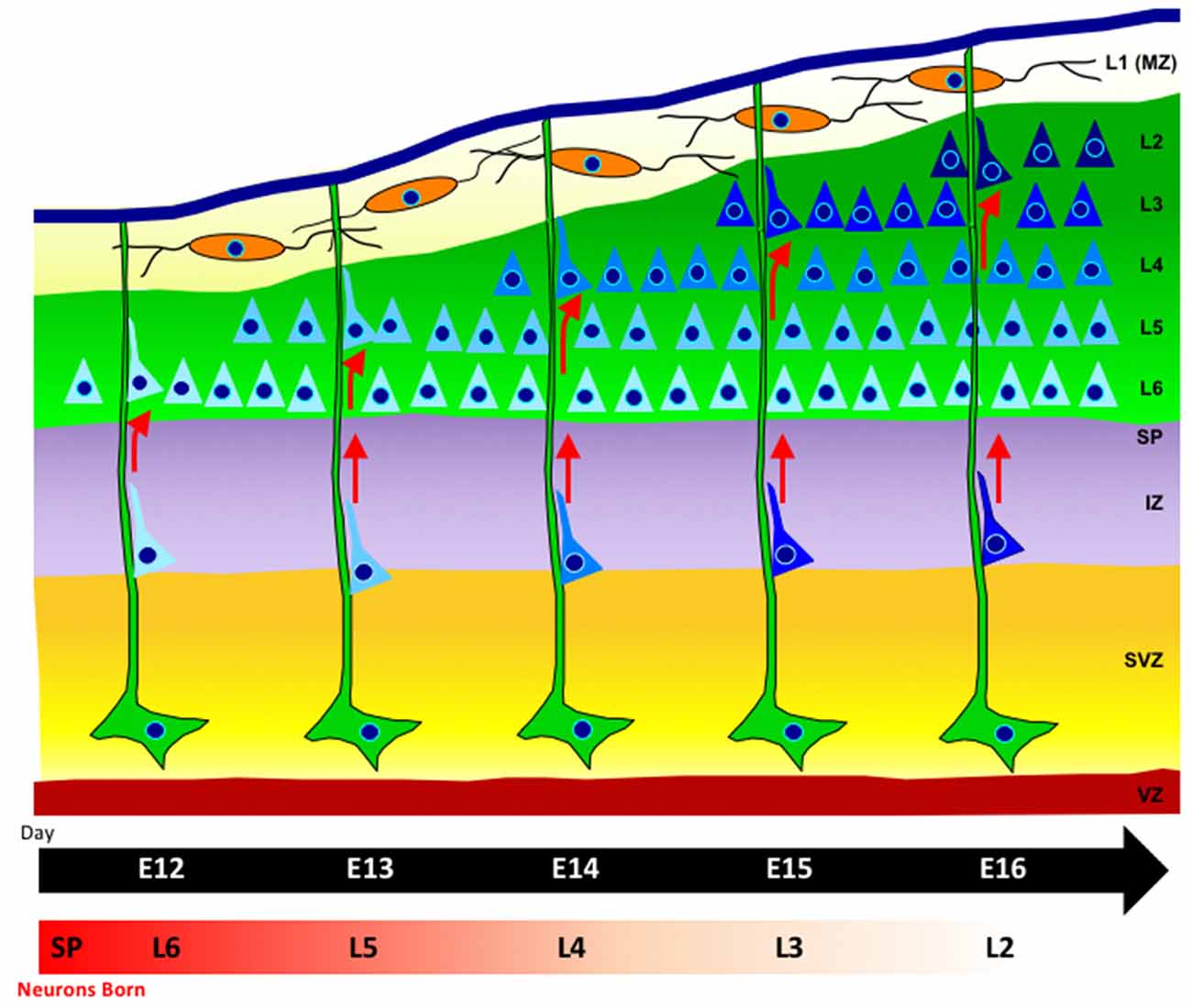
Figure 1. Radial-glia guided neuronal migration. Radial glia cells (green), extend long basal process to the pial surface, with their somas located in the ventricular zone (VZ). Neurons (blue) are born in the VZ and migrate along radial glia fibers. The cortical plate is formed in an inside-out fashion such that later-born neurons that will reside in the upper layers pass through earlier-born neurons in deeper layers (lighter blue shading). The marginal zone contains horizontally-oriented Cajal-Retzius cells (orange) which release the extracellular signaling glycoprotein Reelin.
Studies have shown that defects in neuronal migration are associated with changes in neuron density and soma size, irregular minicolumns and heterotopias (mis-localized neurons; DiCicco-Bloom et al., 2006; Uppal et al., 2014; Chen et al., 2015; Reiner et al., 2016). In humans for example, two separate studies have demonstrated evidence of aberrant cell patterning in the boundary of cortical gray–white matter, suggesting defects in neuronal migration (Hutsler et al., 2007; Avino and Hutsler, 2010). In another study, an abnormal lamination of neurons, but not glia, was detected in the cortex of adolescent ASD brain samples (Stoner et al., 2014).
Reelin is one of the best known regulators involved in neuronal migration (Folsom and Fatemi, 2013; Sekine et al., 2014). Reelin is a glycoprotein that is released from Cajal–Retzius cells in the outer marginal zone. Reelin binds to the receptors Apoer2 and Vldlr on the cell membrane of target neurons, thereby inducing tyrosine phosphorylation of the adaptor protein Dab1, which subsequently initiates signaling cascades (Forster et al., 2006; Pardo and Eberhart, 2007). Reelin is involved in the termination of migration, proper neuronal layering, as well as transition to a bipolar morphology prior to neuronal migration along radial glia. Studies using postmortem human samples suggest aberrant Reelin signaling in ASD patients (Persico et al., 2001; Bonora et al., 2003; Fatemi et al., 2005). Reelin mutations in humans produce disruptions in neuron migration and connectivity, lissencephaly and cerebellar hypoplasia (Hong et al., 2000). In the well-studied Reelin knockout (KO) mice, neuronal migration abnormalities result in an inversion of cortical layering (Falconer, 1951; D’Arcangelo et al., 1995; Hirotsune et al., 1995). Behavioral studies in Reelin KO mice show aggressive behavior, an abnormal gait, social aggression, and impairments in learning and memory (Salinger et al., 2003). T-Box Brain Protein 1 (TBR1) encodes a brain-specific T-box transcription factor, which plays an important role in neurodevelopment and migration and has been identified as a causative gene for ASD (Neale et al., 2012; Deriziotis et al., 2014; De Rubeis et al., 2014). Tbr1 is involved in the differentiation of neurons from intermediate progenitors early in the development of the neocortex (Dwyer and O’Leary, 2001; Han et al., 2011; Willsey et al., 2013). Tbr1 regulates the expression of many ASD-linked genes (Chuang et al., 2015), including the activation of Autism Susceptibility Candidate 2 (AUTS2), which has been identified as an ASD and ID associated gene (Bedogni et al., 2010; Srinivasan et al., 2012). Interestingly, TBR1 KO mice do not express subplate, layer 6 or Cajal-Retzius cell markers and show a large decrease in Reelin expression, however upper cortical layers are typically normal (Hevner et al., 2001).
Multiple mutations within AUTS2 have been identified in patients, strongly linking it as a causative factor for ASD (Sultana et al., 2002; Huang et al., 2010; Pinto et al., 2010; Talkowski et al., 2012; Cheng et al., 2013; Liu et al., 2015). The Auts2 protein is predominantly nuclear localized and evidence suggests a role for Auts2 in regulating gene expression during brain development (Bedogni et al., 2010; Srinivasan et al., 2012; Gao et al., 2014). Binding of Auts2 with polycomb repressive complex 1 (PRC1) inhibits PRC1 activity leading to activation of gene transcription (Gao et al., 2014). In addition to the nuclear component, Auts2 protein is also found in the cytoplasm, where it lays a role in regulating cortical neuronal migration and neurite growth (Hori et al., 2014).
In a cohort of Amish children, mutations within contactin-associated protein-like 2 gene (CNTNAP2) were found to be implicated in ASD and epilepsy (Strauss et al., 2006). CNTNAP2 is a scaffolding protein that’s part of the Neurexin family, whose members have previously been associated with multiple ASD-linked proteins (Jamain et al., 2003; Comoletti et al., 2004). Ectopic neurons were identified in patients with CNTNAP2 mutations, suggesting its involvement in the regulation of neuronal migration (Strauss et al., 2006). Additionally, CNTNAP2 mutations are linked to ADHD as well as epilepsy and seizures, conditions that are commonly co-morbid with ASD (Jackman et al., 2009; Elia et al., 2010; Mefford et al., 2010). Additionally, disruptions in CNTNAP2 are associated with impairments in sociability and language processing (Whalley et al., 2011; Toma et al., 2013; Condro and White, 2014). CNTNAP2 KO mice show an abnormal localization of neurons in the corpus callosum and mis-localization of the Cux1-positive upper layer neurons into the deeper layers, V–VI (Peñagarikano et al., 2011). CNTNAP2 is part of a neuron-glia adhesion complex with contactin 2, therefore it may play a key role in radial glia-guided neuronal migration (Poliak et al., 1999; Denaxa et al., 2001).
Astrotactin 1 (ASTN1) is a neuronal cell surface antigen that regulates neuron-glia interactions that plays a major role in neuron migration (Edmondson et al., 1988; Zheng et al., 1996). Astn1 KO mice have slower cerebellar granule cell migration, aberrant Purkinje cell morphology, decreased glia-neuron interactions and impairments in coordination compared to normal mice (Adams et al., 2002). Astn1 interacts with Astn2 and can regulate its expression at membrane surface, which ultimately regulates neuron-glia adhesion during migration along radial glia (Wilson et al., 2010). Genome wide association studies identified ASTN2 as an ASD candidate gene (Lesch et al., 2008). Patients with ASTN2 deletions are often classified with ASD or other co-morbid diagnoses such as ADHD, obsessive compulsive disorder and delayed language development (Lionel et al., 2014).
NudE nuclear distribution E homolog (NDE1) is involved with the regulation of neuron proliferation, migration, and intracellular transport as part of the Lis1/Nde/Ndel1/cytoplasmic dynein complex (Feng et al., 2000; Kitagawa et al., 2000; Niethammer et al., 2000; Sasaki et al., 2000; McKenney et al., 2010). Lissencephaly 1 (LIS1) was the first gene identified with an involvement in disrupted neuron migration (Reiner et al., 1993) via its interactions with cytoplasmic dynein, Nde1, Ndel1 and cytoplasmic linker protein 170 (CLIP-170; Reiner, 2000; Coquelle et al., 2002). Multiple studies have shown a major role for Lis1 in regulating neuronal migration (Cahana et al., 2001; Tsai et al., 2007; Hippenmeyer et al., 2010) and a cross between an Nde1 KO mouse with Lis1 heterozygous mouse produced a severe disruption of the morphology of the VZ progenitors and radial glia, as well as a significant decrease in brain size (Pawlisz et al., 2008). This was shown to be regulated via stabilization of the dystrophin/dystroglycan glycoprotein complex (Pawlisz and Feng, 2011) and an alteration of the MAPK scaffold protein Kinase Suppressor of Ras (KSR), subsequently producing hyperactivation of MAPK/ERK pathway (Lanctot et al., 2013). In support of these findings, additional studies have shown that dysregulation of the MAPK/ERK pathway affects social behavior in mice and produces ASD phenotypes, while multiple links between ASD and hyperactivation of the Ras signaling have been reported (Levitt and Campbell, 2009; Rauen et al., 2010; Crepel et al., 2011; Faridar et al., 2014).
Inhibitory Neuron Migration
Interneurons, which are primarily inhibitory, utilize tangential migration to migrate into the neocortex and across the plane of radial glia fibers (Lavdas et al., 1999; Marín and Rubenstein, 2001; Nery et al., 2002; Kriegstein and Noctor, 2004; Ayala et al., 2007). Upon reaching the cerebral cortex, interneurons will migrate along radial glia fibers to their final location (de Carlos et al., 1996; Wichterle et al., 2001; Polleux et al., 2002; Poluch and Juliano, 2007). Therefore, when radial glia-mediated migration becomes disrupted, interneurons will also be become mis-localized.
Distal-less homeobox (Dlx) genes are part of the homeodomain transcription factor family which is related to the Drosophila distal-less (Dll) gene (Panganiban and Rubenstein, 2002). Dlx1/2 are found largely in the ganglionic eminences (GE) of the developing brain and play an important role in regulating the migration of inhibitory interneurons from the medial GE into the cortex. Dlx1 KO mice exhibit a decrease in GABAergic neurons and present with epilepsy, a commonly observed pathology in ASD patients (Cobos et al., 2005). Dlx1/Dlx2 double KO mice show major aberrations in the migratory stream of GABAergic neurons and also an accumulation of neuronal precursor cells in the GE (Anderson et al., 1997; Ghanem et al., 2007). Arx, an X-linked homeobox gene and immediate downstream target of Dlx, may regulate Dlx’s role in tangential neuronal migration (Colasante et al., 2008). Patients identified with mutations in Arx display ID, autistic features and epilepsy (Strømme et al., 2002; Turner et al., 2002).
Alterations in Neurite Growth and Spine Formation in ASD
Neurons are highly specialized cells with distinct morphologies comprised of three distinct sections: the soma which contains the nucleus and the majority of the cellular organelles; a long axonal process to transmit information; and a complex dendritic arbor to receive information from neighboring neurons. The dendrites are highly branched and elaborate and therefore occupy a large area within neural tissues (Tahirovic and Bradke, 2009).
Dendrite growth can be viewed as discreet steps. After birth from neural progenitors, neurons start as a simple round soma and must first undergo cellular polarization. Neurons first adopt a multipolar morphology with the extension of minor neurites. The movement of migrating neurons is achieved through the formation, maintenance and constant transformation of microtubules in response to extracellular cues and intracellular polarity signals. During migration, the cell first extends a leading process. Stabilization of a single neurite is required for newly generated neurons to exit the multipolar stage to enter the cortical plate. This stabilization ultimately results in formation of the leading process while the trailing process eventually develops into the future axon (Shim et al., 2008; Witte and Bradke, 2008). Next, in order to move forward, the nucleus undergoes a translocation into the stabilized leading neurite. During this process, termed nucleokinesis, the attachment of microtubules from the centrosome to the nuclear envelope exerts a traction force, pulling the nucleus into the leading neurite attached to a radial glia process (Bellion et al., 2005; Tahirovic and Bradke, 2009).
Once neurons reached their destined cortical layers, substantial dendritic outgrowth is undertaken to form dendritic arbors characteristic of a neuron subtype. At this stage, neurons are tasked with the processes of growing each branch to the correct size, initiating a new branch at the right site to complete a specific branching pattern, and directing each branch’s growth into an appropriate spatial location (Tahirovic and Bradke, 2009; Jan and Jan, 2010). The early postnatal stages of neurite growth are depicted in Figure 2.
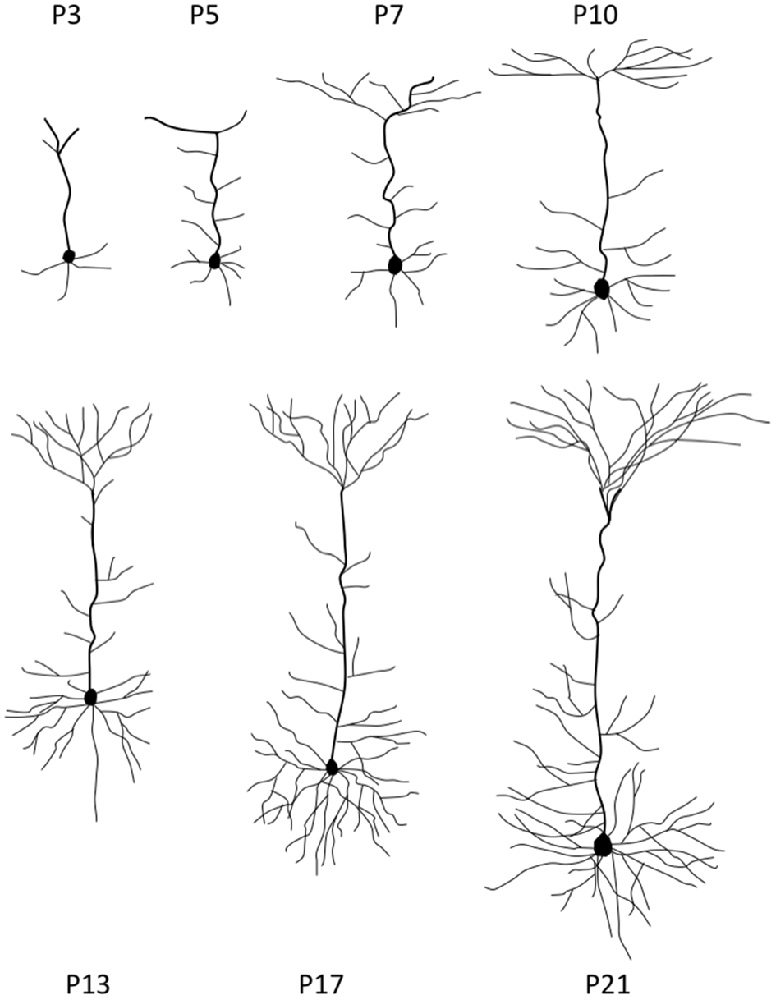
Figure 2. Stages of neurite growth. Representative neocortical mouse pyramidal neuron morphologies at different early postnatal time points during development. P = postnatal.
Dendrites are vastly different from axons in their ultimate morphology, function and developmental processes. In mammalian neurons, a notable distinction is the manner in which the microtubules are organized. Initially all neurites contain microtubules oriented with their plus-end localized distally from the soma. The neurite specified to become the axon will maintain this distal plus-end orientation, while neurites that become dendrites will adopt a mixed orientation (Baas et al., 1988, 1989; Burton, 1988). Additionally, in comparison to axons, dendrites have an enrichment of cellular organelles that are transported with the aid of microtubule motors like dynein, such as Golgi outposts and mitochondria, which are utilized to supply the necessary for cellular materials required for growth (Horton and Ehlers, 2004; Horton et al., 2005; Kapitein et al., 2010).
The morphology of the dendritic arbor is largely developed early during the embryonic period; however, dendrites are highly dynamic and they maintain the overall morphology with various mechanisms into adulthood. Disruptions in dendritic growth, or breakdown of the mechanisms to maintain their morphology can be deleterious, resulting in aberrant network function. Abnormalities in neuronal connectivity between the higher-order association areas have been considered one of the major defects in ASD (Geschwind and Levitt, 2007). MRI studies have shown both large and small changes in connectivity of neuronal networks; however, the neurobiological basis for this disconnectivity remains to be fully elucidated (Casanova and Casanova, 2014; Maximo et al., 2014). A large number of autism studies have focused on dendritic development, although mostly relating to spine morphology and synaptic function, which will be discussed in detail below (Persico and Bourgeron, 2006; Kelleher and Bear, 2008; Bourgeron, 2009). However, the current body of research suggests that this is not the only morphological defect, as children with autism are frequently identified with large-scale anatomical abnormalities, suggesting dysregulation in dendritic growth and development. For example, both macrocephaly and microcephaly are identified in ASD adolescents, with 15% of ASD patients presenting with macrocephaly, while 20% display microcephaly (Lainhart et al., 1997; Fombonne et al., 1999; Cody et al., 2002; Pardo and Eberhart, 2007).
One of the proposed causes leading to macrocephaly has been suggested to be a result of increased dendrite number and size, which may be a product of excessive dendrite arborization, and/or decreased dendrite pruning (Jan and Jan, 2010). Other factors can play a role in macrocephaly, such as increased numbers of neurons and glia, however the number of dendrites may better explain certain forms of macrocephaly. Brain volumes in infants diagnosed with ASD are typically normal, however they display aberrant overgrowth as development progresses. This later onset of increased brain volume should not result from an increase in neuronal number, as the largest areas of the brain have mostly completed neurogenesis prior to birth, except for areas like the hippocampus where smaller amounts of neurogenesis occur later in life (Ming and Song, 2005; Zhao et al., 2008). The timing of increase brain size can be explained by aberrant dendrite growth and branching, which largely occurs postnatally. In humans, neurons grow and continue to elaborate their dendritic arbors, axons and form new synaptic connections until the age of five, with experience-based remodeling of synapses until 20 years of age (Stiles and Jernigan, 2010; Tau and Peterson, 2010; Pescosolido et al., 2012). Dendrites continue to extensively grow after birth, in an activity-dependent manner. Therefore, a large contribution to the increase in brain size postnatally likely results from increased dendritic growth (Redmond et al., 2002).
Previous genetics studies have described multiple high-risk genes for autism that play roles in diverse functions, including synaptic connectivity and synapse function, dendritic and axonal growth, trafficking, transcription and translation (Volders et al., 2011; de Anda et al., 2012; Miao et al., 2013; Bakos et al., 2015). Recently, estimates have suggested that 88% of the genes that are considered to be high-risk for ASD play a role in early neurodevelopmental functions such as neurogenesis and differentiation of neuroblasts. Importantly, ~80% of these genes are involved in later phases of neurodevelopment and regulate processes involved in neurite growth and synapse formation (Casanova and Casanova, 2014).
Dendrites from an individual neuron can have a thousand or more spines, with each spine forming an excitatory synaptic connection upon maturing. Spines and synapses are produced in excess numbers during development, but synaptic numbers are later fine-tuned through activity-dependent stabilization or elimination (Changeux and Danchin, 1976). Spines are highly dynamic and undergo constant turnover and morphological plasticity with a dependency on both developmental stage and activity.
Dendritic spines are typically classified as thin, stubby and mushroom, with the latter considered more mature. Mushroom and stubby morphologies are more permanent and form strong excitatory connections (Trachtenberg et al., 2002; Kasai et al., 2003). Alternatively, thin spines are highly dynamic, shorter lasting, and form weak synaptic connections or no connection at all. Within the spine head, actin becomes enriched helps play a key role in spine formation and structural dynamics (Chazeau and Giannone, 2016). Previous studies have shown that spine formation maintenance are a major cellular processes affected in ASD (Kelleher and Bear, 2008; Bourgeron, 2009; Phillips and Pozzo-Miller, 2015). Using Golgi staining, postmortem analysis of cortical neurons from ASD brain samples showed an increase in dendritic spine density compared to normal patients (Hutsler and Zhang, 2010). Some of the high-risk autism genes implicated in dendritic growth and branching spine formation and synapse maturation are described below.
Methyl CpG binding protein 2 (MECP2) is an X-linked gene that codes for a protein that functions as a transcriptional repressor and an activator (Chahrour et al., 2008). Mutations in MeCP2 were initially linked to Rett syndrome (RTT), a neurodevelopmental condition that presents with motor and speech impairments, cognitive deficits and autism (Amir et al., 1999). RTT is typically caused by loss-of-function mutations in MeCP2; however, there are rare cases that are also caused by MeCP2 duplications.
Using Golgi staining in brain slices from MeCP2 KO mice, reductions in dendritic growth and branching have previously been reported in both apical and basal arbors of motor cortical neurons (Kishi and Macklis, 2004; Stuss et al., 2012). Additionally, to investigate MeCP2 duplication, mice over-expressing the human MeCP2 gene have shown excessive dendritic branching, indicating that MeCP2 over-expression can induce dendritic overgrowth (Jiang et al., 2013). It is possible that in subtypes of neurons MeCP2 can act as a repressor while acting as an activator in others, or it can change roles at different periods in neuronal development. For instance, MeCP2 can activate genes involved in early dendrite growth and repress genes later during dendritic remodeling. An important aspect to these findings is that MeCP2 overexpression affects the dendrites, having no impact on axon growth, suggesting a defined role for MeCP2 in dendritic development. Recent work has provided evidence for the role MeCP2 plays in dendritic development, showing that MeCP2 also regulates the expression of genes post-transcriptionally. These findings show that MeCP2 regulates microRNA (miRNA) processing via a direct interaction with DiGeorge syndrome critical region 8 (DGCR8), which plays a role in mediating the genesis of miRNAs thought to regulate dendrite morphogenesis (Gregory et al., 2004; Cheng et al., 2014).
RTT animal models also show changes in excitatory hippocampal synapse numbers. The loss of the X-linked RTT protein MeCP2 has also been show to result in abnormal dendritic spine morphology and a decrease in spine density (Zhou et al., 2006; Chapleau et al., 2012; Stuss et al., 2012). Additionally, MeCP2 over-expression produces dendritic overgrowth in mice and these animals show a greater rate of spine turnover, with a bias toward spine removal (Jugloff et al., 2005; Jiang et al., 2013). Taken together, MeCP2, the established factor underlying RTT with autism, has a clear role in dendritic morphogenesis and synapse formation, both of which should play a major function in the cognitive impairments seen in RTT patients.
Fragile X mental retardation gene 1 (FMR1) is the gene underlying the disorder Fragile X syndrome (FXS), which results in ID with 15%–30% of patients also displaying ASD phenotypes (Krawczun et al., 1985; Persico and Bourgeron, 2006; Kelleher and Bear, 2008; Santoro et al., 2012). FXS is usually results from an expansion of a CGG triplet in the 5′-UTR region of the FMR1 gene, however a few missense mutations and deletions have been identified (Santoro et al., 2012). Fragile X mental retardation protein (FMRP), the FMR1 gene product, plays a key role in negatively regulating translation, especially local translation at the synapse (Santoro et al., 2012). In terms of FMRP’s role in dendritic growth, mouse studies have shown somewhat contradictory results. In visual cortex pyramidal neurons of FMR1 KO mice, one study has shown defects in dendritic spines, with no observable changes in dendritic morphology (Irwin et al., 2002). Conversely, multiple other studies have shown that FMRP is critical for dendritic growth and branching. In FMR1 KO mice, visual cortex pyramidal neurons show reduced basal dendrite length and branching (Restivo et al., 2005). Using neural stem cells isolated from FMR1 KO mice, or from postmortem tissues of FXS human fetuses, differentiated neurons showed fewer and less complex neurites with smaller somas (Castrén et al., 2005). An important caveat to these studies was that they involved loss-of-function or deletion of FMRP; however, FXS in humans is rarely caused by deletions or missense mutations in the FMR1 gene, but rather via an expansion of the CGG triplet repeat. This has been addressed in studies using transgenic mice with a FXS knock-in mutation consisting of 120–140 CGG repeats. Indeed, these animals display significantly impaired dendritic morphogenesis in addition to alterations in dendritic spine density and morphology (Berman et al., 2012). The studies from Drosophila have elucidated the mechanistic function of FMRP in dendritic growth and have shown that FMRP is involved with transportation of the mRNA for Ras-related C3 botulinum toxin substrate 1 (Rac1), a GTPase. In agreement with this, Rac1 was found to bind FMRP and affect dendrite arborization, indicating that FMRP’s role in dendrite morphogenesis could in part be through its interaction with Rac1 (Lee et al., 2003). These findings strongly support a role for FMRP in dendritic arbor morphogenesis.
FMRP is localized within dendrites and in addition to abnormal dendritic growth and branching, brains from FXS patients display an immature synaptic phenotype (Rudelli et al., 1985). FXS patients have an increased spine density on both apical and basal dendrites in neocortex and more spines characterized by an immature morphology. Additionally, Golgi studies from human FXS patients reveal a significant increase in long spines with less shorter spines compared to controls in multiple cortical areas (Hinton et al., 1991; Irwin et al., 2001). In FMR1 KO mouse models, spine phenotypes correlate with those observed in humans with FXS as these mice have an increase in longer spines and a corresponding decrease in shorter spines. Additionally, dendritic spines in FXS mice display a more immature morphology with fewer mushroom and stubby spines present (Irwin et al., 2001, 2002; McKinney et al., 2005). In addition, changes in synaptic proteins such as postsynaptic density-95 kDa (PSD-95) have been observed in FXS (Ifrim et al., 2015).
Although FMRP’s role in impaired spine formation and morphology in FXS has not been completely elucidated, FMRP has been identified to interact with multiple proteins that are linked to dendritic and spine regulation. Cytoplasmic FMRP-interacting protein 1 (CYFIP1), a binding partner of FMRP, is a protein that has recently generated interest as its genetic locus is chr15q11.2, a susceptibility area in ASD. When bound to FMRP, CYFIP1 can inhibit translation and regulates actin dynamics. This can therefore regulate the growth and removal, as well as the labiality and morphology of spines (De Rubeis et al., 2013). A down-regulation of CYFIP1 mRNA has been detected in subgroups of FXS patients with ASD (Nowicki et al., 2007). Conversely, over-expression of CYFIP1 results in higher dendritic complexity in vitro, while neurons that are haploinsufficient for CYFIP1 show decreased dendritic complexity and an increase in the relative levels of immature to mature dendritic spines (Pathania et al., 2014).
Pten is a phosphatase that de-phosphorylates PIP3, which serves to inhibit PI3K/AKT/mTOR signaling (Kwon et al., 2006; Ogawa et al., 2007; Garcia-Junco-Clemente and Golshani, 2014). Mice lacking Pten selectively in the central nervous system show an increase in activation of the AKT/mTOR/S6K pathway, an increase in neuron size, macrocephaly, as well as decreased and disorganized dendritic and axonal growth (Kwon et al., 2006). Pten KO during early development, as well as in adult mice disrupts neuron morphogenesis, suggesting that Pten plays an important role in dendritic growth and maintenance in adulthood in addition to its function during early neurodevelopment (Kwon et al., 2006; Chow et al., 2009). Therefore, Pten, a protein with a clear link to autism, has a definitive role in dendrite morphogenesis.
TSC1 and TSC2 are tumor-suppressing genes that have been linked to brain tumors in tuberous sclerosis complex (TSC; Huang and Manning, 2008). However, their involvement in neurodevelopmental disorders including epilepsy, autism and ID has become increasingly studied (Kwiatkowski and Manning, 2005; Persico and Bourgeron, 2006). In pheochromocytoma 12 (PC12) cells, an inducible neuron-like cell line, transfection of TSC1 antisense oligonucleotides was shown to increase neurite outgrowth through RhoA activation, while knockdown of Tsc2 decreased neurite growth (Floricel et al., 2007). In another study, overexpression of Tsc1/Tsc2 was found to impair axon formation. Knockdown of Tsc1/Tsc2 in vitro induced multiple axons, while genetic deletion in vivo in the mouse induced ectopic axons (Choi et al., 2008). There is strong evidence that TSC genes are linked to ASD risk and play a major role in neurite growth, therefore it will be important to further elucidate their mechanistic role in neuron morphogenesis and ASD in future research.
Mammalian target of rapamycin (mTOR is a serine/threonine kinase that regulates cellular growth and is induced by growth factors and environmental cues (Laplante and Sabatini, 2012). Via regulation of the cytoskeleton, mTOR plays an important function in regulating dendritic outgrowth, and proteins known to inhibit mTOR signaling have been associated with aberrant dendrite and spine development in ASD (Thomanetz et al., 2013; Skalecka et al., 2016), including Pten and Tsc 1/2 (Weston et al., 2014). Pten serves to inhibit the PI3K/AKT/mTOR signaling pathway, thereby affecting growth and protein translation, and Pten mutations have been discovered in multiple individuals diagnosed with ASD. Additionally, mice carrying PTEN mutations or genetic deletions show impaired social interactions and increased sensory responses. Loss of Pten produces an increase in dendritic growth, synaptic connectivity and disorganized dendritic and axonal processes (Kwon et al., 2006; Orloff et al., 2013). Conversely, knockdown of Pten in the amygdala decreases spine density, with an increase in mushroom spines and decrease in thin protrusions (Haws et al., 2014). Additionally, it has been previously shown that the TSC pathway plays an important role in regulating synaptic function, and in hippocampal neurons, loss of Tsc 2 expression produces an expansion of neuronal somas as well as spines (Tavazoie et al., 2005). These findings provide strong evidence that mTOR, and its interacting proteins, are involved in regulating both dendritic and spine development in ASD.
Neurofibromatosis is a condition in which tumors grow in the nervous system. Mutations in Neurofibromatosis-1 (NF1) produce neurofibromas and between 30% and 65% of children with NF1 mutations display learning disabilities and display significantly higher rates of autism, suggesting a causal relationship to autism (Rosser and Packer, 2003; Garg et al., 2013). Neurofibromin, the protein product of the NF1 gene, is a GTPase activating protein that negatively regulates the Ras signaling pathway (Costa and Silva, 2003). NF1 conditional KO mice display impaired dendritic morphogenesis and Golgi staining from NF1 KO mouse brains reveals shorter apical dendrites in pyramidal neurons. NF1’s function in dendrite morphogenesis has been shown to act through cAMP/PKA/Rho/ROCK signaling (Brown et al., 2012). RhoA plays a major role in actin dynamics and neurite growth and branching. With the inhibition of RhoA, an increase in the branching of neuronal processes is observed, and with the activation of RhoA, a decrease in the length and complexity of processes is observed (Li et al., 2000; Nakayama et al., 2000; Wong et al., 2000).
Loss of expression of KIAA2022/KIDLIA was previously identified by our group and others as the causative protein for severe ID and autistic behavior in multiple families (Cantagrel et al., 2004, 2009; Ishikawa et al., 2012; Van Maldergem et al., 2013; Charzewska et al., 2015; Kuroda et al., 2015). Clinical examinations of patients with KIDLIA mutations or a loss of expression, display enlarged ventricles, Virchow-Robin spaces, a thin corpus callosum and small cerebellar vermis. Additionally, strabismus has been observed in patients, with some dysmorphic features including a round face during early postnatal periods, febrile seizures, severely impaired or no language, stereotypical hand movements and delayed acquisition of motor milestones (Cantagrel et al., 2004, 2009). Interestingly, a patient with decreased expression of KIDLIA had only mild cognitive deficits with a significant delay in language acquisition as well as repetitive and stereotyped behaviors, indicating that the effects may be gene-dosage dependent (Van Maldergem et al., 2013).
With shRNA-mediated knockdown of KIDLIA in rat hippocampal neurons in culture, significant impairments in dendritic growth and branching are observed (Van Maldergem et al., 2013; Gilbert and Man, 2016). Mechanistically, we’ve recently reported that loss of KIDLIA significantly impairs actin dynamics and produces an aberrant increase in total and membrane-localized N-cadherin. N-cadherin was found to bind much greater levels of δ-catenin, thereby releasing the latter’s inhibition on downstream RhoA (Gilbert et al., 2016). Additionally, microcephaly has been reported in human patients with a loss of KIDLIA expression (Cantagrel et al., 2004; Van Maldergem et al., 2013; Kuroda et al., 2015). These findings suggest that the regulation of neuronal morphogenesis through dendrite growth and synapse formation are major underlying factors contributing to the cognitive impairments observed in patients with genetic deletions or functional mutations in KIDLIA.
Ubiquitin protein ligase E3A (UBE3A) is the causative gene for Angelman syndrome, a developmental disorder characterized by language impairments, ataxia, ID and hyperactivity (Williams et al., 2006). Individuals identified with Angelman syndrome are often co-diagnosed with autism (Steffenburg et al., 1996; Peters et al., 2004). In addition, Ube3A over-expression leads to autism in humans and in animal models (Bucan et al., 2009; Smith et al., 2011; Noor et al., 2015). UBE3A is an imprinted gene that encodes an E3 ubiquitin ligase, that typically expresses from both maternal and paternal alleles in most tissues, but is only expressed from the maternal allele in brain (Mabb et al., 2011). Previous studies have shown that Ube3A is required for normal dendritic morphogenesis in the mouse (Miao et al., 2013) and shRNA-mediated knockdown of Ube3A in vivo impairs dendritic growth cortical pyramidal neurons in the mouse (Miao et al., 2013). Additionally, studies in Drosophila strongly support a role for Ube3A in dendritic growth and branching. Both over-expression and loss of the Ube3A Drosophila homolog have been shown to result in decreased dendrite branching in larval sensory neurons (Lu et al., 2009). These studies suggest that Ube3A plays a significant role in dendrite growth and support that dysregulated neuron morphogenesis may underlie developmental disorders like Angelman syndrome and ASD.
Thousand-and-one amino acid kinase 2 (TAOK2) belongs to the family of MAP kinase kinase kinases (MAPKKK) and is located in chromosome 16, an area associated with increased ASD-risk (Weiss et al., 2008) and schizophrenia (McCarthy et al., 2009). Both Taok1 and Taok2 activate mitogen-activated protein kinase (MAPK) pathways via JNK and p38 which results in regulation of gene transcription (Chen and Cobb, 2001; Chen et al., 2003). Additionally, FMRP regulates Taok2 mRNA (Darnell et al., 2011), the protein underlying FXS, providing an additional link to its involvement in neurodevelopmental disorders. A recent study in vivo has shown that knockdown or overexpression of Taok2 showed opposing effects on basal dendrite development in the neocortex (de Anda et al., 2012). Specifically, primary dendrite numbers were reduced after Taok2 knockdown, while the number of primary dendrite numbers was increased with over-expression. Additionally, this effect was preferential for the basal dendrites on basal dendrites, as Taok2 knockdown produced no changes in apical dendrite morphology on the same neurons. The role of Taok2 on dendrite growth was dependent on interactions with Neuropilin 1, a membrane receptor that binds Semaphorin 3A, which leads to initiation of the JNK cascade (de Anda et al., 2012). Interestingly, this finding shows that Taok2 expression selectively affects specific dendritic areas, i.e., the basal dendrites, providing evidence that specialized molecular pathways are used for the formation of different dendritic areas.
Reelin, as discussed previously for its role in neuron migration, seems to also play a significant function in dendrite arborization in hippocampal and cortical neurons (Niu et al., 2004; Jossin and Goffinet, 2007; MacLaurin et al., 2007; Chameau et al., 2009; Hoe et al., 2009; Matsuki et al., 2010). Reelin, and its downstream signaling pathway through Vldrl/Apoer2-Dab1, serves to promote dendrite development. Reeler mice contain a loss-of-function mutation in RELN, and display decreased dendrite branching in the hippocampus (Niu et al., 2004). Additionally, Reelin has also been shown to affect cortical dendritic growth in vivo (Hoe et al., 2009) and Reelin application to Reelin KO mouse brain slices can promote dendritic growth (Nichols and Olson, 2010). The role of alterations in neurite growth and branching and the associated ASD-linked genes, are described in detail later.
Alterations in Synaptic Components in ASD
Synapses are highly specialized structures required for signal transduction and plasticity within neuronal networks, making up the functional contact sites between neurons. A synapse is composed of the axon terminal, the presynapse, a synaptic cleft which contains adhesion proteins and the extracellular matrix, and the postsynaptic density with receptors on a target neuron’s dendrites. The presynapse contains mitochondria and is characterized by a pool of synaptic vesicles filled with neurotransmitters. Action potentials arriving at the axon terminal mediate calcium influx, which causes the vesicles to fuse with specialized regions of the plasma membrane called active zones to release their content into the synaptic cleft. A precise coupling between the electrical stimulus (action potential) and release of neurotransmitter is crucial for proper signal transmission to the postsynaptic neuron. The postsynapse is characterized by the presence of receptors that bind neurotransmitters released from the presynapse, which initiates signaling cascades that ultimately propagate the electrical signal in postsynaptic neuron. Receptors are clustered in a region called the postsynaptic density (PSD) in the excitatory synapse.
Synapses are classified as excitatory and inhibitory, depending on whether they use glutamate or γ-aminobutyric acid (GABA) as their main neurotransmitter, respectively. Additionally, the formation and structure of excitatory and inhibitory synapses is uniquely different. Whereas excitatory synapses made on dendritic spines, inhibitory synapses are formed directly on the dendritic shaft. Additionally, both excitatory and inhibitory synapses have distinct proteomic profiles to specialize each synapse with the appropriate receptors and signaling molecules. Synaptic dysfunctions, whether they arise from functional mutations in pre- or postsynaptic proteins, are a common underlying pathology in ASD and are discussed in detail below.
Presynaptic Proteins Linked to ASD
Neurexins (NRXN) are synaptic adhesion proteins that localize to the membrane of the presynapse and bind Neuroligins (NLGNs), which are localized on the postsynaptic membrane. There are three genes within the NRXN family (NRXN1, NRXN2, and NRXN3) with multiple mutations or copy number variations having been identified in NRXN family members in ASD diagnoses (Feng et al., 2006; Autism Genome Project Consortium et al., 2007; Bremer et al., 2011; Yangngam et al., 2014). NRXN1 mutations have also been identified in multiple neuropsychiatric disorders, including Tourette syndrome, schizophrenia, ADHD and bipolar disorder (Stone et al., 2008; Guilmatre et al., 2009; Zhang et al., 2009; Sundaram et al., 2010; Lionel et al., 2011).
NRXN1α KO mice exhibit reduced excitatory synaptic strength, with a decrease in the input–output relationship of evoked postsynaptic potentials and miniature excitatory postsynaptic current (mEPSC) frequency (Etherton et al., 2009). In behavioral tests, NRXN1 KO mice display an increase in grooming behavior, however no change in spatial learning or social behavior (Etherton et al., 2009). In α-NRXN triple KO mice, where NRXN1α/2α/3α were deleted, synapse formation was normal but α-Nrxns were required to couple Ca2+ channels to vesicle exocytosis (Missler et al., 2003).
The synapsins (SYN) are a family of presynaptic proteins involved in vesicle-mediated neurotransmitter release and neurite outgrowth (Rosahl et al., 1995). In mammals, the synapsin family of proteins contains three members (synapsin 1, synapsin 2, and synapsin 3). Primary neuron cultures from SYN1/2/3 triple KO mice display severely altered synaptic vesicle localization and a significant decrease in the number of synaptic vesicles (Fornasiero et al., 2012). Additionally, mutations in SYN1 (A51G, A550T, Q555X and T567A) have been identified in a family with epilepsy and autistic phenotypes, suggesting its link as a causative factor for ASD (Fassio et al., 2011). Additionally, the nonsense mutation, Q555X, reduces CaMKII and MAPK/ERK activity, which regulates synaptic vesicle trafficking and neurite growth, and the A550T and T567A missense mutations have been shown to impair synapsin localization to the presynapse (Fassio et al., 2011).
In synapsin KO mice, studies have shown behavioral deficits in social novelty and avoidance behavior in social approach as well as epileptic activity, all of which are typical in ASD (Greco et al., 2013; Ketzef and Gitler, 2014). SYN1/3 double KO mice show an impairment in social transmission of food preference, while SYN1/2 double KO mice have a decrease in environmental interest. Additionally, SYN2 KO mice have impairments in social recognition tests and display an increase in repetitive self grooming (Greco et al., 2013). These findings suggest that synapsins play an important role in the underlying pathology leading to behavioral phenotypes typical in ASD.
Postsynaptic Proteins Linked to ASD
Neuroligins (NLGN) are cell adhesion proteins that localize to the postsynaptic membrane and play an integral function in synapse formation via binding their presynaptic partners, NRXNs. In humans NLGN family is composed of five genes (NLGN1/2/3/4/4Y). The large extracellular domain of Nlgns have a high sequence similarity to acetylcholinesterase and is required to bind to β-Nrxn during synapse formation (Dean and Dresbach, 2006). Nlgns-1, -3 and -4 are localized to excitatory synapses, while Nlgn2 is found at inhibitory synapses (Missler et al., 2003) Previous studies have shown that overexpression of Nlgn1 can increase excitatory synaptic strength and the synaptic NMDAR/AMPAR ratio both in vitro and in vivo (Schnell et al., 2012). Conversely, Nlgn2 overexpression increases inhibitory synaptic strength. Additionally, Nlgn1 expression can promote synapse maturity but does not induce synapse formation of glutamatergic synapses (Chubykin et al., 2007; Schnell et al., 2012).
The Shank family are scaffolding proteins localized to the postsynapse and interact with NMDA receptors as well as Nlgn-Nrxn complexes. The Shank protein can interact with multiple important postsynaptic proteins including the actin cytoskeleton via ankyrin repeats, Ca2+ signaling via calpain/calmodulin and the glutamate receptor-interacting protein (GRIP) through its SH3 domain (Lim et al., 1999; Yoo et al., 2014). SHANK1, SHANK2 and SHANK3 comprise the Shank family of proteins and they are found throughout the brain, however each isoform varies in its distribution.
SHANK3 was the first gene in the Shank family of synaptic scaffolding proteins to be linked to ASD. During spine formation, Shank3 is an important scaffolding protein and loss of Shank3 in vitro results in a decrease in both the length and density of spines. Conversely, over-expression of Shank3 results in more mature and larger spines (Betancur et al., 2009). SHANK3 resides on chromosome 22q13.3, and ASD-linked region with deletions linked to Phelan-McDermid syndrome. This syndrome is characterized by developmental delay, severely impaired language, ASD and ID (Phelan, 2008).
Transgenic mice harboring various SHANK mutations or deletions have elucidated Shank’s in synapse formation, function and its role in ASD. Transgenic mice with genetic KO of the longest isoform of SHANK3, have been shown to recapitulate the phenotypes observed from SHANK3 mutations in humans. Although social impairments in these mice have varied, they have all shown increased repetitive grooming, a behavior typical in ASD mouse models (Bozdagi et al., 2010; Peça et al., 2011; Wang et al., 2011; Yang et al., 2012).
Postsynaptic density protein-95 (PSD-95, DLG4) is an important postsynaptic scaffolding protein that localizes to excitatory synapses. PSD-95 is composed of three PDZ domains that target it to the postsynapse, an SH3 domain, and a guanylate kinase domain on its C-terminal. Nlgn, NMDARs and potassium channels all interact with the PDZ domains on PSD-95. PSD-95 localizes to spine heads in excitatory synapses and has been shown to promote synapse stabilization. The E3 ubiquitin ligase, murine double minute-2 (Mdm2) ubiquitinates PSD-95 and subsequently binds with protocadherin-10 (Pcdh10), sending it to the proteasome to be degraded. Pcdh10 is an ASD-linked gene (Morrow et al., 2008) and its expression is regulated via the interaction of myocyte enhancer factor-2 (MEF2) and FMRP (Tsai N. P. et al., 2012). Additionally, in FMRP KO neurons, dysregulated Mdm2 prevents MEF2-induced PSD-95 ubiquitination and synapse elimination (Tsai N. P. et al., 2012), providing evidence for altered activity-dependent synapse elimination in an ASD model. PSD-95 KO mice (DLG4 KO) show multiple behavioral and molecular abnormalities that are linked to ASD pathology. DLG4 KO mice display impaired communication and social interactions, decreased motor coordination, as well as increased anxiety and repetitive behaviors. Additionally, DLG4 KO mice display defects in dendritic spine morphology in the amygdala and aberrant expression of multiple synapse-related genes in the forebrain (Feyder et al., 2010).
Gephyrin is a key postsynaptic scaffolding protein in inhibitory synapses. Gephyrin’s three major domains include a N-terminal G-domain, a C-terminal E-domain, with a large linker region connecting the two domains. Gephyrin interacts with glycine receptors with high affinity and the α and β subunits of the GABAA receptor. Nlgn2 binds to gephyrin via a cytoplasmic motif region and activates collybistin, and the Nlgn2/gephyrin/collybistin complex is necessary for clustering of inhibitory receptors (Poulopoulos et al., 2009). In gephyrin KO mice, there is a decrease in GABAA and glycine receptor clustering, whereas glutamate receptor localization remains normal (Kneussel et al., 1999; Grosskreutz et al., 2003).
Rare exonic deletions within the gephyrin (GPHN) gene have been reported in subsets of patients with ASD (Lionel et al., 2013). A de novo 273 kb deletion in GPHN displayed developmental delay, cyclical seizures, repetitive behaviors increased anxiety and obsessive compulsive disorders, in one family (Lionel et al., 2013).
Adhesion Complexes Involved in ASD
CNTNAP2 is a member of the Nrxn superfamily and plays an important role regulating the clustering of potassium channels and neuron-glia interactions. CNTNAP2 was first linked to ASD Amish children presenting with developmental disorders, seizures, and impaired language were found to have a homozygous mutation in CNTNAP2 (Strauss et al., 2006). After genetic deletion of CNTNAP2, mice show significantly decreased numbers of dendritic spines as well as a decrease in the GluA1 subunit of AMPARs. Additionally, an aggregation of GluA1 was observed in the cytoplasm, suggesting that synaptic deficits may be in part due to a trafficking problem (Varea et al., 2015). CNTNAP2 KO mice show common ASD behavioral phenotypes, including repetitive movements as well as social deficits (Peñagarikano et al., 2011).
Cadherins (CDH) are transmembrane proteins that function in cell-cell adhesion and play roles in neuron migration, dendritic growth, spine morphology, synaptogenesis and synapse remodeling (Tan et al., 2010; Redies et al., 2012; Bian et al., 2015; Egusa et al., 2016; Gilbert et al., 2016). In genome-wide association studies, common variants in CDH9 and CDH10 genes on chromosome 5p14.1 have been identified to link with ASD (Wang et al., 2009). Additionally, deletions within 16q23 in CDH13 have also been identified in ASD patients (Sanders et al., 2011). Neural cadherin (N-cadherin) is a calcium dependent cell-cell adhesion glycoprotein that plays important roles in neurodevelopment (Garcia-Castro et al., 2000). δ-catenin, a neuron specific member of the p120 family of catenins, is a known interactor of the cytoplasmic juxtamembrane region of N-cadherin (Lu et al., 1999). The δ-catenin gene, CTNND2, is candidate gene in ASD and functions within a protein network that has a major role dendritic and spine growth and dynamics (Bian et al., 2015; Turner et al., 2015; Yuan et al., 2015). Over-expression of δ-catenin induces dendritic and spine growth in primary neuron cultures (Martinez et al., 2003; Arikkath et al., 2009), while knockdown of δ-catenin serves to impair dendritic growth (Elia et al., 2006; Arikkath et al., 2008). Figure 3 depicts key synaptic proteins and signaling pathways linked to ASD.
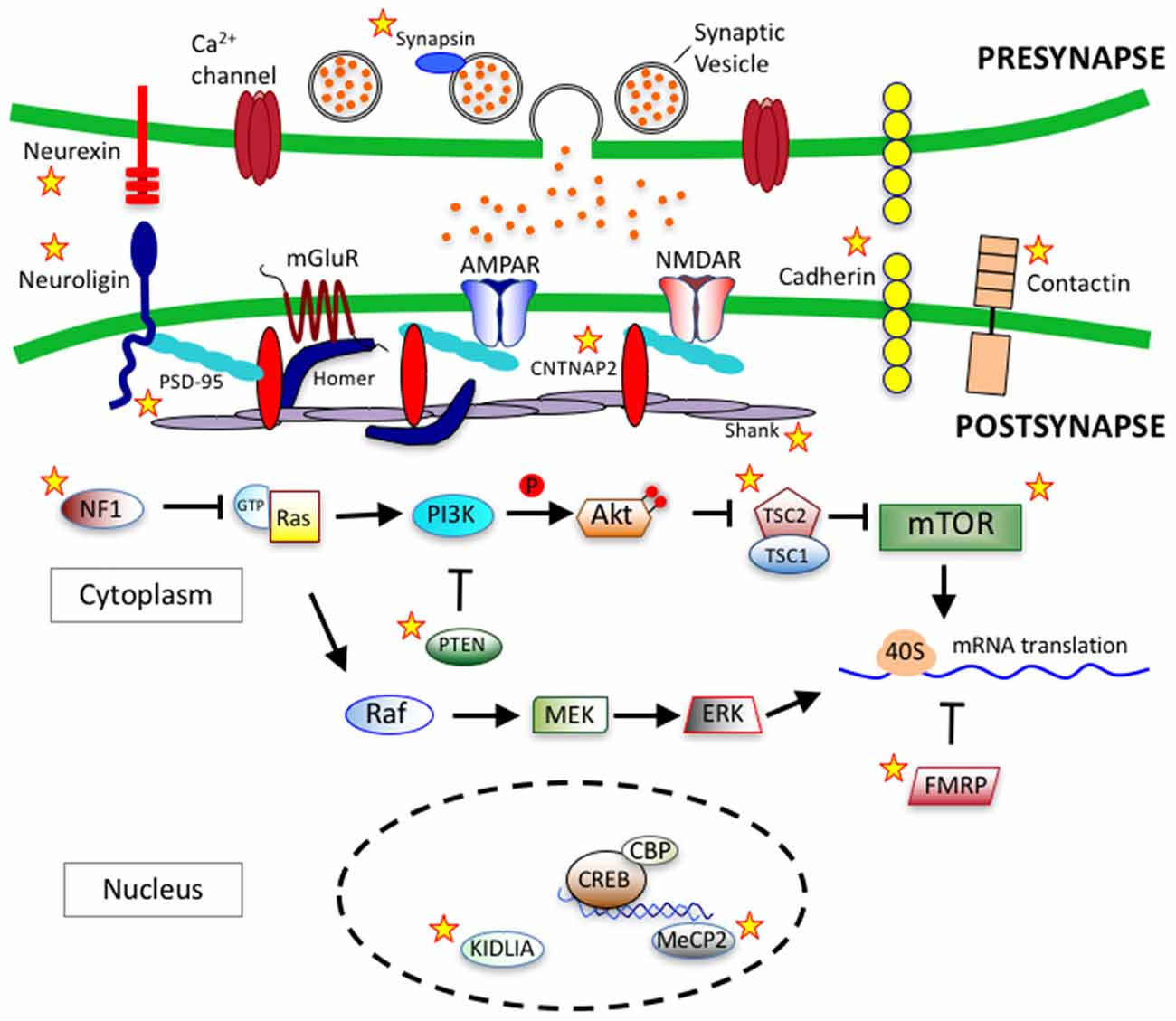
Figure 3. Synaptic proteins and signaling pathways linked to autism spectrum disorder (ASD). ASD-linked synaptic proteins and signaling pathways that relate to synaptogeneis and synaptic function. Stars mark ASD-linked proteins discussed in this review article. Abbreviations: AMPAR, AMPA receptor; NMDAR, NMDA receptor; mGluR, metabotropic glutamate receptor; PSD-95, postsynaptic scaffolding protein 95 kDa; CNTNAP2, contactin-associated protein-like 2 gene; PI3K, phosphoinositide-3 kinase; Ras, RhoGTPase; GTP, Guanosine-5′-triphosphate; NF1, Neurofibromatosis type 1; PTEN, phosphatase and tensin homolog; Akt, serine/threonine specific kinase; TSC, tuberous sclerosis complex; mTOR, mammalian target of rapamycin; Raf, Rapidly accelerated fibrosarcoma serine threnonine kinase, MEK, Mitogen-activated protein kinase kinase; ERK, extracellular signal–regulated kinase; 40S, ribosomal subunit; FMRP, fragile-X mental retardation protein; CREB, cAMP response element-binding protein; CBP, CREB binding protein; MeCP2, methyl CpG binding protein 2; KIDLIA, KIAA2022 gene with intellectual disability (ID) and language impairment in autism; P, phosphate group.
Dysregulation of Synaptic Plasticity and Neuronal Activity in ASD
Based on the different ASD-linked pathways discussed previously, it can be inferred that changes in synaptic plasticity via altered synaptic strength and/or number, may be an underlying pathology in ASD patients and mouse models. Interestingly, many ASD-linked mutation results in altered gene transcription and protein synthesis of synaptic related transcripts, effects which can also be observed with changes in neuronal activity (Kelleher and Bear, 2008; Akins et al., 2009; Darnell et al., 2011; Qiu et al., 2012; Gilbert and Man, 2014).
Initial findings from FMR1 mutant mice, the mouse model of FXS, did not show impairments in LTP using a standard HFS paradigm (Godfraind et al., 1996; Li et al., 2002). However, using a low threshold stimulation protocol, LTP induction was reduced in multiple brain areas, including the hippocampus and somatosensory cortex (Larson et al., 2005; Zhao et al., 2005; Lauterborn et al., 2007). Additionally, mGluR-LTP was impaired in the basolateral amygdala and visual cortex (Wilson and Cox, 2007; Suvrathan et al., 2010). These findings provide evidence that different brain regions or synapse-specific deficits occur in mice with loss of FMRP. The most prominent change in synaptic plasticity has been observed with enhanced mGluR-LTD the cerebellum and in CA1 of the hippocampus (Huber et al., 2002; Koekkoek et al., 2005). An enhancement of LTD in the CA1 region has led to the theory that synaptic loss of FMRP produces increased signaling through mGluRs (Huber et al., 2002; Bear et al., 2004; Osterweil et al., 2010). Additionally, genetic reduction, or mGluR5 inhibition, rescues behavioral deficits in FMR1 mice (Dolen et al., 2007; Michalon et al., 2012). These rescue experiments therefore suggest the ability to reverse some of the impairments in FXS patients. The different impairments in synaptic function found across various brain regions in FMR1 mice provide evidence of a correlation between impaired synaptic plasticity and behavioral deficits in ASD. These studies also provide evidence that the FMR1 KO mouse may be a good model to explore the pathophysiology of ASD and investigate possible treatment strategies (Bhakar et al., 2012).
Synaptic plasticity has been studied in great detail in the UBE3A maternally deficient mice, the Angelman syndrome mouse model. In the CA1, HFS-LTP is reduced in transgenic mice and the reduction in CA1 LTP could be rescued with stronger stimulation protocols (Weeber et al., 2003). These findings indicate that Ube3a’s role in synaptic plasticity may be as a modulator of LTP, not necessarily required for LTP induction. In the same study, a reduction in calmodulin-dependent protein kinase II (CaMKII) activity was observed in transgenic mice, and genetic reduction of CaMKII’s inhibitory autophosphorylation rescued deficits in LTP and learning and memory tasks in transgenic animals (van Woerden et al., 2007). These findings provide evidence that altered CaMKII activity mediates the impairment in synaptic plasticity in UBE3A deficient mice; however, it remains to be elucidated how loss of UBE3A alters the activity of CaMKII.
Both of the TSC1 or TSC2 KO mice are embryonic lethal; however, mice harboring TSC1 or TSC2 heterozygous mutations display synaptic dysfunction and cognitive impairments (Kobayashi et al., 2001; von der Brelie et al., 2006; Ehninger et al., 2008). In TSC2 heterozygous rats, LFS-LTD was decreased and L-LTP was enhanced in the CA1 of the hippocampus; however, E-LTP was unaffected (von der Brelie et al., 2006; Ehninger et al., 2008), suggesting changes in protein synthesis pathways necessary for L-LTP expression. Additionally, in TSC2 heterozygous mice, mGluR-LTD was decreased but LFS-LTD was unchanged (Auerbach et al., 2011). A reduction in mGluR-LTD in TSC2 heterozygous animals opposes what is observed in the FMR1 KO mice; however, mGluR-LTD in both animals don’t show sensitivity to protein synthesis inhibitors (Auerbach et al., 2011). An additional study using mice with a specific deletion of TSC1 in cerebellar Purkinje cells displayed deficits in social interactions, an increase repetitive behavior as wells as defects in ultrasonic vocalizations. These animals however were not investigated for changes in synaptic plasticity in the cerebellum (Tsai P. T. et al., 2012). These findings suggest that impaired synaptic plasticity is a major pathology in TSC mouse models of autism, underlying the observed deficits in social interaction in both in TSC1 and TSC2 heterozygous animals.
In MECP2 KO mice, reductions in LTP and LFS-LTD in CA1 of the hippocampus have been reported. It’s interesting to note that younger mice (~3–5 weeks of age) show no impairments in synaptic plasticity, indicating that impaired synaptic plasticity correlates with the delayed deficits observed in human RTT patients (Chen et al., 2001; Asaka et al., 2006). A mouse model containing a truncation of MeCP2 (MeCP2308/Y) showed a reduction in LTP as well as paired-pulse stimulation, but no changes in mGLuR-LTD (Shahbazian et al., 2002; Weng et al., 2011). Interestingly, another study has shown that impaired LTP in CA1 of the hippocampus could be rescued by genetically reintroducing MeCP2 (Guy et al., 2007; Weng et al., 2011).
Shank proteins regulate levels and modulate the signaling of both metabotropic and ionotropic glutamate receptors at the synapse, and synaptic plasticity has been studied in multiple Shank3 mouse models (Tu et al., 1999; Uchino et al., 2006). Investigation of mEPSCs, paired pulse ratio (PPF and PPD), input/output curves and population spikes have indicated different abnormalities in synaptic transmission in hippocampal CA1 synapses of mice with different Shank3 mutations (Wang et al., 2011). Additionally, hippocampal LTP is reduced in the CA1 of the hippocampus in subsets of mice with Shank3 truncations, as well as LFS- LTD and mGLuR-LTD (Bozdagi et al., 2010; Bangash et al., 2011; Wang et al., 2011).
It has been hypothesized that disruptions in neuronal circuits involved with language and social behavior in subtypes of autism may be caused by unbalanced high levels of excitation, or disproportionately weak inhibition (Rubenstein and Merzenich, 2003; Gao and Penzes, 2015). With a more excitable cortex, the brain would have wide-ranging abnormalities in perception, memory, cognition and motor control, and would be highly susceptible to epilepsy (Rubenstein and Merzenich, 2003). In support of this theory, a decrease in glutamate decarboxylase 67 (GAD67) mRNA in autistic cerebellar Purkinje cells has been identified in human autism samples (Yip et al., 2007). Additionally, another study has shown a decrease in GABAergic inhibition in autism patient samples (Hussman, 2001), indicating that decreased GABAergic inhibition may disrupt the excitation/inhibition balance within neuronal networks in autism.
As a novel form of synaptic regulation opposing the Hebbian type plasticity, homeostatic synaptic plasticity (HSP) is a negative feedback response that serves to compensate for changes in network activity (Turrigiano et al., 1998; Hou et al., 2008, 2011, 2015; Yu and Goda, 2009; Pozo and Goda, 2010; Wang et al., 2012; Gilbert et al., 2016). In response to global decreases or increases in network activity from a homeostatic set-point, synaptic strengths are scaled up or down, respectively (Turrigiano et al., 1998). HSP may be important for maintaining network activity homeostasis and avoiding potential epileptogenic states and sleep may be necessary for synaptic homeostasis (Tononi and Cirelli, 2003; Kuhn et al., 2016). The studies in Drosophila for example, have shown that the size and number of synapses decrease after sleep and increase within a few hours of waking (Gilestro et al., 2009). Interestingly, Nlgns regulate levels of glutamatergic and GABAergic currents after sleep deprivation, indicating they play an important role in this sleep-dependent HSP (Huber et al., 2004; Gilestro et al., 2009; El Helou et al., 2013) and Nlgn defects could result in disturbances in sleep and circadian rhythms, a common disorder found in ASD patients (Bourgeron, 2007). FMRP, the RNA-binding protein involved with regulating dendritic protein translation, has also been shown to be required for increases in synaptic strength after neuronal activity blockade or application of retinoic acid in the mouse hippocampus, indicating that some symptoms of FXS may be due to impaired HSP (Soden and Chen, 2010). Release of brain-derived neurotrophic factor (BDNF) from postsynaptic neurons has previously been indentified to be required for a retrograde homeostatic up-regulation of presynaptic function. Increased BDNF levels in ASD patient blood samples has been observed as well as higher plasma levels of serotonin and N-acetylserotonin (NAS), a potent agonist of the BDNF receptor tyrosine receptor kinase B (TrkB; Jang et al., 2010; Halepoto et al., 2014; Kasarpalkar et al., 2014; Pagan et al., 2014). Excess levels of NAS could therefore increase TrkB-induced PI3K signaling, resulting in increased protein translation, similar to findings observed with mutations in components of the mTOR pathway.
Final Conclusions
ASD is diagnosed at the behavioral level with the presentation of its core phenotypes of impaired social interactions, restrictive interests and repetitive behaviors. Although the etiological factors of ASD are highly heterogeneous, recent research has strongly pointed to common cellular events that are impaired in ASD, including neurogenesis, morphogenesis, synapse maturation and synaptic plasticity. In this regard, loss-, or gain-, of-function mutations in single genes that are causative for ASD have given researchers unique opportunities to make important mechanistic insights. A common theme emerging within the field is that in the developing brain, alterations in dendritic growth, synapse formation and synaptic function result in neuronal network dysfunction, ultimately leading to complex social and cognitive dysfunction. Further elucidation into these pathways, as well as advancements in gene therapies and targeted drugs to modulate these processes, could provide exciting and promising new therapies for the treatment of ASDs.
Author Contributions
JG and H-YM wrote and edited the manuscript.
Funding
This work was supported by NIH grant MH079407 (H-YM).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abrahams, B. S., Arking, D. E., Campbell, D. B., Mefford, H. C., Morrow, E. M., Weiss, L. A., et al. (2013). SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4:36. doi: 10.1186/2040-2392-4-36
Adams, N. C., Tomoda, T., Cooper, M., Dietz, G., and Hatten, M. E. (2002). Mice that lack astrotactin have slowed neuronal migration. Development 129, 965–972.
Akins, M. R., Berk-Rauch, H. E., and Fallon, J. R. (2009). Presynaptic translation: stepping out of the postsynaptic shadow. Front. Neural Circuits 3:17. doi: 10.3389/neuro.04.017.2009
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Anderson, S. A., Eisenstat, D. D., Shi, L., and Rubenstein, J. L. (1997). Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278, 474–476. doi: 10.1126/science.278.5337.474
Angevine, J. B. Jr., and Sidman, R. L. (1961). Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature 192, 766–768. doi: 10.1038/192766b0
Arikkath, J., Israely, I., Tao, Y., Mei, L., Liu, X., and Reichardt, L. F. (2008). Erbin controls dendritic morphogenesis by regulating localization of delta-catenin. J. Neurosci. 28, 7047–7056. doi: 10.1523/JNEUROSCI.0451-08.2008
Arikkath, J., Peng, I. F., Ng, Y. G., Israely, I., Liu, X., Ullian, E. M., et al. (2009). Delta-catenin regulates spine and synapse morphogenesis and function in hippocampal neurons during development. J. Neurosci. 29, 5435–5442. doi: 10.1523/JNEUROSCI.0835-09.2009
Asaka, Y., Jugloff, D. G., Zhang, L., Eubanks, J. H., and Fitzsimonds, R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 21, 217–227. doi: 10.1016/j.nbd.2005.07.005
Auerbach, B. D., Osterweil, E. K., and Bear, M. F. (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68. doi: 10.1038/nature10658
Autism Genome Project Consortium, Szatmari, P., Paterson, A. D., Zwaigenbaum, L., Roberts, W., Brian, J., et al. (2007). Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 39, 319–328. doi: 10.1038/ng1985
Avino, T. A., and Hutsler, J. J. (2010). Abnormal cell patterning at the cortical gray-white matter boundary in autism spectrum disorders. Brain Res. 1360, 138–146. doi: 10.1016/j.brainres.2010.08.091
Ayala, R., Shu, T., and Tsai, L. H. (2007). Trekking across the brain: the journey of neuronal migration. Cell 128, 29–43. doi: 10.1016/j.cell.2006.12.021
Azevedo, F. A., Carvalho, L. R., Grinberg, L. T., Farfel, J. M., Ferretti, R. E., Leite, R. E., et al. (2009). Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 513, 532–541. doi: 10.1002/cne.21974
Baas, P. W., Black, M. M., and Banker, G. A. (1989). Changes in microtubule polarity orientation during the development of hippocampal neurons in culture. J. Cell Biol. 109, 3085–3094. doi: 10.1083/jcb.109.6.3085
Baas, P. W., Deitch, J. S., Black, M. M., and Banker, G. A. (1988). Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc. Natl. Acad. Sci. U S A 85, 8335–8339. doi: 10.1073/pnas.85.21.8335
Bailey, A., Luthert, P., Dean, A., Harding, B., Janota, I., Montgomery, M., et al. (1998). A clinicopathological study of autism. Brain 121, 889–905. doi: 10.1093/brain/121.5.889
Bakhtiari, R., Zürcher, N. R., Rogier, O., Russo, B., Hippolyte, L., Granziera, C., et al. (2012). Differences in white matter reflect atypical developmental trajectory in autism: a Tract-based Spatial Statistics study. Neuroimage Clin. 1, 48–56. doi: 10.1016/j.nicl.2012.09.001
Bakos, J., Bacova, Z., Grant, S. G., Castejon, A. M., and Ostatnikova, D. (2015). Are molecules involved in neuritogenesis and axon guidance related to autism pathogenesis? Neuromolecular Med. 17, 297–304. doi: 10.1007/s12017-015-8357-7
Banerjee-Basu, S., and Packer, A. (2010). SFARI Gene: an evolving database for the autism research community. Dis. Model. Mech. 3, 133–135. doi: 10.1242/dmm.005439
Bangash, M. A., Park, J. M., Melnikova, T., Wang, D., Jeon, S. K., Lee, D., et al. (2011). Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell 145, 758–772. doi: 10.1016/j.cell.2011.03.052
Bauman, M., and Kemper, T. L. (1985). Histoanatomic observations of the brain in early infantile autism. Neurology 35, 866–874. doi: 10.1212/WNL.35.6.866
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. doi: 10.1016/j.tins.2004.04.009
Bedogni, F., Hodge, R. D., Nelson, B. R., Frederick, E. A., Shiba, N., Daza, R. A., et al. (2010). Autism susceptibility candidate 2 (Auts2) encodes a nuclear protein expressed in developing brain regions implicated in autism neuropathology. Gene Expr. Patterns 10, 9–15. doi: 10.1016/j.gep.2009.11.005
Bellion, A., Baudoin, J. P., Alvarez, C., Bornens, M., and Metin, C. (2005). Nucleokinesis in tangentially migrating neurons comprises two alternating phases: forward migration of the Golgi/centrosome associated with centrosome splitting and myosin contraction at the rear. J. Neurosci. 25, 5691–5699. doi: 10.1523/JNEUROSCI.1030-05.2005
Ben-Itzchak, E., Aviva, B., and Zachor, D. A. (2013a). Are special abilities in autism spectrum disorder associated with a distinct clinical presentation? Res. Autism Spec. Dis. 7, 1122–1128. doi: 10.1016/j.rasd.2013.05.003
Ben-Itzchak, E., Ben-Shachar, S., and Zachor, D. A. (2013b). Specific neurological phenotypes in autism spectrum disorders are associated with sex representation. Autism Res. 6, 596–604. doi: 10.1002/aur.1319
Ben-Itzchak, E., and Zachor, D. A. (2007). The effects of intellectual functioning and autism severity on outcome of early behavioral intervention for children with autism. Res. Dev. Disabil. 28, 287–303. doi: 10.1016/j.ridd.2006.03.002
Berman, R. F., Murray, K. D., Arque, G., Hunsaker, M. R., and Wenzel, H. J. (2012). Abnormal dendrite and spine morphology in primary visual cortex in the CGG knock-in mouse model of the fragile X premutation. Epilepsia 53, 150–160. doi: 10.1111/j.1528-1167.2012.03486.x
Bernier, R., Golzio, C., Xiong, B., Stessman, H. A., Coe, B. P., Penn, O., et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. doi: 10.1016/j.cell.2014.06.017
Betancur, C., Sakurai, T., and Buxbaum, J. D. (2009). The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 32, 402–412. doi: 10.1016/j.tins.2009.04.003
Bhakar, A. L., Dölen, G., and Bear, M. F. (2012). The pathophysiology of fragile, X. (and what it teaches us about synapses). Annu. Rev. Neurosci. 35, 417–443. doi: 10.1146/annurev-neuro-060909-153138
Bian, W. J., Miao, W. Y., He, S. J., Qiu, Z., and Yu, X. (2015). Coordinated spine pruning and maturation mediated by inter-spine competition for cadherin/catenin complexes. Cell 162, 808–822. doi: 10.1016/j.cell.2015.07.018
Bilgüvar, K., Oztürk, A. K., Louvi, A., Kwan, K. Y., Choi, M., Tatli, B., et al. (2010). Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467, 207–210. doi: 10.1038/nature09327
Bond, J., Scott, S., Hampshire, D. J., Springell, K., Corry, P., Abramowicz, M. J., et al. (2003). Protein-truncating mutations in ASPM cause variable reduction in brain size. Am. J. Hum. Genet. 73, 1170–1177. doi: 10.1086/379085
Bonora, E., Beyer, K. S., Lamb, J. A., Parr, J. R., Klauck, S. M., Benner, A., et al. (2003). Analysis of reelin as a candidate gene for autism. Mol. Psychiatry 8, 885–892. doi: 10.1038/sj.mp.4001310
Bourgeron, T. (2007). The possible interplay of synaptic and clock genes in autism spectrum disorders. Cold Spring Harb. Symp. Quant. Biol. 72, 645–654. doi: 10.1101/sqb.2007.72.020
Bourgeron, T. (2009). A synaptic trek to autism. Curr. Opin. Neurobiol. 19, 231–234. doi: 10.1016/j.conb.2009.06.003
Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D. L., Takahashi, N., et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction and social communication. Mol. Autism 1:15. doi: 10.1186/2040-2392-1-15
Bremer, A., Giacobini, M., Eriksson, M., Gustavsson, P., Nordin, V., Fernell, E., et al. (2011). Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156, 115–124. doi: 10.1002/ajmg.b.31142
Brown, J. A., Diggs-Andrews, K. A., Gianino, S. M., and Gutmann, D. H. (2012). Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol. Cell. Neurosci. 49, 13–22. doi: 10.1016/j.mcn.2011.08.008
Bucan, M., Abrahams, B. S., Wang, K., Glessner, J. T., Herman, E. I., Sonnenblick, L. I., et al. (2009). Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 5:e1000536. doi: 10.1371/journal.pgen.1000536
Buchman, J. J., Durak, O., and Tsai, L. H. (2011). ASPM regulates Wnt signaling pathway activity in the developing brain. Genes Dev. 25, 1909–1914. doi: 10.1101/gad.16830211
Burton, P. R. (1988). Dendrites of mitral cell neurons contain microtubules of opposite polarity. Brain Res. 473, 107–115. doi: 10.1016/0006-8993(88)90321-6
Butler, M. G., Dasouki, M. J., Zhou, X. P., Talebizadeh, Z., Brown, M., Takahashi, T. N., et al. (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 42, 318–321. doi: 10.1136/jmg.2004.024646
Buxbaum, J. D., Cai, G., Chaste, P., Nygren, G., Goldsmith, J., Reichert, J., et al. (2007). Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 484–491. doi: 10.1002/ajmg.b.30493
Cahana, A., Escamez, T., Nowakowski, R. S., Hayes, N. L., Giacobini, M., von Holst, A., et al. (2001). Targeted mutagenesis of Lis1 disrupts cortical development and LIS1 homodimerization. Proc. Natl. Acad. Sci. U S A 98, 6429–6434. doi: 10.1073/pnas.101122598
Cantagrel, V., Haddad, M. R., Ciofi, P., Andrieu, D., Lossi, A. M., Maldergem, L., et al. (2009). Spatiotemporal expression in mouse brain of Kiaa2022, a gene disrupted in two patients with severe mental retardation. Gene Expr. Patterns 9, 423–429. doi: 10.1016/j.gep.2009.06.001
Cantagrel, V., Lossi, A. M., Boulanger, S., Depetris, D., Mattei, M. G., Gecz, J., et al. (2004). Disruption of a new X linked gene highly expressed in brain in a family with two mentally retarded males. J. Med. Genet. 41, 736–742. doi: 10.1136/jmg.2004.021626
Carper, R. A., Moses, P., Tigue, Z. D., and Courchesne, E. (2002). Cerebral lobes in autism: early hyperplasia and abnormal age effects. Neuroimage 16, 1038–1051. doi: 10.1006/nimg.2002.1099
Casanova, E. L., and Casanova, M. F. (2014). Genetics studies indicate that neural induction and early neuronal maturation are disturbed in autism. Front. Cell. Neurosci. 8:397. doi: 10.3389/fncel.2014.00397
Castrén, M., Tervonen, T., Kärkkäinen, V., Heinonen, S., Castrén, E., Larsson, K., et al. (2005). Altered differentiation of neural stem cells in fragile X syndrome. Proc. Natl. Acad. Sci. U S A 102, 17834–17839. doi: 10.1073/pnas.0508995102
Caviness, V. S. Jr., Nowakowski, R. S., and Bhide, P. G. (2009). Neocortical neurogenesis: morphogenetic gradients and beyond. Trends Neurosci. 32, 443–450. doi: 10.1016/j.tins.2009.05.003
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chameau, P., Inta, D., Vitalis, T., Monyer, H., Wadman, W. J., and van Hooft, J. A. (2009). The N-terminal region of reelin regulates postnatal dendritic maturation of cortical pyramidal neurons. Proc. Natl. Acad. Sci. U S A 106, 7227–7232. doi: 10.1073/pnas.0810764106
Changeux, J. P., and Danchin, A. (1976). Selective stabilisation of developing synapses as a mechanism for the specification of neuronal networks. Nature 264, 705–712. doi: 10.1038/264705a0
Chapleau, C. A., Boggio, E. M., Calfa, G., Percy, A. K., Giustetto, M., and Pozzo-Miller, L. (2012). Hippocampal CA1 pyramidal neurons of Mecp2 mutant mice show a dendritic spine phenotype only in the presymptomatic stage. Neural Plast. 2012:976164. doi: 10.1155/2012/976164
Charzewska, A., Rzońca, S., Janeczko, M., Nawara, M., Smyk, M., Bal, J., et al. (2015). A duplication of the whole KIAA2022 gene validates the gene role in the pathogenesis of intellectual disability and autism. Clin. Genet. 88, 297–299. doi: 10.1111/cge.12528
Chazeau, A., and Giannone, G. (2016). Organization and dynamics of the actin cytoskeleton during dendritic spine morphological remodeling. Cell. Mol. Life Sci. 73, 3053–3073. doi: 10.1007/s00018-016-2214-1
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Chen, Z., and Cobb, M. H. (2001). Regulation of stress-responsive mitogen-activated protein (MAP) kinase pathways by TAO2. J. Biol. Chem. 276, 16070–16075. doi: 10.1074/jbc.M100681200
Chen, J. A., Penagarikano, O., Belgard, T. G., Swarup, V., and Geschwind, D. H. (2015). The emerging picture of autism spectrum disorder: genetics and pathology. Annu. Rev. Pathol. 10, 111–144. doi: 10.1146/annurev-pathol-012414-040405
Chen, Z., Raman, M., Chen, L., Lee, S. F., Gilman, A. G., and Cobb, M. H. (2003). TAO (thousand-and-one amino acid) protein kinases mediate signaling from carbachol to p38 mitogen-activated protein kinase and ternary complex factors. J. Biol. Chem. 278, 22278–22283. doi: 10.1074/jbc.M301173200
Chen, J. F., Zhang, Y., Wilde, J., Hansen, K. C., Lai, F., and Niswander, L. (2014). Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nat. Commun. 5:3885. doi: 10.1038/ncomms4885
Cheng, Y., Quinn, J. F., and Weiss, L. A. (2013). An eQTL mapping approach reveals that rare variants in the SEMA5A regulatory network impact autism risk. Hum. Mol. Genet. 22, 2960–2972. doi: 10.1093/hmg/ddt150
Cheng, T. L., Wang, Z., Liao, Q., Zhu, Y., Zhou, W. H., Xu, W., et al. (2014). MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev. Cell 28, 547–560. doi: 10.1016/j.devcel.2014.01.032
Choi, Y. J., Di Nardo, A., Kramvis, I., Meikle, L., Kwiatkowski, D. J., Sahin, M., et al. (2008). Tuberous sclerosis complex proteins control axon formation. Genes Dev. 22, 2485–2495. doi: 10.1101/gad.1685008
Chow, D. K., Groszer, M., Pribadi, M., Machniki, M., Carmichael, S. T., Liu, X., et al. (2009). Laminar and compartmental regulation of dendritic growth in mature cortex. Nat. Neurosci. 12, 116–118. doi: 10.1038/nn.2255
Chuang, H. C., Huang, T. N., and Hsueh, Y. P. (2015). T-brain-1—A potential master regulator in autism spectrum disorders. Autism Res. 8, 412–426. doi: 10.1002/aur.1456
Chubykin, A. A., Atasoy, D., Etherton, M. R., Brose, N., Kavalali, E. T., Gibson, J. R., et al. (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931. doi: 10.1016/j.neuron.2007.05.029
Cobos, I., Calcagnotto, M. E., Vilaythong, A. J., Thwin, M. T., Noebels, J. L., Baraban, S. C., et al. (2005). Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 8, 1059–1068. doi: 10.1038/nn1499
Cody, H., Pelphrey, K., and Piven, J. (2002). Structural and functional magnetic resonance imaging of autism. Int. J. Dev. Neurosci. 20, 421–438. doi: 10.1016/S0736-5748(02)00053-9
Colasante, G., Collombat, P., Raimondi, V., Bonanomi, D., Ferrai, C., Maira, M., et al. (2008). Arx is a direct target of Dlx2 and thereby contributes to the tangential migration of GABAergic interneurons. J. Neurosci. 28, 10674–10686. doi: 10.1523/JNEUROSCI.1283-08.2008
Comoletti, D., De Jaco, A., Jennings, L. L., Flynn, R. E., Gaietta, G., Tsigelny, I., et al. (2004). The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J. Neurosci. 24, 4889–4893. doi: 10.1523/JNEUROSCI.0468-04.2004
Condro, M. C., and White, S. A. (2014). Distribution of language-related Cntnap2 protein in neural circuits critical for vocal learning. J. Comp. Neurol. 522, 169–185. doi: 10.1002/cne.23394
Coquelle, F. M., Caspi, M., Cordelieres, F. P., Dompierre, J. P., Dujardin, D. L., Koifman, C., et al. (2002). LIS1, CLIP-170’s key to the dynein/dynactin pathway. Mol. Cell. Biol. 22, 3089–3102. doi: 10.1128/mcb.22.9.3089-3102.2002
Costa, R. M., and Silva, A. J. (2003). Mouse models of neurofibromatosis type I: bridging the GAP. Trends Mol. Med. 9, 19–23. doi: 10.1016/s1471-4914(02)00008-4
Courchesne, E., Mouton, P. R., Calhoun, M. E., Semendeferi, K., Ahrens-Barbeau, C., Hallet, M. J., et al. (2011). Neuron number and size in prefrontal cortex of children with autism. JAMA 306, 2001–2010. doi: 10.1001/jama.2011.1638
Courchesne, E., and Pierce, K. (2005). Why the frontal cortex in autism might be talking only to itself: local over-connectivity but long-distance disconnection. Curr. Opin. Neurobiol. 15, 225–230. doi: 10.1016/j.conb.2005.03.001
Crepel, A., Steyaert, J., De la Marche, W., De Wolf, V., Fryns, J. P., Noens, I., et al. (2011). Narrowing the critical deletion region for autism spectrum disorders on 16p11.2. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156, 243–245. doi: 10.1002/ajmg.b.31163
D’Arcangelo, G., Miao, G. G., Chen, S. C., Soares, H. D., Morgan, J. I., and Curran, T. (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723. doi: 10.1038/374719a0
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
de Anda, F. C., Rosario, A. L., Durak, O., Tran, T., Graff, J., Meletis, K., et al. (2012). Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat. Neurosci. 15, 1022–1031. doi: 10.1038/nn.3141
de Carlos, J. A., López-Mascaraque, L., and Valverde, F. (1996). Dynamics of cell migration from the lateral ganglionic eminence in the rat. J. Neurosci. 16, 6146–6156.
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
De Rubeis, S., Pasciuto, E., Li, K. W., Fernandez, E., Di Marino, D., Buzzi, A., et al. (2013). CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 79, 1169–1182. doi: 10.1016/j.neuron.2013.06.039
De Zio, D., Giunta, L., Corvaro, M., Ferraro, E., and Cecconi, F. (2005). Expanding roles of programmed cell death in mammalian neurodevelopment. Semin. Cell Dev. Biol. 16, 281–294. doi: 10.1016/j.semcdb.2004.12.003
Dean, C., and Dresbach, T. (2006). Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 29, 21–29. doi: 10.1016/j.tins.2005.11.003
Denaxa, M., Chan, C. H., Schachner, M., Parnavelas, J. G., and Karagogeos, D. (2001). The adhesion molecule TAG-1 mediates the migration of cortical interneurons from the ganglionic eminence along the corticofugal fiber system. Development 128, 4635–4644.
Deriziotis, P., O’Roak, B. J., Graham, S. A., Estruch, S. B., Dimitropoulou, D., Bernier, R. A., et al. (2014). De novo TBR1 mutations in sporadic autism disrupt protein functions. Nat. Commun. 5:4954. doi: 10.1038/ncomms5954
DiCicco-Bloom, E., Lord, C., Zwaigenbaum, L., Courchesne, E., Dager, S., Schmitz, C., et al. (2006). The developmental neurobiology of autism spectrum disorder. J. Neurosci. 26, 6897–6906. doi: 10.1523/JNEUROSCI.1712-06.2006
Dolen, G., Osterweil, E., Rao, B. S., Smith, G. B., Auerbach, B. D., Chattarji, S., et al. (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. doi: 10.1016/j.neuron.2007.12.001
Dwyer, N. D., and O’Leary, D. D. (2001). Tbr1 conducts the orchestration of early cortical development. Neuron 29, 309–311. doi: 10.1016/s0896-6273(01)00202-1
Edmondson, J. C., Liem, R. K., Kuster, J. E., and Hatten, M. E. (1988). Astrotactin: a novel neuronal cell surface antigen that mediates neuron-astroglial interactions in cerebellar microcultures. J. Cell Biol. 106, 505–517. doi: 10.1083/jcb.106.2.505
Egusa, S. F., Inoue, Y. U., Asami, J., Terakawa, Y. W., Hoshino, M., and Inoue, T. (2016). Classic cadherin expressions balance postnatal neuronal positioning and dendrite dynamics to elaborate the specific cytoarchitecture of the mouse cortical area. Neurosci. Res. 105, 49–64. doi: 10.1016/j.neures.2015.09.006
Ehninger, D., Han, S., Shilyansky, C., Zhou, Y., Li, W., Kwiatkowski, D. J., et al. (2008). Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 14, 843–848. doi: 10.1038/nm1788
El Helou, J., Belanger-Nelson, E., Freyburger, M., Dorsaz, S., Curie, T., La Spada, F., et al. (2013). Neuroligin-1 links neuronal activity to sleep-wake regulation. Proc. Natl. Acad. Sci. U S A 110, 9974–9979. doi: 10.1073/pnas.1221381110
Elia, J., Gai, X., Xie, H. M., Perin, J. C., Geiger, E., Glessner, J. T., et al. (2010). Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry 15, 637–646. doi: 10.1038/mp.2009.57
Elia, L., Yamamoto, M., Zang, K., and Reichardt, L. (2006). p120 catenin regulates dendritic spine and synapse development through Rho-family GTPases and cadherins. Neuron 51, 43–56. doi: 10.1016/j.neuron.2006.05.018
Etherton, M. R., Blaiss, C. A., Powell, C. M., and Südhof, T. C. (2009). Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc. Natl. Acad. Sci. U S A 106, 17998–18003. doi: 10.1073/pnas.0910297106
Falconer, D. S. (1951). Two new mutants, ‘trembler’ and ‘reeler’, with neurological actions in the house mouse (Mus musculus L.). J. Genet. 50, 192–201. doi: 10.1007/bf02996215
Faridar, A., Jones-Davis, D., Rider, E., Li, J., Gobius, I., Morcom, L., et al. (2014). Mapk/Erk activation in an animal model of social deficits shows a possible link to autism. Mol. Autism 5:57. doi: 10.1186/2040-2392-5-57
Fassio, A., Patry, L., Congia, S., Onofri, F., Piton, A., Gauthier, J., et al. (2011). SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum. Mol. Genet. 20, 2297–2307. doi: 10.1093/hmg/ddr122
Fatemi, S. H., Snow, A. V., Stary, J. M., Araghi-Niknam, M., Reutiman, T. J., Lee, S., et al. (2005). Reelin signaling is impaired in autism. Biol. Psychiatry 57, 777–787. doi: 10.1016/j.biopsych.2004.12.018
Feng, Y., Olson, E. C., Stukenberg, P. T., Flanagan, L. A., Kirschner, M. W., and Walsh, C. A. (2000). LIS1 regulates CNS lamination by interacting with mNudE, a central component of the centrosome. Neuron 28, 665–679. doi: 10.1016/s0896-6273(00)00145-8
Feng, J., Schroer, R., Yan, J., Song, W., Yang, C., Bockholt, A., et al. (2006). High frequency of neurexin 1β signal peptide structural variants in patients with autism. Neurosci. Lett. 409, 10–13. doi: 10.1016/j.neulet.2006.08.017
Feyder, M., Karlsson, R. M., Mathur, P., Lyman, M., Bock, R., Momenan, R., et al. (2010). Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams’ syndrome. Am. J. Psychiatry 167, 1508–1517. doi: 10.1176/appi.ajp.2010.10040484
Filimonenko, M., Isakson, P., Finley, K. D., Anderson, M., Jeong, H., Melia, T. J., et al. (2010). The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 38, 265–279. doi: 10.1016/j.molcel.2010.04.007
Fish, J. L., Kosodo, Y., Enard, W., Pääbo, S., and Huttner, W. B. (2006). Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc. Natl. Acad. Sci. U S A 103, 10438–10443. doi: 10.1073/pnas.0604066103
Floricel, F., Higaki, K., Maki, H., Nanba, E., Ninomiya, H., and Ohno, K. (2007). Antisense suppression of TSC1 gene product, hamartin, enhances neurite outgrowth in NGF-treated PC12h cells. Brain Dev. 29, 502–509. doi: 10.1016/j.braindev.2007.01.007
Folsom, T. D., and Fatemi, S. H. (2013). The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 68, 122–135. doi: 10.1016/j.neuropharm.2012.08.015
Fombonne, E., Roge, B., Claverie, J., Courty, S., and Fremolle, J. (1999). Microcephaly and macrocephaly in autism. J. Autism Dev. Disord. 29, 113–119. doi: 10.1023/A:1023036509476
Fornasiero, E. F., Raimondi, A., Guarnieri, F. C., Orlando, M., Fesce, R., Benfenati, F., et al. (2012). Synapsins contribute to the dynamic spatial organization of synaptic vesicles in an activity-dependent manner. J. Neurosci. 32, 12214–12227. doi: 10.1523/JNEUROSCI.1554-12.2012
Forster, E., Jossin, Y., Zhao, S., Chai, X., Frotscher, M., and Goffinet, A. M. (2006). Recent progress in understanding the role of Reelin in radial neuronal migration, with specific emphasis on the dentate gyrus. Eur. J. Neurosci. 23, 901–909. doi: 10.1111/j.1460-9568.2006.04612.x
Friedman, S. D., Shaw, D. W., Artru, A. A., Dawson, G., Petropoulos, H., and Dager, S. R. (2006). Gray and white matter brain chemistry in young children with autism. Arch. Gen. Psychiatry 63, 786–794. doi: 10.1001/archpsyc.63.7.786
Gao, Z., Lee, P., Stafford, J. M., von Schimmelmann, M., Schaefer, A., and Reinberg, D. (2014). An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516, 349–354. doi: 10.1038/nature13921
Gao, R., and Penzes, P. (2015). Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 15, 146–167. doi: 10.2174/1566524015666150303003028
Gao, P., Sultan, K. T., Zhang, X. J., and Shi, S. H. (2013). Lineage-dependent circuit assembly in the neocortex. Development 140, 2645–2655. doi: 10.1242/dev.087668
Garcia-Castro, M. I., Vielmetter, E., and Bronner-Fraser, M. (2000). N-Cadherin, a cell adhesion molecule involved in establishment of embryonic left-right asymmetry. Science 288, 1047–1051. doi: 10.1126/science.288.5468.1047
Garcia-Junco-Clemente, P., and Golshani, P. (2014). PTEN: a master regulator of neuronal structure, function, and plasticity. Commun. Integr. Biol. 7:e28358. doi: 10.4161/cib.28358
Garg, S., Lehtonen, A., Huson, S. M., Emsley, R., Trump, D., Evans, D. G., et al. (2013). Autism and other psychiatric comorbidity in neurofibromatosis type 1: evidence from a population-based study. Dev. Med. Child Neurol. 55, 139–145. doi: 10.1111/dmcn.12043
Gasquoine, P. G. (2014). Contributions of the insula to cognition and emotion. Neuropsychol. Rev. 24, 77–87. doi: 10.1007/s11065-014-9246-9
Geschwind, D., and Levitt, P. (2007). Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol. 17, 103–111. doi: 10.1016/j.conb.2007.01.009
Ghanem, N., Yu, M., Long, J., Hatch, G., Rubenstein, J. L., and Ekker, M. (2007). Distinct cis-regulatory elements from the Dlx1/Dlx2 locus mark different progenitor cell populations in the ganglionic eminences and different subtypes of adult cortical interneurons. J. Neurosci. 27, 5012–5022. doi: 10.1523/JNEUROSCI.4725-06.2007
Gilbert, J., and Man, H. Y. (2014). Translational dysregulation in autism. Cell Dev. Biol. 3:e124. doi: 10.4172/2168-9296.1000e124
Gilbert, J., and Man, H. Y. (2016). The X-linked autism protein KIAA2022/KIDLIA regulates neurite outgrowth via N-cadherin and delta-catenin signaling. eNeuro 3:ENEURO.0238-16.2016. doi: 10.1523/ENEURO.0238-16.2016
Gilbert, J., Shu, S., Yang, X., Lu, Y., Zhu, L. Q., and Man, H. Y. (2016). β-Amyloid triggers aberrant over-scaling of homeostatic synaptic plasticity. Acta Neuropathol. Commun. 4:131. doi: 10.1186/s40478-017-0423-y
Gilestro, G. F., Tononi, G., and Cirelli, C. (2009). Widespread changes in synaptic markers as a function of sleep and wakefulness in Drosophila. Science 324, 109–112. doi: 10.1126/science.1166673
Gillberg, C., and de Souza, L. (2002). Head circumference in autism, Asperger syndrome and ADHD: a comparative study. Dev. Med. Child Neurol. 44, 296–300. doi: 10.1017/s0012162201002110
Godfraind, J. M., Reyniers, E., De Boulle, K., D’Hooge, R., De Deyn, P. P., Bakker, C. E., et al. (1996). Long-term potentiation in the hippocampus of fragile X knockout mice. Am. J. Med. Genet. 64, 246–251. doi: 10.1002/(SICI)1096-8628(19960809)64:2<246::AID-AJMG2>3.0.CO;2-S
Goffin, A., Hoefsloot, L. H., Bosgoed, E., Swillen, A., and Fryns, J. P. (2001). PTEN mutation in a family with Cowden syndrome and autism. Am. J. Med. Genet. 105, 521–524. doi: 10.1002/ajmg.1477
Gotts, S. J., Simmons, W. K., Milbury, L. A., Wallace, G. L., Cox, R. W., and Martin, A. (2012). Fractionation of social brain circuits in autism spectrum disorders. Brain 135, 2711–2725. doi: 10.1093/brain/aws160
Greco, B., Manago, F., Tucci, V., Kao, H. T., Valtorta, F., and Benfenati, F. (2013). Autism-related behavioral abnormalities in synapsin knockout mice. Behav. Brain Res. 251, 65–74. doi: 10.1016/j.bbr.2012.12.015
Gregory, R. I., Yan, K. P., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N., et al. (2004). The Microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240. doi: 10.1038/nature03120
Grosskreutz, Y., Betz, H., and Kneussel, M. (2003). Rescue of molybdenum cofactor biosynthesis in gephyrin-deficient mice by a Cnx1 transgene. Biochem. Biophys. Res. Commun. 301, 450–455. doi: 10.1016/s0006-291x(02)03062-0
Gruber, R., Zhou, Z., Sukchev, M., Joerss, T., Frappart, P. O., and Wang, Z. Q. (2011). MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat. Cell Biol. 13, 1325–1334. doi: 10.1038/ncb2342
Guilmatre, A., Dubourg, C., Mosca, A. L., Legallic, S., Goldenberg, A., Drouin-Garraud, V., et al. (2009). Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch. Gen. Psychiatry 66, 947–956. doi: 10.1001/archgenpsychiatry.2009.80
Gupta, A., Tsai, L. H., and Wynshaw-Boris, A. (2002). Life is a journey: a genetic look at neocortical development. Nat. Rev. Genet. 3, 342–355. doi: 10.1038/nrg799
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389
Halepoto, D. M., Bashir, S., and A L-Ayadhi, L. (2014). Possible role of brain-derived neurotrophic factor (BDNF) in autism spectrum disorder: current status. J. Coll. Physicians Surg. Pak. 24, 274–278.
Han, W., Kwan, K. Y., Shim, S., Lam, M. M., Shin, Y., Xu, X., et al. (2011). TBR1 directly represses Fezf2 to control the laminar origin and development of the corticospinal tract. Proc. Natl. Acad. Sci. U S A 108, 3041–3046. doi: 10.1073/pnas.1016723108
Happe, F., Ronald, A., and Plomin, R. (2006). Time to give up on a single explanation for autism. Nat. Neurosci. 9, 1218–1220. doi: 10.1038/nn1770
Hatten, M. E. (1999). Central nervous system neuronal migration. Annu. Rev. Neurosci. 22, 511–539. doi: 10.1146/annurev.neuro.22.1.511
Haws, M. E., Jaramillo, T. C., Espinosa, F., Widman, A. J., Stuber, G. D., Sparta, D. R., et al. (2014). PTEN knockdown alters dendritic spine/protrusion morphology, not density. J. Comp. Neurol. 522, 1171–1190. doi: 10.1002/cne.23488
Hazlett, H. C., Poe, M., Gerig, G., Smith, R. G., Provenzale, J., Ross, A., et al. (2005). Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Arch. Gen. Psychiatry 62, 1366–1376. doi: 10.1001/archpsyc.62.12.1366
Hevner, R. F., Shi, L., Justice, N., Hsueh, Y., Sheng, M., Smiga, S., et al. (2001). Tbr1 regulates differentiation of the preplate and layer 6. Neuron 29, 353–366. doi: 10.1016/s0896-6273(01)00211-2
Higgins, J., Midgley, C., Bergh, A. M., Bell, S. M., Askham, J. M., Roberts, E., et al. (2010). Human ASPM participates in spindle organisation, spindle orientation and cytokinesis. BMC Cell Biol. 11:85. doi: 10.1186/1471-2121-11-85
Hinton, V. J., Brown, W. T., Wisniewski, K., and Rudelli, R. D. (1991). Analysis of neocortex in three males with the fragile X syndrome. Am. J. Med. Genet. 41, 289–294. doi: 10.1002/ajmg.1320410306
Hippenmeyer, S., Youn, Y. H., Moon, H. M., Miyamichi, K., Zong, H., Wynshaw-Boris, A., et al. (2010). Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron 68, 695–709. doi: 10.1016/j.neuron.2010.09.027
Hirotsune, S., Takahara, T., Sasaki, N., Hirose, K., Yoshiki, A., Ohashi, T., et al. (1995). The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat. Genet. 10, 77–83. doi: 10.1038/ng0595-77
Hoe, H. S., Lee, K. J., Carney, R. S., Lee, J., Markova, A., Lee, J. Y., et al. (2009). Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J. Neurosci. 29, 7459–7473. doi: 10.1523/JNEUROSCI.4872-08.2009
Hong, S. E., Shugart, Y. Y., Huang, D. T., Shahwan, S. A., Grant, P. E., Hourihane, J. O., et al. (2000). Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 26, 93–96. doi: 10.1038/79246
Hori, K., Nagai, T., Shan, W., Sakamoto, A., Taya, S., Hashimoto, R., et al. (2014). Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep. 9, 2166–2179. doi: 10.1016/j.celrep.2014.11.045
Horton, A. C., and Ehlers, M. D. (2004). Secretory trafficking in neuronal dendrites. Nat. Cell Biol. 6, 585–591. doi: 10.1038/ncb0704-585
Horton, A., Rácz, B., Monson, E., Lin, A., Weinberg, R., and Ehlers, M. (2005). Polarized secretory trafficking directs cargo for asymmetric dendrite growth and morphogenesis. Neuron 48, 757–771. doi: 10.1016/j.neuron.2005.11.005
Hou, Q., Gilbert, J., and Man, H. Y. (2011). Homeostatic regulation of AMPA receptor trafficking and degradation by light-controlled single-synaptic activation. Neuron 72, 806–818. doi: 10.1016/j.neuron.2011.10.011
Hou, Q., Ruan, H., Gilbert, J., Wang, G., Ma, Q., Yao, W. D., et al. (2015). MicroRNA miR124 is required for the expression of homeostatic synaptic plasticity. Nat. Commun. 6:10045. doi: 10.1038/ncomms10045
Hou, Q., Zhang, D., Jarzylo, L., Huganir, R. L., and Man, H. Y. (2008). Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc. Natl. Acad. Sci. U S A 105, 775–780. doi: 10.1073/pnas.0706447105
Huang, Z. (2009). Molecular regulation of neuronal migration during neocortical development. Mol. Cell. Neurosci. 42, 11–22. doi: 10.1016/j.mcn.2009.06.003
Huang, J., and Manning, B. D. (2008). The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J. 412, 179–190. doi: 10.1042/BJ20080281
Huang, X. L., Zou, Y. S., Maher, T. A., Newton, S., and Milunsky, J. M. (2010). A de novo balanced translocation breakpoint truncating the autism susceptibility candidate 2 (AUTS2) gene in a patient with autism. Am. J. Med. Genet. A 152A, 2112–2114. doi: 10.1002/ajmg.a.33497
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U S A 99, 7746–7750. doi: 10.1073/pnas.122205699
Huber, R., Ghilardi, M. F., Massimini, M., and Tononi, G. (2004). Local sleep and learning. Nature 430, 78–81. doi: 10.1038/nature02663
Huguet, G., Ey, E., and Bourgeron, T. (2013). The genetic landscapes of autism spectrum disorders. Annu. Rev. Genomics Hum. Genet. 14, 191–213. doi: 10.1146/annurev-genom-091212-153431
Hussman, J. P. (2001). Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J. Autism Dev. Disord. 31, 247–248. doi: 10.1023/A:1010715619091
Hutsler, J. J., Love, T., and Zhang, H. (2007). Histological and magnetic resonance imaging assessment of cortical layering and thickness in autism spectrum disorders. Biol. Psychiatry 61, 449–457. doi: 10.1016/j.biopsych.2006.01.015
Hutsler, J. J., and Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. doi: 10.1016/j.brainres.2009.09.120
Ifrim, M. F., Williams, K. R., and Bassell, G. J. (2015). Single-molecule imaging of PSD-95 mRNA translation in dendrites and its dysregulation in a mouse model of Fragile X syndrome. J. Neurosci. 35, 7116–7130. doi: 10.1523/JNEUROSCI.2802-14.2015
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Irwin, S. A., Idupulapati, M., Gilbert, M. E., Harris, J. B., Chakravarti, A. B., Rogers, E. J., et al. (2002). Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am. J. Med. Genet. 111, 140–146. doi: 10.1002/ajmg.10500
Irwin, S. A., Patel, B., Idupulapati, M., Harris, J. B., Crisostomo, R. A., Larsen, B. P., et al. (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med. Genet. 98, 161–167. doi: 10.1002/1096-8628(20010115)98:2<161::AID-AJMG1025>3.3.CO;2-2
Ishikawa, T., Miyata, S., Koyama, Y., Yoshikawa, K., Hattori, T., Kumamoto, N., et al. (2012). Transient expression of Xpn, an XLMR protein related to neurite extension, during brain development and participation in neurite outgrowth. Neuroscience 214, 181–191. doi: 10.1016/j.neuroscience.2012.04.030
Jackman, C., Horn, N. D., Molleston, J. P., and Sokol, D. K. (2009). Gene associated with seizures, autism and hepatomegaly in an Amish girl. Pediatr. Neurol. 40, 310–313. doi: 10.1016/j.pediatrneurol.2008.10.013
Jackson, A. P., Eastwood, H., Bell, S. M., Adu, J., Toomes, C., Carr, I. M., et al. (2002). Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 71, 136–142. doi: 10.1086/341283
Jackson, A. P., McHale, D. P., Campbell, D. A., Jafri, H., Rashid, Y., Mannan, J., et al. (1998). Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am. J. Hum. Genet. 63, 541–546. doi: 10.1086/301966
Jamain, S., Quach, H., Betancur, C., Råstam, M., Colineaux, C., Gillberg, I., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Jan, Y. N., and Jan, L. Y. (2010). Branching out: mechanisms of dendritic arborization. Nat. Rev. Neurosci. 11, 316–328. doi: 10.1038/nrn2836
Jang, S. W., Liu, X., Pradoldej, S., Tosini, G., Chang, Q., Iuvone, P. M., et al. (2010). N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. U S A 107, 3876–3881. doi: 10.1073/pnas.0912531107
Jiang, M., Ash, R. T., Baker, S. A., Suter, B., Ferguson, A., Park, J., et al. (2013). Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J. Neurosci. 33, 19518–19533. doi: 10.1523/JNEUROSCI.1745-13.2013
Jossin, Y., and Goffinet, A. M. (2007). Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol. Cell. Biol. 27, 7113–7124. doi: 10.1128/mcb.00928-07
Jugloff, D. G., Jung, B. P., Purushotham, D., Logan, R., and Eubanks, J. H. (2005). Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol. Dis. 19, 18–27. doi: 10.1016/j.nbd.2004.11.002
Kaindl, A. M., Passemard, S., Kumar, P., Kraemer, N., Issa, L., Zwirner, A., et al. (2010). Many roads lead to primary autosomal recessive microcephaly. Prog. Neurobiol. 90, 363–383. doi: 10.1016/j.pneurobio.2009.11.002
Kapitein, L. C., Schlager, M. A., Kuijpers, M., Wulf, P. S., van Spronsen, M., MacKintosh, F. C., et al. (2010). Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr. Biol. 20, 290–299. doi: 10.1016/j.cub.2009.12.052
Kasai, H., Matsuzaki, M., Noguchi, J., Yasumatsu, N., and Nakahara, H. (2003). Structure-stability-function relationships of dendritic spines. Trends Neurosci. 26, 360–368. doi: 10.1016/s0166-2236(03)00162-0
Kasarpalkar, N. J., Kothari, S. T., and Dave, U. P. (2014). Brain-derived neurotrophic factor in children with autism spectrum disorder. Ann. Neurosci. 21, 129–133. doi: 10.5214/ans.0972.7531.210403
Katayama, Y., Nishiyama, M., Shoji, H., Ohkawa, Y., Kawamura, A., Sato, T., et al. (2016). CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 537, 675–679. doi: 10.1038/nature19357
Kelleher, R., and Bear, M. (2008). The autistic neuron: troubled translation? Cell 135, 401–406. doi: 10.1016/j.cell.2008.10.017
Ketzef, M., and Gitler, D. (2014). Epileptic synapsin triple knockout mice exhibit progressive long-term aberrant plasticity in the entorhinal cortex. Cereb. Cortex 24, 996–1008. doi: 10.1093/cercor/bhs384
Kishi, N., and Macklis, J. D. (2004). MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 27, 306–321. doi: 10.1016/j.mcn.2004.07.006
Kitagawa, M., Umezu, M., Aoki, J., Koizumi, H., Arai, H., and Inoue, K. (2000). Direct association of LIS1, the lissencephaly gene product, with a mammalian homologue of a fungal nuclear distribution protein, rNUDE. FEBS Lett. 479, 57–62. doi: 10.1016/s0014-5793(00)01856-1
Kneussel, M., Brandstatter, J. H., Laube, B., Stahl, S., Muller, U., and Betz, H. (1999). Loss of postsynaptic GABAA receptor clustering in gephyrin-deficient mice. J. Neurosci. 19, 9289–9297.
Kobayashi, T., Minowa, O., Sugitani, Y., Takai, S., Mitani, H., Kobayashi, E., et al. (2001). A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc. Natl. Acad. Sci. U S A 98, 8762–8767. doi: 10.1073/pnas.151033798
Koekkoek, S. K., Yamaguchi, K., Milojkovic, B. A., Dortland, B. R., Ruigrok, T. J., Maex, R., et al. (2005). Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 47, 339–352. doi: 10.1016/j.neuron.2005.07.005
Krawczun, M. S., Jenkins, E. C., and Brown, W. T. (1985). Analysis of the fragile-X chromosome: localization and detection of the fragile site in high resolution preparations. Hum. Genet. 69, 209–211. doi: 10.1007/bf00293026
Kriegstein, A. R., and Noctor, S. C. (2004). Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 27, 392–399. doi: 10.1016/j.tins.2004.05.001
Kuhn, M., Wolf, E., Maier, J. G., Mainberger, F., Feige, B., Schmid, H., et al. (2016). Sleep recalibrates homeostatic and associative synaptic plasticity in the human cortex. Nat. Commun. 7:12455. doi: 10.1038/ncomms12455
Kuroda, Y., Ohashi, I., Naruto, T., Ida, K., Enomoto, Y., Saito, T., et al. (2015). Delineation of the KIAA2022 mutation phenotype: two patients with X-linked intellectual disability and distinctive features. Am. J. Med. Genet. A 167, 1349–1353. doi: 10.1002/ajmg.a.37002
Kwiatkowski, D. J., and Manning, B. D. (2005). Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum. Mol. Genet. 14, R251–R258. doi: 10.1093/hmg/ddi260
Kwon, C.-H., Luikart, B., Powell, C., Zhou, J., Matheny, S., Zhang, W., et al. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. doi: 10.1016/j.neuron.2006.03.023
Lainhart, J. E., Piven, J., Wzorek, M., Landa, R., Santangelo, S. L., Coon, H., et al. (1997). Macrocephaly in children and adults with autism. J. Am. Acad. Child Adolesc. Psychiatry 36, 282–290. doi: 10.1097/00004583-199702000-00019
Lanctot, A. A., Peng, C. Y., Pawlisz, A. S., Joksimovic, M., and Feng, Y. (2013). Spatially dependent dynamic MAPK modulation by the Nde1-Lis1-Brap complex patterns mammalian CNS. Dev. Cell 25, 241–255. doi: 10.1016/j.devcel.2013.04.006
Landa, R. J. (2008). Diagnosis of autism spectrum disorders in the first 3 years of life. Nat. Clin. Pract. Neurol. 4, 138–147. doi: 10.1038/ncpneuro0731
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Larson, J., Jessen, R. E., Kim, D., Fine, A. K., and du Hoffmann, J. (2005). Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J. Neurosci. 25, 9460–9469. doi: 10.1523/jneurosci.2638-05.2005
Lauterborn, J. C., Rex, C. S., Kramár, E., Chen, L. Y., Pandyarajan, V., Lynch, G., et al. (2007). Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J. Neurosci. 27, 10685–10694. doi: 10.1523/jneurosci.2624-07.2007
Lavdas, A. A., Grigoriou, M., Pachnis, V., and Parnavelas, J. G. (1999). The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J. Neurosci. 19, 7881–7888.
Lee, A., Li, W., Xu, K., Bogert, B. A., Su, K., and Gao, F. B. (2003). Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development 130, 5543–5552. doi: 10.1242/dev.00792
Lesch, K. P., Timmesfeld, N., Renner, T. J., Halperin, R., Roser, C., Nguyen, T. T., et al. (2008). Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J. Neural Transm. 115, 1573–1585. doi: 10.1007/s00702-008-0119-3
Letinic, K., Zoncu, R., and Rakic, P. (2002). Origin of GABAergic neurons in the human neocortex. Nature 417, 645–649. doi: 10.1038/nature00779
Levitt, P., and Campbell, D. B. (2009). The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J. Clin. Invest. 119, 747–754. doi: 10.1172/jci37934
Li, J., Pelletier, M. R., Perez Velazquez, J. L., and Carlen, P. L. (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 19, 138–151. doi: 10.1006/mcne.2001.1085
Li, Z., Van Aelst, L., and Cline, H. T. (2000). Rho GTPases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat. Neurosci. 3, 217–225. doi: 10.1038/72920
Lim, S., Naisbitt, S., Yoon, J., Hwang, J. I., Suh, P. G., Sheng, M., et al. (1999). Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J. Biol. Chem. 274, 29510–29518. doi: 10.1074/jbc.274.41.29510
Lionel, A. C., Crosbie, J., Barbosa, N., Goodale, T., Thiruvahindrapuram, B., Rickaby, J., et al. (2011). Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci. Transl. Med. 3:95ra75. doi: 10.1126/scitranslmed.3002464
Lionel, A. C., Tammimies, K., Vaags, A. K., Rosenfeld, J. A., Ahn, J. W., Merico, D., et al. (2014). Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum. Mol. Genet. 23, 2752–2768. doi: 10.1093/hmg/ddt669
Lionel, A. C., Vaags, A. K., Sato, D., Gazzellone, M. J., Mitchell, E. B., Chen, H. Y., et al. (2013). Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum. Mol. Genet. 22, 2055–2066. doi: 10.1093/hmg/ddt056
Liu, Y., Zhao, D., Dong, R., Yang, X., Zhang, Y., Tammimies, K., et al. (2015). De novo exon 1 deletion of AUTS2 gene in a patient with autism spectrum disorder and developmental delay: a case report and a brief literature review. Am. J. Med. Genet. A 167, 1381–1385. doi: 10.1002/ajmg.a.37050
Lu, Q., Paredes, M., Medina, M., and Zhou, J. (1999). Δ-Catenin, an adhesive junction-associated protein which promotes cell scattering. J. Cell Biol. 144, 519–532.
Lu, Y., Wang, F., Li, Y., Ferris, J., Lee, J. A., and Gao, F. B. (2009). The Drosophila homologue of the Angelman syndrome ubiquitin ligase regulates the formation of terminal dendritic branches. Hum. Mol. Genet. 18, 454–462. doi: 10.1093/hmg/ddn373
Mabb, A. M., Judson, M. C., Zylka, M. J., and Philpot, B. D. (2011). Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34, 293–303. doi: 10.1016/j.tins.2011.04.001
MacLaurin, S. A., Krucker, T., and Fish, K. N. (2007). Hippocampal dendritic arbor growth in vitro: regulation by Reelin-Disabled-1 signaling. Brain Res. 1172, 1–9. doi: 10.1016/j.brainres.2007.07.035
Major, M. B., Roberts, B. S., Berndt, J. D., Marine, S., Anastas, J., Chung, N., et al. (2008). New regulators of Wnt/β-catenin signaling revealed by integrative molecular screening. Sci. Signal. 1:ra12. doi: 10.1126/scisignal.2000037
Marín, O., and Rubenstein, J. L. (2001). A long, remarkable journey: tangential migration in the telencephalon. Nat. Rev. Neurosci. 2, 780–790. doi: 10.1038/35097509
Martinez, M., Ochiishi, T., Majewski, M., and Kosik, K. S. (2003). Dual regulation of neuronal morphogenesis by a δ-catenin-cortactin complex and Rho. J. Cell Biol. 162, 99–111. doi: 10.1083/jcb.200211025
Matsuki, T., Matthews, R. T., Cooper, J. A., van der Brug, M. P., Cookson, M. R., Hardy, J. A., et al. (2010). Reelin and stk25 have opposing roles in neuronal polarization and dendritic Golgi deployment. Cell 143, 826–836. doi: 10.1016/j.cell.2010.10.029
Maximo, J. O., Cadena, E. J., and Kana, R. K. (2014). The implications of brain connectivity in the neuropsychology of autism. Neuropsychol. Rev. 24, 16–31. doi: 10.1007/s11065-014-9250-0
McCarthy, S. E., Makarov, V., Kirov, G., Addington, A. M., McClellan, J., Yoon, S., et al. (2009). Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 41, 1223–1227. doi: 10.1038/ng.474
McKenney, R. J., Vershinin, M., Kunwar, A., Vallee, R. B., and Gross, S. P. (2010). LIS1 and NudE induce a persistent dynein force-producing state. Cell 141, 304–314. doi: 10.1016/j.cell.2010.02.035
McKinney, B. C., Grossman, A. W., Elisseou, N. M., and Greenough, W. T. (2005). Dendritic spine abnormalities in the occipital cortex of C57BL/6 Fmr1 knockout mice. Am. J. Med. Genet. B Neuropsychiatr. Genet. 136B, 98–102. doi: 10.1002/ajmg.b.30183
Mefford, H. C., Muhle, H., Ostertag, P., von Spiczak, S., Buysse, K., Baker, C., et al. (2010). Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 6:e1000962. doi: 10.1371/journal.pgen.1000962
Miao, S., Chen, R., Ye, J., Tan, G. H., Li, S., Zhang, J., et al. (2013). The Angelman syndrome protein Ube3a is required for polarized dendrite morphogenesis in pyramidal neurons. J. Neurosci. 33, 327–333. doi: 10.1523/jneurosci.2509-12.2013
Michalon, A., Sidorov, M., Ballard, T. M., Ozmen, L., Spooren, W., Wettstein, J. G., et al. (2012). Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56. doi: 10.1016/j.neuron.2012.03.009
Miles, J. H., Takahashi, T. N., Bagby, S., Sahota, P. K., Vaslow, D. F., Wang, C. H., et al. (2005). Essential versus complex autism: definition of fundamental prognostic subtypes. Am. J. Med. Genet. A 135, 171–180. doi: 10.1002/ajmg.a.30590
Ming, G. L., and Song, H. (2005). Adult neurogenesis in the mammalian central nervous system. Annu. Rev. Neurosci. 28, 223–250. doi: 10.1146/annurev.neuro.28.051804.101459
Missler, M., Zhang, W., Rohlmann, A., Kattenstroth, G., Hammer, R. E., Gottmann, K., et al. (2003). α-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423, 939–948. doi: 10.1038/nature01755
Morgan, J. T., Barger, N., Amaral, D. G., and Schumann, C. M. (2014). Stereological study of amygdala glial populations in adolescents and adults with autism spectrum disorder. PLoS One 9:e110356. doi: 10.1371/journal.pone.0110356
Morrow, E. M., Yoo, S. Y., Flavell, S. W., Kim, T. K., Lin, Y., Hill, R. S., et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223. doi: 10.1126/science.1157657
Nadarajah, B., and Parnavelas, J. G. (2002). Modes of neuronal migration in the developing cerebral cortex. Nat. Rev. Neurosci. 3, 423–432. doi: 10.1038/nrn845
Nakayama, A. Y., Harms, M. B., and Luo, L. (2000). Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J. Neurosci. 20, 5329–5338.
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Nery, S., Fishell, G., and Corbin, J. G. (2002). The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat. Neurosci. 5, 1279–1287. doi: 10.1038/nn971
Nichols, A. J., and Olson, E. C. (2010). Reelin promotes neuronal orientation and dendritogenesis during preplate splitting. Cereb. Cortex 20, 2213–2223. doi: 10.1093/cercor/bhp303
Niethammer, M., Smith, D. S., Ayala, R., Peng, J., Ko, J., Lee, M. S., et al. (2000). NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 28, 697–711. doi: 10.1016/s0896-6273(00)00147-1
Niu, S., Renfro, A., Quattrocchi, C. C., Sheldon, M., and D’Arcangelo, G. (2004). Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron 41, 71–84. doi: 10.1016/s0896-6273(03)00819-5
Noor, A., Dupuis, L., Mittal, K., Lionel, A. C., Marshall, C. R., Scherer, S. W., et al. (2015). 15q11.2 Duplication encompassing only the UBE3A gene is associated with developmental delay and neuropsychiatric phenotypes. Hum. Mutat. 36, 689–693. doi: 10.1002/humu.22800
Nowicki, S. T., Tassone, F., Ono, M. Y., Ferranti, J., Croquette, M. F., Goodlin-Jones, B., et al. (2007). The Prader-Willi phenotype of fragile X syndrome. J. Dev. Behav. Pediatr. 28, 133–138. doi: 10.1097/01.DBP.0000267563.18952.c9
Ogawa, S., Kwon, C. H., Zhou, J., Koovakkattu, D., Parada, L. F., and Sinton, C. M. (2007). A seizure-prone phenotype is associated with altered free-running rhythm in Pten mutant mice. Brain Res. 1168, 112–123. doi: 10.1016/j.brainres.2007.06.074
Orloff, M. S., He, X., Peterson, C., Chen, F., Chen, J. L., Mester, J. L., et al. (2013). Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am. J. Hum. Genet. 92, 76–80. doi: 10.1016/j.ajhg.2012.10.021
Orosco, L. A., Ross, A. P., Cates, S. L., Scott, S. E., Wu, D., Sohn, J., et al. (2014). Loss of Wdfy3 in mice alters cerebral cortical neurogenesis reflecting aspects of the autism pathology. Nat. Commun. 5:4692. doi: 10.1038/ncomms5692
Osterweil, E. K., Krueger, D. D., Reinhold, K., and Bear, M. F. (2010). Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 30, 15616–15627. doi: 10.1523/jneurosci.3888-10.2010
Ozgen, H. M., van Daalen, E., Bolton, P. F., Maloney, V. K., Huang, S., Cresswell, L., et al. (2009). Copy number changes of the microcephalin 1 gene (MCPH1) in patients with autism spectrum disorders. Clin. Genet. 76, 348–356. doi: 10.1111/j.1399-0004.2009.01254.x
Pagan, C., Delorme, R., Callebert, J., Goubran-Botros, H., Amsellem, F., Drouot, X., et al. (2014). The serotonin-N-acetylserotonin-melatonin pathway as a biomarker for autism spectrum disorders. Transl. Psychiatry 4:e479. doi: 10.1038/tp.2014.120
Panganiban, G., and Rubenstein, J. L. (2002). Developmental functions of the Distal-less/Dlx homeobox genes. Development 129, 4371–4386.
Pardo, C. A., and Eberhart, C. G. (2007). The neurobiology of autism. Brain Pathol. 17, 434–447. doi: 10.1111/j.1750-3639.2007.00102.x
Parikshak, N. N., Luo, R., Zhang, A., Won, H., Lowe, J. K., Chandran, V., et al. (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. doi: 10.1016/j.cell.2013.10.031
Passemard, S., Titomanlio, L., Elmaleh, M., Afenjar, A., Alessandri, J. L., Andria, G., et al. (2009). Expanding the clinical and neuroradiologic phenotype of primary microcephaly due to ASPM mutations. Neurology 73, 962–969. doi: 10.1212/wnl.0b013e3181b8799a
Passemard, S., Verloes, A., Billette de Villemeur, T., Boespflug-Tanguy, O., Hernandez, K., Laurent, M., et al. (2016). Abnormal spindle-like microcephaly-associated (ASPM) mutations strongly disrupt neocortical structure but spare the hippocampus and long-term memory. Cortex 74, 158–176. doi: 10.1016/j.cortex.2015.10.010
Pathania, M., Davenport, E. C., Muir, J., Sheehan, D. F., López-Doménech, G., and Kittler, J. T. (2014). The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl. Psychiatry 4:e374. doi: 10.1038/tp.2014.36
Pawlisz, A. S., and Feng, Y. (2011). Three-dimensional regulation of radial glial functions by Lis1-Nde1 and dystrophin glycoprotein complexes. PLoS Biol. 9:e1001172. doi: 10.1371/journal.pbio.1001172
Pawlisz, A. S., Mutch, C., Wynshaw-Boris, A., Chenn, A., Walsh, C. A., and Feng, Y. (2008). Lis1-Nde1-dependent neuronal fate control determines cerebral cortical size and lamination. Hum. Mol. Genet. 17, 2441–2455. doi: 10.1093/hmg/ddn144
Peça, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Pelphrey, K. A., and Carter, E. J. (2008). Brain mechanisms for social perception: lessons from autism and typical development. Ann. N Y Acad. Sci. 1145, 283–299. doi: 10.1196/annals.1416.007
Peñagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H., et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. doi: 10.1016/j.cell.2011.08.040
Persico, A., and Bourgeron, T. (2006). Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 29, 349–358. doi: 10.1016/j.tins.2006.05.010
Persico, A. M., D’Agruma, L., Maiorano, N., Totaro, A., Militerni, R., Bravaccio, C., et al. (2001). Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry 6, 150–159. doi: 10.1038/sj.mp.4000850
Pescosolido, M. F., Yang, U., Sabbagh, M., and Morrow, E. M. (2012). Lighting a path: genetic studies pinpoint neurodevelopmental mechanisms in autism and related disorders. Dialogues Clin. Neurosci. 14, 239–252.
Peters, S. U., Beaudet, A. L., Madduri, N., and Bacino, C. A. (2004). Autism in Angelman syndrome: implications for autism research. Clin. Genet. 66, 530–536. doi: 10.1111/j.1399-0004.2004.00362.x
Phelan, M. C. (2008). Deletion 22q13.3 syndrome. Orphanet J. Rare Dis. 3:14. doi: 10.1186/1750-1172-3-14
Phillips, M., and Pozzo-Miller, L. (2015). Dendritic spine dysgenesis in autism related disorders. Neurosci. Lett. 601, 30–40. doi: 10.1016/j.neulet.2015.01.011
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi: 10.1038/nature09146
Poliak, S., Gollan, L., Martinez, R., Custer, A., Einheber, S., Salzer, J. L., et al. (1999). Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24, 1037–1047. doi: 10.1016/s0896-6273(00)81049-1
Polleux, F., Whitford, K. L., Dijkhuizen, P. A., Vitalis, T., and Ghosh, A. (2002). Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development 129, 3147–3160.
Poluch, S., and Juliano, S. L. (2007). A normal radial glial scaffold is necessary for migration of interneurons during neocortical development. Glia 55, 822–830. doi: 10.1002/glia.20488
Poulopoulos, A., Aramuni, G., Meyer, G., Soykan, T., Hoon, M., Papadopoulos, T., et al. (2009). Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 63, 628–642. doi: 10.1016/j.neuron.2009.08.023
Pozo, K., and Goda, Y. (2010). Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351. doi: 10.1016/j.neuron.2010.04.028
Pulvers, J. N., Bryk, J., Fish, J. L., Wilsch-Bräuninger, M., Arai, Y., Schreier, D., et al. (2010). Mutations in mouse Aspm (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline. Proc. Natl. Acad. Sci. U S A 107, 16595–16600. doi: 10.1073/pnas.1010494107
Qiu, Z., Sylwestrak, E. L., Lieberman, D. N., Zhang, Y., Liu, X. Y., and Ghosh, A. (2012). The Rett syndrome protein MeCP2 regulates synaptic scaling. J. Neurosci. 32, 989–994. doi: 10.1523/jneurosci.0175-11.2012
Rakic, P. (1972). Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol. 145, 61–83. doi: 10.1002/cne.901450105
Rakic, S., and Zecevic, N. (2000). Programmed cell death in the developing human telencephalon. Eur. J. Neurosci. 12, 2721–2734. doi: 10.1046/j.1460-9568.2000.00153.x
Rauen, K. A., Schoyer, L., McCormick, F., Lin, A. E., Allanson, J. E., Stevenson, D. A., et al. (2010). Proceedings from the 2009 genetic syndromes of the Ras/MAPK pathway: from bedside to bench and back. Am. J. Med. Genet. A 152A, 4–24. doi: 10.1002/ajmg.a.33183
Redcay, E. (2008). The superior temporal sulcus performs a common function for social and speech perception: implications for the emergence of autism. Neurosci. Biobehav. Rev. 32, 123–142. doi: 10.1016/j.neubiorev.2007.06.004
Redies, C., Hertel, N., and Hubner, C. A. (2012). Cadherins and neuropsychiatric disorders. Brain Res. 1470, 130–144. doi: 10.1016/j.brainres.2012.06.020
Redmond, L., Kashani, A. H., and Ghosh, A. (2002). Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34, 999–1010. doi: 10.1016/s0896-6273(02)00737-7
Reiner, O. (2000). LIS1. Let’s interact sometimes… (part 1). Neuron 28, 633–636. doi: 10.1016/S0896-6273(00)00142-2
Reiner, O., Carrozzo, R., Shen, Y., Wehnert, M., Faustinella, F., Dobyns, W. B., et al. (1993). Isolation of a Miller-Dieker lissencephaly gene containing G protein β-subunit-like repeats. Nature 364, 717–721. doi: 10.1136/jmg.30.11.972-c
Reiner, O., Karzbrun, E., Kshirsagar, A., and Kaibuchi, K. (2016). Regulation of neuronal migration, an emerging topic in autism spectrum disorders. J. Neurochem. 136, 440–456. doi: 10.1111/jnc.13403
Restivo, L., Ferrari, F., Passino, E., Sgobio, C., Bock, J., Oostra, B. A., et al. (2005). Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc. Natl. Acad. Sci. U S A 102, 11557–11562. doi: 10.1073/pnas.0504984102
Richards, M. W., Leung, J. W., Roe, S. M., Li, K., Chen, J., and Bayliss, R. (2010). A pocket on the surface of the N-terminal BRCT domain of Mcph1 is required to prevent abnormal chromosome condensation. J. Mol. Biol. 395, 908–915. doi: 10.1016/j.jmb.2009.11.029
Roberts, E., Hampshire, D. J., Pattison, L., Springell, K., Jafri, H., Corry, P., et al. (2002). Autosomal recessive primary microcephaly: an analysis of locus heterogeneity and phenotypic variation. J. Med. Genet. 39, 718–721. doi: 10.1136/jmg.39.10.718
Rosahl, T. W., Spillane, D., Missler, M., Herz, J., Selig, D. K., Wolff, J. R., et al. (1995). Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375, 488–493. doi: 10.1038/375488a0
Rosser, T. L., and Packer, R. J. (2003). Neurocognitive dysfunction in children with neurofibromatosis type 1. Curr. Neurol. Neurosci. Rep. 3, 129–136. doi: 10.1007/s11910-003-0064-3
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183x.2003.00037.x
Rudelli, R. D., Brown, W. T., Wisniewski, K., Jenkins, E. C., Laure-Kamionowska, M., Connell, F., et al. (1985). Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol. 67, 289–295. doi: 10.1007/bf00687814
Sacco, R., Militerni, R., Frolli, A., Bravaccio, C., Gritti, A., Elia, M., et al. (2007). Clinical, morphological, and biochemical correlates of head circumference in autism. Biol. Psychiatry 62, 1038–1047. doi: 10.1016/j.biopsych.2007.04.039
Salinger, W. L., Ladrow, P., and Wheeler, C. (2003). Behavioral phenotype of the reeler mutant mouse: effects of RELN gene dosage and social isolation. Behav. Neurosci. 117, 1257–1275. doi: 10.1037/0735-7044.117.6.1257
Sanders, S. J., Ercan-Sencicek, A. G., Hus, V., Luo, R., Murtha, M. T., Moreno-De-Luca, D., et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7Q12.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. doi: 10.1016/j.neuron.2011.05.002
Santoro, M. R., Bray, S. M., and Warren, S. T. (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 7, 219–245. doi: 10.1146/annurev-pathol-011811-132457
Sasaki, S., Shionoya, A., Ishida, M., Gambello, M. J., Yingling, J., Wynshaw-Boris, A., et al. (2000). A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron 28, 681–696. doi: 10.1016/s0896-6273(00)00146-x
Schnell, E., Bensen, A. L., Washburn, E. K., and Westbrook, G. L. (2012). Neuroligin-1 overexpression in newborn granule cells in vivo. PLoS One 7:e48045. doi: 10.1371/journal.pone.0048045
Schumann, C. M., Bloss, C. S., Barnes, C. C., Wideman, G. M., Carper, R. A., Akshoomoff, N., et al. (2010). Longitudinal magnetic resonance imaging study of cortical development through early childhood in autism. J. Neurosci. 30, 4419–4427. doi: 10.1523/jneurosci.5714-09.2010
Sekine, K., Kubo, K., and Nakajima, K. (2014). How does Reelin control neuronal migration and layer formation in the developing mammalian neocortex? Neurosci. Res. 86, 50–58. doi: 10.1016/j.neures.2014.06.004
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., et al. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254. doi: 10.1016/s0896-6273(02)00768-7
Shim, S. Y., Wang, J., Asada, N., Neumayer, G., Tran, H. C., Ishiguro, K., et al. (2008). Protein 600 is a microtubule/endoplasmic reticulum-associated protein in CNS neurons. J. Neurosci. 28, 3604–3614. doi: 10.1523/jneurosci.5278-07.2008
Skalecka, A., Liszewska, E., Bilinski, R., Gkogkas, C., Khoutorsky, A., Malik, A. R., et al. (2016). mTOR kinase is needed for the development and stabilization of dendritic arbors in newly born olfactory bulb neurons. Dev. Neurobiol. 76, 1308–1327. doi: 10.1002/dneu.22392
Smith, S. E., Zhou, Y. D., Zhang, G., Jin, Z., Stoppel, D. C., and Anderson, M. P. (2011). Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 3:103ra97. doi: 10.1126/scitranslmed.3002627
Soden, M. E., and Chen, L. (2010). Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J. Neurosci. 30, 16910–16921. doi: 10.1523/jneurosci.3660-10.2010
Srinivasan, K., Leone, D. P., Bateson, R. K., Dobreva, G., Kohwi, Y., Kohwi-Shigematsu, T., et al. (2012). A network of genetic repression and derepression specifies projection fates in the developing neocortex. Proc. Natl. Acad. Sci. U S A 109, 19071–19078. doi: 10.1073/pnas.1216793109
Steffenburg, S., Gillberg, C. L., Steffenburg, U., and Kyllerman, M. (1996). Autism in Angelman syndrome: a population-based study. Pediatr. Neurol. 14, 131–136. doi: 10.1016/0887-8994(96)00011-2
Stiles, J., and Jernigan, T. L. (2010). The basics of brain development. Neuropsychol. Rev. 20, 327–348. doi: 10.1007/s11065-010-9148-4.
Stoner, R., Chow, M. L., Boyle, M. P., Sunkin, S. M., Mouton, P. R., Roy, S., et al. (2014). Patches of disorganization in the neocortex of children with autism. N Engl J. Med. 370, 1209–1219. doi: 10.1056/NEJMoa1307491
Stone, J. L., O’Donovan, M. C., Gurling, H., Kirov, G. K., Blackwood, D. H. R., Corvin, A., et al. (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241. doi: 10.1038/nature07239
Strauss, K. A., Puffenberger, E. G., Huentelman, M. J., Gottlieb, S., Dobrin, S. E., Parod, J. M., et al. (2006). Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J. Med. 354, 1370–1377. doi: 10.1056/NEJMoa052773
Strømme, P., Mangelsdorf, M. E., Scheffer, I. E., and Gecz, J. (2002). Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev. 24, 266–268. doi: 10.1016/s0387-7604(02)00079-7
Stuss, D. P., Boyd, J. D., Levin, D. B., and Delaney, K. R. (2012). MeCP2 mutation results in compartment-specific reductions in dendritic branching and spine density in layer 5 motor cortical neurons of YFP-H mice. PLoS One 7:e31896. doi: 10.1371/journal.pone.0031896
Sultana, R., Yu, C. E., Yu, J., Munson, J., Chen, D., Hua, W., et al. (2002). Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics 80, 129–134. doi: 10.1006/geno.2002.6810
Sundaram, S. K., Huq, A. M., Wilson, B. J., and Chugani, H. T. (2010). Tourette syndrome is associated with recurrent exonic copy number variants. Neurology 74, 1583–1590. doi: 10.1212/wnl.0b013e3181e0f147
Suvrathan, A., Hoeffer, C. A., Wong, H., Klann, E., and Chattarji, S. (2010). Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. U S A 107, 11591–11596. doi: 10.1073/pnas.1002262107
Tahirovic, S., and Bradke, F. (2009). Neuronal polarity. Cold Spring Harb. Perspect. Biol. 1:a001644. doi: 10.1101/cshperspect.a001644
Talkowski, M. E., Rosenfeld, J. A., Blumenthal, I., Pillalamarri, V., Chiang, C., Heilbut, A., et al. (2012). Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149, 525–537. doi: 10.1016/j.cell.2012.03.028
Tan, Z.-J., Peng, Y., Song, H.-L., Zheng, J.-J., and Yu, X. (2010). N-cadherin-dependent neuron-neuron interaction is required for the maintenance of activity-induced dendrite growth. Proc. Natl. Acad. Sci. U S A 107, 9873–9878. doi: 10.1073/pnas.1003480107
Tau, G. Z., and Peterson, B. S. (2010). Normal development of brain circuits. Neuropsychopharmacology 35, 147–168. doi: 10.1038/npp.2009.115
Tavazoie, S. F., Alvarez, V. A., Ridenour, D. A., Kwiatkowski, D. J., and Sabatini, B. L. (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734. doi: 10.1038/nn1566
Thomanetz, V., Angliker, N., Cloetta, D., Lustenberger, R. M., Schweighauser, M., Oliveri, F., et al. (2013). Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 201, 293–308. doi: 10.1083/jcb.201205030
Thornton, G. K., and Woods, C. G. (2009). Primary microcephaly: do all roads lead to Rome? Trends Genet. 25, 501–510. doi: 10.1016/j.tig.2009.09.011
Toma, C., Hervás, A., Torrico, B., Balmaña, N., Salgado, M., Maristany, M., et al. (2013). Analysis of two language-related genes in autism: a case-control association study of FOXP2 and CNTNAP2. Psychiatr. Genet. 23, 82–85. doi: 10.1097/ypg.0b013e32835d6fc6
Tononi, G., and Cirelli, C. (2003). Sleep and synaptic homeostasis: a hypothesis. Brain Res. Bull. 62, 143–150. doi: 10.1016/j.brainresbull.2003.09.004
Toro, R., Konyukh, M., Delorme, R., Leblond, C., Chaste, P., Fauchereau, F., et al. (2010). Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 26, 363–372. doi: 10.1016/j.tig.2010.05.007
Trachtenberg, J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E., et al. (2002). Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794. doi: 10.1038/nature01273
Tsai, J. W., Bremner, K. H., and Vallee, R. B. (2007). Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat. Neurosci. 10, 970–979. doi: 10.1038/nn1934
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. doi: 10.1038/nature11310
Tsai, N. P., Wilkerson, J. R., Guo, W., Maksimova, M. A., DeMartino, G. N., Cowan, C. W., et al. (2012). Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 151, 1581–1594. doi: 10.1016/j.cell.2012.11.040
Tu, J. C., Xiao, B., Naisbitt, S., Yuan, J. P., Petralia, R. S., Brakeman, P., et al. (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592. doi: 10.1016/s0896-6273(00)80810-7
Tunca, Y., Vurucu, S., Parma, J., Akin, R., Désir, J., Baser, I., et al. (2006). Prenatal diagnosis of primary microcephaly in two consanguineous families by confrontation of morphometry with DNA data. Prenat. Diagn. 26, 449–453. doi: 10.1002/pd.1434
Turner, G., Partington, M., Kerr, B., Mangelsdorf, M., and Gecz, J. (2002). Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identical ARX gene mutation. Am. J. Med. Genet. 112, 405–411. doi: 10.1002/ajmg.10714
Turner, T. N., Sharma, K., Oh, E. C., Liu, Y. P., Collins, R. L., Sosa, M. X., et al. (2015). Loss of delta-catenin function in severe autism. Nature 520, 51–56. doi: 10.1038/nature14186
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896. doi: 10.1038/36103
Uchino, S., Wada, H., Honda, S., Nakamura, Y., Ondo, Y., Uchiyama, T., et al. (2006). Direct interaction of post-synaptic density-95/Dlg/ZO-1 domain-containing synaptic molecule Shank3 with GluR1 α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor. J. Neurochem. 97, 1203–1214. doi: 10.1111/j.1471-4159.2006.03831.x
Uppal, N., Wicinski, B., Buxbaum, J. D., Heinsen, H., Schmitz, C., and Hof, P. R. (2014). Neuropathology of the anterior midcingulate cortex in young children with autism. J. Neuropathol. Exp. Neurol. 73, 891–902. doi: 10.1097/nen.0000000000000108
Valiente, M., and Marin, O. (2010). Neuronal migration mechanisms in development and disease. Curr. Opin. Neurobiol. 20, 68–78. doi: 10.1016/j.conb.2009.12.003
van Kooten, I. A., Palmen, S. J., von Cappeln, P., Steinbusch, H. W., Korr, H., Heinsen, H., et al. (2008). Neurons in the fusiform gyrus are fewer and smaller in autism. Brain 131, 987–999. doi: 10.1093/brain/awn033
Van Maldergem, L., Hou, Q., Kalscheuer, V. M., Rio, M., Doco-Fenzy, M., Medeira, A., et al. (2013). Loss of function of KIAA2022 causes mild to severe intellectual disability with an autism spectrum disorder and impairs neurite outgrowth. Hum. Mol. Genet. 22, 3306–3314. doi: 10.1093/hmg/ddt187
van Woerden, G. M., Harris, K. D., Hojjati, M. R., Gustin, R. M., Qiu, S., de Avila Freire, R., et al. (2007). Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of αCaMKII inhibitory phosphorylation. Nat. Neurosci. 10, 280–282. doi: 10.1038/nn1845
Varea, O., Martin-de-Saavedra, M. D., Kopeikina, K. J., Schurmann, B., Fleming, H. J., Fawcett-Patel, J. M., et al. (2015). Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proc. Natl. Acad. Sci. U S A 112, 6176–6181. doi: 10.1073/pnas.1423205112
Varga, E. A., Pastore, M., Prior, T., Herman, G. E., and McBride, K. L. (2009). The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet. Med. 11, 111–117. doi: 10.1097/gim.0b013e31818fd762
Volders, K., Nuytens, K., and Creemers, J. W. (2011). The autism candidate gene Neurobeachin encodes a scaffolding protein implicated in membrane trafficking and signaling. Curr. Mol. Med. 11, 204–217. doi: 10.2174/156652411795243432
von der Brelie, C., Waltereit, R., Zhang, L., Beck, H., and Kirschstein, T. (2006). Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur. J. Neurosci. 23, 686–692. doi: 10.1111/j.1460-9568.2006.04594.x
Wang, G., Gilbert, J., and Man, H. Y. (2012). AMPA receptor trafficking in homeostatic synaptic plasticity: functional molecules and signaling cascades. Neural Plast. 2012:825364. doi: 10.1155/2012/825364
Wang, X., McCoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533. doi: 10.1038/nature07999
Weeber, E. J., Jiang, Y. H., Elgersma, Y., Varga, A. W., Carrasquillo, Y., Brown, S. E., et al. (2003). Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J. Neurosci. 23, 2634–2644.
Wegiel, J., Kuchna, I., Nowicki, K., Imaki, H., Wegiel, J., Marchi, E., et al. (2010). The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 119, 755–770. doi: 10.1007/s00401-010-0655-4
Weiss, L. A., Shen, Y., Korn, J. M., Arking, D. E., Miller, D. T., Fossdal, R., et al. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J. Med. 358, 667–675. doi: 10.1056/NEJMoa075974
Weng, S. M., McLeod, F., Bailey, M. E., and Cobb, S. R. (2011). Synaptic plasticity deficits in an experimental model of rett syndrome: long-term potentiation saturation and its pharmacological reversal. Neurosci 180, 314–321. doi: 10.1016/j.neuroscience.2011.01.061
Weston, M. C., Chen, H., and Swann, J. W. (2014). Loss of mTOR repressors Tsc1 or Pten has divergent effects on excitatory and inhibitory synaptic transmission in single hippocampal neuron cultures. Front. Mol. Neurosci. 7:1. doi: 10.3389/fnmol.2014.00001
Whalley, H. C., O’Connell, G., Sussmann, J. E., Peel, A., Stanfield, A. C., Hayiou-Thomas, M. E., et al. (2011). Genetic variation in CNTNAP2 alters brain function during linguistic processing in healthy individuals. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B, 941–948. doi: 10.1002/ajmg.b.31241
Wichterle, H., Turnbull, D. H., Nery, S., Fishell, G., and Alvarez-Buylla, A. (2001). in utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development 128, 3759–3771.
Williams, C. A., Beaudet, A. L., Clayton-Smith, J., Knoll, J. H., Kyllerman, M., Laan, L. A., et al. (2006). Angelman syndrome 2005: updated consensus for diagnostic criteria. Am. J. Med. Genet. A 140, 413–418. doi: 10.1002/ajmg.a.31074
Willsey, A. J., Sanders, S. J., Li, M., Dong, S., Tebbenkamp, A. T., Muhle, R. A., et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. doi: 10.1016/j.cell.2013.10.020
Wilson, B. M., and Cox, C. L. (2007). Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc. Natl. Acad. Sci. U S A 104, 2454–2459. doi: 10.1073/pnas.0610875104
Wilson, P. M., Fryer, R. H., Fang, Y., and Hatten, M. E. (2010). Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J. Neurosci. 30, 8529–8540. doi: 10.1523/JNEUROSCI.0032-10.2010
Witte, H., and Bradke, F. (2008). The role of the cytoskeleton during neuronal polarization. Curr. Opin. Neurobiol. 18, 479–487. doi: 10.1016/j.conb.2008.09.019
Wong, W. T., Faulkner-Jones, B. E., Sanes, J. R., and Wong, R. O. (2000). Rapid dendritic remodeling in the developing retina: dependence on neurotransmission and reciprocal regulation by Rac and Rho. J. Neurosci. 20, 5024–5036.
Yamaguchi, Y., and Miura, M. (2015). Programmed cell death in neurodevelopment. Dev. Cell 32, 478–490. doi: 10.1016/j.devcel.2015.01.019
Yang, M., Bozdagi, O., Scattoni, M. L., Wöhr, M., Roullet, F. I., Katz, A. M., et al. (2012). Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J. Neurosci. 32, 6525–6541. doi: 10.1523/JNEUROSCI.6107-11.2012
Yangngam, S., Plong-On, O., Sripo, T., Roongpraiwan, R., Hansakunachai, T., Wirojanan, J., et al. (2014). Mutation screening of the neurexin 1 gene in thai patients with intellectual disability and autism spectrum disorder. Genet. Test. Mol. Biomarkers 18, 510–515. doi: 10.1089/gtmb.2014.0003
Yip, J., Soghomonian, J. J., and Blatt, G. J. (2007). Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol. 113, 559–568. doi: 10.1007/s00401-006-0176-3
Yoo, J., Bakes, J., Bradley, C., Collingridge, G. L., and Kaang, B. K. (2014). Shank mutant mice as an animal model of autism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130143. doi: 10.1098/rstb.2013.0143
Yu, L., and Goda, Y. (2009). Dendritic signalling and homeostatic adaptation. Curr. Opin. Neurobiol. 19, 327–335. doi: 10.1016/j.conb.2009.07.002
Yu, T. W., Mochida, G. H., Tischfield, D. J., Sgaier, S. K., Flores-Sarnat, L., Sergi, C. M., et al. (2010). Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet. 42, 1015–1020. doi: 10.1038/ng.683
Yuan, L., Seong, E., Beuscher, J. L., and Arikkath, J. (2015). Delta-catenin regulates spine architecture via cadherin and PDZ-dependent interactions. J. Biol. Chem. 290, 10947–10957. doi: 10.1074/jbc.m114.632679
Zhang, D., Cheng, L., Qian, Y., Alliey-Rodriguez, N., Kelsoe, J. R., Greenwood, T., et al. (2009). Singleton deletions throughout the genome increase risk of bipolar disorder. Mol. Psychiatry 14, 376–380. doi: 10.1038/mp.2008.144
Zhao, C., Deng, W., and Gage, F. H. (2008). Mechanisms and functional implications of adult neurogenesis. Cell 132, 645–660. doi: 10.1016/j.cell.2008.01.033
Zhao, H., Dupont, J., Yakar, S., Karas, M., and LeRoith, D. (2004). PTEN inhibits cell proliferation and induces apoptosis by downregulating cell surface IGF-IR expression in prostate cancer cells. Oncogene 23, 786–794. doi: 10.1038/sj.onc.1207162
Zhao, M. G., Toyoda, H., Ko, S. W., Ding, H. K., Wu, L. J., and Zhuo, M. (2005). Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J. Neurosci. 25, 7385–7392. doi: 10.1523/JNEUROSCI.1520-05.2005
Zheng, C., Heintz, N., and Hatten, M. E. (1996). CNS gene encoding astrotactin, which supports neuronal migration along glial fibers. Science 272, 417–419. doi: 10.1126/science.272.5260.417
Keywords: autism, ASD, developmental neurobiological disorders, neurogenesis, dendrite growth, neuron morphogenesis, synapse, synaptic plasticity
Citation: Gilbert J and Man H-Y (2017) Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell. Neurosci. 11:359. doi: 10.3389/fncel.2017.00359
Received: 07 September 2017; Accepted: 31 October 2017;
Published: 20 November 2017.
Edited by:
Maria Cristina D’Adamo, University of Malta, MaltaReviewed by:
Alba Di Pardo, Centre for Neurogenetics and Rare Diseases, ItalyPirta Elina Hotulainen, Minerva Institute for Medical Research, Finland
Copyright © 2017 Gilbert and Man. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heng-Ye Man, hman@bu.edu