 Sagrario Manzano
Sagrario Manzano Luis Agüera2†
Luis Agüera2† Javier Olazarán
Javier Olazarán- 1Department of Neurology, Infanta Leonor Hospital, Madrid, Spain
- 2Centro de Investigación Biomédica en Red de Salud Mental (CIBERSAM), Instituto de Investigación Hospital 12 de Octubre (i+12), Madrid, Spain
- 3Unit of Neurodegenerative Diseases DOMUS-Vi, Department of Neurology - Àptima Terrassa, Barcelona, Spain
- 4Memory Disorders Unit, HM Hospitals & Neurology Service, Gregorio Marañón Hospital, Madrid, Spain
Alzheimer's disease (AD) is the most prevalent neurodegenerative condition, especially among elderly people. The presence of cortical β-amyloid deposition, together with tau phosphorylation and intracellular accumulation of neurofibrillary tangles (NFT) is the main neuropathologic criteria for AD diagnosis. Additionally, a role of inflammatory, mitochondrial, and metabolic factors has been suggested. Tramiprosate binds to soluble amyloid, thus inhibiting its aggregation in the brain. It reduced oligomeric and fibrillar (plaque) amyloid, diminished hippocampal atrophy, improved cholinergic transmission, and stabilized cognition in preclinical and clinical studies. In this narrative review, current information on the efficacy and safety of tramiprosate, both in AD and in other neurocognitive disorders, is presented. Possible directions for future studies with tramiprosate are also discussed.
Introduction
Alzheimer's disease (AD) has become the leading cause of cognitive impairment in an increasingly aging society (1). The slow but progressive course of cognitive deterioration, together with the lack of treatments that may halt the disease entails important functional dependence, particularly in the advanced stages of dementia. According to the World Health Organization (WHO) and Alzheimer Disease International (ADI) estimations, a 3-fold increase in the prevalence of dementia is expected by 2050, with 132 million people affected all over the world (2), which will produce enormous burden for healthcare systems and society (3, 4). Clearly, treatments are urgently needed, in order to stop, or at least delay, the course of cognitive deterioration and functional dependence in AD and other aging-associated neurocognitive disorders.
The aim of this paper is to review current information on the efficacy and safety of tramiprosate, in AD and in other neurocognitive disorders. In addition, possible directions for future studies with tramiprosate will be commented.
Pathogenesis of Alzheimer's Disease
Amyloid Pathology
AD is defined by progressive cognitive impairment and functional dependence, along with frequent behavioral abnormalities. Amyloid deposition is the earliest manifestation of the disease, giving a unique chemical and pathological signature to AD (5). The oligomerization of the amyloid peptide is postulated to drive the hyperphosphorylation of microtubule associated (tau) protein via tau misfolding, aggregation, and transynaptic transmission. While amyloid deposition develops very slowly, tau pathology is necessary for the occurrence of synaptic dysfunction and neuronal degeneration. In addition, a pivotal role of infectious agents and glial reaction has been suggested in the pathogenesis of AD (6, 7).
According to the amyloid hypothesis, selective enzymatic cleavage of the Aβ precursor protein (APP) generates Aβ peptide, oligomers, and fibrils, which form amyloid plaques and are eventually accumulated in the brain (5, 8). This accumulation results from an excessive production or a reduced clearance of Aβ, leading to a pathologic imbalance of the protein (9), being the 42-amino acid amyloid peptide (Aβ42) the main isoform found. Conformational transition from random-coil to β-pleated sheet shows greater tendency to aggregate and allows excessive deposition that eventually leads to neurotoxicity (10).
Since Aβ is crucial in AD pathogenesis, anti-amyloid treatment strategies are currently being investigated (11).
Tau Pathology
Amyloid deposition in AD is commonly associated to an abnormal tau phosphorylation or aggregation. Tau is a protein that helps in maintaining the neuronal cell microarchitecture, promoting microtubule assembly and stabilization. In patients with AD, the hyperphosphorylation of tau increases its activity and promotes its accumulation, leading to neurofibrillary tangles (NFT) formation. In turn, NFT cause synapses loss, axonal transport impairment, and mitochondrial and cytoskeletal dysfunction (12). Additionally, tau misfolding and propagation through the synapses would also be crucial in the development of AD (6).
Inflammation
In the last years, several authors have suggested a pivotal role of the immune system in AD pathogenesis; however, the relationship between immune response and amyloid accumulation is not completely elucidated. Innate immune response is localized at amyloid deposits and at NFT formation sites, promoting formation and aggregation of Aβ and NFT. Amyloid deposits are surrounded by astrocytes and microglial cells, involving complement cascade and microglia activation, release of pro-inflammatory mediators, and neuronal damage.
It is hypothesized that in AD, sustained immune response activation triggers a loop of continued signals that exacerbates amyloid deposit and tau pathology in a rapid and irreversible way (13, 14).
Apolipoprotein E
Apolipoprotein E (ApoE) genotype is the major genetic risk factor for late-onset AD. The APOE ε4 allele confers a 4- to 12-fold higher risk of AD and lowers the age of onset by ~10–15 years (15, 16). The ApoE ε4 allele may also play a role in early-onset AD, delaying the onset of symptoms (17), and in other neurodegenerative diseases, where APOE ε4 status is a risk factor for co-pathology and poor evolution (18). ApoE protein binds to soluble Aβ and affects aggregation, transport, and clearance of amyloid within the central nervous system (CNS), increasing Aβ oligomers in the brain, which would contribute to the loss of dendritic spines thus accelerating memory impairment and leading to earlier cognitive decline in AD (16, 19, 20). Besides, preclinical studies showed that the expression of ApoE4 is associated with activation of a pro-inflammatory pathway in pericytes and blood-brain barrier breakdown, leading to neuronal uptake of neurotoxic proteins, as well as reductions in the blood flow (21).
In addition to APOE, over 20 risk loci have been identified in genome-wide association studies of late-onset AD or dementia, which are related to immunity, lipid metabolism, tau binding proteins, and amyloid precursor protein metabolism, showing the genetic and pathophysiological complexity of the disease and also highlighting the importance of comorbid or pleiotropic associations, gender differences, maternal history, and epigenetic factors (22).
AD Biomarkers
AD prodromal biomarkers are crucial to establish early diagnosis and therefore early treatment. The diagnosis criteria of the National Institute on Aging and the Alzheimer's Association (NIA-AA) 2011 proposed Aβ42 and tau determination in CSF for AD diagnosis (23). Reduced cerebrospinal fluid (CSF) Aβ42 levels indicate brain deposition, which can be determined by CSF Aβ42/Aβ40 ratio or by amyloid positron emission tomography (PET) (24). Tau accumulation is associated with tangle formation and can be determined through Aβ/tau ratio in CSF and also by PET imaging (6, 25). Additional imaging biomarkers of downstream neuronal degeneration or injury include reduced 18-fluorodeoxyglucose (FDG) uptake in temporo-parietal cortex determined by PET and atrophy in the temporal lobe and medial parietal cortex determined by magnetic resonance imaging (MRI) (23).
Moreover, it is important to correlate biomarkers with the risk of dementia derived from AD, in particular, correlating biomarkers with low, intermediate, or high risk patients. In a cross-sectional study, Eliassen et al. researched the biomarker profile in patients with subjective cognitive decline (SCD), amnestic (aMCI), and non-amnestic (naMCI) mild cognitive impairment (MCI) patients. They observed elevated total-tau in aMCI and SCD groups. In addition, cortical glucose metabolism was found to be lower in aMCI, and naMCI patients showed a tendency for lower glucose metabolism. Finally, in aMCI and SCD groups was recorded a thinner entorhinal cortex (ERC). The authors concluded that naMCI and SCD patients showed a comparable biomarker profile, while aMCI showed the most pathologic biomarker burden (47.2% of patients with ≥2 biomarkers). Thus, aMCI showed more frequently AD pathological biomarkers representing a risk group (26).
Although exists an armamentarium of biomarkers to identify AD, additional biomarkers are still needed. A tiered approach has been proposed to screen candidate patients for biomarker assessment so that patients who do not show signs of AD pathophysiology may be excluded from cost biomarker performance. Clearly, the implementation of blood amyloid biomarkers in clinical practice would facilitate AD early detection, diagnosis, and follow-up in the future (27). Recently, neurofilament light chain (NfL) has been described as a sensible serum marker for the presymptomatic stages of AD (28).
Tramiprosate
Tramiprosate is an orally administered small aminosulfonate compound that binds to Lys16, Lys28, and Asp23 of Aβ42. This binding results in the stabilization of Aβ42 monomers, thus reducing oligomeric and fibrillar (plaque) amyloid aggregation. The inhibition of oligomer formation and elongation provides neuroprotection against Aβ-induced subsequent deposition (10, 29–31).
Mechanisms of action of tramiprosate include effects on amyloid, but also anti-inflammatory effects as demonstrated in MCI patients (32, 33). A third mechanism of action is linked to the GABA-A receptor (GABA-AR). The molecular structure of tramiprosate is related to the neurotransmitter γ-amino butyric acid (GABA) structure, and it acts as a functional agonist (34, 35). The clinical effect of tramiprosate in cognitive and functional areas was mainly observed in AD patients which were homozygous for the ε4 allele of APOE (15, 16).
Although tramiprosate is a safe and usually well-tolerated drug, it is not yet authorized as a new AD drug. Besides, new tramiprosate prodrugs and metabolites are also being developed. ALZ-801 is an oral-administered tramiprosate prodrug with significantly improved pharmacokinetic variability and gastrointestinal tolerance. The phase I program reported good safety and tolerability results in healthy volunteers (36). On the other hand, 3-sulfopropanoic acid (3-SPA), the main metabolite of tramiprosate and ALZ-801, is an endogenous molecule present in the brain of patients with AD and other neurodegenerative disorders which has demonstrated anti-Aβ aggregation activity in vitro, with an efficacy comparable to tramiprosate. Thus, clinical improvements observed with tramiprosate or ALZ-801 may be partially due to the potential protective role of 3-SPA in the brain (36, 37).
Literature Search Criteria
For this review, preclinical and clinical studies on tramiprosate were searched. For the selection of studies with relevant information on homotaurine, the following databases were consulted: Medline, Dimensions.ai, Google Scholar, and Cochrane Library, with the search completion date 01/05/2019. Search strings were performed on the above databases constructed from the term homotaurine and/or tramiprosate and all their equivalents or synonyms as input terms into the MeSH (Medical Subject Headings) database, the NLM controlled vocabulary thesaurus. These terms were conjugated with other terms derived from their pharmacological mechanism of action: “GABA Agonists,” “GABA receptor agonist,” or “glycosaminoglycan mimetic.” For screening, a restriction was made to those papers with IMRAD structure, published since 2006 (years of the first publication of tramiprosate phase II studies) and preferably in English. In the selection of studies, priority was given to prospective studies and reviews with adequate methodological quality. In addition, a secondary manual search of the bibliography of the studies finally selected was carried out to detect possible omissions that could be of interest for this work.
Preclinical Studies on Tramiprosate
In preclinical studies, tramiprosate reduced oligomer formation and fibrillar (plaque) amyloid deposition in mouse model of AD. Treatment with tramiprosate induced a decrease of soluble amyloid protein levels and its deposition in the brain (plaque). Also, plasma Aβ levels declined in a dose-dependent manner indicating a role of tramiprosate in brain metabolism of Aβ or in its transport (10).
One preclinical study observed that tramiprosate (3-APS) favored tau polymerization in fibrillar aggregates, but these tau aggregates were not toxic in neuronal cultures. 3-APS also did not affect the binding of tau to microtubules and it promoted the decrease of tau-actin complexes that could be toxic for the cells (38).
In addition, GABA-dependent activity of tramiprosate has been reported. In rat primary neurons, tramiprosate binds with high affinity to the GABA-A receptors inducing caspase 3/7 activation. Accordingly, this effect is significantly decreased when neurons are pretreated with GABA-AR antagonists (35). In primary neurons treated with Aβ42, tramiprosate reduced caspase 3/7 and caspase 9 activities, both basal and Aβ42-induced. Also, in organotypic hippocampal slice cultures (OHCs) it decreased Aβ42-induced cellular mortality. Thus, in primary neurons, tramiprosate entails neuroprotective mechanisms against activation of caspases due to Aβ, and cellular mortality in OHCs; however, its effect on DNA damage is independent of the activation of GABA-AR (34).
Aβ induces long-term potentiation (LTP) inhibition in rat brain, and tramiprosate has shown no reverse this effect (39). Besides, it also inhibits Aβ42-induced extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) activation by a GABA-A-independent mechanism (40). Overall, preclinical studies of tramiprosate demonstrate neuroprotective mechanisms involving both GABAergic and non-GABAergic pathways. A summary of preclinical studies on tramiprosate is showed in Table 1.
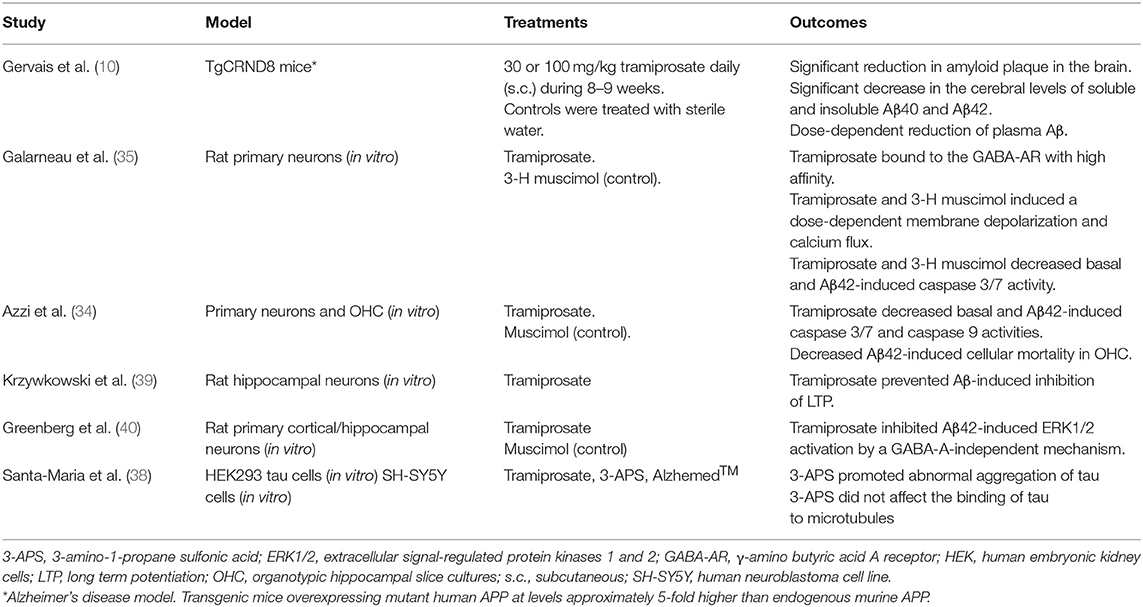
Table 1. Preclinical studies on trampirosate.
Clinical Studies with Tramiprosate in AD and other Neurocognitive Disorders
Results from a phase II trial demonstrate that tramiprosate safely reduces Aβ42 levels in the CSF of patients with mild-to-moderate AD. This CSF Aβ42 levels reduction together with clinical evaluations at long term indicate a role of tramiprosate in disease-modification. In addition, 3-months administration of tramiprosate was safe and well-tolerated (41). In the subsequent phase III study (Alphase study), tramiprosate did not show significant differences, but results were confounded by unexplained variance. In fact, a trend was observed for a treatment effect on the Alzheimer Disease Assessment Scale-cognitive subscale (ADAS-cog) (42). Moreover, pooled analysis of the two phase III trials (n = 2,025 patients with mild to moderate AD) considering ApoE4 allele distribution showed significant differences in ADAS-cog scores and a positive tendency on Clinical Dementia Rating Scale-Sum of Boxes (CDR-SB) in homozygote patients on 150 mg bid. ApoE4 heterozygotes showed an intermediate level of efficacy and non-ApoE4 patients did not show clinical benefits (29). Finally, subsequent re-analyses revealed most efficacy in the homozygote patients which were at the mildest clinical stage of disease (Mini-Mental State Examination 22–26). In those patients, tramiprosate showed benefits on ADAS-cog, CDR-SB, and DAD (Disability Assessment for Dementia) compared to placebo. Cognitive stabilization was observed over 78 weeks in the ADAS-cog, while both cognitive (ADAS-cog) and functional (DAD) effects increased over time (43).
The effect of tramiprosate on hippocampal volume was evaluated in a subgroup of patients (n = 312) from the Alphase study. In the final model analyses was demonstrated a significant link between tramiprosate dose and the reduction in hippocampus volume change (44, 45). Overall, the results of the phase III trials suggest a disease modifying effect of tramiprosate in AD, particular for the ApoE4/4 patients, at the earliest clinical stages of disease (46).
In patients with aMCI, which is very often an early clinical manifestation of AD, the effects of tramiprosate have been evaluated. Patients who met the criteria for aMCI (47, 48) and had a Clinical Dementia Rating (CDR) score of 0.5 (49) on tramiprosate showed less hippocampus and temporal lobe volume loss, which entails an improvement in short-term memory. Thus, tramiprosate supplementation protects against hippocampus atrophy and improves episodic short-term memory (33). In this line, a recent study evaluating tramiprosate administration in aMCI patients also showed improved short-term episodic memory performance in ApoE4/4 carriers. In addition to neuropsychological and functional assessments, cytokine levels were performed at baseline and after 1 year, and a significant decrease in IL-18 serum levels was observed, suggesting a drug-related anti-inflammatory effect (32). Another study in patients with symptomatic MCI showed an even earlier response. In patients with MCI according to Petersen criteria (48), 1-year administration of tramiprosate showed significant improvements from baseline expressed as MMSE (Mini Mental State Examination) score at months 8 and 12 in patients with aMCI, and at month 4 in those with naMCI (50). Finally, one study highlighted that tramiprosate could also modulate mechanisms of synaptic plasticity in aMCI patients, again diagnosed according to Petersen criteria (48). Treatment with 100 mg tramiprosate during 4 weeks showed measurable changes of short latency afferent inhibition (SLAI) suggesting a function in enhancing acetylcholine transmission, through the modulation of inhibitory cortical activity (51).
Tramiprosate efficacy has also been evaluated in Parkinson's disease (PD). One single-blind randomized controlled study that evaluated the safety and efficacy of tramiprosate in patients with PD and cognitive impairment showed that patients on treatment improved in the non-motor symptoms of the Unified Parkinson's Disease Rating Scale and presented a beneficial effect on excessive sleepiness measured by the Epworth Sleepiness Scale after 6 months, compared to controls (52).
In subjects with lobar intracerebral hemorrhage, a phase II double-blinded trial illustrated the pharmacokinetics and demonstrated the safety and tolerability of three different doses of tramiprosate. Nevertheless, testing of neurological function (measured by NIHSS), daily functioning (Barthel Index), cognition (ADAS-cog), and executive function (EXIT25) did not show significant changes among subjects during 12 weeks of treatment, compared to baseline (53). A summary of clinical studies with tramiprosate in AD and other neurocognitive disorders is showed in Table 2.
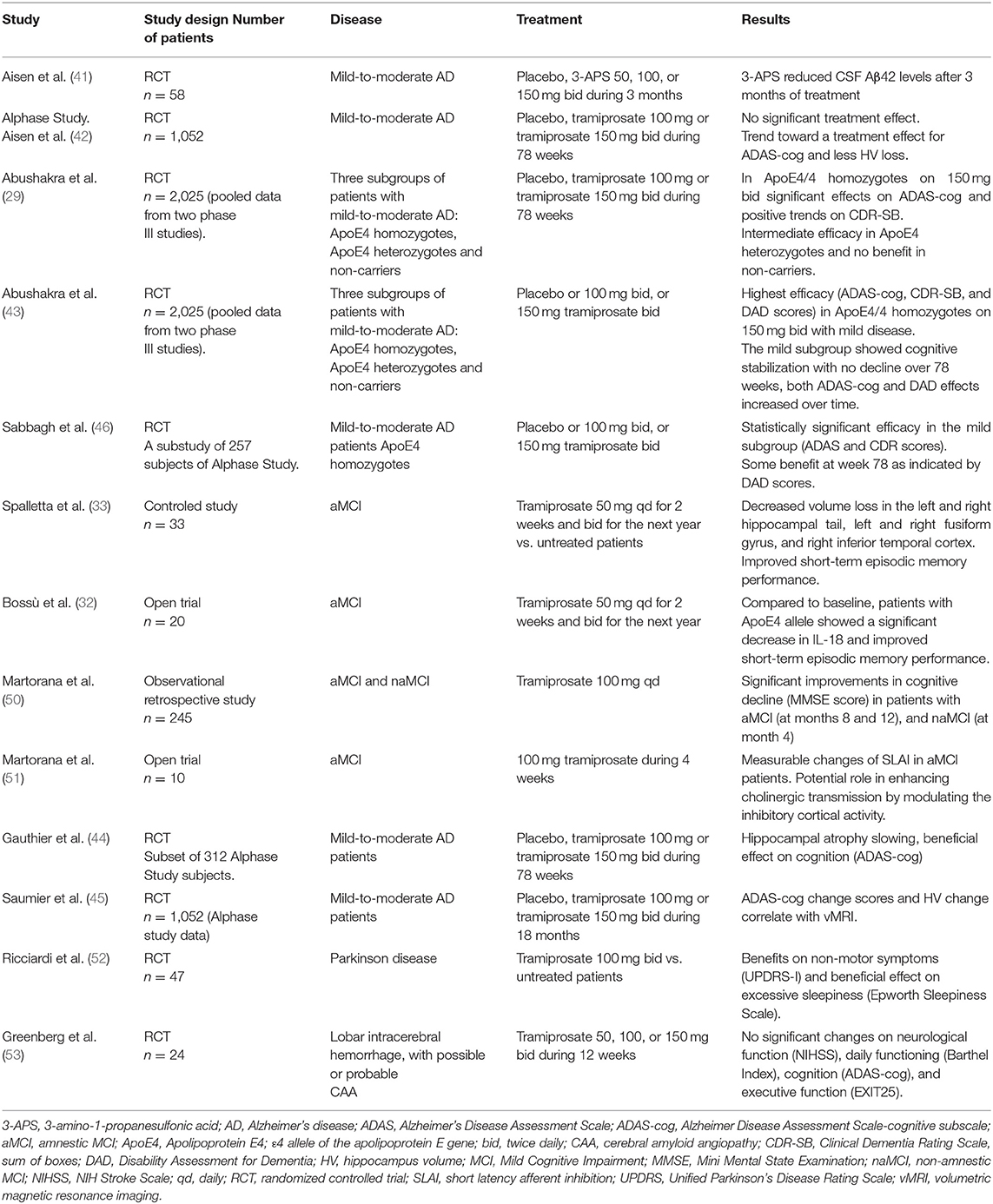
Table 2. Clinical studies on trampirosate.
Safety of tramiprosate was excellent in all the clinical studies, with acceptable tolerability. In fact, mortality rate in the Alphase study was higher in the placebo group (4.0%) compared to the 100 mg bid (2.8%) and 150 mg bid (2.3%) groups. Patients with at least one adverse event for the placebo, 100 mg bid, and 150 mg bid groups were 92.1, 95.2, and 94.8%, respectively. Gastrointestinal disorders were the most common adverse events that led to discontinuation (3.6% of the patients). Nausea, vomiting, syncope, and weight loss seem to be dose-dependent; however, the incidence of those adverse events in the 100 mg bid group did not show significant differences compared to placebo. The ApoE4 patients displayed similar safety and tolerability profile compared to the complete sample and no amyloid-related imaging abnormalities (ARIAs) were observed (41, 43).
Limitations of Tramiprosate Studies
Previous studies demonstrated that alteration of tau correlates better than amyloid deposition with the cognitive and behavioral manifestations of Alzheimer's patients (54, 55). Hence, it could be argued that, since tramiprosate mainly prevents amyloid aggregation, a clinical effect should not be expected. However, the efficacy of tramiprosate should be considered as multimodal, through different mechanisms, the reduction of amyloid deposition, the improvement of cholinergic transmission, an anti-inflammatory effect, and a probable neuroprotective effect, not favoring an abnormal aggregation of tau. Moreover, there is agreement regarding the convenience of early action on amyloid deposition and other mechanisms of AD to prevent tau pathology and cognitive deterioration (6, 56).
Tramiprosate studies have some limitations that could interfere in the clinical outcomes and study conclusions, mainly suboptimal study design (outcome measurements, lack or inadequate biomarkers used) and small sample size. Most importantly, time point for treatment inception, related to disease development, could also interfere in the study outcomes. Available data from failed phase III studies suggest that drug therapy should be started early, since patients with mild to moderate AD could be in advanced and irreversible stages of neurodegeneration at that point, being too late to improve their outcomes (41–43).
Future Studies with Tramiprosate
Although tramiprosate is safe and well-tolerated, large controlled trials are still needed, particularly studies focusing on MCI and AD. Given the clinical heterogeneity of AD, the trials should be conducted in patients which are well-defined for the amyloid presence, tau protein, and neurodegeneration biomarkers (57). The heterogeneity of AD and MCI (clinical aspects, diagnosis, correlation with neuropathology, co-pathology in late onset AD, etc.) should be considered in future trials. Clearly, tramiprosate effects in neurodegeneration, inflammation, and hippocampus atrophy could be useful in patients at the early stages of AD to delay the onset of dementia (15).
Additionally, a major challenge will be to identify subgroups of highly respondent patients as well as the clinical and biological predictors of response. Nowadays, the main challenges for the management of neurocognitive disorders are to develop disease-modifying treatments and to improve symptomatic treatments. Tramiprosate has showed promising results in this line, and future directions with this drug are discussed below.
Tramiprosate as a Disease-Modifying Agent
The pooled results of both phase III studies demonstrated the efficacy of tramiprosate in the treatment of ApoE4/4 carriers with mild AD through Aβ binding. These patients showed a stabilization of cognitive and functional performance, supporting the potential of this drug as a disease modifier (30, 43). Thus, future phase IV studies enriched with ApoE4/4 carriers focusing on aMCI and mild-moderate AD patients would be of interest.
A recent study established the ALZ-801 dose that provides bioequivalent exposures to 150 mg bid tramiprosate (ALZ-801 265 mg bid), which demonstrated cognitive and functional improvements in ApoE4/4 homozygotes. These data reinforced the development of phase III studies with ALZ-801 in ApoE4/4 carriers with AD or MCI (43, 58, 59).
Lewy body disease (LBD), Parkinson's disease dementia (PDD), vascular dementia (VaD) and frontotemporal dementia (FTD) patients are less frequently ApoE4 carriers than AD patients (60); however, these diseases are frequently associated with AD (61, 62). Hence, the subgroup of non-AD, but ApoE4/4 carriers, dementia patients would be candidates to receive tramiprosate or ALZ-801 treatment (60, 63–67).
Since clinical improvements observed with tramiprosate or ALZ-801 may be partially due to 3-SPA, it would be interesting to elucidate its role in ApoE4/4 carriers in future studies (36, 37). A summary of future investigations with tramiprosate, ALZ-801 and 3-SPA as disease modifiers on different neurocognitive disorders is given in Table 3.

Table 3. Future lines of research with tramiprosate, ALZ-801, and 3-APS.
Tramiprosate as a Symptomatic Treatment (Central Cholinergic Dysfunction)
Frequently, cognitive, behavioral, and motor disabilities are related to cholinergic dysfunction (68). Tramiprosate is a central GABA partial receptor agonist that modulates inhibitory cortical activity and improves cortical cholinergic transmissions in AD patients (51). In this line, tramiprosate or ALZ-801 could be useful for aMCI patients and mild/moderate AD patients to manage disease symptoms. Besides, other neurological entities with acetylcholine deficit could also benefit from treatment with tramiprosate.
Central cholinergic dysfunction has been reported in Parkinson's disease (PD) patients with early gait disturbances (69), visual hallucinations (70), REM sleep behavior disorder (71), dysphagia (72), cognitive decline (73), and olfactory impairment (74, 75). Thus, treatment with tramiprosate (or ALZ-801) alone or associated with cholinesterase inhibitors (CEI) may be considered in these PD subtypes to improve cholinergic transmission.
In VaD patients, deficits in the acetylcholine transmission or in the cortical and subcortical function are commonly present. Multiple factors influence production of vascular lesions in the basal forebrain cholinergic neuronal system (arterial hypertension, sustained hypoperfusion, cerebral small vessel disease, inflammatory reactions, oxidative stress, cerebral amyloid angiopathy damage, and ischemic cerebrovascular disease, among others). In these patients treatment with CEI may be useful (76), so exploring the potential benefit of tramiprosate (or ALZ-801), alone o combined, could also be interesting (66).
Traumatic brain injury (TBI) reduces cholinergic neurotransmission, decreases evoked release of acetylcholine, and alters cholinergic receptor levels. Treatment with CEI (galantamine) can reduce TBI pathology and improve cognitive function. Hence, TBI may be another candidate that could benefit from tramiprosate treatment (77).
In addition, it would be helpful to explore tramiprosate benefits in other neurodegenerative disorders which present dysfunction in cholinergic neurons and cholinergic transmission in the brain, such as Huntington's disease (78). The proposed future investigations with tramiprosate, ALZ-801, and 3-SPA on central cholinergic dysfunction are shown in Table 3.
Other non-AD neurodegenerative disorders with loss of episodic memory and selective hippocampal atrophy could also benefit from tramiprosate treatment. Thus, future studies with tramiprosate should be directed toward entities such as primary age-related tauopathy (PART) (79) and limbic-predominant age-related TDP-43 encephalopathy (LATE) (80).
Finally, since treatment combinations with different targets and pathophysiological mechanisms have been successful in other complex disorders such as HIV, this might also orientate future treatment in AD (81). Thus, it would be helpful to analyze the effects of combination therapies in the early stages, for example, treatment with tramiprosate and monoclonal anti-amyloid antibodies or CEI.
Conclusions
Timely management of cognitive deterioration is of outmost importance in our aging society. Tramiprosate has been shown safe and effective in several neurocognitive disorders, particularly AD and MCI, where the results are consistent with a disease-modifying effect. Due to the heterogeneity of AD and MCI, definition of subgroups of responders should be crucial to implement future treatments. Clearly, large controlled trials, including well-characterized patients in terms of biomarkers and risk factors, are needed to confirm and extend the promising results of tramiprosate and 3-APS in the treatment of AD and other neurocognitive disorders.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This paper was financed by Neuraxpharm Spain SL. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Editor declares he has received honoraria from Neuraxpharm, and declares no further competing interests that would impact the handling of this manuscript.
References
1. Alzheimer's Association. 2016 Alzheimer's disease facts and figures. Alzheimers Dement J Alzheimers Assoc. (2016) 12:459–509. doi: 10.1016/j.jalz.2016.03.001
2. Prince M, Wimo A, Guerchet M, Ali GC, Wu YTPM. World Alzheimer Report 2015. The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends (2015).
3. Olazarán J, Agüera-Ortiz L, Argimón JM, Reed C, Ciudad A, Andrade P, et al. Costs and quality of life in community-dwelling patients with Alzheimer's disease in Spain: results from the GERAS II observational study. Int Psychogeriatr. (2017) 29:2081–93. doi: 10.1017/S1041610217001211
4. Santana I, Farinha F, Freitas S, Rodrigues V, Carvalho Á. The epidemiology of dementia and Alzheimer disease in Portugal: estimations of prevalence and treatment-costs. Acta Med Port. (2015) 28:182–8. doi: 10.20344/amp.6025
5. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. (1992) 256:184–5. doi: 10.1126/science.1566067
6. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. (2019) 179:312–39. doi: 10.1016/j.cell.2019.09.001
7. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc. (2018) 14:1602–14. doi: 10.1016/j.jalz.2018.06.3040
8. Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. (2005) 115:1121–9. doi: 10.1172/JCI25100
9. Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, Schneider T, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement J Alzheimers Assoc. (2017) 13:841–9. doi: 10.1016/j.jalz.2017.06.2266
10. Gervais F, Paquette J, Morissette C, Krzywkowski P, Yu M, Azzi M, et al. Targeting soluble Aβ peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol Aging. (2007) 28:537–47. doi: 10.1016/j.neurobiolaging.2006.02.015
11. Aisen PS, Briand R, Saumier D, Laurin J, Duong A, Garceau D. Targeting amyloid with tramiprosate in patients with mild-to-moderate Alzheimer disease. Prog Neurother Neuropsychopharmacol. (2008) 3:111–25. doi: 10.1017/S1748232107000171
12. Gao Y, Tan L, Yu J-T, Tan L. Tau in Alzheimer's disease: mechanisms and therapeutic strategies. Curr Alzheimer Res. (2018) 15:283–300. doi: 10.2174/1567205014666170417111859
13. Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer's disease. Nat Rev Immunol. (2006) 6:404–16. doi: 10.1038/nri1843
14. Felsky D, Roostaei T, Nho K, Risacher SL, Bradshaw EM, Petyuk V, et al. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat Commun. (2019) 10:409. doi: 10.1038/s41467-018-08279-3
15. Breitner JCS, Wyse BW, Anthony JC, Welsh-Bohmer KA, Steffens DC, Norton MC, et al. APOE-epsilon4 count predicts age when prevalence of AD increases, then declines: the Cache County Study. Neurology. (1999) 53:321–31. doi: 10.1212/WNL.53.2.321
16. Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. (2004) 25:641–50. doi: 10.1016/j.neurobiolaging.2003.12.023
17. De Luca V, Orfei MD, Gaudenzi S, Caltagirone C, Spalletta G. Inverse effect of the APOE epsilon4 allele in late- and early-onset Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci. (2016) 266:599–606. doi: 10.1007/s00406-015-0663-4
18. Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. (2018) 141:2181–93. doi: 10.1093/brain/awy146
19. Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. (2012) 2:a006312. doi: 10.1101/cshperspect.a006312
20. Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu. Rev. Med. (1996) 47, 387–400. doi: 10.1146/annurev.med.47.1.387
21. Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. (2012) 485:512–6. doi: 10.1038/nature11087
22. Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. (2019) 51:414–30. doi: 10.1038/s41588-019-0358-2
23. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. J Alzheimers Assoc. (2011) 7:263–9. doi: 10.1016/j.jalz.2011.03.005
24. Lladó Plarrumaní A. Biomarcadores de líquido cefalorraquídeo en la Enfermedad de Alzheimer. Inf. Psiquiátr. (2018) 232:13–9.
25. Wren MC, Lashley T, Årstad E, Sander K. Large inter- and intra-case variability of first generation tau PET ligand binding in neurodegenerative dementias. Acta Neuropathol Commun. (2018) 6:34. doi: 10.1186/s40478-018-0535-z
26. Eliassen CF, Reinvang I, Selnes P, Grambaite R, Fladby T, Hessen E. Biomarkers in subtypes of mild cognitive impairment and subjective cognitive decline. Brain Behav. (2017) 7:e00776. doi: 10.1002/brb3.776
27. Hampel H, O'Bryant SE, Molinuevo JL, Zetterberg H, Masters CL, Lista S, et al. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. (2018) 14:639–52. doi: 10.1038/s41582-018-0079-7
28. Preische O, Schultz SA, Apel A, Kuhle J, Kaeser SA, Barro C, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer's disease. Nat Med. (2019) 25:277–83. doi: 10.1038/s41591-018-0304-3
29. Abushakra S, Porsteinsson A, Vellas B, Cummings J, Gauthier S, Hey JA, et al. Clinical benefits of tramiprosate in Alzheimer's disease are associated with higher number of APOE4 alleles: the “APOE4 gene-dose effect.” J Prev Alzheimers Dis. (2016) 3:219–28. doi: 10.14283/jpad.2016.115
30. Kocis P, Tolar M, Yu J, Sinko W, Ray S, Blennow K, et al. Elucidating the Aβ42 anti-aggregation mechanism of action of tramiprosate in Alzheimer's disease: integrating molecular analytical methods, pharmacokinetic and clinical Data. CNS Drugs. (2017) 31:495–509. doi: 10.1007/s40263-017-0434-z
31. Tolar M, Abushakra S, Sabbagh M. The path forward in Alzheimer's disease therapeutics: reevaluating the amyloid cascade hypothesis. Alzheimers Dement. (2019) 1–8. doi: 10.1016/j.jalz.2019.09.075
32. Bossù P, Salani F, Ciaramella A, Sacchinelli E, Mosca A, Banaj N, et al. Anti-inflammatory effects of homotaurine in patients with amnestic mild cognitive impairment. Front. Aging Neurosci. (2018) 10:285. doi: 10.3389/fnagi.2018.00285
33. Spalletta G, Cravello L, Gianni W, Piras F, Iorio M. Homotaurine effects on hippocampal volume loss and episodic memory in amnestic mild cognitive impairment. J Alzheimers Dis. (2016) 50:807–16. doi: 10.3233/JAD-150484
34. Azzi M, Morissette S, Fallon L, Martin R, Galarneau A, Sebastiani G, et al. Involvement of both gaba-dependent and - independent pathways in tramiprosate neuroprotective effects against amyloid- βeta toxicity. Neurodegener Dis. (2007) 4(Suppl. 1):284–5. doi: 10.1159/000102531
35. Galarneau A, Lacoste M-C, Morissette S, Delorme D, Greenberg BD. P-190: GABA-dependent pathways in the neuroprotective effect of tramiprosate against amyloid-toxicity. Alzheimers Dement J Alzheimers Assoc. (2007) 3:S158–9. doi: 10.1016/j.jalz.2007.04.153
36. Hey JA, Yu JY, Versavel M, Abushakra S, Kocis P, Power A, et al. Clinical pharmacokinetics and safety of ALZ-801, a novel prodrug of tramiprosate in development for the treatment of Alzheimer's disease. Clin Pharmacokinet. (2018) 57:315–33. doi: 10.1007/s40262-017-0608-3
37. Hey JA, Kocis P, Hort J, Abushakra S, Power A, Vyhnálek M, et al. Discovery and identification of an endogenous metabolite of tramiprosate and its prodrug ALZ-801 that inhibits beta amyloid oligomer formation in the human brain. CNS Drugs. (2018) 32:849–61. doi: 10.1007/s40263-018-0554-0
38. Santa-Maria I, Hernández F, Del Rio J, Moreno FJ, Avila J. Tramiprosate, a drug of potential interest for the treatment of Alzheimer's disease, promotes an abnormal aggregation of tau. Mol Neurodegener. (2007) 2:17. doi: 10.1186/1750-1326-2-17
39. Krzywkowski P, Sebastiani G, Williams S. Tramiprosate Prevents Amyloid Beta-induced Inhibition of Long-term Potentiation in Rat Hippocampal Slices.In: 8th Int. Conf. AD/PD. Salzburg (2007).
40. Greenberg BD, Fallon L, Lagacé C, Sebastiani G, Paquette J, Delorme D. P-192: tramiprosate decreases amyloid-beta; induced ERK1/2 activity in primary rat neurons by a GABA-independent pathway. Alzheimer's Dement. J. Alzheimer's Assoc. (2007) 3:S159. doi: 10.1016/j.jalz.2007.04.155
41. Aisen PS, Saumier D, Briand R, Laurin J, Gervais F, Tremblay P, et al. A Phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology. (2006) 67:1757–63. doi: 10.1212/01.wnl.0000244346.08950.64
42. Aisen PS, Gauthier S, Ferris SH, Saumier D, Haine D, Garceau D, et al. Tramiprosate in mild-to-moderate Alzheimer's disease multi-centre study (the Alphase Study). Arch Med Sci. (2011) 7:102–11. doi: 10.5114/aoms.2011.20612
43. Abushakra S, Porsteinsson A, Scheltens P, Sadowsky C, Vellas B, Gauthier S, et al. Clinical effects of tramiprosate in APOE4/4 homozygous patients with mild Alzheimer's disease suggest disease modification potential. J Prev Alzheimers Dis. (2017) 4:149–56. doi: 10.14283/jpad.2017.26
44. Gauthier S, Aisen PS, Ferris SH, Saumier D, Duong A, Haine D, et al. Effect of tramiprosate in patients with mild-to-moderate Alzheimer's disease: exploratory analyses of the MRI sub-group of the Alphase study. J Nutr Heal Aging. (2009) 13:550–7. doi: 10.1007/s12603-009-0106-x
45. Saumier D, Aisen PS, Gauthier S, Vellas B, Ferris SH, Duong A, et al. Lessons learned in the use of volumetric MRI in therapeutic trials in Alzheimer's disease: the ALZHEMEDTM (tramiprosate) experience. J Nutr Heal Aging. (2009) 13:370–2. doi: 10.1007/s12603-009-0047-4
46. Sabbagh MN. Clinical effects of oral tramiprosate in APOE4/4 homozygous patients with mild Alzheimer's disease suggest disease modification. J Prev Alzheimers Dis. (2017) 4:136–7. doi: 10.14283/jpad.2017.24
47. Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. (2011) 7:270–9. doi: 10.1016/j.jalz.2011.03.008
48. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. (2004) 256:183–94. doi: 10.1111/j.1365-2796.2004.01388.x
49. Morris JC. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr. (1997) 9:177–8. doi: 10.1017/S1041610297004870
50. Martorana A, Motta C, Koch G, Massaia M, Mondino S, Raniero I, et al. Effect of homotaurine in patients with cognitive impairment: results from an Italian observational retrospective study. J Gerontol Geriatr. (2018) 66:15–20.
51. Martorana A, Di Lorenzo F, Manenti G, Semprini R, Koch G. Homotaurine induces measurable changes of short latency afferent inhibition in a group of mild cognitive impairment individuals. Front Aging Neurosci. (2014) 6:254. doi: 10.3389/fnagi.2014.00254
52. Ricciardi L, Nigris F, De Specchia A, Fasano A. Homotaurine in Parkinson's disease. Neurol. Sci. (2015) 36:1581–7. doi: 10.1007/s10072-015-2201-6
53. Greenberg SM, Rosand J, Schneider AT, Creed Pettigrew L, Gandy SE, Rovner B, et al. A phase 2 study of tramiprosate for cerebral amyloid angiopathy. Alzheimer Dis Assoc Disord. (2006) 20:269–74. doi: 10.1097/01.wad.0000213845.28624.f4
54. Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. (1992) 13:179–89. doi: 10.1016/0197-4580(92)90027-U
55. Price JL, McKeel DWJ, Buckles VD, Roe CM, Xiong C, Grundman M, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. (2009) 30:1026–36. doi: 10.1016/j.neurobiolaging.2009.04.002
56. Morris JC, Price JL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer's disease. J Mol Neurosci. (2001) 17:101–18. doi: 10.1385/jmn:17:2:101
57. Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. (2018) 14:535–62. doi: 10.1016/j.jalz.2018.02.018
58. Caltagirone C, Ferrannini L, Marchionni N, Nappi G, Scapagnini G, Trabucchi M. The potential protective effect of trampirosate (homotaurine) against Alzheimer's disease: a review. Aging Clin Exp Res. (2012) 24:580–7. doi: 10.3275/8585
59. Tsolaki M. Future strategies of management of Alzheimer's Disease. The role of homotaurine Hell J Nucl Med. (2019) 22:82–94.
60. Nielsen AS, Ravid R, Kamphorst W, Jørgensen OS. Apolipoprotein E ε4 in an autopsy series of various dementing disorders. J Alzheimers Dis. (2003) 5:119–25. doi: 10.3233/JAD-2003-5206
61. Chung EJ, Babulal GM, Monsell SE, Cairns NJ, Roe CM, Morris JC. Clinical features of Alzheimer disease with and without lewy bodies. JAMA Neurol. (2015) 72:789–96. doi: 10.1001/jamaneurol.2015.0606
62. Compta Y, Parkkinen L, O'Sullivan SS, Vandrovcova J, Holton JL, Collins C, et al. Lewy- and Alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain. (2011) 134:1493–505. doi: 10.1093/brain/awr031
63. Banach M, Rizzo M, Nikolic D, Howard G, Howard V, Mikhailidis D. Intensive LDL-cholesterol lowering therapy and neurocognitive function. Pharmacol Ther. (2017) 170:181–91. doi: 10.1016/j.pharmthera.2016.11.001
64. Helisalmi S, Linnaranta K, Lehtovirta M, Mannermaa A, Heinonen O, Ryynänen M, et al. Apolipoprotein E polymorphism in patients with different neurodegenerative disorders. Neurosci Lett. (1996) 205:61–4. doi: 10.1016/0304-3940(96)12373-9
65. Larsson V, Torisson G, Londos E. Relative survival in patients with dementia with Lewy bodies and Parkinson's disease dementia. PLoS ONE. (2018) 13:e0202044. doi: 10.1371/journal.pone.0202044
66. Liu Y, Dong Y-H, Lyu P-Y, Chen W-H, Li R. Hypertension-induced cerebral small vessel disease leading to cognitive impairment. Chin Med J. (2018) 131:615–9. doi: 10.4103/0366-6999.226069
67. Marder K, Maestre G, Cote L, Mejia H, Alfaro B, Halim A, et al. The apolipoprotein? 4 allele in Parkinson's disease with and without dementia. Neurology. (1994) 44:1330–1.
68. Tata AM, Velluto L, Reale CDM. Cholinergic system dysfunction and neurodegenerative diseases: cause or effect? CNS Neurol Disord Drug Targets. (2014) 13:1294–303. doi: 10.2174/1871527313666140917121132
69. Rochester L, Yarnall AJ, Baker MR, David RV, Lord S, Galna B, et al. Cholinergic dysfunction contributes to gait disturbance in early Parkinson's disease. Brain. (2012) 135:2779–88. doi: 10.1093/brain/aws207
70. Dauwan M, Hoff JI, Vriens EM, Hillebrand A, Stam CJ, Sommer IE. Aberrant resting-state oscillatory brain activity in Parkinson's disease patients with visual hallucinations: an MEG source-space study. Neuroimage Clin. (2019) 22:101752. doi: 10.1016/j.nicl.2019.101752
71. Nardone R, Brigo F, Versace V, Höller Y, Tezzon F, Saltuari L, et al. Cortical afferent inhibition abnormalities reveal cholinergic dysfunction in Parkinson's disease: a reappraisal. J Neural Transm. (2017) 124:1417–29. doi: 10.1007/s00702-017-1775-y
72. Lee KD, Koo JH, Song SH, Jo KD, Lee MK, Jang W. Central cholinergic dysfunction could be associated with oropharyngeal dysphagia in early Parkinson's disease. J Neural Transm. (2015) 122:1553–61. doi: 10.1007/s00702-015-1427-z
73. Pagano G, Rengo G, Pasqualetti G, Femminella GD, Monzani F, Ferrara N, et al. Cholinesterase inhibitors for Parkinson's disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. (2015) 86:767–73. doi: 10.1136/jnnp-2014-308764
74. Oh E, Park J, Youn J, Kim JS, Park S, Jang W. Olfactory dysfunction in early Parkinson's disease is associated with short latency afferent inhibition reflecting central cholinergic dysfunction. Clin Neurophysiol. (2017) 128:1061–8. doi: 10.1016/j.clinph.2017.03.011
75. Versace V, Langthaler PB, Sebastianelli L, Höller Y, Brigo F, Orioli A, et al. Impaired cholinergic transmission in patients with Parkinson's disease and olfactory dysfunction. J Neurol Sci. (2017) 377:55–61. doi: 10.1016/j.jns.2017.03.049
76. Román GC, Kalaria RN. Vascular determinants of cholinergic deficits in Alzheimer disease and vascular dementia. Neurobiol Aging. (2006) 27:1769–85. doi: 10.1016/j.neurobiolaging.2005.10.004
77. Zhao J, Hylin MJ, Kobori N, Hood KN, Moore AN, Dash PK. Post-injury administration of galantamine reduces traumatic brain injury pathology and improves outcome. J Neurotrauma. (2018) 35:362–74. doi: 10.1089/neu.2017.5102
78. D'Souza GX, Waldvogel HJ. Targeting the cholinergic system to develop a novel therapy for Huntington's disease. J Huntingtons Dis. (2016) 5:333–42. doi: 10.3233/JHD-160200
79. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. (2014) 128:755–66. doi: 10.1007/s00401-014-1349-0
80. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. (2019) 142:1503–27. doi: 10.1093/brain/awz099
Keywords: homotaurine, tramiprosate, amyloid, Alzheimer's disease, neurocognitive disorders, neurodegenerative diseases
Citation: Manzano S, Agüera L, Aguilar M and Olazarán J (2020) A Review on Tramiprosate (Homotaurine) in Alzheimer's Disease and Other Neurocognitive Disorders. Front. Neurol. 11:614. doi: 10.3389/fneur.2020.00614
Received: 25 November 2019; Accepted: 26 May 2020;
Published: 07 July 2020.
Edited by:
Gianfranco Spalletta, Santa Lucia Foundation (IRCCS), ItalyReviewed by:
Francesca Assogna, Santa Lucia Foundation (IRCCS), ItalyKarelle Leroy, Université libre de Bruxelles, Belgium
Copyright © 2020 Manzano, Agüera, Aguilar and Olazarán. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javier Olazarán, javier@mariawolff.es
†These authors have contributed equally to this work