 Jiaming Zhang1
Jiaming Zhang1 Jia-Da Li
Jia-Da Li- 1Center for Reproductive Medicine, The First Affiliated Hospital, University of South China, Hengyang, China
- 2Hunan Key Laboratory of Animal Models for Human Diseases, School of Life Sciences, Central South University, Changsha, China
- 3Hunan Key Laboratory of Medical Genetics, Center for Medical Genetics, Central South University, Changsha, China
Parkinson’s disease is the second most common neurodegenerative disorder. Although the pathogenesis of Parkinson’s disease is not entirely clear, the aberrant aggregation of α-synuclein has long been considered as an important risk factor. Elucidating the mechanisms that influence the aggregation of α-synuclein is essential for developing an effective diagnostic, preventative and therapeutic strategy to treat this devastating disease. The aggregation of α-synuclein is influenced by several post-translational modifications. Here, we summarized the major post-translational modifications (phosphorylation, ubiquitination, truncation, nitration, O-GlcNAcylation) of α-synuclein and the effect of these modifications on α-synuclein aggregation, which may provide potential targets for future therapeutics.
Introduction
Parkinson’s disease (PD), the second most common neurodegenerative disorder, manifests with resting tremor, bradykinesia, rigidity, postural instability, and gait impairment (Auluck et al., 2010; Tysnes and Storstein, 2017). PD is characterized by loss of dopaminergic neuronal cells in the substantia nigra pars compacta (SNpc) and cytoplasmic deposition of amyloid-like aggregates termed Lewy Bodies (LB) (Forno, 1996; Braak et al., 2003; Shulman et al., 2011).
The major component of LB is α-synuclein aggregates (Spillantini et al., 1997). Furthermore, duplications, triplications, or point mutations in α-synuclein also contribute to some autosomal dominant early-onset PDs and sporadic PDs (Golbe et al., 1990; Polymeropoulos et al., 1997; Kruger et al., 1998; Singleton et al., 2003, 2004; Farrer et al., 2004; Zarranz et al., 2004; Hoffman-Zacharska et al., 2013; Proukakis et al., 2013; Pasanen et al., 2014; Ysselstein et al., 2017). Golbe et al. (1990) identified the α-synuclein A53T mutation in a PD patient. Several other mutations have been identified since then, such as A30P, A18T, A29S, E46K, H50Q, G51D, and A53E.
The contribution of α-synuclein in the pathogenesis of PD has been extensively studied in a variety of animal models, including mice, Drosophila, and Caenorhabditis elegans. Transgenic mice or flies overexpressing WT, A30P or A53T α-synuclein show motor deficits and neuronal inclusions (Feany and Bender, 2000; Kahle et al., 2000; Masliah et al., 2000; van der Putten et al., 2000; Maguire-Zeiss et al., 2005; Lelan et al., 2011; Lin et al., 2012). The α-synuclein aggregates in dopaminergic neurons are found in WT, A30P, or A53T human α-synuclein transgenic nematodes C. elegans (Kuwahara et al., 2006). Overexpression of human α-synuclein in C. elegans causes age- and dose-dependent dopaminergic neurodegeneration (Cao et al., 2005; Hamamichi et al., 2008).
α-Synuclein also undergoes extensive post-translational modification (PTM), which influence the aggregation and/or cytotoxicity. PTMs may mediate the environmental factors on the pathogenesis. In this review, we will summarize physiological and pathological roles of α-synuclein, emphasizing the involvement of PTMs.
Structure of α-Synuclein
In humans, α-synuclein is a member of synuclein family, which includes α-synuclein, β-synuclein, and γ-synuclein (Lashuel et al., 2013). α-Synuclein, a 140-amino acid protein, is composed of three distinct domains. The N-terminus (1–60 residues) contains four imperfect KTKEGV motif repeats. The central hydrophobic domain of α-synuclein (61–95 residues), also known as the non-amyloid component (NAC), is crucial for its aggregation (Giasson et al., 2001). The C-terminus (96–140 residues) is enriched in acidic residues and is the major phosphorylation site (Uversky and Eliezer, 2009).
α-Synucleins purified from bacterial or mouse tissues under denaturing conditions are ‘natively unfolded’ monomers of about 14 kDa (Weinreb et al., 1996). It may acquire α-helical secondary structure upon binding to lipid vesicles (Davidson et al., 1998; Eliezer et al., 2001). Bartels et al. (2011) found that endogenous α-synuclein under non-denaturing conditions form a folded tetramer and non-crosslinked monomer in all cells, plus some putative dimers in the HeLa, HEK, and red blood cells. They further showed that very few native human α-synuclein tetramers form aggregation, whereas recombinantly expressed monomers readily aggregated into amyloid-like fibrils in vitro (Bartels et al., 2011).
Function of α-Synuclein
α-Synuclein is mainly expressed at presynaptic terminals and has been implicated in numerous cellular processes (Adamczyk et al., 2005). However, the exact physiological function of α-synuclein is still unclear. Under physiological conditions, α-synuclein may be involved in the compartmentalization, storage, and recycling of neurotransmitters (Allen Reish and Standaert, 2015).
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins are crucial for release of neurotransmitters at the neuronal synapse, vesicle recycling, and synaptic integrity (Goda, 1997; Gerst, 1999). Burre et al. (2010) demonstrated that α-synuclein acts as a molecular chaperone to assist the folding and refolding of SNARE proteins. α-Synuclein directly binds to the SNARE protein synaptobrevin-2 and promote the formation of SNARE-complex (Burre et al., 2010). Moreover, α-synuclein is also involved in the dynamics of synaptic vesicles (SVs) trafficking to control the amount of vesicles docked at the synapses during neurotransmitter release (Burre, 2015). As a result, α-synuclein null mice exhibit accelerated recovery of neurotransmitter release when presented with multiple stimuli. Depletion of α-synuclein from rodent hippocampal neurons also induces a significant loss of undocked SVs (Cabin et al., 2002).
Aggregation of α-Synuclein
Aggregates of α-synuclein are the major component of Lewy body, the pathological marker of PD, dementia with Lewy bodies and Lewy body variant of Alzheimer’s disease (Spillantini et al., 1997, 1998). The aggregation of α-synuclein is formed in three steps. The first step is the rate-limiting step, in which the soluble unstructured monomeric species were converted into partially soluble oligomers when nucleation-dependent chain polymerization occurs. Then, the oligomers aggregate into insoluble mature fibrils. At last, the amyloid fibrillar aggregates are formed (Harper et al., 1997; Walsh et al., 1997; Lambert et al., 1998). Miake et al. (2002) have shown that α-synuclein filaments assembled in vitro or extracted from multiple system atrophy (MSA) brains are insoluble to detergents and partially resistant to proteinase K (PK) digestion. Variable amounts of neuritic PK-resistant α-synuclein have been detected in the striatum of all the LB disease cases. PK resistance of α-synuclein may be useful for the development of biomarkers of LB diseases (Neumann et al., 2004).
Both fibrils and oligomers have been shown to display toxicity. Peelaerts et al. (2015) showed that α-synuclein fibrils can lead to progressive motor impairment and cell death. Lots of studies have suggested that amyloids associated with neurodegenerative diseases spread in a prion-like fashion. Fibrillar α-synuclein assemblies seed the aggregation of monomeric α-synuclein in vitro and spread from one cell to another in cell cultures and animal models (Wood et al., 1999; Desplats et al., 2009; Hansen et al., 2011). Multiple lines of evidence have also suggested that oligomeric species of α-synuclein are toxic. In this review, we mainly summarized the evidence supporting the toxicity of α-synuclein oligomers in PD and possible mechanisms for this toxicity.
Toxicity of α-Synuclein
α-Synuclein aggregates may cause cytotoxicity through several pathways, such as mitochondrial dysfunction, endoplasmic reticulum (ER) stress, proteasome system dysfunction, phagocytosis and inflammatory response in microglia, membrane damage, and synaptic dysfunction.
Mitochondrial Dysfunction
The loss of dopaminergic neurons is a major pathological feature of PD patient. Dopaminergic neurons are particularly sensitive to mitochondrial dysfunction due to their high energy demands and increased oxidative stress (Ryan et al., 2015). Both the monomer and oligomer of α-synuclein show toxicity to mitochondria. The translocase of the outer membrane (TOM) 20 receptors are important for the mitochondrial protein import. α-Synuclein can inhibit the protein import of mitochondria by binding to TOM20 (Di Maio et al., 2016). The voltage-dependent anion channel (VDAC) is the major channel of the mitochondrial outer membrane, which controls most of the metabolite fluxes in and out of the mitochondria. Rostovtseva et al. (2015) showed that monomeric α-synuclein reversibly block VDAC in a highly voltage-dependent manner.
α-Synuclein oligomers cause mitochondria fragmentation in a dopaminergic cell line SH-SY5Y (Plotegher et al., 2014). α-Synuclein oligomers decreased the retention time of exogenously added calcium, promoted calcium-induced mitochondrial swelling and depolarization. α-Synuclein oligomers also accelerated cytochrome C release, which cause the apoptosis of dopaminergic neurons (Luth et al., 2014).
Endoplasmic Reticulum Stress
Endoplasmic reticulum is responsible for the synthesis, modification, and delivery of proteins to their target sites within the secretory pathway and the extracellular space. Disruption of any of these processes may cause ER stress (Hampton, 2000). The folding-incompetent proteins can cause ER stress and an ER stress response, called unfolded protein response (UPR). UPR is the biochemical basis for many ER storage diseases (Schroder and Kaufman, 2005). Castillo-Carranza et al. (2012) showed that α-synuclein oligomers induced ER stress in SH-SY5Y cells. Colla et al. (2012) found that accumulation of the toxic α-synuclein oligomers are temporally and spatially linked to the induction of chronic ER stress in the α-synuclein transgenic mice.
Proteasome System Dysfunction
Ubiquitin proteasome system is a highly regulated mechanism of intracellular protein degradation and turnover (Tanaka and Chiba, 1998). PD patients have a vulnerable proteasomal function in the substantia nigra, which may be due to the inhibition of α-synuclein oligomers on the proteasomal system (McNaught and Jenner, 2001; McNaught et al., 2001, 2002, 2003). Indeed, α-synuclein is co-localized with ubiquitin and 20S proteasomal components in Lewy bodies. The α-synuclein oligomers may directly bind to the 20S proteasome. Binding of α-synuclein oligomers to the proteasome inhibits the chymotrypsin-like proteasomal activity of the 20S proteolytic particle (Lindersson et al., 2004). Interestingly, A53T α-synuclein oligomers impaired the proteasomal activity in PC12 cells, which can be reversed by Congo Red, an inhibitor of α-synuclein oligomerization (Emmanouilidou et al., 2010).
Phagocytosis and Inflammatory Response in Microglia
Microglia are the resident macrophage cells in the central nervous system (CNS), involved in chemotaxis, phagocytosis, and secretion of a variety of cytokines and proteases. Microglia have a close relationship with the pathogenesis of PD (Sanchez-Guajardo et al., 2015; Ferreira and Romero-Ramos, 2018). Park et al. (2008) found that microglial phagocytosis is enhanced by extracellular monomeric α-synuclein but inhibited by the aggregated α-synuclein. The inflammatory response in microglia is activated by Toll-like receptor 2 (TLR2) (Stirling et al., 2014). Kim et al. (2013) showed that extracellular α-synuclein released from neuronal cells is an endogenous agonist for TLR2.
Membrane Damage
The cell membrane is an important barrier to prevent extracellular substances from entering the cell, which ensures the relative stability of the intracellular environment and enables various biochemical reactions to run in an orderly manner. Membrane integrity is essential for the basic function of all cell types. Dysfunctional membranes can also lead to abnormal calcium homeostasis. α-Synuclein has been shown to undergo accelerated aggregation at membrane surfaces when incubated with synthetic or natural phospholipid vesicles or supported lipid bilayers, presumably because the two dimensional surface of the membrane increases the probability of molecular interactions needed for oligomerization (Haque et al., 2010). Danzer et al. (2007) showed some types of α-synuclein oligomers induced cell death via disruption of cellular calcium influx by a presumably pore-forming mechanism. Angelova et al. (2016) further confirmed that α-synuclein interacts with membranes to affect Ca2+ signaling in a structure-specific manner and the oligomeric β-sheet-rich α-synuclein species ultimately leads to Ca2+ dysregulation.
Several approaches have been developed to alleviate the α-synuclein-induced membrane damage. Endosulfine-α, which can bind specifically to membrane-associated α-synuclein, alleviates dopaminergic cell death by interfering with the formation of neurotoxic α-synuclein oligomers at the membrane surface (Ysselstein et al., 2017). A novel compound NPT100-18A, which can displace α-synuclein from the membrane, can also reduce a-synuclein toxicity (Wrasidlo et al., 2016).
Synaptic Dysfunction
Synaptic dysfunction is an early pathological feature of PD (Schulz-Schaeffer, 2010). SNARE complex is required for SV fusion. α-Synuclein oligomers prevent the formation of the SNARE complex by binding to synaptobrevin (Choi et al., 2013).
Axonal transport, which relies on the microtubule (MT) network, is fundamental for the maintenance of neuronal homeostasis (Goldstein et al., 2008). Prots et al. (2013) showed that α-synuclein oligomers significantly inhibited MT assembly. 3,4-Dihydroxyphenylacetaldehyde (DOPAL) is a catabolite generated from dopamine by monoamine oxidase (Burke et al., 2003; Goldstein et al., 2011). It has been shown that DOPAL can cause α-synuclein oligomerization in vitro and in cell models (Burke et al., 2008; Lima et al., 2018). Plotegher et al. (2017) showed that this kind of α-synuclein-DOPAL oligomers can permeabilize cholesterol-containing lipid membranes mimicking SVs in vitro, which suggests that the synergistic effect of α-synuclein and DOPAL accumulation in DA neurons may lead to the formation of oligomers, negatively impacting the structure and function of SVs.
Vesicles for synaptic release are produced by the Golgi apparatus. The dysfunction of Golgi can lead to abnormity of synaptic function. Gosavi et al. (2002) showed that over-expression of α-synuclein in COS-7 cells caused Golgi fragmentation. As mentioned previously, pore-like oligomers of α-synuclein could also rupture SVs, leading to decreased neurotransmitter release, as well as permeabilization of cell membranes, which could result in Ca2+ influx and excitotoxicity (Danzer et al., 2007).
Post-Translational Modifications of α-Synuclein
α-Synuclein is subjected to extensive post-transcriptional modifications (PTMs), including phosphorylation, ubiquitination, nitration, truncation, and O-GlcNAcylation. PTMs of α-synuclein may influence its toxicity and aggregation.
Phosphorylation
α-Synuclein within LB can be phosphorylated at serine 129 and 87 (S129-P, S87-P) (Hasegawa et al., 2002; Anderson et al., 2006; Paleologou et al., 2010). S129-P has emerged as a defining hallmark of PD and related synucleinopathies. Feany and Bender (2000) showed that α-synuclein is also phosphorylated at tyrosine 125, 133, and 136 (Y125-P, Y133-P, and Y136-P) (Ellis et al., 2001; Nakamura et al., 2001; Ahn et al., 2002; Negro et al., 2002; Takahashi et al., 2003). The kinases that mediate phosphorylation at Y125 of α-synuclein are still unknown. Hejjaoui et al. (2011) have developed a semi-synthetic strategy that enables the site-specific introduction of single phosphorylation at Y125. They showed that phosphorylation at Y125 does not affect the fibrillization of α-synuclein (Burai et al., 2015). The impact of the phosphorylation at tyrosine 133 and 135 on α-synuclein aggregation is still unknown.
A number of kinases have been shown to phosphorylate α-synuclein at S129 in vitro, including casein kinase I (CKI), casein kinase II (CKII), the G protein-coupled receptor kinases (GRK), LRRK2, and polo-like kinases (PLK) (Okochi et al., 2000; Pronin et al., 2000; Inglis et al., 2009).
Fujiwara et al. (2002) showed that phosphorylation of S129 in α-synuclein by CKII promotes in vitro fibrillation. Smith et al. (2005) indicated that phosphorylation at S-129 by CKII promotes the formation of cytoplasmic inclusions in some cell culture models.
Phosphorylation of α-synuclein at S-129 by GRK2 was reported to be toxic. Feany and Bender (2000) have studied the phosphorylation of α-synuclein in Drosophila. They showed that co-expression of Drosophila GRK2 with α-synuclein enhances the formation of α-synuclein oligomers and accelerates neuronal loss, as compared to the Drosophila expressing α-synuclein alone (Feany and Bender, 2000).
α-Synuclein phosphorylation at S129 is largely reduced in PLK2-/- transgenic mice, supporting the involvement of PLK in α-synuclein phosphorylation in vivo (Inglis et al., 2009). PLK2-induced phosphorylation has no effect on the aggregation of α-synuclein. Nevertheless, Oueslati et al. (2013) showed that PLK2 binds directly to α-synuclein in an ATP-dependent manner and regulates α-synuclein selective clearance via the lysosome–autophagic degradation pathway, which suggests a neuroprotective role of PLK2 against PD pathology.
Ubiquitination and Sumoylation
The core of LBs is immunoreactive for both α-synuclein and ubiquitin proteins and is surrounded by a rim of α-synuclein (Gomez-Tortosa et al., 2000). However, the major α-synuclein species in LBs is mono-, di-, and tri-ubiquitinated, suggesting the involvement of ubiquitination in the pathophysiologic properties of α-synuclein (Hasegawa et al., 2002; Sampathu et al., 2003; Tofaris et al., 2003; Nonaka et al., 2005). The ubiquitination of α-synuclein is correlated with three E3 ubiquitin-protein ligases: C-terminal U-box domain of co-chaperone Hsp70-interacting protein (CHIP), seven in absentia homolog (SIAH) and neuronal precursor cell-expressed, developmentally down-regulated gene 4 (Nedd4) (Liani et al., 2004; Shin et al., 2005; Tofaris et al., 2011).
The mammalian homologs of Drosophila seven in absentia (SIAH-1 and SIAH-2) gene have been characterized as a family of RING-type E3 ligases (Wheeler et al., 2002). Both in vivo and in vitro data showed that ubiquitination of α-synuclein by SIAH promotes the formation of inclusions. Rott et al. (2008) showed that ubiquitination of α-synuclein in vitro by SIAH promotes the formation of higher molecular weight α-synuclein. They then used electron microscopy to show that α-synuclein ubiquitinated by SIAH formed more aggregates (Rott et al., 2008). Lee et al. (2008) also showed that SIAH-1 or SIAH-2-mediated ubiquitination enhances the aggregation of α-synuclein and formation of α-synuclein-positive inclusion in PC12 cells and SH-SY5Y human neuroblastoma (Lee et al., 2008).
CHIP is a multidomain chaperone, utilizing both a tetratricopeptide/Hsp70 binding domain and a U-box/ubiquitin ligase domain to recognize misfolded proteins (Demand et al., 2001; Murata et al., 2001). CHIP is able to mono- and poly-ubiquitinate α-synuclein (Kalia et al., 2011). Shin et al. (2005) showed that CHIP colocalizes with α-synuclein in Lewy bodies and also in a cell culture model of α-synuclein inclusions. Overexpression of CHIP inhibits α-synuclein aggregation and increases α-synuclein degradation in cell culture. Interestingly, they also indicated that CHIP can regulate α-synuclein degradation both via the proteasomal degradation pathway and the lysosomal degradation pathway (Shin et al., 2005). A study from Tetzlaff et al. (2008) showed that CHIP selectively reduced α-synuclein oligomerization and toxicity in a tetratricopeptide domain-dependent, U-box independent manner by specifically degrading toxic α-synuclein oligomers.
Nedd4 is a HECT-domain E3 that functions at the plasma membrane in the turnover of a number of membrane-associated proteins. Tofaris et al. (2011) showed that Nedd4 can act as an E3 for α-synuclein. They demonstrated that Nedd4 directly binds to α-synuclein in brain and cell extracts and promotes the degradation of endogenous α-synuclein by lysosomes (Tofaris et al., 2011). They further found that Nedd4-mediated degradation protects against α-synuclein-induced toxicity in the Drosophila and rodent models of Parkinson’s disease (Davies et al., 2014). Nedd4-1-linked Lys-63 ubiquitination was demonstrated to specify the fate of extrinsic and de novo synthesized α-synuclein by facilitating their targeting to endosomes (Sugeno et al., 2014). In yeast ubiquitin ligase, the Nedd4 ortholog Rsp5 is a key enzyme involved in the degradation of abnormal or unfavorable proteins. Wijayanti et al. (2015) have isolated novel hyperactive forms of Rsp5 that alleviate α-synuclein toxicity, by enhancing the clearance of α-synuclein, including the processes of interaction, ubiquitination, and degradation.
The site-specific effects of ubiquitination on aggregation and clearance have been studied using a semi-synthetic strategy (Hejjaoui et al., 2011; Abeywardana et al., 2013). Monomeric ubiquitination of α-synuclein at K6 was shown to resist fibril formation when compared to unmodified protein (Hejjaoui et al., 2011); Ubiquitination of α-synuclein at K10 and K23 readily form fibrils; Ubiquitination of α-synuclein at K6, K12, and K21 moderately inhibit the formation of fibrils; Ubiquitination of α-synuclein at K32, K34, K43, and K96 displayed no fibril formation, suggesting a strong inhibitory effect (Meier et al., 2012). Haj-Yahya et al. (2013) have incorporated K48-linked di- or tetra-Ub chains onto the side chain of Lys12 of α-synuclein and demonstrated that the length of the Ub chain plays an important role in regulating α-synuclein fibril formation and clearance.
α-Synuclein is also conjugated to small ubiquitin-like modifier (SUMO) at lysines. Rott et al. (2017) demonstrated that α-synuclein is SUMOylated by PIAS2, and SUMOylated α-synuclein and PIAS2 are markedly elevated in the substantia nigra of PD brains. Further, Lewy bodies are positive for both SUMO1 and PIAS2. They found that SUMOylation increases α-synuclein aggregation by two self-reinforcing mechanisms. First, SUMOylation by PIAS2 directly promotes the aggregation of α-synuclein. Second, SUMOylation impairs α-synuclein ubiquitination and prevents α-synuclein degradation. Therefore, SUMOylation blockers may provide a strategy to prevent intracellular α-synuclein aggregation (Rott et al., 2017). However, Krumova et al. (2011) showed that sumoylation inhibits α-synuclein aggregation and toxicity. In vitro study demonstrated that SUMOylation at K102 of α-synuclein results in more pronounced inhibition of aggregation than the corresponding modification at K96 (Abeywardana and Pratt, 2015).
Truncation
Besides full-length α-synuclein, there exist small amounts of various truncated species with apparent molecular masses of 10–15 kDa in the LBs (Baba et al., 1998; Crowther et al., 1998; Campbell et al., 2001). It is estimated that about 15% α-synuclein in LBs is truncated. And the C-terminally truncated α-synuclein may act as effective seeds to accelerate the aggregation of the full-length protein.
The carboxyl-terminal-truncated α-synuclein produced by aberrant proteolysis, is found in association with α-synuclein aggregates (Tofaris et al., 2003). Tofaris et al. (2003) investigated the effects of truncation by generating both transgenic Drosophila and transgenic mice expressing human α-synuclein. They found that the truncated form of α-synuclein (1–120) increased accumulation of high molecular weight α-synuclein species, and enhanced neurotoxicity in vivo (Periquet et al., 2007). They showed that the striatal dopamine levels are reduced and the transgenic mice showed a progressive reduction in spontaneous locomotion and an increased response to amphetamine (Tofaris et al., 2006). Hoyer et al. (2004) used recombinant proteins and showed that the fragments (1–110; 1–119; 110–140) promoting nucleation seed the aggregation of full-length α-synuclein. Murray et al. (2003) also showed that the truncated α-synuclein variants, 1–89, 1–102, 1–110, 1–120, and 1–130 aggregated more rapidly than the full-length protein.
Several enzymes have been implicated in the truncation of α-synuclein, including calpain I, Neurosin, Cathepsin D, and Matrix metalloproteinase 3 (Iwata et al., 2003; Mishizen-Eberz et al., 2005; Sevlever et al., 2008; Choi et al., 2011).
Since α-synuclein is predominantly localized to the pre-synaptic terminal, it may be a substrate for soluble or membrane-associated proteases such as the calcium-activated neutral protease calpain I. Mishizen-Eberz et al. (2003) demonstrated that Calpain I cleaves wild-type α-synuclein predominantly after amino acid 57 and within the NAC region (73, 74, and 83). Calpain-mediated processing of soluble α-synuclein inhibits fibrillization, while processing of fibrillar α-synuclein promotes further aggregation (Mishizen-Eberz et al., 2005).
Neurosin, a serine protease predominantly expressed in the CNS, is presumed to play an important role in the degradation of α-synuclein (Iwata et al., 2003). Neurosin cleaves α-synuclein after amino acid 80 and 97. Cleavage of α-synuclein after 80 by neurosin may inhibit the polymerization; however, the fragment cleaved after 97 has a stronger propensity to polymerize than non-processed α-synuclein (Kasai et al., 2008).
Nitration
Oxidative injury has been implicated in the pathogenesis of PD (Schapira and Jenner, 2011; Bose and Beal, 2016). The action of oxygen and nitric oxide and their products, especially peroxynitrite, leads to the nitration of tyrosine residues in proteins. Giasson et al. (2000) first showed that a-synuclein is nitrated when present in the major filamentous and in the insoluble fractions of affected brain regions of synucleinopathies. All four tyrosine residues in α-synuclein (Y39, Y125, Y133, and Y136) are susceptible to nitration (Sevcsik et al., 2011; Burai et al., 2015).
Danielson et al. (2009) showed that nitration of Y-39 accelerates the oligomerization of α-synuclein, and a mutation in this residue leads to high levels of fibrilization.
Hodara et al. (2004) showed that monomeric or dimeric forms of nitrated α-synuclein accelerate the fibril formation and seed the fibrillation of non-modified α-synuclein. On the other hand, nitrated α-synuclein oligomers inhibit the fibril formation (Hodara et al., 2004).
Through site-specific incorporation of 3-nitrotyrosine at different regions of α-synuclein, Burai et al. (2015) indicated that different site-specifically nitrated α-synuclein species exhibit distinct aggregation properties. They further showed that intermolecular interactions between the N- and C-terminal regions of α-synuclein play critical roles in mediating nitration-induced α-synuclein oligomerization (Burai et al., 2015).
O-GlcNAcylation
O-GlcNAcylation is a dynamic biochemical process, in which N-acetylglucosamine (GlcNAc) from uridine 5′-diphospho-N-acetylglucosamine (UDP-GlcNAc) is transferred to the serine and threonine residues of proteins by O-GlcNAc transferase (OGT) and removed by O-GlcNAcase (OGA) (Hart et al., 2007). More than 1,000 proteins can be modified by O-GlcNAc, including molecular chaperones, transcription factors, RNA polymerase II, nucleoporin, RNA binding proteins, kinases, and cytoskeletal proteins (Hardiville and Hart, 2014). O-GlcNAcylation has identified threonine (T) residue 33, 34, 54, 59, 64, 72, 75, 81, and 87 of α-synuclein isolated from mouse and human samples (Wang et al., 2009, 2010, 2017; Alfaro et al., 2012; Morris et al., 2015).
To understand the effect of O-GlcNAcylation on the aggregation of α-synuclein, Marotta et al. (2015) synthesized a peptide of α-synuclein comprising residues 68–77, in which the T72 is O-GlcNAcylated. As compared with the unmodified peptide, the O-GlcNAcylated peptide inhibits full-length α-synuclein fibrillization. They further synthesized a full-length α-synuclein, with O-GlcNAcylation at T72. O-GlcNAcylation at T72 completely blocks the formation of both fiber and oligomer aggregates in vitro. They synthesized a full-length α-synuclein with O-GlcNAcylation at S87, which still aggregates but with slower kinetics than the unmodified protein (Lewis et al., 2017). Recently, Levine et al. (2019) showed that several of the O-GlcNAc sites inhibit the toxicity of extracellular α-synuclein fibers that are likely culprits in the spread of PD. They also demonstrated that O-GlcNAcylation can inhibit the aggregation of an aggressive mutant of α-synuclein.
To study the functional consequences of enzymatic O-GlcNAcylation of α-synuclein, we co-expressed a shorter form of OGT (sOGT) and α-synuclein in bacteria and got enzymatically O-GlcNAcylated α-synuclein. The enzymatic O-GlcNAcylation also significantly blocked α-synuclein aggregation (Zhang et al., 2017).
Conclusion and Perspective
In this review, we have summarized the major PTMs of α-synuclein (Figure 1). Since the presence of PTMs in α-synuclein is able to influence its aggregation and toxicity (Table 1), targeting PTMs may be used to develop novel therapeutic approaches for PD. However, it should be noted that most of the effect of PTMs on the α-synuclein aggregation are carried out in vitro; the in vivo effect is still elusive. Furthermore, α-synuclein may have multiple different PTMs at the same time in vivo; however, the current researches regarding PTMs of α-synuclein are studied individually. The interaction of PTMs of α-synuclein has been widely studied. Phosphorylated α-synuclein has been reported to be one of the target proteins for ubiquitination in synucleinopathies (Hasegawa et al., 2002). Shahpasandzadeh et al. (2014), for the first time, demonstrated an interplay between α-synuclein sumoylation and phosphorylation to control protein turnover. They showed that sumoylation exhibits a protective role against α-synuclein toxicity and inclusion formation in yeast cells (Shahpasandzadeh et al., 2014). There is a complex and dynamic interplay between O-GlcNAcylation and phosphorylation (Hart et al., 2007). The interplay between α-synuclein O-GlcNAcylation and phosphorylation is still unknown. As mentioned before, neurosin is one enzyme that mediates the truncation of α-synuclein. Kasai et al. (2008) showed that phosphorylated α-synuclein was more resistant to degradation by neurosin than non-phosphorylated α-synuclein. A reciprocal reaction may also occur between other PTMs, which still need further study. Third, it is interesting that, in some cases, the same modification may have a different effect. For instance, phosphorylation at S129 by CKII may promote aggregation, but phosphorylation at S129 by PLK2 promotes degradation. More studies are required to understand the underlying mechanisms for the discrepancy. Lastly, although it is well known that PTMs are regulated by the environmental stimuli, few studies have attempted to use PTMs to link environmental factors and α-synuclein toxicity. Thus, future studies on the PTMs of α-synuclein in vivo will help to address these concerns, and improve our understanding surrounding the role of the gene-environment interaction in PD pathogenesis.
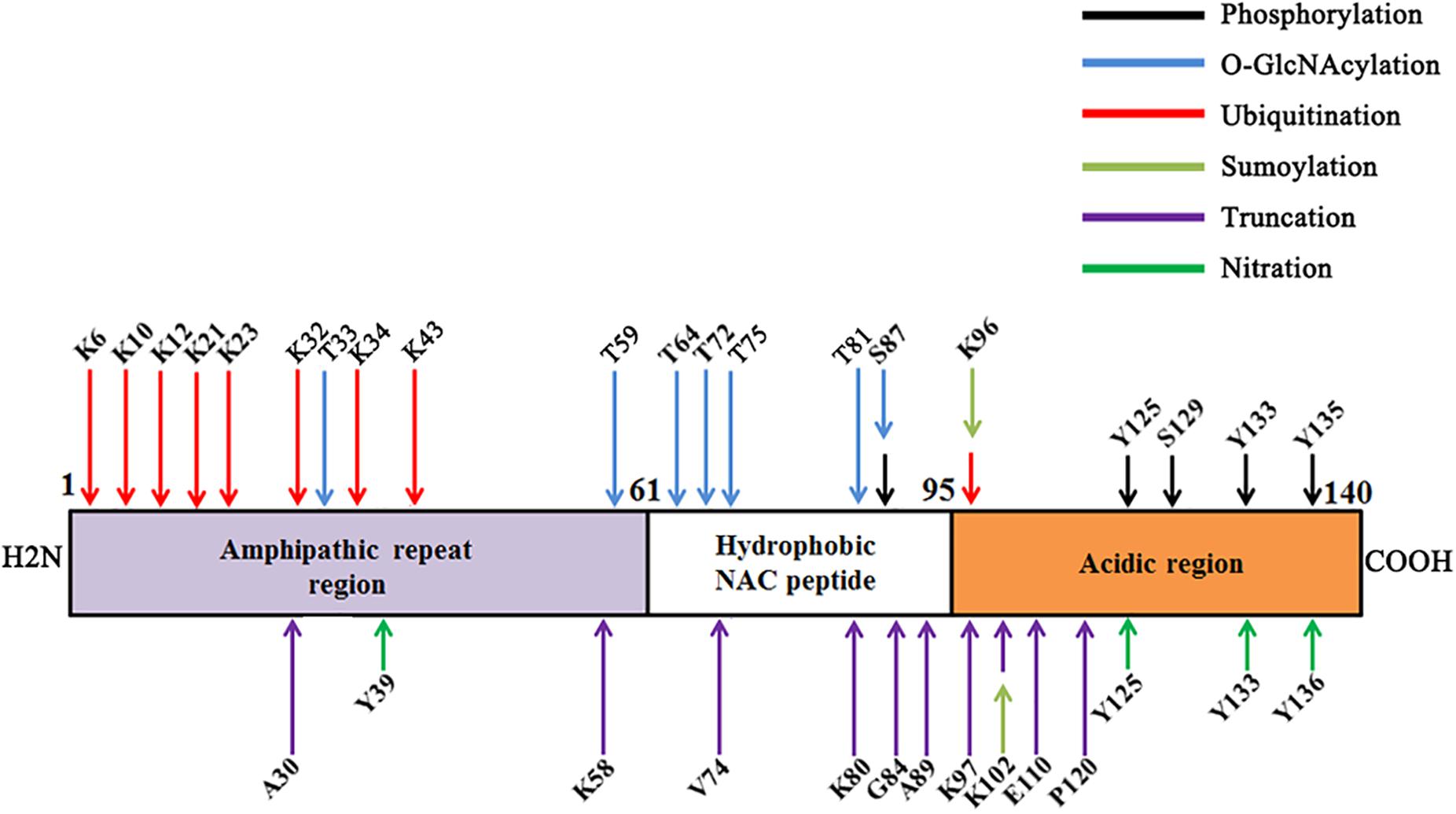
Figure 1. Major post-translational modifications (PTMs) on various amino acids of α-synuclein.
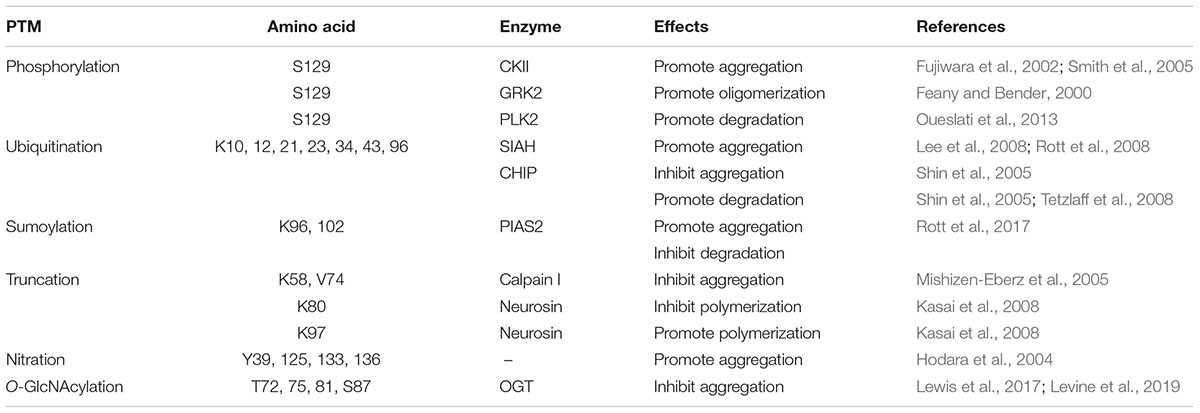
Table 1. Functional consequences of the major PTMs on α-synuclein.
Author Contributions
All authors participated in designing the concept of this manuscript.
Funding
This project was financially supported by National Natural Science Foundation of China (Grant Nos. 81770780 and 81728013), the Key Research and Development Programs from Hunan Province (Grant Nos. 2018DK2010 and 2018DK2013).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to the authors whose work could not be cited in this review due to space limitations but appreciate all of the many contributions to the large body of literature we have tried to summarize here.
References
Abeywardana, T., Lin, Y. H., Rott, R., Engelender, S., and Pratt, M. R. (2013). Site-specific differences in proteasome-dependent degradation of monoubiquitinated alpha-synuclein. Chem. Biol. 20, 1207–1213. doi: 10.1016/j.chembiol.2013.09.009
Abeywardana, T., and Pratt, M. R. (2015). Extent of inhibition of alpha-synuclein aggregation in vitro by SUMOylation is conjugation site- and SUMO isoform-selective. Biochemistry 54, 959–961. doi: 10.1021/bi501512m
Adamczyk, A., Solecka, J., and Strosznajder, J. B. (2005). Expression of alpha-synuclein in different brain parts of adult and aged rats. J. Physiol. Pharmacol. 56, 29–37.
Ahn, S., Kim, J., Lucaveche, C. L., Reedy, M. C., Luttrell, L. M., Lefkowitz, R. J., et al. (2002). Src-dependent tyrosine phosphorylation regulates dynamin self-assembly and ligand-induced endocytosis of the epidermal growth factor receptor. J. Biol. Chem. 277, 26642–26651. doi: 10.1074/jbc.M201499200
Alfaro, J. F., Gong, C. X., Monroe, M. E., Aldrich, J. T., Clauss, T. R., Purvine, S. O., et al. (2012). Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. U.S.A. 109, 7280–7285. doi: 10.1073/pnas.1200425109
Allen Reish, H. E., and Standaert, D. G. (2015). Role of alpha-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Parkinsons Dis. 5, 1–19. doi: 10.3233/JPD-140491
Anderson, J. P., Walker, D. E., Goldstein, J. M., de Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Angelova, P. R., Ludtmann, M. H., Horrocks, M. H., Negoda, A., Cremades, N., Klenerman, D., et al. (2016). Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. J. Cell Sci. 129, 1792–1801. doi: 10.1242/jcs.180737
Auluck, P. K., Caraveo, G., and Lindquist, S. (2010). alpha-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 26, 211–233. doi: 10.1146/annurev.cellbio.042308.113313
Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M., et al. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Bose, A., and Beal, M. F. (2016). Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 139(Suppl. 1), 216–231. doi: 10.1111/jnc.13731
Braak, H., Del Tredici, K., Rub, U., de Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
Burai, R., Ait-Bouziad, N., Chiki, A., and Lashuel, H. A. (2015). Elucidating the role of site-specific nitration of alpha-synuclein in the pathogenesis of Parkinson’s disease via protein semisynthesis and mutagenesis. J. Am. Chem. Soc. 137, 5041–5052. doi: 10.1021/ja5131726
Burke, W. J., Kumar, V. B., Pandey, N., Panneton, W. M., Gan, Q., Franko, M. W., et al. (2008). Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 115, 193–203. doi: 10.1007/s00401-007-0303-9
Burke, W. J., Li, S. W., Williams, E. A., Nonneman, R., and Zahm, D. S. (2003). 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson’s disease pathogenesis. Brain Res. 989, 205–213. doi: 10.1016/S0006-8993(03)03354-7
Burre, J. (2015). The synaptic function of alpha-synuclein. J. Parkinsons Dis. 5, 699–713. doi: 10.3233/JPD-150642
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Cabin, D. E., Shimazu, K., Murphy, D., Cole, N. B., Gottschalk, W., McIlwain, K. L., et al. (2002). Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 22, 8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002
Campbell, B. C., McLean, C. A., Culvenor, J. G., Gai, W. P., Blumbergs, P. C., Jakala, P., et al. (2001). The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J. Neurochem. 76, 87–96. doi: 10.1046/j.1471-4159.2001.00021.x
Cao, S., Gelwix, C. C., Caldwell, K. A., and Caldwell, G. A. (2005). Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J. Neurosci. 25, 3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005
Castillo-Carranza, D. L., Zhang, Y., Guerrero-Munoz, M. J., Kayed, R., Rincon-Limas, D. E., and Fernandez-Funez, P. (2012). Differential activation of the ER stress factor XBP1 by oligomeric assemblies. Neurochem. Res. 37, 1707–1717. doi: 10.1007/s11064-012-0780-7
Choi, B. K., Choi, M. G., Kim, J. Y., Yang, Y., Lai, Y., Kweon, D. H., et al. (2013). Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. U.S.A. 110, 4087–4092. doi: 10.1073/pnas.1218424110
Choi, D. H., Kim, Y. J., Kim, Y. G., Joh, T. H., Beal, M. F., and Kim, Y. S. (2011). Role of matrix metalloproteinase 3-mediated alpha-synuclein cleavage in dopaminergic cell death. J. Biol. Chem. 286, 14168–14177. doi: 10.1074/jbc.M111.222430
Colla, E., Coune, P., Liu, Y., Pletnikova, O., Troncoso, J. C., Iwatsubo, T., et al. (2012). Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J. Neurosci. 32, 3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012
Crowther, R. A., Jakes, R., Spillantini, M. G., and Goedert, M. (1998). Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 436, 309–312. doi: 10.1016/S0014-5793(98)01146-6
Danielson, S. R., Held, J. M., Schilling, B., Oo, M., Gibson, B. W., and Andersen, J. K. (2009). Preferentially increased nitration of alpha-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson’s disease. Anal. Chem. 81, 7823–7828. doi: 10.1021/ac901176t
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27, 9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007
Davidson, W. S., Jonas, A., Clayton, D. F., and George, J. M. (1998). Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. doi: 10.1074/jbc.273.16.9443
Davies, S. E., Hallett, P. J., Moens, T., Smith, G., Mangano, E., Kim, H. T., et al. (2014). Enhanced ubiquitin-dependent degradation by NEDD4 protects against alpha-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol. Dis. 64, 79–87. doi: 10.1016/j.nbd.2013.12.011
Demand, J., Alberti, S., Patterson, C., and Hohfeld, J. (2001). Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 11, 1569–1577. doi: 10.1016/S0960-9822(01)00487-0
Desplats, P., Lee, H. J., Bae, E. J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015. doi: 10.1073/pnas.0903691106
Di Maio, R., Barrett, P. J., Hoffman, E. K., Barrett, C. W., Zharikov, A., Borah, A., et al. (2016). alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 8:342ra78. doi: 10.1126/scitranslmed.aaf3634
Eliezer, D., Kutluay, E., Bussell, R. Jr., and Browne, G. (2001). Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 307, 1061–1073. doi: 10.1006/jmbi.2001.4538
Ellis, C. E., Schwartzberg, P. L., Grider, T. L., Fink, D. W., and Nussbaum, R. L. (2001). alpha-synuclein is phosphorylated by members of the Src family of protein-tyrosine kinases. J. Biol. Chem. 276, 3879–3884. doi: 10.1074/jbc.M010316200
Emmanouilidou, E., Stefanis, L., and Vekrellis, K. (2010). Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 31, 953–968. doi: 10.1016/j.neurobiolaging.2008.07.008
Farrer, M., Kachergus, J., Forno, L., Lincoln, S., Wang, D. S., Hulihan, M., et al. (2004). Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 55, 174–179. doi: 10.1002/ana.10846
Feany, M. B., and Bender, W. W. (2000). A Drosophila model of Parkinson’s disease. Nature 404, 394–398. doi: 10.1038/35006074
Ferreira, S. A., and Romero-Ramos, M. (2018). Microglia response during Parkinson’s disease: alpha-synuclein intervention. Front. Cell. Neurosci. 12:247. doi: 10.3389/fncel.2018.00247
Forno, L. S. (1996). Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 55, 259–272. doi: 10.1097/00005072-199603000-00001
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Gerst, J. E. (1999). SNAREs and SNARE regulators in membrane fusion and exocytosis. Cell. Mol. Life Sci. 55, 707–734. doi: 10.1007/s000180050328
Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. doi: 10.1126/science.290.5493.985
Giasson, B. I., Murray, I. V., Trojanowski, J. Q., and Lee, V. M. (2001). A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 276, 2380–2386. doi: 10.1074/jbc.M008919200
Goda, Y. (1997). SNAREs and regulated vesicle exocytosis. Proc. Natl. Acad. Sci. U.S.A. 94, 769–772. doi: 10.1073/pnas.94.3.769
Golbe, L. I., Di Iorio, G., Bonavita, V., Miller, D. C., and Duvoisin, R. C. (1990). A large kindred with autosomal dominant Parkinson’s disease. Ann. Neurol. 27, 276–282. doi: 10.1002/ana.410270309
Goldstein, A. Y., Wang, X., and Schwarz, T. L. (2008). Axonal transport and the delivery of pre-synaptic components. Curr. Opin. Neurobiol. 18, 495–503. doi: 10.1016/j.conb.2008.10.003
Goldstein, D. S., Sullivan, P., Holmes, C., Kopin, I. J., Basile, M. J., and Mash, D. C. (2011). Catechols in post-mortem brain of patients with Parkinson disease. Eur. J. Neurol. 18, 703–710. doi: 10.1111/j.1468-1331.2010.03246.x
Gomez-Tortosa, E., Newell, K., Irizarry, M. C., Sanders, J. L., and Hyman, B. T. (2000). alpha-Synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol. 99, 352–357. doi: 10.1007/s004010051135
Gosavi, N., Lee, H. J., Lee, J. S., Patel, S., and Lee, S. J. (2002). Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 277, 48984–48992. doi: 10.1074/jbc.M208194200
Haj-Yahya, M., Fauvet, B., Herman-Bachinsky, Y., Hejjaoui, M., Bavikar, S. N., Karthikeyan, S. V., et al. (2013). Synthetic polyubiquitinated alpha-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc. Natl. Acad. Sci. U.S.A. 110, 17726–17731. doi: 10.1073/pnas.1315654110
Hamamichi, S., Rivas, R. N., Knight, A. L., Cao, S., Caldwell, K. A., and Caldwell, G. A. (2008). Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. U.S.A. 105, 728–733. doi: 10.1073/pnas.0711018105
Hampton, R. Y. (2000). ER stress response: getting the UPR hand on misfolded proteins. Curr. Biol. 10, R518–R521. doi: 10.1016/S0960-9822(00)00583-2
Hansen, C., Angot, E., Bergstrom, A. L., Steiner, J. A., Pieri, L., Paul, G., et al. (2011). alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725. doi: 10.1172/JCI43366
Haque, F., Pandey, A. P., Cambrea, L. R., Rochet, J. C., and Hovis, J. S. (2010). Adsorption of alpha-synuclein on lipid bilayers: modulating the structure and stability of protein assemblies. J. Phys. Chem. B 114, 4070–4081. doi: 10.1021/jp1006704
Hardiville, S., and Hart, G. W. (2014). Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 20, 208–213. doi: 10.1016/j.cmet.2014.07.014
Harper, J. D., Lieber, C. M., and Lansbury, P. T. Jr. (1997). Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-beta protein. Chem. Biol. 4, 951–959. doi: 10.1016/S1074-5521(97)90303-3
Hart, G. W., Housley, M. P., and Slawson, C. (2007). Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022. doi: 10.1038/nature05815
Hasegawa, M., Fujiwara, H., Nonaka, T., Wakabayashi, K., Takahashi, H., Lee, V. M., et al. (2002). Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076. doi: 10.1074/jbc.M208046200
Hejjaoui, M., Haj-Yahya, M., Kumar, K. S., Brik, A., and Lashuel, H. A. (2011). Towards elucidation of the role of ubiquitination in the pathogenesis of Parkinson’s disease with semisynthetic ubiquitinated alpha-synuclein. Angew. Chem. Int. Ed. Engl. 50, 405–409. doi: 10.1002/anie.201005546
Hodara, R., Norris, E. H., Giasson, B. I., Mishizen-Eberz, A. J., Lynch, D. R., Lee, V. M., et al. (2004). Functional consequences of alpha-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 279, 47746–47753. doi: 10.1074/jbc.M408906200
Hoffman-Zacharska, D., Koziorowski, D., Ross, O. A., Milewski, M., Poznanski, J. A., Jurek, M., et al. (2013). Novel A18T and pA29S substitutions in alpha-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism Relat. Disord. 19, 1057–1060. doi: 10.1016/j.parkreldis.2013.07.011
Hoyer, W., Cherny, D., Subramaniam, V., and Jovin, T. M. (2004). Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry 43, 16233–16242. doi: 10.1021/bi048453u
Inglis, K. J., Chereau, D., Brigham, E. F., Chiou, S. S., Schobel, S., Frigon, N. L., et al. (2009). Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J. Biol. Chem. 284, 2598–2602. doi: 10.1074/jbc.C800206200
Iwata, A., Maruyama, M., Akagi, T., Hashikawa, T., Kanazawa, I., Tsuji, S., et al. (2003). Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum. Mol. Genet. 12, 2625–2635. doi: 10.1093/hmg/ddg283
Kahle, P. J., Neumann, M., Ozmen, L., Muller, V., Jacobsen, H., Schindzielorz, A., et al. (2000). Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 20, 6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000
Kalia, L. V., Kalia, S. K., Chau, H., Lozano, A. M., Hyman, B. T., and McLean, P. J. (2011). Ubiquitinylation of alpha-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLoS One 6:e14695. doi: 10.1371/journal.pone.0014695
Kasai, T., Tokuda, T., Yamaguchi, N., Watanabe, Y., Kametani, F., Nakagawa, M., et al. (2008). Cleavage of normal and pathological forms of alpha-synuclein by neurosin in vitro. Neurosci. Lett. 436, 52–56. doi: 10.1016/j.neulet.2008.02.057
Kim, C., Ho, D. H., Suk, J. E., You, S., Michael, S., Kang, J., et al. (2013). Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4:1562. doi: 10.1038/ncomms2534
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Krumova, P., Meulmeester, E., Garrido, M., Tirard, M., Hsiao, H. H., Bossis, G., et al. (2011). Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 194, 49–60. doi: 10.1083/jcb.201010117
Kuwahara, T., Koyama, A., Gengyo-Ando, K., Masuda, M., Kowa, H., Tsunoda, M., et al. (2006). Familial Parkinson mutant alpha-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J. Biol. Chem. 281, 334–340. doi: 10.1074/jbc.M504860200
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Lashuel, H. A., Overk, C. R., Oueslati, A., and Masliah, E. (2013). The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48. doi: 10.1038/nrn3406
Lee, J. T., Wheeler, T. C., Li, L., and Chin, L. S. (2008). Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 17, 906–917. doi: 10.1093/hmg/ddm363
Lelan, F., Boyer, C., Thinard, R., Remy, S., Usal, C., Tesson, L., et al. (2011). Effects of human alpha-synuclein A53T-A30P mutations on SVZ and local olfactory bulb cell proliferation in a transgenic rat model of Parkinson disease. Parkinsons Dis. 2011:987084. doi: 10.4061/2011/987084
Levine, P. M., Galesic, A., Balana, A. T., Mahul-Mellier, A. L., Navarro, M. X., De Leon, C. A., et al. (2019). alpha-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 116, 1511–1519. doi: 10.1073/pnas.1808845116
Lewis, Y. E., Galesic, A., Levine, P. M., De Leon, C. A., Lamiri, N., Brennan, C. K., et al. (2017). O-GlcNAcylation of alpha-synuclein at serine 87 reduces aggregation without affecting membrane binding. ACS Chem. Biol. 12, 1020–1027. doi: 10.1021/acschembio.7b00113
Liani, E., Eyal, A., Avraham, E., Shemer, R., Szargel, R., Berg, D., et al. (2004). Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 5500–5505. doi: 10.1073/pnas.0401081101
Lima, V. A., do Nascimento, L. A., Eliezer, D., and Follmer, C. (2018). Role of Parkinson’s disease-linked mutations and N-terminal acetylation on the oligomerization of alpha-synuclein induced by 3,4-dihydroxyphenylacetaldehyde. ACS Chem. Neurosci. doi: 10.1021/acschemneuro.8b00498 [Epub ahead of print].
Lin, X., Parisiadou, L., Sgobio, C., Liu, G., Yu, J., Sun, L., et al. (2012). Conditional expression of Parkinson’s disease-related mutant alpha-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J. Neurosci. 32, 9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012
Lindersson, E., Beedholm, R., Hojrup, P., Moos, T., Gai, W., Hendil, K. B., et al. (2004). Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 279, 12924–12934. doi: 10.1074/jbc.M306390200
Luth, E. S., Stavrovskaya, I. G., Bartels, T., Kristal, B. S., and Selkoe, D. J. (2014). Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 289, 21490–21507. doi: 10.1074/jbc.M113.545749
Maguire-Zeiss, K. A., Short, D. W., and Federoff, H. J. (2005). Synuclein, dopamine and oxidative stress: co-conspirators in Parkinson’s disease? Brain Res. Mol. Brain Res. 134, 18–23. doi: 10.1016/j.molbrainres.2004.09.014
Marotta, N. P., Lin, Y. H., Lewis, Y. E., Ambroso, M. R., Zaro, B. W., Roth, M. T., et al. (2015). O-GlcNAc modification blocks the aggregation and toxicity of the protein alpha-synuclein associated with Parkinson’s disease. Nat. Chem. 7, 913–920. doi: 10.1038/nchem.2361
Masliah, E., Rockenstein, E., Veinbergs, I., Mallory, M., Hashimoto, M., Takeda, A., et al. (2000). Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269. doi: 10.1126/science.287.5456.1265
McNaught, K. S., Belizaire, R., Isacson, O., Jenner, P., and Olanow, C. W. (2003). Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 179, 38–46. doi: 10.1006/exnr.2002.8050
McNaught, K. S., Belizaire, R., Jenner, P., Olanow, C. W., and Isacson, O. (2002). Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci. Lett. 326, 155–158. doi: 10.1016/S0304-3940(02)00296-3
McNaught, K. S., and Jenner, P. (2001). Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 297, 191–194. doi: 10.1016/S0304-3940(00)01701-8
McNaught, K. S., Olanow, C. W., Halliwell, B., Isacson, O., and Jenner, P. (2001). Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat. Rev. Neurosci. 2, 589–594. doi: 10.1038/35086067
Meier, F., Abeywardana, T., Dhall, A., Marotta, N. P., Varkey, J., Langen, R., et al. (2012). Semisynthetic, site-specific ubiquitin modification of alpha-synuclein reveals differential effects on aggregation. J. Am. Chem. Soc. 134, 5468–5471. doi: 10.1021/ja300094r
Miake, H., Mizusawa, H., Iwatsubo, T., and Hasegawa, M. (2002). Biochemical characterization of the core structure of alpha-synuclein filaments. J. Biol. Chem. 277, 19213–19219. doi: 10.1074/jbc.M110551200
Mishizen-Eberz, A. J., Guttmann, R. P., Giasson, B. I., Day, G. A. III, Hodara, R., Ischiropoulos, H., et al. (2003). Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro. J. Neurochem. 86, 836–847. doi: 10.1046/j.1471-4159.2003.01878.x
Mishizen-Eberz, A. J., Norris, E. H., Giasson, B. I., Hodara, R., Ischiropoulos, H., Lee, V. M., et al. (2005). Cleavage of alpha-synuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry 44, 7818–7829. doi: 10.1021/bi047846q
Morris, M., Knudsen, G. M., Maeda, S., Trinidad, J. C., Ioanoviciu, A., Burlingame, A. L., et al. (2015). Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 18, 1183–1189. doi: 10.1038/nn.4067
Murata, S., Minami, Y., Minami, M., Chiba, T., and Tanaka, K. (2001). CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2, 1133–1138. doi: 10.1093/embo-reports/kve246
Murray, I. V., Giasson, B. I., Quinn, S. M., Koppaka, V., Axelsen, P. H., Ischiropoulos, H., et al. (2003). Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42, 8530–8540. doi: 10.1021/bi027363r
Nakamura, T., Yamashita, H., Takahashi, T., and Nakamura, S. (2001). Activated Fyn phosphorylates alpha-synuclein at tyrosine residue 125. Biochem. Biophys. Res. Commun. 280, 1085–1092. doi: 10.1006/bbrc.2000.4253
Negro, A., Brunati, A. M., Donella-Deana, A., Massimino, M. L., and Pinna, L. A. (2002). Multiple phosphorylation of alpha-synuclein by protein tyrosine kinase Syk prevents eosin-induced aggregation. FASEB J. 16, 210–212. doi: 10.1096/fj.01-0517fje
Neumann, M., Muller, V., Kretzschmar, H. A., Haass, C., and Kahle, P. J. (2004). Regional distribution of proteinase K-resistant alpha-synuclein correlates with Lewy body disease stage. J. Neuropathol. Exp. Neurol. 63, 1225–1235. doi: 10.1093/jnen/63.12.1225
Nonaka, T., Iwatsubo, T., and Hasegawa, M. (2005). Ubiquitination of alpha-synuclein. Biochemistry 44, 361–368. doi: 10.1021/bi0485528
Okochi, M., Walter, J., Koyama, A., Nakajo, S., Baba, M., Iwatsubo, T., et al. (2000). Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J. Biol. Chem. 275, 390–397. doi: 10.1074/jbc.275.1.390
Oueslati, A., Schneider, B. L., Aebischer, P., and Lashuel, H. A. (2013). Polo-like kinase 2 regulates selective autophagic alpha-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, E3945–E3954. doi: 10.1073/pnas.1309991110
Paleologou, K. E., Oueslati, A., Shakked, G., Rospigliosi, C. C., Kim, H. Y., Lamberto, G. R., et al. (2010). Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 30, 3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010
Park, J. Y., Paik, S. R., Jou, I., and Park, S. M. (2008). Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: implications for Parkinson’s disease. Glia 56, 1215–1223. doi: 10.1002/glia.20691
Pasanen, P., Myllykangas, L., Siitonen, M., Raunio, A., Kaakkola, S., Lyytinen, J., et al. (2014). Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 35, 2180.e1–2180.e5. doi: 10.1016/j.neurobiolaging.2014.03.024
Peelaerts, W., Bousset, L., Van der Perren, A., Moskalyuk, A., Pulizzi, R., Giugliano, M., et al. (2015). alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344. doi: 10.1038/nature14547
Periquet, M., Fulga, T., Myllykangas, L., Schlossmacher, M. G., and Feany, M. B. (2007). Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 27, 3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007
Plotegher, N., Berti, G., Ferrari, E., Tessari, I., Zanetti, M., Lunelli, L., et al. (2017). DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function. Sci. Rep. 7:40699. doi: 10.1038/srep40699
Plotegher, N., Gratton, E., and Bubacco, L. (2014). Number and Brightness analysis of alpha-synuclein oligomerization and the associated mitochondrial morphology alterations in live cells. Biochim. Biophys. Acta 1840, 2014–2024. doi: 10.1016/j.bbagen.2014.02.013
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Pronin, A. N., Morris, A. J., Surguchov, A., and Benovic, J. L. (2000). Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 275, 26515–26522. doi: 10.1074/jbc.M003542200
Prots, I., Veber, V., Brey, S., Campioni, S., Buder, K., Riek, R., et al. (2013). alpha-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 288, 21742–21754. doi: 10.1074/jbc.M113.451815
Proukakis, C., Dudzik, C. G., Brier, T., MacKay, D. S., Cooper, J. M., Millhauser, G. L., et al. (2013). A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. doi: 10.1212/WNL.0b013e31828727ba
Rostovtseva, T. K., Gurnev, P. A., Protchenko, O., Hoogerheide, D. P., Yap, T. L., Philpott, C. C., et al. (2015). alpha-synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J. Biol. Chem. 290, 18467–18477. doi: 10.1074/jbc.M115.641746
Rott, R., Szargel, R., Haskin, J., Shani, V., Shainskaya, A., Manov, I., et al. (2008). Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 283, 3316–3328. doi: 10.1074/jbc.M704809200
Rott, R., Szargel, R., Shani, V., Hamza, H., Savyon, M., Abd Elghani, F., et al. (2017). SUMOylation and ubiquitination reciprocally regulate alpha-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. U.S.A. 114, 13176–13181. doi: 10.1073/pnas.1704351114
Ryan, B. J., Hoek, S., Fon, E. A., and Wade-Martins, R. (2015). Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem. Sci. 40, 200–210. doi: 10.1016/j.tibs.2015.02.003
Sampathu, D. M., Giasson, B. I., Pawlyk, A. C., Trojanowski, J. Q., and Lee, V. M. (2003). Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am. J. Pathol. 163, 91–100. doi: 10.1016/S0002-9440(10)63633-4
Sanchez-Guajardo, V., Tentillier, N., and Romero-Ramos, M. (2015). The relation between alpha-synuclein and microglia in Parkinson’s disease: recent developments. Neuroscience 302, 47–58. doi: 10.1016/j.neuroscience.2015.02.008
Schapira, A. H., and Jenner, P. (2011). Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 26, 1049–1055. doi: 10.1002/mds.23732
Schroder, M., and Kaufman, R. J. (2005). ER stress and the unfolded protein response. Mutat. Res. 569, 29–63. doi: 10.1016/j.mrfmmm.2004.06.056
Schulz-Schaeffer, W. J. (2010). The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 120, 131–143. doi: 10.1007/s00401-010-0711-0
Sevcsik, E., Trexler, A. J., Dunn, J. M., and Rhoades, E. (2011). Allostery in a disordered protein: oxidative modifications to alpha-synuclein act distally to regulate membrane binding. J. Am. Chem. Soc. 133, 7152–7158. doi: 10.1021/ja2009554
Sevlever, D., Jiang, P., and Yen, S. H. (2008). Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47, 9678–9687. doi: 10.1021/bi800699v
Shahpasandzadeh, H., Popova, B., Kleinknecht, A., Fraser, P. E., Outeiro, T. F., and Braus, G. H. (2014). Interplay between sumoylation and phosphorylation for protection against alpha-synuclein inclusions. J. Biol. Chem. 289, 31224–31240. doi: 10.1074/jbc.M114.559237
Shin, Y., Klucken, J., Patterson, C., Hyman, B. T., and McLean, P. J. (2005). The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 280, 23727–23734. doi: 10.1074/jbc.M503326200
Shulman, J. M., De Jager, P. L., and Feany, M. B. (2011). Parkinson’s disease: genetics and pathogenesis. Annu. Rev. Pathol. 6, 193–222. doi: 10.1146/annurev-pathol-011110-130242
Singleton, A., Gwinn-Hardy, K., Sharabi, Y., Li, S. T., Holmes, C., Dendi, R., et al. (2004). Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain 127(Pt 4), 768–772. doi: 10.1093/brain/awh081
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Smith, W. W., Margolis, R. L., Li, X., Troncoso, J. C., Lee, M. K., Dawson, V. L., et al. (2005). Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 25, 5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stirling, D. P., Cummins, K., Mishra, M., Teo, W., Yong, V. W., and Stys, P. (2014). Toll-like receptor 2-mediated alternative activation of microglia is protective after spinal cord injury. Brain 137(Pt 3), 707–723. doi: 10.1093/brain/awt341
Sugeno, N., Hasegawa, T., Tanaka, N., Fukuda, M., Wakabayashi, K., Oshima, R., et al. (2014). Lys-63-linked ubiquitination by E3 ubiquitin ligase NEDD4-1 facilitates endosomal sequestration of internalized alpha-synuclein. J. Biol. Chem. 289, 18137–18151. doi: 10.1074/jbc.M113.529461
Takahashi, M., Kanuka, H., Fujiwara, H., Koyama, A., Hasegawa, M., Miura, M., et al. (2003). Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci. Lett. 336, 155–158. doi: 10.1016/S0304-3940(02)01258-2
Tanaka, K., and Chiba, T. (1998). The proteasome: a protein-destroying machine. Genes Cells 3, 499–510. doi: 10.1046/j.1365-2443.1998.00207.x
Tetzlaff, J. E., Putcha, P., Outeiro, T. F., Ivanov, A., Berezovska, O., Hyman, B. T., et al. (2008). CHIP targets toxic alpha-Synuclein oligomers for degradation. J. Biol. Chem. 283, 17962–17968. doi: 10.1074/jbc.M802283200
Tofaris, G. K., Garcia Reitbock, P., Humby, T., Lambourne, S. L., O’Connell, M., Ghetti, B., et al. (2006). Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J. Neurosci. 26, 3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006
Tofaris, G. K., Kim, H. T., Hourez, R., Jung, J. W., Kim, K. P., and Goldberg, A. L. (2011). Ubiquitin ligase NEDD4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 17004–17009. doi: 10.1073/pnas.1109356108
Tofaris, G. K., Razzaq, A., Ghetti, B., Lilley, K. S., and Spillantini, M. G. (2003). Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 278, 44405–44411. doi: 10.1074/jbc.M308041200
Tysnes, O. B., and Storstein, A. (2017). Epidemiology of Parkinson’s disease. J. Neural Transm. 124, 901–905. doi: 10.1007/s00702-017-1686-y
Uversky, V. N., and Eliezer, D. (2009). Biophysics of Parkinson’s disease: structure and aggregation of alpha-synuclein. Curr. Protein Pept. Sci. 10, 483–499. doi: 10.2174/138920309789351921
van der Putten, H., Wiederhold, K. H., Probst, A., Barbieri, S., Mistl, C., Danner, S., et al. (2000). Neuropathology in mice expressing human alpha-synuclein. J. Neurosci. 20, 6021–6029. doi: 10.1523/JNEUROSCI.20-16-06021.2000
Walsh, D. M., Lomakin, A., Benedek, G. B., Condron, M. M., and Teplow, D. B. (1997). Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 272, 22364–22372. doi: 10.1074/jbc.272.35.22364
Wang, S., Yang, F., Petyuk, V. A., Shukla, A. K., Monroe, M. E., Gritsenko, M. A., et al. (2017). Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J. Pathol. 243, 78–88. doi: 10.1002/path.4929
Wang, Z., Park, K., Comer, F., Hsieh-Wilson, L. C., Saudek, C. D., and Hart, G. W. (2009). Site-specific GlcNAcylation of human erythrocyte proteins: potential biomarker(s) for diabetes. Diabetes 58, 309–317. doi: 10.2337/db08-0994
Wang, Z., Udeshi, N. D., O’Malley, M., Shabanowitz, J., Hunt, D. F., and Hart, G. W. (2010). Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 9, 153–160. doi: 10.1074/mcp.M900268-MCP200
Weinreb, P. H., Zhen, W., Poon, A. W., Conway, K. A., and Lansbury, P. T. Jr. (1996). NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35, 13709–13715. doi: 10.1021/bi961799n
Wheeler, T. C., Chin, L. S., Li, Y., Roudabush, F. L., and Li, L. (2002). Regulation of synaptophysin degradation by mammalian homologues of seven in absentia. J. Biol. Chem. 277, 10273–10282. doi: 10.1074/jbc.M107857200
Wijayanti, I., Watanabe, D., Oshiro, S., and Takagi, H. (2015). Isolation and functional analysis of yeast ubiquitin ligase Rsp5 variants that alleviate the toxicity of human alpha-synuclein. J. Biochem. 157, 251–260. doi: 10.1093/jb/mvu069
Wood, S. J., Wypych, J., Steavenson, S., Louis, J. C., Citron, M., and Biere, A. L. (1999). alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J. Biol. Chem. 274, 19509–19512. doi: 10.1074/jbc.274.28.19509
Wrasidlo, W., Tsigelny, I. F., Price, D. L., Dutta, G., Rockenstein, E., Schwarz, T. C., et al. (2016). A de novo compound targeting alpha-synuclein improves deficits in models of Parkinson’s disease. Brain 139(Pt 12), 3217–3236. doi: 10.1093/brain/aww238
Ysselstein, D., Dehay, B., Costantino, I. M., McCabe, G. P., Frosch, M. P., George, J. M., et al. (2017). Endosulfine-alpha inhibits membrane-induced alpha-synuclein aggregation and protects against alpha-synuclein neurotoxicity. Acta Neuropathol. Commun. 5:3. doi: 10.1186/s40478-016-0403-7
Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Keywords: Parkinson’s disease, α-synuclein, toxicity, post-translational modifications, aggregation
Citation: Zhang J, Li X and Li J-D (2019) The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 13:381. doi: 10.3389/fnins.2019.00381
Received: 24 January 2019; Accepted: 02 April 2019;
Published: 18 April 2019.
Edited by:
Andrei Surguchov, University of Kansas Medical Center, United StatesReviewed by:
George K. Tofaris, University of Oxford, United KingdomMatthew Robert Pratt, University of Southern California, United States
Kunikazu Tanji, Hirosaki University, Japan
Copyright © 2019 Zhang, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia-Da Li, lijiada@sklmg.edu.cn