Parkinson’s disease-implicated kinases in the brain; insights into disease pathogenesis
 Nicolas Dzamko
Nicolas Dzamko Jinxia Zhou1,2
Jinxia Zhou1,2  Yue Huang
Yue Huang Glenda M. Halliday
Glenda M. Halliday- 1School of Medical Sciences, University of New South Wales, Kensington, NSW, Australia
- 2Neuroscience Research Australia, Randwick, NSW, Australia
Substantial evidence implicates abnormal protein kinase function in various aspects of Parkinson’s disease (PD) etiology. Elevated phosphorylation of the PD-defining pathological protein, α-synuclein, correlates with its aggregation and toxic accumulation in neurons, whilst genetic missense mutations in the kinases PTEN-induced putative kinase 1 and leucine-rich repeat kinase 2, increase susceptibility to PD. Experimental evidence also links kinases of the phosphoinositide 3-kinase and mitogen-activated protein kinase signaling pathways, amongst others, to PD. Understanding how the levels or activities of these enzymes or their substrates change in brain tissue in relation to pathological states can provide insight into disease pathogenesis. Moreover, understanding when and where kinase dysfunction occurs is important as modulation of some of these signaling pathways can potentially lead to PD therapeutics. This review will summarize what is currently known in regard to the expression of these PD-implicated kinases in pathological human postmortem brain tissue.
Introduction
Studies of the postmortem human brain have been invaluable in gaining insights into the etiology of Parkinson’s disease (PD), an increasingly common movement disorder resulting from the early selective loss of dopamine producing neurons in the substantia nigra. Dr. Fritz Lewy discovered the eponymous intracellular inclusion bodies synonymous with PD (Rodrigues et al., 2010). Drs. Tretiakoff, Hassler, and others were able to demonstrate degeneration of the substantia nigra in PD (Hassler, 1938). Postmortem brain studies were integral to the experiments of Carlsson and colleagues and their discoveries on the therapeutic potential of exogenous dopamine treatment for PD patients (Carlsson, 1959) and their discoveries on the therapeutic potential of exogenous dopamine treatment for PD patients and more recently, the work of Braak and others has suggested that PD spreads through the brain in a predictable or staged fashion (Braak et al., 2003; Halliday et al., 2008).
The Braak staging hypothesis is modeled on the toxic spread and accumulation of α-synuclein, a 17–18 kDa presynaptic protein encoded by the SNCA gene. Point mutations in, or multiplications of, the SNCA gene cause familial PD in an autosomal-dominant fashion (Polymeropoulos et al., 1997), whilst genome-wide association studies conclude that common variations in the SNCA gene increase the risk of sporadic PD (Pihlstrom and Toft, 2011). Moreover, α-synuclein is the predominant component of Lewy bodies, where it accumulates in an aggregated form (Spillantini et al., 1997). Hence, α-synuclein is proposed as a key protein in the pathogenesis of PD. Accumulating evidence suggests that α-synuclein acts in a prion-like manner, inducing the aggregation of healthy α-synuclein and propagating the spread of PD from neuron to neuron (Olanow and Brundin, 2013). The aggregated and proposed toxic form of α-synuclein is hyperphosphorylated (Oueslati et al., 2010). In disease free conditions only 4% of total α-synuclein is phosphorylated in brain, but in PD and related synucleinopathies, >90% of α-synuclein deposited in Lewy bodies is phosphorylated (Fujiwara et al., 2002; Anderson et al., 2006). In particular, phosphorylation of pathological α-synuclein on serine 129 (S129) is prevalent in PD postmortem brain (Fujiwara et al., 2002; Anderson et al., 2006; Zhou et al., 2011; Lue et al., 2012; Walker et al., 2013). Although the biological consequences of α-synuclein phosphorylation remain inconclusive, there is much interest in the identification of the kinases mediating this event. A number of candidate kinases, including members of the polo-like kinase (PLK), casein kinase (CK), and G protein coupled receptor kinase (GRK) families have subsequently been identified.
In addition to the hyperphosphorylation of α-synuclein, kinase dysfunction is also genetically linked to PD. In particular, missense mutations in the leucine-rich repeat kinase 2 (LRRK2) are causal for autosomal-dominant familial PD (Paisan-Ruiz et al., 2004; Zimprich et al., 2004), whilst multiple mutations in the PTEN-induced putative kinase 1 (PINK1) protein are causative for familial PD in a recessive fashion (Valente et al., 2004). Moreover, common polymorphisms identified by genome-wide association in loci encoding cyclin G-associated kinase (GAK) and serine/threonine kinase 39 [STK39, more commonly known as STE20-related proline alanine-rich kinase (SPAK)], have implicated these kinases as susceptibility enzymes for sporadic PD (Pankratz et al., 2009; Nalls et al., 2011; Sharma et al., 2012).
Finally a myriad of laboratory studies have focused on kinase signaling in PD. Kinases remain attractive targets for the treatment of many human diseases. Kinases of the MAPK and PI3K signaling pathways including extracellular signal related protein kinase (ERK), c-Jun N-terminal kinase (JNK), p38, protein kinase B (PKB), and mammalian target of rapamycin (mTOR) make particularly attractive targets for PD through their ability to coordinate and regulate cell survival, apoptosis, inflammation, and autophagy.
Kinases Mediating α-Synuclein S129 Phosphorylation in PD
The exact mechanism resulting in the pathological accumulation of S129 phosphorylated α-synuclein is unclear. A number of kinases phosphorylate α-synuclein at this residue in vitro with accumulating evidence for a role in vivo (Figure 1). Understanding how this major pathological protein becomes hyperphosphorylated and the extent to which post-translational modifications impact upon the aggregation and prion-like spread of α-synuclein could provide key insight into PD etiology.
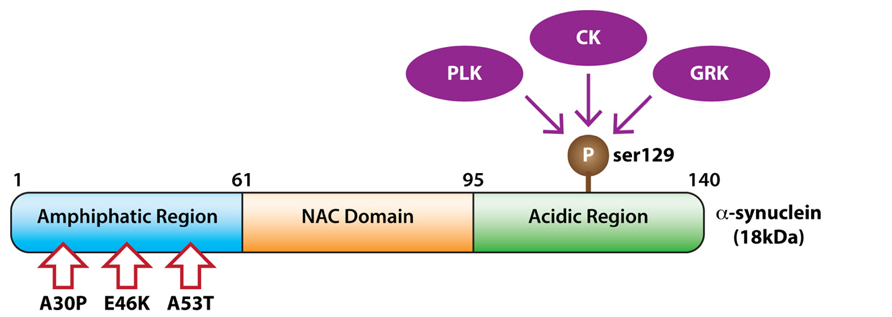
FIGURE 1. Kinases phosphorylating α-synuclein. The domain structure of α-synuclein showing phosphorylation at serine 129 by members of the polo-like kinase (PLK), casein kinase (CK), and G protein coupled receptor kinase (GRK) families. Pathogenic α-synuclein missense mutations are indicated with arrows.
Polo-Like Kinases (PLKs)
Polo-like kinases (PLKs) comprise a serine/threonine kinase family containing an N-terminal kinase catalytic domain and a C-terminal polo-box domain (PBD) that is involved in substrate binding and regulation of kinase activity. Five mammalian PLK family members from three subfamilies have been identified, including the PLK1 subfamily, the PLK4 subfamily, and the PLK2 subfamily (containing PLK2, PLK3, and PLK5; de Carcer et al., 2011b). The study of PLKs has focused primarily on their critical roles in the cell cycle (Winkles and Alberts, 2005); however, recent studies suggest PLKs also have important roles in terminally differentiated cells of the nervous system (Seeburg et al., 2005).
In particular, PLKs 1–3 are capable of phosphorylating α-synuclein (de Carcer et al., 2011a, b). Comparative studies suggest that PLK2 and PLK3 directly phosphorylate α-synuclein at Ser129 in vitro with high stoichiometry, whilst PLK4 is unable to phosphorylate α-synuclein at this residue (Anderson et al., 2006; Inglis et al., 2009). The low kinase activity of PLK4 against α-synuclein, and other substrates, is partially explained by its unique structure, with only a single polo-box in the PBD, resulting in a much-reduced electropositive environment in its substrate-binding site (Mbefo et al., 2010). Human PLK5 lacks a functional kinase domain due to a premature stop codon in exon 6 and is therefore unable to phosphorylate α-synuclein.
Increasing PLK2 or PLK3 significantly up-regulates α-synuclein Ser129 phosphorylation (Mbefo et al., 2010; Waxman and Giasson, 2011), whilst their inhibition or reduction remarkably decreases α-synuclein phosphorylation in both cell and animal models (Inglis et al., 2009; Waxman and Giasson, 2011). This has led to efforts to generate small molecule PLK inhibitors for potential therapeutic use (Bowers et al., 2013; Fitzgerald et al., 2013; Bergeron et al., 2014). The utility of such compounds, however, has been questioned by a recent study showing that Ser129 phosphorylation by PLK2 is required for autophagic degradation of α-synuclein (Oueslati et al., 2013). In this study overexpression of PLK2, as opposed to inhibition, prevented the toxic accumulation of α-synuclein in rodent models, suggesting more work is required to delineate the exact role of PLKs in α-synuclein pathology.
In addition, studies investigating the association of PLKs with α-synuclein pathology / phosphorylation in human brain are lacking. The central nervous system has relatively high levels of PLK2, 3, and 5, low levels of PLK1 and seems to lack PLK4 (Winkles and Alberts, 2005; de Carcer et al., 2011a, b). PLK2 and PLK3 are expressed in most regions of the brain, but surprisingly there is almost no expression of either PLK2 or PLK3 in the cerebellum (Winkles and Alberts, 2005; de Carcer et al., 2011b). Whether PLK2 or PLK3, the main family members that can phosphorylate α-synuclein are increased, or indeed more active, in PD brain remains to be determined. The recent identification of autophosphorylation sites on PLK2 (Rozeboom and Pak, 2012) and other potential selective substrates (Salvi et al., 2012) could allow readouts of PLK2 activity to be examined in PD brain. It would be of interest to determine if the phosphorylation of such substrates correlated to levels of α-synuclein phosphorylation in pathology rich brain regions in PD cases.
Casein Kinases (CKs)
Casein kinases (CKs) comprise a ubiquitously expressed serine/threonine kinase family (Peters et al., 1999) containing two members, CK1 and CK2, which differ substantially in terms of structure, localization and function (Perez et al., 2011). CK1 consists of a small N-terminal lobe, a large C-terminal lobe and a catalytic cleft where ATP and substrates bind (Cheong and Virshup, 2011). To date, at least seven CK1 isoforms (α, β, γ1–3, δ, and ∊) and their various splice variants, ranging from 22 to 55 kDa, have been localized within the membrane, nucleus, and cytoplasm of eukaryote cells, and additionally in the mitotic spindles of mammalian cells (Fish et al., 1995). All CK1 isoforms are highly homologous in their kinase domains (Knippschild et al., 2005), presenting a strong preference for “primed,” pre-phosphorylated substrate. However, they can also phosphorylate related unprimed sites under certain conditions (Cheong and Virshup, 2011).
In contrast to CK1, CK2 is a tetrameric enzyme assembled from two catalytic subunits (CK2α and CK2α′) and a regulatory subunit (CK2β dimer). The two catalytic subunits α and α′ share 90% sequence homology in their N-terminal region, but the regulatory subunit β does not have any similarity to the other two subunits. CK2 is found in many organisms and tissues and nearly every subcellular compartment. It can phosphorylate more than 300 substrate proteins (Meggio and Pinna, 2003) involved in diverse cellular processes including cell division, proliferation, apoptosis, and DNA repair.
Both CK1 and CK2 can constitutively phosphorylate α-synuclein at Ser129 in vitro (Okochi et al., 2000; Waxman and Giasson, 2008), and inhibition of CK1 or CK2 reduces α-synuclein Ser129 phosphorylation in vivo (Okochi et al., 2000; Ishii et al., 2007; Waxman and Giasson, 2008), with CK2 inhibition seemingly more efficient at reducing phosphorylation (Ishii et al., 2007). However, at least one study failed to find an effect of CK1 inhibition on α-synuclein Ser129 phosphorylation in a cellular model (Waxman and Giasson, 2011). This discrepancy could result from, at least partially, the specificity of CK1 inhibitors and more studies are needed to define the relationship between CKs and α-synuclein.
In pathological human brain CK1δ co-localizes predominantly with tau-containing inclusions such as neurofibrillary tangles, and does not co-localize with α-synuclein in Lewy bodies in PD (Schwab et al., 2000). In contrast, CK2β regulatory subunits are present in the halo region of Lewy bodies in PD substantia nigra (Ryu et al., 2008), suggesting that CK2 may be more pathologically relevant to PD. More work is required to determine any correlations between CK isoforms and the pathological accumulation of phosphorylated α-synuclein in PD.
G Protein Coupled Receptor Kinases (GRKs)
G Protein coupled receptor kinases comprise a serine/threonine kinase family that regulate G protein-coupled receptors (GPCRs) by phosphorylating their intracellular domains after their associated G proteins have been released and activated (Gurevich et al., 2012). Structurally, GRKs contain a central catalytic domain flanked by an N-terminus containing a regulator of G protein signaling homology domain and a variable length C-terminal end. Based on sequence homology and tissue expression, GRKs are further classified into three subfamilies: the rhodopsin kinase or visual GRK subfamily (GRK1 and GRK7), the β-adrenergic receptor kinases subfamily (GRK2 and GRK3), and the GRK4 subfamily (GRK4, GRK5, and GRK6; Gurevich et al., 2012; Kamal et al., 2012).
Exactly which GRK isoforms phosphorylate α-synuclein under pathological conditions is unclear. In vitro GRK2 preferentially phosphorylates α and β synuclein isoforms while GRK5 prefers α-synuclein as a substrate (Pronin et al., 2000). However, knockdown of either GRK5 or GRK2 failed to diminish the phosphorylation of α-synuclein in cell models (Sakamoto et al., 2009; Liu et al., 2010). In contrast, knockdown of GRK3 or GRK6 significantly decreased α-synuclein Ser129 phosphorylation levels (Sakamoto et al., 2009), suggesting further work is required to verify the role of GRK isoforms in phosphorylating α-synuclein.
G protein coupled receptor kinase isoforms, 2, 3, 5, and 6, are highly expressed in the human brain. In PD brain, however, GRK protein levels tend to be lower than controls (Bychkov et al., 2008) with conflicting reports regarding the co-localization of GRK5 in Lewy bodies (Arawaka et al., 2006; Takahashi et al., 2006).
Overview of Kinases Involved in α-Synuclein S129 Phosphorylation in PD
Understanding events that promote α-synuclein pathology is increasingly important as evidence suggests a pathogenic prion-like spread of α-synuclein in PD (Olanow and Brundin, 2013; Recasens et al., 2014). There are now multiple human brain tissue studies using the methods developed by Braak and colleagues to observe the progression of pathology in PD brain showing that substantial α-synuclein S129 phosphorylation precedes the aggregation of α-synuclein in Lewy bodies (Zhou et al., 2011; Lue et al., 2012; Walker et al., 2013). Stoichiometrically, PLK2 seemingly contributes most to such α-synuclein S129 phosphorylation; however, studies with PLK2 knockout mice show that other kinases also contribute (Bergeron et al., 2014). Information from other PD models, however, remains controversial on the role α-synuclein S129 phosphorylation plays in disease pathogenesis, with some studies suggesting that S129 phosphorylation promotes α-synuclein oligomerization and/or toxicity (Chen and Feany, 2005; Febbraro et al., 2013) whilst others suggest that phosphorylation reduces toxicity or has no effect (McFarland et al., 2009; Oueslati et al., 2010; Sato et al., 2013; Escobar et al., 2014). This makes determining other relevant kinases difficult and information on any differences between species and models (acute versus chronic) will need further consideration. More data from informative staged human brain studies as well as from primate models with acute and chronic phases is likely to assist with clarifying the role of α-synuclein S129 phosphorylation over the course of PD. It is also important to note that other post-translation modifications of α-synuclein, such as ubiquitylation or nitrosylation, may equally contribute to the pathological process (Oueslati et al., 2010), with similar staged human brain and primate model data on the relative contributions of different protein modifications yet to be published.
Kinases Genetically Implicated in PD
Monogenetic causes of PD presently account for less than 10% of all cases (Gasser, 2009). However, the identification of genetic causes has invigorated PD research by providing new avenues of mechanistic investigation and therapeutic treatment. Missense mutations in LRRK2 and PINK1 cause PD in an autosomal-dominant or recessive manner, respectively, whilst common variations in the LRRK2 and loci encoding the GAK and STK39 genes have been implicated as risk factors for PD (Sharma et al., 2012). Understanding how mutations in these kinases alters their function and the biological processes they regulate has great potential for uncovering initiating events leading to the onset of PD.
Leucine-Rich Repeat Kinase 2 (LRRK2)
Mutations in the LRRK2 gene were discovered as causal for PD in 2004 (Paisan-Ruiz et al., 2004; Zimprich et al., 2004). Subsequently, some 40 missense mutations have been described across the LRRK2 protein with six of these demonstrated as pathogenic (Paisan-Ruiz, 2009). Collectively these LRRK2 mutations account for the majority of autosomal-dominantly inherited PD (Kett and Dauer, 2012). LRRK2-associated PD is largely clinically and pathologically indistinguishable from sporadic PD (Healy et al., 2008), suggesting that understanding LRRK2 function has implications for all forms of PD. Moreover, large-scale genome-wide association studies show that common variations in non-coding regions of the LRRK2 gene also confer greater risk for sporadic PD (Satake et al., 2009; Simon-Sanchez et al., 2009; Sharma et al., 2012).
Leucine-rich repeat kinase 2 is a 286 kDa multi-domain containing member of the receptor interacting protein kinase (RIPK) family. LRRK2 has N-terminal ankyrin repeats, leucine-rich repeats, a ras of complex (ROC) GTPase domain with adjoining C-terminal of ROC (COR) domain, a serine/threonine protein kinase domain and C-terminal WD40 repeats. Intriguingly, the majority of the pathogenic mutations lie in the catalytic domains of LRRK2. The most common mutation results in the substitution of glycine to serine (G2019S) in the activation loop of the protein kinase domain resulting in a constitutive threefold increase in LRRK2 kinase activity (West et al., 2005; Jaleel et al., 2007). The next most common mutations, substitution of arginine to either histidine (R1141H), cysteine (R1441C), or glycine (R1441G) lie in the GTPase domain. Some evidence suggests that these mutations also increase kinase activity (Sheng et al., 2012), potentially by trapping LRRK2 in a GTP bound active state (Liao et al., 2014). It has previously been shown that GTP binding is required for LRRK2 kinase activity (Taymans et al., 2011) and a complex relationship exists between the two domains (Taymans, 2012). The increase in catalytic kinase activity with LRRK2 mutations has led to the development of LRRK2 kinase inhibitors as potential PD therapeutics (Deng et al., 2012) and much interest has focused on determining the targets of LRRK2 kinase activity (Dzamko and Halliday, 2013). One such robust effect for LRRK2 kinase activity is to mediate the phosphorylation-dependent interaction of LRRK2 with isoforms of the 14-3-3 adaptor protein (Dzamko et al., 2010); however, consensus regarding the PD-relevant physiological functions of LRRK2 has remained largely elusive.
LRRK2 mRNA expression shows a widespread neuronal localization in human brain; however, intriguingly, only weak levels are detected in the substantia nigra (Higashi et al., 2007; Sharma et al., 2011). Moreover, decreased LRRK2 mRNA was found in certain non-nigral regions of PD brain (cerebellum, amygdala, frontal cortex, and cingulate gyrus; Sharma et al., 2011), suggesting a pathogenic role for LRRK2 outside of nigral neurons. This contrasts with the increased levels of LRRK2 protein reported in PD brain regions with pathological accumulation of α-synuclein (Cho et al., 2013; Guerreiro et al., 2013). The exact nature of the relationship between LRRK2 and α-synuclein is somewhat unclear as postmortem localization studies have produced conflicting results. Some studies have demonstrated localization of LRRK2 to α-synuclein pathology (Miklossy et al., 2006; Alegre-Abarrategui et al., 2008; Qing et al., 2009; Sharma et al., 2011) whilst others have not (Giasson et al., 2006; Higashi et al., 2007; Melrose et al., 2007; Waxman et al., 2009). Studies comparing different LRRK2 antibodies have shown that discrepancies in LRRK2 tissue localization likely occurs through use of antibodies unsuitable for immunohistochemistry (Biskup et al., 2007; Melrose et al., 2007; Davies et al., 2013). Indeed, recent data using more rigorous methods shows LRRK2 and α-synuclein co-localize in a small proportion of PD pathologies (Guerreiro et al., 2013). Despite such data, further work is required to define the relationship between LRRK2 expression and protein levels, between LRRK2 and α-synuclein increases and aggregation, and indeed determine if these proteins interact in the same molecular pathway.
As LRRK2 is also expressed by glial cells in normal human brain (Miklossy et al., 2006) and in tissue culture, and its expression in glia is increased by interferon gamma (Gardet et al., 2010) and bacterial lipopolysaccharide (LPS; Moehle et al., 2012), the neuroinflammation prevalent in PD affected regions may promote the expression changes observed for LRRK2 specifically in microglia rather than neurons. It will also be important to correlate any changes in LRRK2 expression to cell type. The recent demonstration that there is a primate specific LRRK2 promoter that differentiates primate expression of the protein in the brain from that observed in rodents (West et al., 2014) underlies the requirement for further observations in staged human tissue specimens in order to determine the role of LRRK2 kinase function in PD pathogenesis.
PTEN-Induced Putative Kinase 1 (PINK1)
Homozygous missense mutations in the PINK1 gene were identified as a cause of familial PD in 2004 (Valente et al., 2004). Around 50 missense mutations have subsequently been identified across the PINK1 protein in a number of populations (Kawajiri et al., 2011). Mutations in PINK1 are the second most common cause of recessive PD (following mutations in the ubiquitin ligase Parkin) and are thought to contribute to 1–8% of familial PD (Kawajiri et al., 2011). Unlike LRRK2, PINK1 mutations reduce kinase activity and cause an atypical form of PD characterized by an early age of onset and slower clinical progression (Abou-Sleiman et al., 2006; Woodroof et al., 2011).
The PINK1 protein comprises a serine/threonine protein kinase domain, a N-terminal mitochondrial targeting motif and a trans-membrane domain located between the two. The mitochondrial targeting motif is required for recruitment of PINK1 to mitochondrial membranes. Following recruitment in healthy mitochondria, PINK1 is enzymatically cleaved to produce a shorter fragment, which is degraded by the proteasome (Narendra et al., 2010). In this way, PINK1 is maintained at very low levels. Pharmacological uncoupling of the mitochondrial membrane leading to a loss of membrane potential; however, results in inhibition of PINK1 cleavage and its accumulation on depolarized mitochondrial membranes (Matsuda et al., 2010; Narendra et al., 2010). The kinase activity of PINK1 is also increased under these conditions with PINK1 undergoing autophosphorylation (Kondapalli et al., 2012). Intriguingly, Parkin is then also recruited to depolarized mitochondria where it is phosphorylated and activated by PINK1 (Matsuda et al., 2010; Kondapalli et al., 2012). Parkin then mediates the degradation of dysfunctional mitochondria by mitophagy (mitochondrial autophagy). Therefore, the two proteins responsible for the majority of familial early onset PD appear to function in the same pathway important for the regulation of mitochondrial quality control. Whether this is the pathway ultimately responsible for loss of neurons in PD is still unclear (Grenier et al., 2013) and it should be noted that PINK1 has also been implicated in other biological processes such as neurite maintenance (Dagda et al., 2014) and inflammation (Lee and Chung, 2012; Kim et al., 2013) among others.
Detailed in-situ hybridization studies using rodent brain demonstrate that PINK1 mRNA is expressed throughout the brain with the strongest signal in neurons of the olfactory bulb, neocortex, prefrontal cortex, piriform cortex, hippocampus, amygdala, brainstem, and cerebellar Purkinje cells (Taymans et al., 2006). Similarly, in human brain, PINK1 mRNA is widely expressed in neurons with highest signals recorded for the temporal cortex, amygdala, substantia nigra, cerebellar Purkinje cells and the dentate nucleus (Blackinton et al., 2007). PINK1 mRNA is undetectable in glial cells (Taymans et al., 2006; Blackinton et al., 2007) and is not different in the substantia nigra of sporadic PD patients compared to controls (Blackinton et al., 2007). The mRNA distribution of PINK1 has been largely confirmed at the protein level with the exception that PINK1 immunoreactivity was also observed in glia, albeit with weak staining compared to neuronal staining (Gandhi et al., 2006). PINK1 is predominantly localized to mitochondria and does not change in amount or localization in the brain of patients with idiopathic PD, although PINK1 immunoreactivity is detected in ~10% of brainstem Lewy bodies (Gandhi et al., 2006). Interestingly, a case report describing the neuropathology of an early onset PINK1 homozygous mutation patient showed a pattern of Lewy body pathology with atypical Braak Lewy body staging (Samaranch et al., 2010). This was due to the absence of Lewy bodies, and indeed cell loss in the locus coeruleus, potentially helping to explain the longer disease duration of PINK1-associated PD (Samaranch et al., 2010). Collectively these studies suggest that alterations in PINK1 function, rather than protein levels, likely contribute to PD. This is consistent with observations that the majority of described PINK1 mutations result in a loss of kinase activity (Woodroof et al., 2011). Whether PINK1 autophosphorylation or PINK1-induced Parkin phosphorylation are altered in sporadic PD, and how this correlates to mitochondrial health and/or neuronal loss, would be interesting to explore.
Cyclin G-Associated Kinase (GAK)
The Ser/Thr protein kinase GAK was originally identified via its interaction with cyclin G and cyclin-dependent kinase 5 (CDK5; Kanaoka et al., 1997). The kinase domain is located at the N-terminus and a leucine-zipper region is located at the C-terminus. The majority of the protein comprises a TAG domain that has 80% identity to the auxilin protein (Kanaoka et al., 1997). Both auxilin and GAK play key roles in the uncoating of clatherin-coated vesicles and the regulation of clatherin-mediated endocytosis (Eisenberg and Greene, 2007). The latter is completely blocked in GAK deficient mouse embryonic fibroblasts (Lee et al., 2008). GAK also plays a key role in brain development. Conditional deletion of GAK in mouse brain resulted in marked cell loss and morphological changes in new-born pups, potentially due to a lack of proliferation of neural progenitor cells in the subventricular zone of the hippocampus. Conditional GAK knockout mice die soon after birth whilst conventional GAK knockouts are embryonic lethal (Lee et al., 2008). Moreover transgenic mice expressing kinase inactive GAK die within 30 min of birth due to respiratory dysfunction (Tabara et al., 2011). Respiratory problems are also associated with use of gefitinib (Tabara et al., 2011), an anti-cancer epidermal growth factor receptor (EGFR) inhibitor that also inhibits GAK.
Single nucleotide polymorphisms in the GAK locus were first associated with PD susceptibility following genome-wide association analysis of a large number of familial PD patients (Pankratz et al., 2009). The association has since been robustly replicated in different populations (Rhodes et al., 2011; Sharma et al., 2012). One GAK SNP, rs1564282 is associated with higher expression of α-synuclein in PD brain, and when GAK mRNA was knocked down with siRNA, there was accumulation of α-synuclein in cell culture models (Dumitriu et al., 2011). This provides some biochemical evidence for a role for GAK in PD, although the toxic effects of GAK knockdown/inhibition suggest that GAK is unlikely to be dramatically decreased in PD brain. Moreover, an alternative microarray based study has shown that GAK mRNA expression is increased in the substantia nigra of PD patients (Grunblatt et al., 2004). Intriguingly, GAK has also been proposed to interact with LRRK2 and potentially help co-ordinate the clearance of trans-Golgi derived vesicles (Beilina et al., 2014); however, GAK protein expression in PD brain and any association with PD pathology has been poorly explored.
Serine/Threonine Kinase 39 (STK39)
Serine/threonine kinase is more commonly referred to in the literature as SPAK. The majority of work on SPAK has focused on the enzymes role as a regulator of the Na+/Cl- and Na+/K+/2Cl- ion co-transporters, NCC and NKCC, respectively. In response to osmotic stress SPAK is activated by phosphorylation at T233 in its activation loop by isoforms of WNK (with-no lysine) kinases and in turn phosphorylates NCC/NKCC to promote transporter activity (Richardson and Alessi, 2008). These ion co-transporters are major drug targets of current anti-hypertensive medications and evidence suggests that inhibition of SPAK may also lower blood pressure (Richardson and Alessi, 2008; Glover and O’shaughnessy, 2011). Indeed, variations in the STK39 gene have been implicated in hypertension in the Amish population through genome-wide association, with the resulting non-coding mutations increasing the allelic expression of SPAK (Wang et al., 2009a). This association, however, has failed to reach significance in other populations (Cunnington et al., 2009; Persu and Vikkula, 2011).
Genome-wide association studies have also implicated variations in the STK39 locus with PD. First identified through large-scale meta-analysis (Liu et al., 2011; Nalls et al., 2011), the association of STK39 SNPs with PD has been subsequently replicated in Asian and Caucasian populations (Lill et al., 2012; Sharma et al., 2012). The three reported STK39 SNPs associating with PD differ from the reported SNP for hypertension. Whether these polymorphisms affect SPAK expression is unknown.
Studies using rats show that SPAK is highly expressed in the nervous system, especially brain where it is detected in neurons, Purkinje cells and choroid epithelial cells (Ushiro et al., 1998). Glial cells do not show immunoreactivity for SPAK (Ushiro et al., 1998). In developing brain, SPAK plays a role in the regulation of Cl- concentration and in-turn release of the neurotransmitter GABA (Delpire and Austin, 2010). SPAK has also been suggested to act as a stress-response kinase with its overexpression or activation leading to increased phosphorylation of p38 MAPK (Yan et al., 2007). Whether levels of SPAK protein, phosphorylation of SPAK T233 or phosphorylation of the SPAK ion transporter substrates are altered in PD brain has not been investigated.
Overview of Kinases Genetically Implicated in PD
There is much interest in LRRK2 as both a key to understanding PD pathogenesis and a potential therapeutic target, as PINK1 mutations cause an atypical form of PD and the mechanism/s of increased PD risk due to SPAK and GAK polymorphisms is presently unclear. The mRNA expression of LRRK2 is decreased in PD brain; however, LRRK2 protein is increased, at least in Lewy body-rich regions at end-stage disease. Further work is required to determine if LRRK2 protein is altered earlier in PD pathogenesis and in particular, as LRRK2 expression can be induced with inflammatory agonists in microglia (Moehle et al., 2012), the cell types expressing LRRK2 may be important. Localization studies of LRRK2, and indeed PINK1 have proven difficult, as a number of available antibodies are not optimal for this procedure. Moreover, the kinase activity of LRRK2 has not been explored in PD brain. This is potentially important as kinase inhibiting therapeutics are being targeted toward LRRK2, even though it is unclear if the toxic effects of LRRK2 mutations are kinase-dependent. At least one risk variant reportedly decreases LRRK2 kinase activity (Rudenko et al., 2012b) leading to suggestions that other functions of LRRK2 such as GTPase activity may be important (Rudenko et al., 2012a). The identification of bona fide substrates for PINK1 and LRRK2 will be important for inferring any changes in enzymatic activity in the PD brain.
Kinases Experimentally Implicated in PD
Kinases of the Mitogen Activated Protein Kinase (MAPK) Pathway
The MAPK superfamily of serine/threonine protein kinases consists of three major branches, the JNKs, the p38 kinases and the ERKs (Figure 2; for review see Kyriakis and Avruch, 2012). The three JNK isoforms (JNK1, JNK2, and JNK3) and four p38 isoforms (p38α, p38β, p38γ, and p38δ) are referred to as stress activated protein kinases (SAPKs). In particular, JNK is activated by a number of environmental stresses implicated in PD including, toxins, inflammatory agonists and misfolded protein-induced ER stress. The activation of p38 is more restricted to inflammatory agonists whilst the two ERK isoforms (ERK1 and ERK2) are activated principally in response to mitogens, although a high level of cross-talk exists between the different MAPK branches. Upon activation, JNK, ERK, and p38 phosphorylate a large number of substrates in a proline-directed manner. In some instances substrates can be specific, such as JNK to phosphorylate the AP-1 transcription factor component c-Jun, or ERK to phosphorylate the p90 ribosomal S6 kinase (RSK), or shared, such as ERK and p38 to phosphorylate the mitogen and stress activated kinase (MSK). Biologically, the MAPKs modulate a number of important functions including development, immunity, apoptosis, cell growth and division, autophagy and cell survival.
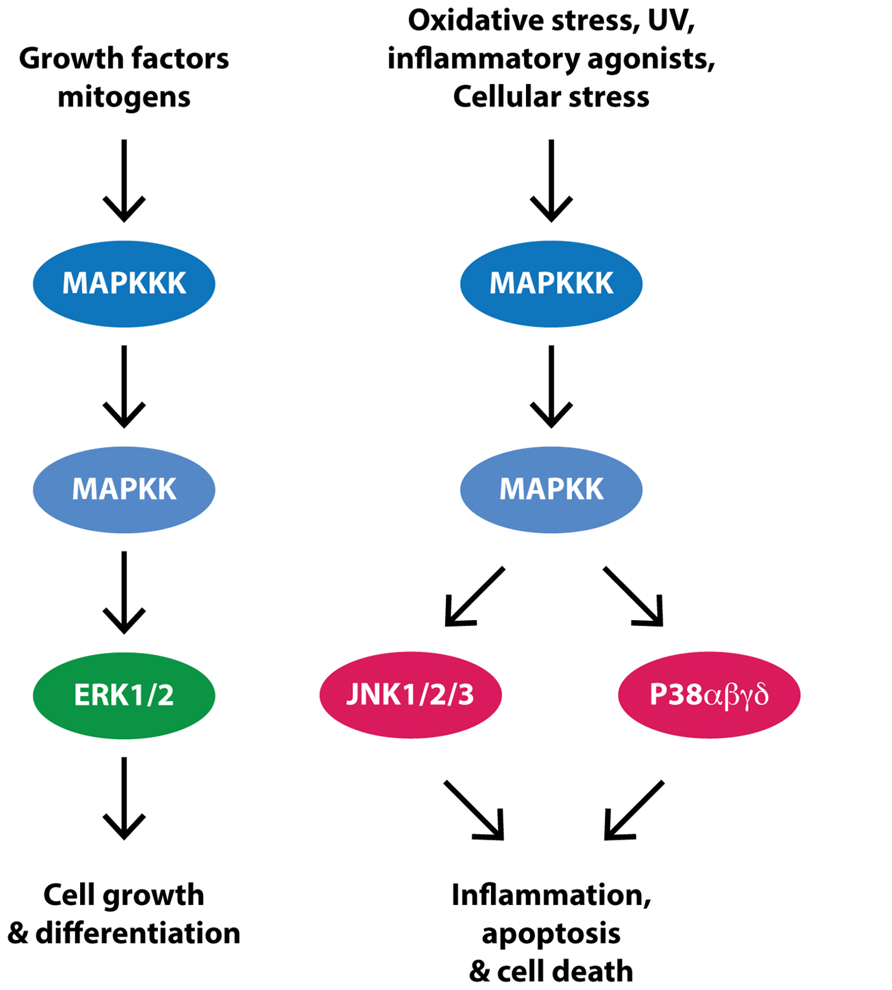
FIGURE 2. Simplifed MAPK signaling. Receptor activation by growth factors or mitogens triggers a signal cascade in which mitogen activated protein kinase kinase kinases (MAPKKK) are activated and in turn activate mitogen activated kinase kinases (MAPKK) and then the mitogen activated protein kinases (MAPK), ERK, JNK, and P38. Evidence suggests that this pathway is upregulated in PD substantia nigra dopaminergic neurons, potentially contributing to cell death.
JNK and p38
c-Jun N-terminal kinase is robustly activated in common toxin models of PD such as LPS, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine (6-OHDA; Choi et al., 1999; Saporito et al., 2000; Xing et al., 2007). Genetic deletion of JNK2 and JNK3 protect against MPTP-induced neurodegeneration in mice (Hunot et al., 2004) and kinase inhibitors of JNK have neuroprotective effects in the MPTP (Saporito et al., 1999; Wang et al., 2004, 2009b; Chambers et al., 2011) and 6-OHDA (Chambers et al., 2013) models of PD. Moreover a host of anti-oxidant and anti-inflammatory compounds offering varying degrees of neuroprotection in these models are thought to have a mechanism of action, at least in part, involving inhibition of JNK activation (Xing et al., 2007; Castro-Caldas et al., 2012; Lee et al., 2013; Zhai et al., 2013). Genetic deletion of the p38 substrate MK2, also protects against MPTP-induced neurodegeneration in rodents, by reducing the neuroinflammation associated with MPTP lesions (Thomas et al., 2008). Both JNK and p38 are also implicated in the death of neuronal cells following treatment with another environmental toxin used to model PD, rotenone (Newhouse et al., 2004; Gao et al., 2013), with inhibition of p38 potentially protective (Choi et al., 2014). Despite the evidence from cellular and animal models, however, a clinical trial of the JNK inhibitor CEP-1347 failed to show benefit in human PD patients (Investigators, 2007), possibly because of an absence of substantial changes in these kinases in patients with chronic PD (Ferrer et al., 2001).
Increased nuclear staining of the JNK substrate, c-Jun, has been observed in the substantia nigra of PD patients (Hunot et al., 2004). Translocation of c-Jun to the nucleus requires JNK phosphorylation and is a surrogate marker of JNK activity. The association between JNK and p38 and α-synuclein pathology has also been explored in the substantia nigra and brainstem regions of control and PD brain. In this study, granular phosphorylated p38 immunoreactivity was observed in association with diffuse α-synuclein pathology, more consistent with Lewy neurites than Lewy bodies in the substantia nigra (Ferrer et al., 2001). In contrast, phosphorylated JNK rarely stained Lewy body containing neurons (Ferrer et al., 2001). There was also no association between phosphorylated JNK immunostaining and apoptosis in PD substantia nigra neurons (Ferrer et al., 2001). These studies suggest a potential early role for p38 in the formation of Lewy bodies whereas JNK appears not to be involved. The protective effects of JNK inhibitors may instead be mediated through glial cells. Further studies could explore how JNK activity in glia relates to PD pathogenesis.
ERK
Extracellular signal related protein kinase is activated following treatment of cells with 6-OHDA and MPTP and inhibitors of ERK provide protection in these PD cell models (Kulich and Chu, 2001; Gomez-Santos et al., 2002). ERK crosstalk also modulates protective effects of neurotrophins and anti-oxidant treatments (Chu et al., 2004; Hetman and Gozdz, 2004).
In neurons in the substantia nigra of PD patients, phosphorylated ERK immunoreactivity shows granular aggregations, distinct from the diffuse cytoplasmic localization of phosphorylated ERK in cortical neurons from control and PD patients (Zhu et al., 2002). The aggregated pattern of phosphorylated ERK staining is also observed in pigmented neurons of the locus coeruleus in PD patients, but is absent in glial cells. The levels of phosphorylated ERK increase in substantia nigra neurons in PD patients, where the granular inclusions partly associate with mitochondria and weakly with endosomes (Zhu et al., 2003). Increased phosphorylation of ERK correlates with increased staining for the ERK substrate RSK1 (Zhu et al., 2002) and total levels of ERK do not differ between control and PD samples (Zhu et al., 2002), collectively demonstrating an increase in ERK activity in PD brain. ERK also associates with Lewy bodies, particularly the halo region (Ferrer et al., 2001; Zhu et al., 2002, 2003). Moreover, granular ERK inclusions are often seen in PD neurons devoid of α-synuclein pathology and sometimes not seen at all in neurons with severe α-synuclein pathology suggesting a potential early role for ERK in PD pathogenesis (Zhu et al., 2002).
Kinases of the Phosphoinositide 3-Kinase (PI3K) Pathway
The PI3K pathway controls cell survival and proliferation and thus has been studied extensively in the context of cancer. PI3K is classically activated by tyrosine kinase receptors following their binding of insulin or insulin like growth factors (e.g., IGF1; Figure 3). Activated PI3K phosphorylates membrane-associated phosphatidylinositol 4,5-bisphosphate (PIP2) to produce phosphatidylinositol 2,4,5-triphosphate (PIP3), which results in recruitment of PKB (also known as AKT; Figure 3). PKB is in turn phosphorylated in its activation loop (at Thr308) by phosphoinositide-dependent kinase 1 (PDK1) and at its C-terminal hydrophobic motif (Ser473) by mTOR complex 2 (mTORC2). Activated PKB phosphorylates a number of substrates including mTOR, to promote protein synthesis and inhibit autophagy, glycogen synthase kinase 3β (GSK3β), to induce glycogen and regulate glucose metabolism and fork head box-O class (FOXO), a transcription factor regulating genes essential for cell growth, proliferation, and survival.
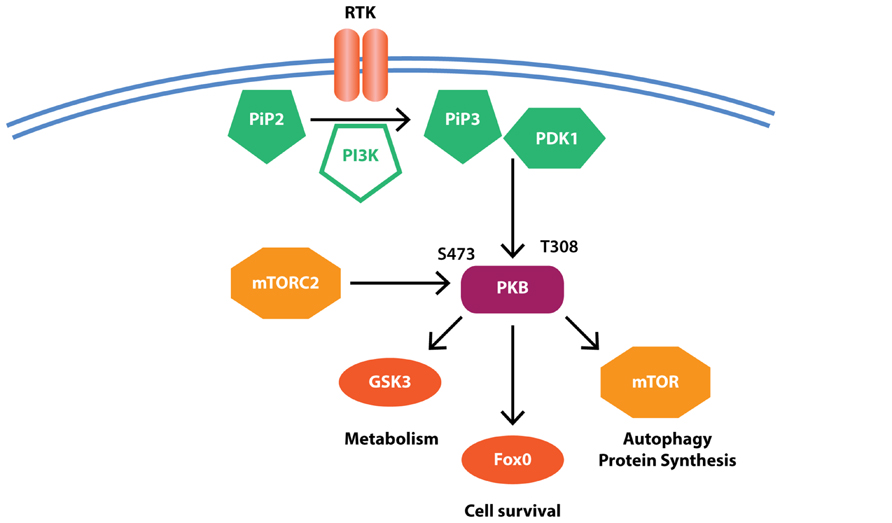
FIGURE 3. Simplified PI3K signaling. Receptor-ligand binding results in the activation of phosphoinositide 3-kinase (PI3K) that in turn mediates the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 2,4,5-triphosphate (PIP3). PIP3 recruits protein kinase B (PKB) where it is activated by phosphorylation at Thr308 by phosphoinositide-dependent kinase 1 (PDK1) and Ser473 by mammalian target of rapamycin (mTOR) complex 2 (mTORC2). PKB then further phosphorylates downstream substrates to regulate cell survival and metabolic pathways. Evidence suggests reduced PKB but increased mTOR and GSK3β activity in PD, potentially contributing to protein accumulation and reduced cell survival.
PKB
Administration of 6-OHDA results in reduced PKB Thr308 and Ser473 phosphorylation and marked loss of PKB activity in cell culture and rodent PD models and a number of compounds that stimulate PKB activity have demonstrated neuroprotection in this model, as well as other PD toxin models, including MPTP and rotenone (for review see Greene et al., 2011). Overexpression of PKB in rodent brain also protects dopaminergic neurons from 6-OHDA-induced cell death (Ries et al., 2006) and activation of PKB likely contributes to the neuroprotective effects of trophic factors such as glial cell line-derived neurotrophic factor (GDNF; Ugarte et al., 2003) and potentially to the effects of the monoamine oxidase B inhibitor and PD drug rasagiline (Mandel et al., 2007; Sagi et al., 2007).
In PD midbrain the phosphorylation of PKB Ser473 is reduced in both cytosolic and membrane fractions (Timmons et al., 2009). PKB and its phosphorylation at Ser473 are also robustly detected in dopaminergic neurons of the substantia nigra and are consequently reduced in PD with loss of these neurons. However, PKB immunoreactivity is still detected in surviving PD dopaminergic neurons (Timmons et al., 2009). A second study has confirmed reduced phosphorylation of PKB at both Thr308 and Ser473 in PD substantia nigra dopaminergic neurons (Malagelada et al., 2008). Results of this study suggest that reduced phosphorylation of PKB is restricted to dopaminergic neurons as non-neuromelanin containing neurons of the midbrain expressed similar levels of PKB and phosphorylated PKB in both control and PD states. Interestingly, a robust increase in PKB and phosphorylated Ser473 PKB was detected in cells with glial morphology in the substantia nigra region in PD (Timmons et al., 2009). Whilst reduced PKB pathway activity in neurons may contribute to their loss in PD, the contribution of increased PKB activity in glia to the progression of PD has not been explored.
GSK3β
The two isoforms of GSK3, GSK3α, and GSK3β, are ubiquitously expressed in the brain where they predominantly act to regulate glucose metabolism. Inhibition of the GSK3β isoform can protect against MPTP, 6-OHDA, and LPS-induced neurotoxicity (Kozikowski et al., 2006; Wang et al., 2007; Morales-Garcia et al., 2013) whilst its activation has been implicated in rotenone toxicity (Hongo et al., 2012). GSK3β has also been implicated in microglial-mediated inflammation (Yuskaitis and Jope, 2009) and the neuroprotective effects of GSK3β inhibitors may be mediated, at least in part, through anti-inflammatory actions (Yuskaitis and Jope, 2009; Morales-Garcia et al., 2013).
In brain, increased total and phosphorylated GSK3β is detected as punctate structures in the cytosol of pigmented neurons in PD substantia nigra (Nagao and Hayashi, 2009). GSK3β and phosphorylated GSK3β partly co-localize to the halo region of Lewy bodies and also to Lewy neurites (Nagao and Hayashi, 2009). GSK3β protein is also significantly increased in the striatum of PD brains where its phosphorylation correlates with both tau and α-synuclein pathology (Wills et al., 2010). This suggests a potential role for GSK3β in promoting the early stages of tau interaction with α-synuclein, leading to α-synuclein pathology in PD. This could be important as genome-wide association studies implicate polymorphisms in the MAPT and SNCA genes as the most robustly reproducible risk factors for sporadic PD (Satake et al., 2009; Simon-Sanchez et al., 2009).
mTOR
The mTOR kinase exists in two complexes termed mTORC1 and mTORC2, with mTORC2 regulating PKB activity and mTORC1 regulating protein synthesis and autophagy (Laplante and Sabatini, 2012). The phosphorylation of mTORC1 by PKB promotes protein synthesis and inhibits autophagy. Reduced phosphorylation of PKB in PD brain may therefore be expected to promote autophagy; however, this process is clearly dysregulated in PD as autophagy markers are also significantly decreased in PD substantia nigra (Chu et al., 2009; Alvarez-Erviti et al., 2010). Indeed, evidence suggests that dysfunctional autophagy pathways play a key role in the pathogenesis of PD (Lynch-Day et al., 2012). Moreover, the mTOR inhibitor (and therefore autophagy inducing) rapamycin prevents MPTP-induced neurodegeneration (Dehay et al., 2010; Liu et al., 2013). Rapamycin also has protective properties in rotenone and α-synuclein PD models (Pan et al., 2009; Spencer et al., 2009; Crews et al., 2010; Xiong et al., 2011) suggesting inhibition of mTORC1 has potential as a treatment for PD.
Protein levels of neuronal mTOR were significantly increased in the temporal cortex of cases with dementia with Lewy bodies, particularly in neurons displaying accumulation of α-synuclein (Crews et al., 2010). In comparison, brain tissue from cases with Alzheimer’s disease had normal levels of mTOR in the temporal cortex (Crews et al., 2010). Up-regulation of mTOR is consistent with a phenotype of increased protein synthesis and reduced autophagy, promoting the accumulation of potentially toxic proteins. In the context of PD, increased mTOR would likely aid the propagation of α-synuclein, however, whether changes in mTOR are associated with the spread of α-synuclein pathology in PD brain is unknown.
Overview of Kinases Experimentally Implicated in PD
It is evident from human tissue studies, particularly those focused on the nigral dopaminergic system, that inflammatory pathways are activated in PD and autophagy pathways are impaired. PD brain tissue samples from different brain regions at different stages of pathology could inform on the order of these events and provide more insight into whether certain kinases are causal for PD pathologies. The discovery that certain toxins (MPTP, rotenone, LPS, 6-OHDA) induce a selective loss of dopaminergic neurons in rodent models has facilitated a wealth of information regarding the order of the biological processes leading to such neuronal death as well as signaling proteins mediating these events. While these toxin-based models do not replicate all the features of sporadic PD, such as an age-dependent phenotype and the presence of α-synuclein pathology, they have implicated a range of kinases as important in the process. It will be important in many instances to use pathologically staged human brain tissue to validate the expression of kinases and their isoforms and any disease-associated changes identified experimentally in mice.
Additional Kinases and Overall Conclusions
In addition to the kinases discussed above, a number of other kinases are emerging as having potential roles in PD pathogenesis and/or potential therapeutic targets. These include CDK5, a kinase whose activity is increased by MPTP treatment and inhibition attenuates MPTP-induced neuronal loss (Smith et al., 2003; Qu et al., 2007). The eIF2alpha kinase (also known as PERK), whose inhibition was recently shown to attenuate neurodegeneration in prion-infected mice (Moreno et al., 2013), and AMP-activated protein kinase (AMPK), a major metabolic regulatory enzyme whose activation has been associated with neuroprotection in a number of PD models (Wu et al., 2011; Bayliss and Andrews, 2013; Dulovic et al., 2014; Li et al., 2014). AMPK is also activated by thiazolidinones (Fryer et al., 2002), compounds that have neuroprotective properties in a number of settings (Carta, 2013). Of these kinases, CDK5 and PERK have been studied in PD brain, with CDK5 localizing to Lewy bodies (Brion and Couck, 1995; Nakamura et al., 1997) and PERK increased in PD substantia nigra neurons (Hoozemans et al., 2007). PERK levels also correlated with α-synuclein deposition (Hoozemans et al., 2007), making PERK in particular a very interesting candidate for further study.
Thus, a number of protein kinases have been implicated in the pathogenesis of PD covering a diverse array of biological functions including oxidative stress, inflammation, and autophagy (Figure 4). However, delineating the exact order by which these biological functions go wrong in PD brain is still a major challenge, despite the staging methods now in more common use. It is likely that some kinases are more important for initiating the disease whilst others are more important for disease propagation. In this regard, the majority of brain tissue work to date has focused on the substantia nigra region in PD, a region mostly at end-stage pathology in patients dying with PD. Thus brain tissue studies using this region are not informative on early pathogenic events, and assessment of this region provides limited information on the cause or consequence of many findings. With a greater understanding of how PD spreads throughout the brain in a staged fashion, brain regions can be selected to determine biochemical responses across the disease spectrum, particularly assessing regions with evidence of only early perturbations indicative of PD. Such an approach should provide some insight into which processes may precede PD pathology and which processes may propagate PD pathology. This is important for determining when potential therapies, such as kinase inhibitors, are likely to exert maximum efficacy. It is also important to recognize potential caveats of postmortem studies such as postmortem delay, comorbidities, and drug regimes, even though many caveats can be controlled with appropriate sample selection. In the absence of animal models that replicate all the cardinal features of PD, human pathological postmortem brain tissue remains an important resource to understand the biochemical details of PD and to verify cell and animal model hypothesis testing.
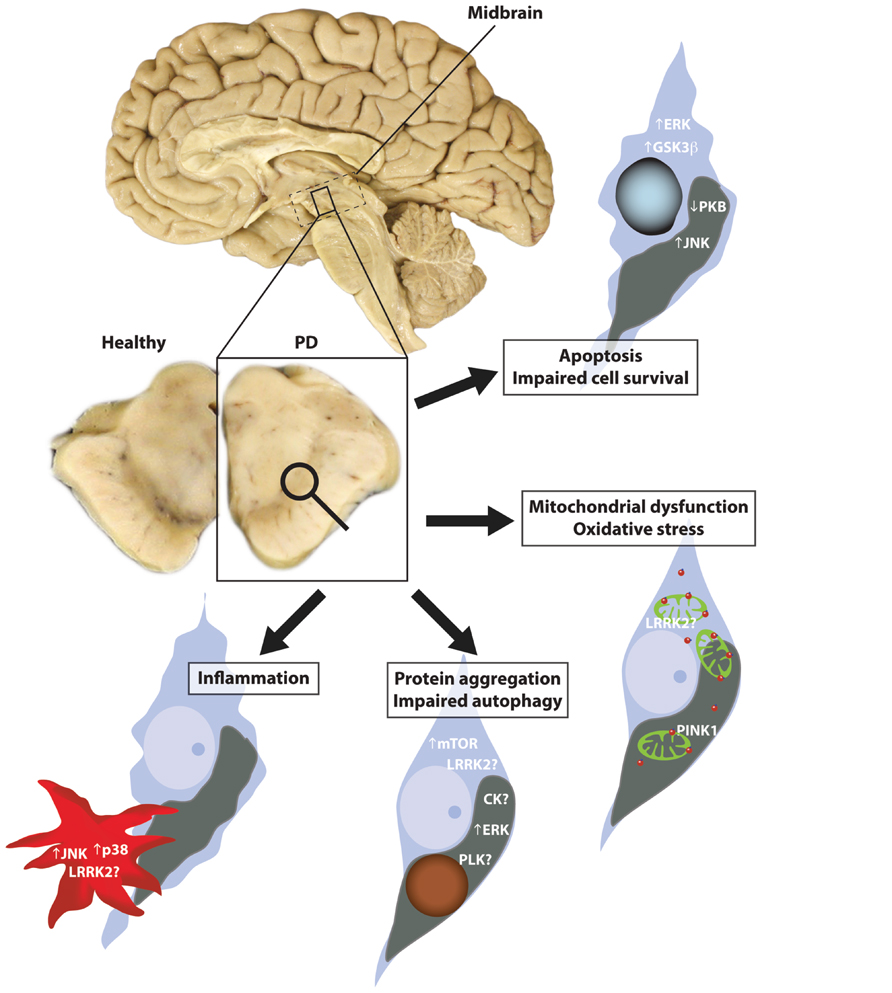
FIGURE 4. Kinases implicated in Parkinson’s disease (PD). PD is characterized by the loss of pigmented dopaminergic neurons in the substantia nigra region of the midbrain. A number of biological processes have been implicated in this neuronal loss including mitochondrial dysfunction, oxidative stress, autophagy, and inflammation. In the substantia nigra, a number of kinases impacted by these processes combine to promote the accumulation of phosphorylated α -synuclein and induce conditions that reduce cell viability.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Nicolas Dzamko and Glenda M. Halliday receive funding from the National Health and Medical Research Council (NHMRC), the Michael J. Fox Foundation for Parkinson’s disease research, Parkinson’s NSW and the Shake it Up Australia Foundation. We thank Heidi Cartwright for generating the figures.
References
Abou-Sleiman, P. M., Muqit, M. M., and Wood, N. W. (2006). Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 7, 207–219. doi: 10.1038/nrn1868
Alegre-Abarrategui, J., Ansorge, O., Esiri, M., and Wade-Martins, R. (2008). LRRK2 is a component of granular alpha-synuclein pathology in the brainstem of Parkinson’s disease. Neuropathol. Appl. Neurobiol. 34, 272–283. doi: 10.1111/j.1365-2990.2007.00888.x
Alvarez-Erviti, L., Rodriguez-Oroz, M. C., Cooper, J. M., Caballero, C., Ferrer, I., Obeso, J. A., et al. (2010). Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 67, 1464–1472. doi: 10.1001/archneurol.2010.198
Anderson, J. P., Walker, D. E., Goldstein, J. M., De Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Arawaka, S., Wada, M., Goto, S., Karube, H., Sakamoto, M., Ren, C. H., et al. (2006). The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson’s disease. J. Neurosci. 26, 9227–9238. doi: 10.1523/JNEUROSCI.0341-06.2006
Bayliss, J. A., and Andrews, Z. B. (2013). Ghrelin is neuroprotective in Parkinson’s disease: molecular mechanisms of metabolic neuroprotection. Ther. Adv. Endocrinol. Metab. 4, 25–36. doi: 10.1177/2042018813479645
Beilina, A., Rudenko, I. N., Kaganovich, A., Civiero, L., Chau, H., Kalia, S. K., et al. (2014). Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 111, 2626–2631. doi: 10.1073/pnas.1318306111
Bergeron, M., Motter, R., Tanaka, P., Fauss, D., Babcock, M., Chiou, S.S., et al. (2014). In vivo modulation of polo-like kinases supports a key role for PLK2 in Ser129 alpha-synuclein phosphorylation in mouse brain. Neuroscience 256, 72–82. doi: 10.1016/j.neuroscience.2013.09.061
Biskup, S., Moore, D. J., Rea, A., Lorenz-Deperieux, B., Coombes, C. E., Dawson, V. L., et al. (2007). Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci. 8:102. doi: 10.1186/1471-2202-8–102
Blackinton, J. G., Anvret, A., Beilina, A., Olson, L., Cookson, M. R., and Galter, D. (2007). Expression of PINK1 mRNA in human and rodent brain and in Parkinson’s disease. Brain Res. 1184, 10–16. doi: 10.1016/j.brainres.2007.09.056
Bowers, S., Truong, A. P., Ye, M., Aubele, D. L., Sealy, J. M., Neitz, R. J., et al. (2013). Design and synthesis of highly selective, orally active Polo-like kinase-2 (Plk-2) inhibitors. Bioorg. Med. Chem. Lett. 23, 2743–2749. doi: 10.1016/j.bmcl.2013.02.065
Braak, H., Del Tredici, K., Rub, U., De Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
Brion, J. P., and Couck, A. M. (1995). Cortical and brainstem-type Lewy bodies are immunoreactive for the cyclin-dependent kinase 5. Am. J. Pathol. 147, 1465–1476.
Bychkov, E. R., Gurevich, V. V., Joyce, J. N., Benovic, J. L., and Gurevich, E. V. (2008). Arrestins and two receptor kinases are upregulated in Parkinson’s disease with dementia. Neurobiol. Aging 29, 379–396. doi: 10.1016/j.neurobiolaging.2006.10.012
Carlsson, A. (1959). The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol. Rev. 11, 490–493.
Carta, A. R. (2013). PPAR-gamma: therapeutic prospects in Parkinson’s disease. Curr. Drug Targets 14, 743–751. doi: 10.2174/1389450111314070004
Castro-Caldas, M., Carvalho, A. N., Rodrigues, E., Henderson, C. J., Wolf, C. R., Rodrigues, C. M., et al. (2012). Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson’s disease. Mol. Neurobiol. 46, 475–486. doi: 10.1007/s12035-012-8295-4
Chambers, J. W., Howard, S., and Lograsso, P. V. (2013). Blocking c-Jun N-terminal kinase (JNK) translocation to the mitochondria prevents 6-hydroxydopamine-induced toxicity in vitro and in vivo. J. Biol. Chem. 288, 1079–1087. doi: 10.1074/jbc.M112.421354
Chambers, J. W., Pachori, A., Howard, S., Ganno, M., Hansen, D. Jr., Kamenecka, T., et al. (2011). Small molecule c-jun-N-terminal kinase (JNK) inhibitors protect dopaminergic neurons in a model of Parkinson’s disease. ACS Chem. Neurosci. 2, 198–206. doi: 10.1021/cn100109k
Chen, L., and Feany, M. B. (2005). Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 8, 657–663. doi: 10.1038/nn1443
Cheong, J. K., and Virshup, D. M. (2011). Casein kinase 1: complexity in the family. Int. J. Biochem. Cell Biol. 43, 465–469. doi: 10.1016/j.biocel.2010.12.004
Cho, H. J., Liu, G., Jin, S. M., Parisiadou, L., Xie, C., Yu, J., et al. (2013). MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 22, 608–620. doi: 10.1093/hmg/dds470
Choi, B. S., Kim, H., Lee, H. J., Sapkota, K., Park, S. E., Kim, S., et al. (2014). Celastrol from ‘Thunder God Vine’ protects SH-SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson’s disease. Neurochem. Res. 39, 84–96. doi: 10.1007/s11064-013-1193-y
Choi, W. S., Yoon, S. Y., Oh, T. H., Choi, E. J., O’malley, K. L., and Oh, Y. J. (1999). Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: role of caspases, ROS, and JNK. J. Neurosci. Res. 57, 86–94. doi: 10.1002/(SICI)1097-4547(19990701)57:1<86::AID-JNR9>3.0.CO;2-E
Chu, C. T., Levinthal, D. J., Kulich, S. M., Chalovich, E. M., and Defranco, D. B. (2004). Oxidative neuronal injury. The dark side of ERK1/2. Eur. J. Biochem. 271, 2060–2066. doi: 10.1111/j.1432-1033.2004.04132.x
Chu, Y., Dodiya, H., Aebischer, P., Olanow, C. W., and Kordower, J. H. (2009). Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol. Dis. 35, 385–398. doi: 10.1016/j.nbd.2009.05.023
Crews, L., Spencer, B., Desplats, P., Patrick, C., Paulino, A., Rockenstein, E., et al. (2010). Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS ONE 5:e9313. doi: 10.1371/journal.pone.0009313
Cunnington, M. S., Kay, C., Avery, P. J., Mayosi, B. M., Koref, M. S., and Keavney, B. (2009). STK39 polymorphisms and blood pressure: an association study in British Caucasians and assessment of cis-acting influences on gene expression. BMC Med. Genet. 10:135. doi: 10.1186/1471–2350-10–135
Dagda, R. K., Pien, I., Wang, R., Zhu, J., Wang, K. Z., Callio, J., et al. (2014). Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J. Neurochem. 128, 864–877. doi: 10.1111/jnc.12494
Davies, P., Hinkle, K. M., Sukar, N. N., Sepulveda, B., Mesias, R., Serrano, G., et al. (2013). Comprehensive characterization and optimization of anti-LRRK2 (leucine-rich repeat kinase 2) monoclonal antibodies. Biochem. J. 453, 101–113. doi: 10.1042/BJ20121742
de Carcer, G., Escobar, B., Higuero, A. M., Garcia, L., Anson, A., Perez, G., et al. (2011a). Plk5, a polo box domain-only protein with specific roles in neuron differentiation and glioblastoma suppression. Mol. Cell. Biol. 31, 1225–1239. doi: 10.1128/MCB.00607-10
de Carcer, G., Manning, G., and Malumbres, M. (2011b). From Plk1 to Plk5: functional evolution of polo-like kinases. Cell Cycle 10, 2255–2262. doi: 10.4161/cc.10.14.16494
Dehay, B., Bove, J., Rodriguez-Muela, N., Perier, C., Recasens, A., Boya, P., et al. (2010). Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 30, 12535–12544. doi: 10.1523/JNEUROSCI.1920–10.2010
Delpire, E., and Austin, T. M. (2010). Kinase regulation of Na+–K+–2Cl- cotransport in primary afferent neurons. J. Physiol. 588, 3365–3373. doi: 10.1113/jphysiol.2010.190769
Deng, X., Choi, H. G., Buhrlage, S. J., and Gray, N. S. (2012). Leucine-rich repeat kinase 2 inhibitors: a patent review (2006–2011). Expert Opin. Ther. Pat. 22, 1415–1426. doi: 10.1517/13543776.2012.729041
Dulovic, M., Jovanovic, M., Xilouri, M., Stefanis, L., Harhaji-Trajkovic, L., Kravic-Stevovic, T., et al. (2014). The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol. Dis. 63, 1–11. doi: 10.1016/j.nbd.2013.11.002
Dumitriu, A., Pacheco, C. D., Wilk, J. B., Strathearn, K. E., Latourelle, J. C., Goldwurm, S., et al. (2011). Cyclin-G-associated kinase modifies alpha-synuclein expression levels and toxicity in Parkinson’s disease: results from the GenePD Study. Hum. Mol. Genet. 20, 1478–1487. doi: 10.1093/hmg/ddr026
Dzamko, N., Deak, M., Hentati, F., Reith, A. D., Prescott, A. R., Alessi, D. R., et al. (2010). Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem. J. 430, 405–413. doi: 10.1042/BJ20100784
Dzamko, N., and Halliday, G. M. (2013). Unlocking the secrets of LRRK2 function with selective kinase inhibitors. Future Neurol. 8, 347–357. doi: 10.2217/fnl.13.9
Eisenberg, E., and Greene, L. E. (2007). Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic 8, 640–646. doi: 10.1111/j.1600-0854.2007.00568.x
Escobar, V. D., Kuo, Y. M., Orrison, B. M., Giasson, B. I., and Nussbaum, R. L. (2014). Transgenic mice expressing S129 phosphorylation mutations in alpha-synuclein. Neurosci. Lett. 563, 96–100. doi: 10.1016/j.neulet.2014.01.033
Febbraro, F., Sahin, G., Farran, A., Soares, S., Jensen, P. H., Kirik, D., et al. (2013). Ser129D mutant alpha-synuclein induces earlier motor dysfunction while S129A results in distinctive pathology in a rat model of Parkinson’s disease. Neurobiol. Dis. 56, 47–58. doi: 10.1016/j.nbd.2013.03.014
Ferrer, I., Blanco, R., Carmona, M., Puig, B., Barrachina, M., Gomez, C., et al. (2001). Active, phosphorylation-dependent mitogen-activated protein kinase (MAPK/ERK), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), and p38 kinase expression in Parkinson’s disease and dementia with Lewy bodies. J. Neural. Transm. 108, 1383–1396. doi: 10.1007/s007020100015
Fish, K. J., Cegielska, A., Getman, M. E., Landes, G. M., and Virshup, D. M. (1995). Isolation and characterization of human casein kinase I epsilon (CKI), a novel member of the CKI gene family. J. Biol. Chem. 270, 14875–14883. doi: 10.1074/jbc.270.25.14875
Fitzgerald, K., Bergeron, M., Willits, C., Bowers, S., Aubele, D. L., Goldbach, E., et al. (2013). Pharmacological inhibition of polo like kinase 2 (PLK2) does not cause chromosomal damage or result in the formation of micronuclei. Toxicol. Appl. Pharmacol. 269, 1–7. doi: 10.1016/j.taap.2013.02.012
Fryer, L. G., Parbu-Patel, A., and Carling, D. (2002). The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 277, 25226–25232. doi: 10.1074/jbc.M202489200
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Gandhi, S., Muqit, M. M., Stanyer, L., Healy, D. G., Abou-Sleiman, P. M., Hargreaves, I., et al. (2006). PINK1 protein in normal human brain and Parkinson’s disease. Brain 129, 1720–1731. doi: 10.1093/brain/awl114
Gao, F., Chen, D., Hu, Q., and Wang, G. (2013). Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS ONE 8:e72046. doi: 10.1371/journal.pone.0072046
Gardet, A., Benita, Y., Li, C., Sands, B. E., Ballester, I., Stevens, C., et al. (2010). LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol. 185, 5577–5585. doi: 10.4049/jimmunol.1000548
Gasser, T. (2009). Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev. Mol. Med. 11:e22. doi: 10.1017/S1462399409001148
Giasson, B. I., Covy, J. P., Bonini, N. M., Hurtig, H. I., Farrer, M. J., Trojanowski, J. Q., et al. (2006). Biochemical and pathological characterization of Lrrk2. Ann. Neurol. 59, 315–322. doi: 10.1002/ana.20791
Glover, M., and O’shaughnessy, K. M. (2011). SPAK and WNK kinases: a new target for blood pressure treatment? Curr. Opin. Nephrol. Hypertens. 20, 16–22. doi: 10.1097/MNH.0b013e32834132bc
Gomez-Santos, C., Ferrer, I., Reiriz, J., Vinals, F., Barrachina, M., and Ambrosio, S. (2002). MPP+ increases alpha-synuclein expression and ERK/MAP-kinase phosphorylation in human neuroblastoma SH-SY5Y cells. Brain Res. 935, 32–39. doi: 10.1016/S0006-8993(02)02422-8
Greene, L. A., Levy, O., and Malagelada, C. (2011). Akt as a victim, villain and potential hero in Parkinson’s disease pathophysiology and treatment. Cell Mol. Neurobiol. 31, 969–978. doi: 10.1007/s10571-011-9671–9678
Grenier, K., Mclelland, G. L., and Fon, E. A. (2013). Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front. Neurol. 4:100. doi: 10.3389/fneur.2013.00100
Grunblatt, E., Mandel, S., Jacob-Hirsch, J., Zeligson, S., Amariglo, N., Rechavi, G., et al. (2004). Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J. Neural. Transm. 111, 1543–1573. doi: 10.1007/s00702-004-0212-1
Guerreiro, P. S., Huang, Y., Gysbers, A., Cheng, D., Gai, W. P., Outeiro, T. F., et al. (2013). LRRK2 interactions with alpha-synuclein in Parkinson’s disease brains and in cell models. J. Mol. Med. (Berl.) 91, 513–522. doi: 10.1007/s00109-012-0984-y
Gurevich, E. V., Tesmer, J. J., Mushegian, A., and Gurevich, V. V. (2012). G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol. Ther. 133, 40–69. doi: 10.1016/j.pharmthera.2011.08.001
Halliday, G., Hely, M., Reid, W., and Morris, J. (2008). The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol. 115, 409–415. doi: 10.1007/s00401-008-0344-8
Hassler, R. (1938). Zur Pathologie der paralysis agitans und des post-enzephalitschen Parkinsonismus. J. Psychol. Neurol. 48, 387–476.
Healy, D. G., Falchi, M., O’sullivan, S. S., Bonifati, V., Durr, A., Bressman, S., et al. (2008). Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case–control study. Lancet Neurol. 7, 583–590. doi: 10.1016/S1474-4422(08)70117-0
Hetman, M., and Gozdz, A. (2004). Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur. J. Biochem. 271, 2050–2055. doi: 10.1111/j.1432-1033.2004.04133.x
Higashi, S., Biskup, S., West, A. B., Trinkaus, D., Dawson, V. L., Faull, R. L., et al. (2007). Localization of Parkinson’s disease-associated LRRK2 in normal and pathological human brain. Brain Res. 1155, 208–219. doi: 10.1016/j.brainres.2007.04.034
Hongo, H., Kihara, T., Kume, T., Izumi, Y., Niidome, T., Sugimoto, H., et al. (2012). Glycogen synthase kinase-3beta activation mediates rotenone-induced cytotoxicity with the involvement of microtubule destabilization. Biochem. Biophys. Res. Commun. 426, 94–99. doi: 10.1016/j.bbrc.2012.08.042
Hoozemans, J. J., Van Haastert, E. S., Eikelenboom, P., De Vos, R. A., Rozemuller, J. M., and Scheper, W. (2007). Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 354, 707–711. doi: 10.1016/j.bbrc.2007.01.043
Hunot, S., Vila, M., Teismann, P., Davis, R. J., Hirsch, E. C., Przedborski, S., et al. (2004). JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 665–670. doi: 10.1073/pnas.0307453101
Inglis, K. J., Chereau, D., Brigham, E. F., Chiou, S. S., Schobel, S., Frigon, N. L., et al. (2009). Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J. Biol. Chem. 284, 2598–2602. doi: 10.1074/jbc.C800206200
Investigators., P. S. G. P. (2007). Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 69, 1480–1490. doi: 10.1212/01.wnl.0000277648.63931.c0
Ishii, A., Nonaka, T., Taniguchi, S., Saito, T., Arai, T., Mann, D., et al. (2007). Casein kinase 2 is the major enzyme in brain that phosphorylates Ser129 of human alpha-synuclein: Implication for alpha-synucleinopathies. FEBS Lett. 581, 4711–4717. doi: 10.1016/j.febslet.2007.08.067
Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A., et al. (2007). LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 405, 307–317. doi: 10.1042/BJ20070209
Kamal, F. A., Travers, J. G., and Blaxall, B. C. (2012). G protein-coupled receptor kinases in cardiovascular disease: why “where” matters. Trends Cardiovasc. Med. 22, 213–219. doi: 10.1016/j.tcm.2012.07.023
Kanaoka, Y., Kimura, S. H., Okazaki, I., Ikeda, M., and Nojima, H. (1997). GAK: a cyclin G associated kinase contains a tensin/auxilin-like domain. FEBS Lett. 402, 73–80. doi: 10.1016/S0014-5793(96)01484-6
Kawajiri, S., Saiki, S., Sato, S., and Hattori, N. (2011). Genetic mutations and functions of PINK1. Trends Pharmacol. Sci. 32, 573–580. doi: 10.1016/j.tips.2011.06.001
Kett, L. R., and Dauer, W. T. (2012). Leucine-rich repeat kinase 2 for beginners: six key questions. Cold Spring Harb. Perspect. Med. 2, a009407. doi: 10.1101/cshperspect.a009407
Kim, J., Byun, J. W., Choi, I., Kim, B., Jeong, H. K., Jou, I., et al. (2013). PINK1 deficiency enhances inflammatory cytokine release from acutely prepared brain slices. Exp. Neurobiol. 22, 38–44. doi: 10.5607/en.2013.22.1.38
Knippschild, U., Gocht, A., Wolff, S., Huber, N., Lohler, J., and Stoter, M. (2005). The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell. Signal. 17, 675–689. doi: 10.1016/j.cellsig.2004.12.011
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2, 120080. doi: 10.1098/rsob.120080.
Kozikowski, A. P., Gaisina, I. N., Petukhov, P. A., Sridhar, J., King, L. T., Blond, S. Y., et al. (2006). Highly potent and specific GSK-3beta inhibitors that block tau phosphorylation and decrease alpha-synuclein protein expression in a cellular model of Parkinson’s disease. Chem. Med. Chem. 1, 256–266. doi: 10.1002/cmdc.200500039
Kulich, S. M., and Chu, C. T. (2001). Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: implications for Parkinson’s disease. J. Neurochem. 77, 1058–1066. doi: 10.1046/j.1471-4159.2001.00304.x
Kyriakis, J. M., and Avruch, J. (2012). Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 92, 689–737. doi: 10.1152/physrev.00028.2011
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Lee, D. W., Zhao, X., Yim, Y. I., Eisenberg, E., and Greene, L. E. (2008). Essential role of cyclin-G-associated kinase (Auxilin-2) in developing and mature mice. Mol. Biol. Cell 19, 2766–2776. doi: 10.1091/mbc.E07-11–1115
Lee, H. J., and Chung, K. C. (2012). PINK1 positively regulates IL-1beta-mediated signaling through Tollip and IRAK1 modulation. J. Neuroinflammation 9, 271. doi: 10.1186/1742-2094-9–271
Lee, K. W., Im, J. Y., Woo, J. M., Grosso, H., Kim, Y. S., Cristovao, A. C., et al. (2013). Neuroprotective and anti-inflammatory properties of a coffee component in the MPTP model of Parkinson’s disease. Neurotherapeutics 10, 143–153. doi: 10.1007/s13311-012-0165-2
Li, X., Geng, J., and Liu, J. (2014). Adiponectin offers protection against L166P mutant DJ-1-induced neuronal cytotoxicity mediated by APPL1-dependent AMPK activation. Int. J. Neurosci. 124, 350–361. doi: 10.3109/00207454.2013.846340
Liao, J., Wu, C. X., Burlak, C., Zhang, S., Sahm, H., Wang, M., et al. (2014). Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc. Natl. Acad. Sci U.S.A. doi: 10.1073/pnas.1323285111.
Lill, C. M., Roehr, J. T., Mcqueen, M. B., Kavvoura, F. K., Bagade, S., Schjeide, B. M., et al. (2012). Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database. PLoS Genet. 8:e1002548. doi: 10.1371/journal.pgen.1002548
Liu, K., Shi, N., Sun, Y., Zhang, T., and Sun, X. (2013). Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem. Res. 38, 201–207. doi: 10.1007/s11064-012-0909-8
Liu, P., Wang, X., Gao, N., Zhu, H., Dai, X., Xu, Y., et al. (2010). G protein-coupled receptor kinase 5, overexpressed in the alpha-synuclein up-regulation model of Parkinson’s disease, regulates bcl-2 expression. Brain Res. 1307, 134–141. doi: 10.1016/j.brainres.2009.10.036
Liu, X., Cheng, R., Verbitsky, M., Kisselev, S., Browne, A., Mejia-Sanatana, H., et al. (2011). Genome-wide association study identifies candidate genes for Parkinson’s disease in an Ashkenazi Jewish population. BMC Med. Genet. 12:104. doi: 10.1186/1471-2350-12–104
Lue, L. F., Walker, D. G., Adler, C. H., Shill, H., Tran, H., Akiyama, H., et al. (2012). Biochemical increase in phosphorylated alpha-synuclein precedes histopathology of Lewy-type synucleinopathies. Brain Pathol. 22, 745–756. doi: 10.1111/j.1750-3639.2012.00585.x
Lynch-Day, M. A., Mao, K., Wang, K., Zhao, M., and Klionsky, D. J. (2012). The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2, a009357. doi: 10.1101/cshperspect.a009357
Malagelada, C., Jin, Z. H., and Greene, L. A. (2008). RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J. Neurosci. 28, 14363–14371. doi: 10.1523/JNEUROSCI.3928-08.2008
Mandel, S. A., Sagi, Y., and Amit, T. (2007). Rasagiline promotes regeneration of substantia nigra dopaminergic neurons in post-MPTP-induced Parkinsonism via activation of tyrosine kinase receptor signaling pathway. Neurochem. Res. 32, 1694–1699. doi: 10.1007/s11064-007-9351-8
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221. doi: 10.1083/jcb.200910140
Mbefo, M. K., Paleologou, K. E., Boucharaba, A., Oueslati, A., Schell, H., Fournier, M., et al. (2010). Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 285, 2807–2822. doi: 10.1074/jbc.M109.081950
McFarland, N. R., Fan, Z., Xu, K., Schwarzschild, M. A., Feany, M. B., Hyman, B. T., et al. (2009). Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J. Neuropathol. Exp. Neurol. 68, 515–524. doi: 10.1097/NEN.0b013e3181a24b53
Meggio, F., and Pinna, L. A. (2003). One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17, 349–368. doi: 10.1096/fj.02-0473rev
Melrose, H. L., Kent, C. B., Taylor, J. P., Dachsel, J. C., Hinkle, K. M., Lincoln, S. J., et al. (2007). A comparative analysis of leucine-rich repeat kinase 2 (Lrrk2) expression in mouse brain and Lewy body disease. Neuroscience 147, 1047–1058. doi: 10.1016/j.neuroscience.2007.05.027
Miklossy, J., Arai, T., Guo, J. P., Klegeris, A., Yu, S., Mcgeer, E. G., et al. (2006). LRRK2 expression in normal and pathologic human brain and in human cell lines. J. Neuropathol. Exp. Neurol. 65, 953–963. doi: 10.1097/01.jnen.0000235121.98052.54
Moehle, M. S., Webber, P. J., Tse, T., Sukar, N., Standaert, D. G., Desilva, T. M., et al. (2012). LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 32, 1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012
Morales-Garcia, J. A., Susin, C., Alonso-Gil, S., Perez, D. I., Palomo, V., Perez, C., et al. (2013). Glycogen synthase kinase-3 inhibitors as potent therapeutic agents for the treatment of Parkinson disease. ACS Chem. Neurosci. 4, 350–360. doi: 10.1021/cn300182g
Moreno, J.A., Halliday, M., Molloy, C., Radford, H., Verity, N., Axten, J.M., et al. (2013). Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5, 206ra138. doi: 10.1126/scitranslmed.3006767
Nagao, M., and Hayashi, H. (2009). Glycogen synthase kinase-3beta is associated with Parkinson’s disease. Neurosci. Lett. 449, 103–107. doi: 10.1016/j.neulet.2008.10.104
Nakamura, S., Kawamoto, Y., Nakano, S., Akiguchi, I., and Kimura, J. (1997). p35nck5a and cyclin-dependent kinase 5 colocalize in Lewy bodies of brains with Parkinson’s disease. Acta Neuropathol. 94, 153–157. doi: 10.1007/s004010050687
Nalls, M. A., Plagnol, V., Hernandez, D. G., Sharma, M., Sheerin, U. M., Saad, M., et al. (2011). Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 377, 641–649. doi: 10.1016/S0140-6736(10)62345-8.
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Newhouse, K., Hsuan, S. L., Chang, S. H., Cai, B., Wang, Y., and Xia, Z. (2004). Rotenone-induced apoptosis is mediated by p38 and JNK MAP kinases in human dopaminergic SH-SY5Y cells. Toxicol. Sci. 79, 137–146. doi: 10.1093/toxsci/kfh089
Okochi, M., Walter, J., Koyama, A., Nakajo, S., Baba, M., Iwatsubo, T., et al. (2000). Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J. Biol. Chem. 275, 390–397. doi: 10.1074/jbc.275.1.390
Olanow, C. W., and Brundin, P. (2013). Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov. Disord. 28, 31–40. doi: 10.1002/mds.25373
Oueslati, A., Fournier, M., and Lashuel, H. A. (2010). Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 183, 115–145. doi: 10.1016/S0079-6123(10)83007-9
Oueslati, A., Schneider, B. L., Aebischer, P., and Lashuel, H. A. (2013). Polo-like kinase 2 regulates selective autophagic alpha-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, E3945–E3954. doi: 10.1073/pnas.1309991110
Paisan-Ruiz, C. (2009). LRRK2 gene variation and its contribution to Parkinson disease. Hum. Mutat. 30, 1153–1160. doi: 10.1002/humu.21038
Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., Van Der Brug, M., et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. doi: 10.1016/j.neuron.2004.10.023
Pan, T., Rawal, P., Wu, Y., Xie, W., Jankovic, J., and Le, W. (2009). Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 164, 541–551. doi: 10.1016/j.neuroscience.2009.08.014
Pankratz, N., Wilk, J. B., Latourelle, J. C., Destefano, A. L., Halter, C., Pugh, E. W., et al. (2009). Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 124, 593–605. doi: 10.1007/s00439-008-0582-9
Perez, D. I., Gil, C., and Martinez, A. (2011). Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med. Res. Rev. 31, 924–954. doi: 10.1002/med.20207
Persu, A., and Vikkula, M. (2011). A genome-wide association study-derived candidate gene seeks replication: STK39. J. Hypertens. 29, 434–436. doi: 10.1097/HJH.0b013e328344b6b3
Peters, J. M., Mckay, R. M., Mckay, J. P., and Graff, J. M. (1999). Casein kinase I transduces Wnt signals. Nature 401, 345–350. doi: 10.1038/43830
Pihlstrom, L., and Toft, M. (2011). Genetic variability in SNCA and Parkinson’s disease. Neurogenetics 12, 283–293. doi: 10.1007/s10048-011-0292-7
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Pronin, A. N., Morris, A. J., Surguchov, A., and Benovic, J. L. (2000). Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 275, 26515–26522. doi: 10.1074/jbc.M003542200
Qing, H., Zhang, Y., Deng, Y., Mcgeer, E. G., and Mcgeer, P. L. (2009). Lrrk2 interaction with alpha-synuclein in diffuse Lewy body disease. Biochem. Biophys. Res. Commun. 390, 1229–1234. doi: 10.1016/j.bbrc.2009.10.126
Qu, D., Rashidian, J., Mount, M. P., Aleyasin, H., Parsanejad, M., Lira, A., et al. (2007). Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron 55, 37–52. doi: 10.1016/j.neuron.2007.05.033
Recasens, A., Dehay, B., Bove, J., Carballo-Carbajal, I., Dovero, S., Perez-Villalba, A., et al. (2014). Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 75, 351–362. doi: 10.1002/ana.24066
Rhodes, S. L., Sinsheimer, J. S., Bordelon, Y., Bronstein, J. M., and Ritz, B. (2011). Replication of GWAS associations for GAK and MAPT in Parkinson’s disease. Ann. Hum. Genet. 75, 195–200. doi: 10.1111/j.1469-1809.2010.00616.x
Richardson, C., and Alessi, D. R. (2008). The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J. Cell Sci. 121, 3293–3304. doi: 10.1242/jcs.029223
Ries, V., Henchcliffe, C., Kareva, T., Rzhetskaya, M., Bland, R., During, M. J., et al. (2006). Oncoprotein Akt/PKB induces trophic effects in murine models of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 103, 18757–18762. doi: 10.1073/pnas.0606401103
Rodrigues E Silva, A. M., Geldsetzer, F., Holdorff, B., Kielhorn, F. W., Balzer-Geldsetzer, M., Oertel, W. H., et al. (2010). Who was the man who discovered the “Lewy bodies”? Mov. Disord. 25, 1765–1773. doi: 10.1002/mds.22956
Rozeboom, A. M., and Pak, D. T. (2012). Identification and functional characterization of polo-like kinase 2 autoregulatory sites. Neuroscience 202, 147–157. doi: 10.1016/j.neuroscience.2011.11.003
Rudenko, I. N., Chia, R., and Cookson, M. R. (2012a). Is inhibition of kinase activity the only therapeutic strategy for LRRK2-associated Parkinson’s disease? BMC Med. 10:20. doi: 10.1186/1741-7015-10–20
Rudenko, I. N., Kaganovich, A., Hauser, D. N., Beylina, A., Chia, R., Ding, J., et al. (2012b). The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson’s disease is a partial loss-of-function mutation. Biochem. J. 446, 99–111. doi: 10.1042/BJ20120637
Ryu, M. Y., Kim, D. W., Arima, K., Mouradian, M. M., Kim, S. U., and Lee, G. (2008). Localization of CKII beta subunits in Lewy bodies of Parkinson’s disease. J. Neurol. Sci. 266, 9–12. doi: 10.1016/j.jns.2007.08.027
Sagi, Y., Mandel, S., Amit, T., and Youdim, M. B. (2007). Activation of tyrosine kinase receptor signaling pathway by rasagiline facilitates neurorescue and restoration of nigrostriatal dopamine neurons in post-MPTP-induced parkinsonism. Neurobiol. Dis. 25, 35–44. doi: 10.1016/j.nbd.2006.07.020
Sakamoto, M., Arawaka, S., Hara, S., Sato, H., Cui, C., Machiya, Y., et al. (2009). Contribution of endogenous G-protein-coupled receptor kinases to Ser129 phosphorylation of alpha-synuclein in HEK293 cells. Biochem. Biophys. Res. Commun. 384, 378–382. doi: 10.1016/j.bbrc.2009.04.130
Salvi, M., Trashi, E., Cozza, G., Franchin, C., Arrigoni, G., and Pinna, L. A. (2012). Investigation on PLK2 and PLK3 substrate recognition. Biochim. Biophys. Acta 1824, 1366–1373. doi: 10.1016/j.bbapap.2012.07.003
Samaranch, L., Lorenzo-Betancor, O., Arbelo, J. M., Ferrer, I., Lorenzo, E., Irigoyen, J., et al. (2010). PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133, 1128–1142. doi: 10.1093/brain/awq051
Saporito, M. S., Brown, E. M., Miller, M. S., and Carswell, S. (1999). CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons in vivo. J. Pharmacol. Exp. Ther. 288, 421–427.
Saporito, M. S., Thomas, B. A., and Scott, R. W. (2000). MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J. Neurochem. 75, 1200–1208. doi: 10.1046/j.1471-4159.2000.0751200.x
Satake, W., Nakabayashi, Y., Mizuta, I., Hirota, Y., Ito, C., Kubo, M., et al. (2009). Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 41, 1303–1307. doi: 10.1038/ng.485
Sato, H., Kato, T., and Arawaka, S. (2013). The role of Ser129 phosphorylation of alpha-synuclein in neurodegeneration of Parkinson’s disease: a review of in vivo models. Rev. Neurosci. 24, 115–123. doi: 10.1515/revneuro-2012-0071
Schwab, C., Demaggio, A. J., Ghoshal, N., Binder, L. I., Kuret, J., and Mcgeer, P. L. (2000). Casein kinase 1 delta is associated with pathological accumulation of tau in several neurodegenerative diseases. Neurobiol. Aging 21, 503–510. doi: 10.1016/S0197-4580(00)00110-X
Seeburg, D. P., Pak, D., and Sheng, M. (2005). Polo-like kinases in the nervous system. Oncogene 24, 292–298. doi: 10.1038/sj.onc.1208277
Sharma, M., Ioannidis, J. P., Aasly, J. O., Annesi, G., Brice, A., Van Broeckhoven, C., et al. (2012). Large-scale replication and heterogeneity in Parkinson disease genetic loci. Neurology 79, 659–667. doi: 10.1212/WNL.0b013e318264e353
Sharma, S., Bandopadhyay, R., Lashley, T., Renton, A. E., Kingsbury, A. E., Kumaran, R., et al. (2011). LRRK2 expression in idiopathic and G2019S positive Parkinson’s disease subjects: a morphological and quantitative study. Neuropathol. Appl. Neurobiol. 37, 777–790. doi: 10.1111/j.1365-2990.2011.01187.x
Sheng, Z., Zhang, S., Bustos, D., Kleinheinz, T., Le Pichon, C. E., Dominguez, S. L., et al. (2012). Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci. Transl. Med. 4, 164ra161. doi: 10.1126/scitranslmed.3004485
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312. doi: 10.1038/ng.487
Smith, P. D., Crocker, S. J., Jackson-Lewis, V., Jordan-Sciutto, K. L., Hayley, S., Mount, M. P., et al. (2003). Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 100, 13650–13655. doi: 10.1073/pnas.2232515100
Spencer, B., Potkar, R., Trejo, M., Rockenstein, E., Patrick, C., Gindi, R., et al. (2009). Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 29, 13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Tabara, H., Naito, Y., Ito, A., Katsuma, A., Sakurai, M. A., Ohno, S., et al. (2011). Neonatal lethality in knockout mice expressing the kinase-dead form of the gefitinib target GAK is caused by pulmonary dysfunction. PLoS ONE 6:e26034. doi: 10.1371/journal.pone.0026034
Takahashi, M., Uchikado, H., Caprotti, D., Weidenheim, K. M., Dickson, D. W., Ksiezak-Reding, H., et al. (2006). Identification of G-protein coupled receptor kinase 2 in paired helical filaments and neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 65, 1157–1169. doi: 10.1097/01.jnen.0000248542.82681.12
Taymans, J. M. (2012). The GTPase function of LRRK2. Biochem. Soc. Trans. 40, 1063–1069. doi: 10.1042/BST20120133
Taymans, J. M., Van Den Haute, C., and Baekelandt, V. (2006). Distribution of PINK1 and LRRK2 in rat and mouse brain. J. Neurochem. 98, 951–961. doi: 10.1111/j.1471-4159.2006.03919.x
Taymans, J. M., Vancraenenbroeck, R., Ollikainen, P., Beilina, A., Lobbestael, E., De Maeyer, M., et al. (2011). LRRK2 kinase activity is dependent on LRRK2 GTP binding capacity but independent of LRRK2 GTP binding. PLoS ONE 6:e23207. doi: 10.1371/journal.pone.0023207
Thomas, T., Timmer, M., Cesnulevicius, K., Hitti, E., Kotlyarov, A., and Gaestel, M. (2008). MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing neuroinflammation–relevance in a mouse model of Parkinson’s disease. J. Neurochem. 105, 2039–2052. doi: 10.1111/j.1471-4159.2008.05310.x
Timmons, S., Coakley, M. F., Moloney, A. M., and O’ Neill, C. (2009). Akt signal transduction dysfunction in Parkinson’s disease. Neurosci. Lett. 467, 30–35. doi: 10.1016/j.neulet.2009.09.055
Ugarte, S. D., Lin, E., Klann, E., Zigmond, M. J., and Perez, R. G. (2003). Effects of GDNF on 6-OHDA-induced death in a dopaminergic cell line: modulation by inhibitors of PI3 kinase and MEK. J. Neurosci. Res. 73, 105–112. doi: 10.1002/jnr.10632
Ushiro, H., Tsutsumi, T., Suzuki, K., Kayahara, T., and Nakano, K. (1998). Molecular cloning and characterization of a novel Ste20-related protein kinase enriched in neurons and transporting epithelia. Arch. Biochem. Biophys. 355, 233–240. doi: 10.1006/abbi.1998.0736
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Walker, D. G., Lue, L. F., Adler, C. H., Shill, H. A., Caviness, J. N., Sabbagh, M. N., et al. (2013). Changes in properties of serine 129 phosphorylated alpha-synuclein with progression of Lewy-type histopathology in human brains. Exp. Neurol. 240, 190–204. doi: 10.1016/j.expneurol.2012.11.020
Wang, W., Shi, L., Xie, Y., Ma, C., Li, W., Su, X., et al. (2004). SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci. Res. 48, 195–202. doi: 10.1016/j.neures.2003.10.012
Wang, W., Yang, Y., Ying, C., Li, W., Ruan, H., Zhu, X., et al. (2007). Inhibition of glycogen synthase kinase-3beta protects dopaminergic neurons from MPTP toxicity. Neuropharmacology 52, 1678–1684. doi: 10.1016/j.neuropharm.2007.03.017
Wang, Y., O’connell, J. R., Mcardle, P. F., Wade, J. B., Dorff, S. E., Shah, S. J., et al. (2009a). From the Cover: Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc. Natl. Acad. Sci. U.S.A. 106, 226–231. doi: 10.1073/pnas.0808358106
Wang, Y., Zhang, Y., Wei, Z., Li, H., Zhou, H., Zhang, Z., et al. (2009b). JNK inhibitor protects dopaminergic neurons by reducing COX-2 expression in the MPTP mouse model of subacute Parkinson’s disease. J. Neurol. Sci. 285, 172–177. doi: 10.1016/j.jns.2009.06.034
Waxman, E. A., Covy, J. P., Bukh, I., Li, X., Dawson, T. M., and Giasson, B. I. (2009). Leucine-rich repeat kinase 2 expression leads to aggresome formation that is not associated with alpha-synuclein inclusions. J. Neuropathol. Exp. Neurol. 68, 785–796. doi: 10.1097/NEN.0b013e3181aaf4fd
Waxman, E. A., and Giasson, B. I. (2008). Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J. Neuropathol. Exp. Neurol. 67, 402–416. doi: 10.1097/NEN.0b013e31816fc995
Waxman, E. A., and Giasson, B. I. (2011). Characterization of kinases involved in the phosphorylation of aggregated alpha-synuclein. J. Neurosci. Res. 89, 231–247. doi: 10.1002/jnr.22537
West, A. B., Cowell, R. M., Daher, J. P., Moehle, M. S., Hinkle, K. M., Melrose, H. L., et al. (2014). Differential LRRK2 expression in the cortex, striatum, and substantia nigra in transgenic and nontransgenic rodents. J. Comp. Neurol. 522, Spc1. doi: 10.1002/cne.23583
West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., et al. (2005). Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 16842–16847. doi: 10.1073/pnas.0507360102
Wills, J., Jones, J., Haggerty, T., Duka, V., Joyce, J. N., and Sidhu, A. (2010). Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp. Neurol. 225, 210–218. doi: 10.1016/j.expneurol.2010.06.017
Winkles, J. A., and Alberts, G. F. (2005). Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues. Oncogene 24, 260–266. doi: 10.1038/sj.onc.1208219
Woodroof, H. I., Pogson, J. H., Begley, M., Cantley, L. C., Deak, M., Campbell, D. G., et al. (2011). Discovery of catalytically active orthologues of the Parkinson’s disease kinase PINK1: analysis of substrate specificity and impact of mutations. Open Biol. 1, 110012. doi: 10.1098/rsob.110012
Wu, Y., Li, X., Zhu, J. X., Xie, W., Le, W., Fan, Z., et al. (2011). Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 19, 163–174. doi: 10.1159/000328516
Xing, B., Liu, M., and Bing, G. (2007). Neuroprotection with pioglitazone against LPS insult on dopaminergic neurons may be associated with its inhibition of NF-kappaB and JNK activation and suppression of COX-2 activity. J. Neuroimmunol. 192, 89–98. doi: 10.1016/j.jneuroim.2007.09.029
Xiong, N., Jia, M., Chen, C., Xiong, J., Zhang, Z., Huang, J., et al. (2011). Potential autophagy enhancers attenuate rotenone-induced toxicity in SH-SY5Y. Neuroscience 199, 292–302. doi: 10.1016/j.neuroscience.2011.10.031
Yan, Y., Nguyen, H., Dalmasso, G., Sitaraman, S. V., and Merlin, D. (2007). Cloning and characterization of a new intestinal inflammation-associated colonic epithelial Ste20-related protein kinase isoform. Biochim. Biophys. Acta 1769, 106–116. doi: 10.1016/j.bbaexp.2007.01.003
Yuskaitis, C. J., and Jope, R. S. (2009). Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 21, 264–273. doi: 10.1016/j.cellsig.2008.10.014
Zhai, A., Zhu, X., Wang, X., Chen, R., and Wang, H. (2013). Secalonic acid A protects dopaminergic neurons from 1-methyl-4-phenylpyridinium (MPP(+))-induced cell death via the mitochondrial apoptotic pathway. Eur. J. Pharmacol. 713, 58–67. doi: 10.1016/j.ejphar.2013.04.029
Zhou, J., Broe, M., Huang, Y., Anderson, J. P., Gai, W. P., Milward, E. A., et al. (2011). Changes in the solubility and phosphorylation of alpha-synuclein over the course of Parkinson’s disease. Acta Neuropathol. 121, 695–704. doi: 10.1007/s00401-011-0815-1
Zhu, J. H., Guo, F., Shelburne, J., Watkins, S., and Chu, C. T. (2003). Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 13, 473–481 doi: 10.1111/j.1750-3639.2003.tb00478.x
Zhu, J. H., Kulich, S. M., Oury, T. D., and Chu, C. T. (2002). Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am. J. Pathol. 161, 2087–2098. doi: 10.1016/S0002-9440(10)64487-2
Keywords: kinase, LRRK2, JNK, PLK, PINK1, GAK, MAPK, brain
Citation: Dzamko N, Zhou J, Huang Y and Halliday GM (2014) Parkinson’s disease-implicated kinases in the brain; insights into disease pathogenesis. Front. Mol. Neurosci. 7:57. doi: 10.3389/fnmol.2014.00057
Received: 31 March 2014; Accepted: 05 June 2014;
Published online: 24 June 2014.
Edited by:
Jean-Marc Taymans, KU Leuven, BelgiumReviewed by:
Mark R. Cookson, National Institutes of Health, USAWilma Dorethea Van De Berg, VU University Medical Center, Netherlands
Copyright © 2014 Dzamko, Zhou, Huang and Halliday. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolas Dzamko and Glenda M. Halliday, School of Medical Sciences, University of New South Wales, Barker Street, Randwick, NSW, Australia e-mail: n.dzamko@neura.edu.au; g.halliday@neura.edu.au