Human DNA Helicase B as a Candidate for Unwinding Secondary CGG Repeat Structures at the Fragile X Mental Retardation Gene
 Gulfem D. Guler
Gulfem D. Guler Zev Rosenwaks2
Zev Rosenwaks2  Jeannine Gerhardt
Jeannine Gerhardt- 1Celgene Quanticel Research, San Francisco, CA, United States
- 2The Ronald O. Perelman and Claudia Cohen Center for Reproductive Medicine, Weill Cornell Medicine, Cornell University, New York, NY, United States
- 3Department of Obstetrics and Gynecology, Weill Cornell Medicine, Cornell University, New York, NY, United States
The fragile X syndrome (FXS) is caused by a CGG repeat expansion at the fragile X mental retardation (FMR1) gene. FMR1 alleles with more than 200 CGG repeats bear chromosomal fragility when cells experience folate deficiency. CGG repeats were reported to be able to form secondary structures, such as hairpins, in vitro. When such secondary structures are formed, repeats can lead to replication fork stalling even in the absence of any additional perturbation. Indeed, it was recently shown that the replication forks stall at the endogenous FMR1 locus in unaffected and FXS cells, suggesting the formation of secondary repeat structures at the FMR1 gene in vivo. If not dealt with properly replication fork stalling can lead to polymerase slippage and repeat expansion as well as fragile site expression. Despite the presence of repeat structures at the FMR1 locus, chromosomal fragility is only expressed under replicative stress suggesting the existence of potential molecular mechanisms that help the replication fork progress through these repeat regions. DNA helicases are known to aid replication forks progress through repetitive DNA sequences. Yet, the identity of the DNA helicase(s) responsible for unwinding the CGG repeats at FMR1 locus is not known. We found that the human DNA helicase B (HDHB) may provide an answer for this question. We used chromatin-immunoprecipitation assay to study the FMR1 region and common fragile sites (CFS), and asked whether HDHB localizes at replication forks stalled at repetitive regions even in unperturbed cells. HDHB was strongly enriched in S-phase at the repetitive DNA at CFS and FMR1 gene but not in the flanking regions. Taken together, these results suggest that HDHB functions in preventing or repairing stalled replication forks that arise in repeat-rich regions even in unperturbed cells. Furthermore, we discuss the importance and potential role of HDHB and other helicases in the resolution of secondary CGG repeat structures.
Introduction
The fragile X syndrome (FXS) is the most common inherited form of intellectual disability. FXS is caused by a CGG repeat expansion on the X chromosome in the 5’UTR of the fragile X mental retardation (FMR1) gene (Nelson et al., 2013). CGG repeat expansion leads to FMR1 gene silencing. FXS is inherited from women carrying a premutation, 55–200 CGG repeats. More than 200 CGG repeats are categorized as a full mutation (FM), resulting in FXS (Kronquist et al., 2008). Furthermore, expansion of CGG repeats in premutation range within FMR1 gene is also associated with other disorders; fragile X-associated primary ovarian insufficiency (FXPOI; Sherman, 2000), fragile X-associated diminished ovarian reserve (DOR; Man et al., 2017) and fragile X-associated tremor ataxia syndrome (FXTAS; Hagerman et al., 2001). In contrast to FXS patients, the FMR1 gene is transcribed in premutation carriers. A toxic RNA gain-of-function and the expression of an abnormal FMRpolyglycin protein is suggested to cause the symptoms in premutation carriers (Hagerman and Hagerman, 2013; Todd et al., 2013).
The expanded repeats at the FMR1 gene locus in FXS cells are characterized as rare fragile sites (RFS). While RFS are only found in some individuals, common fragile sites (CFS; Durkin and Glover, 2007) and early replicating fragile sites (ERFS; Barlow et al., 2013) are found in every individual. In contrast to RFS, CFS and ERFS contain non-expanding repetitive DNA sequences, and are therefore stable under normal conditions. However, RFS as well as CFS and ERFS tend to break upon replicative stress, which could result in chromosomal deletions, translocations and sister chromatid exchanges (Glover and Stein, 1987, 1988; Wang et al., 1997). It was suggested that these chromosomal alterations are consequences of prolonged replication fork stalling at repetitive DNA sequences located at these fragile sites.
It was reported that DNA and RNA containing CGG repeats are able to form secondary structures, such as hairpins (Gacy et al., 1995; Gacy and McMurray, 1998), G-quadruplexes (Fry and Loeb, 1994; Khateb et al., 2004) and R-loops (Groh et al., 2014; Loomis et al., 2014). DNA templates with such secondary structures stall replication forks in vitro and in vivo (Samadashwily et al., 1997; Pelletier et al., 2003; Voineagu et al., 2009; Gerhardt et al., 2014). Consistent with prolonged fork stalling at CGG repeats, replication of the FMR1 locus is delayed in FXS cells compared to unaffected cells (Hansen et al., 1993; Subramanian et al., 1996). Prolonged replication fork stalling and uncompleted DNA replication at this fragile site can lead to genomic instability such as DNA break-induced chromosomal alterations or repeat expansions due to DNA polymerase slippage (Madireddy and Gerhardt, 2017). On the other hand, non-canonical secondary structures may turn CGG repeat containing FMR1 mRNA into toxic RNA, which may be pathogenic through sequestering RNA-binding proteins. Thus, molecular mechanisms that disrupt these secondary structures are crucial for genome stability and cellular function.
Here we discuss mechanisms that may prevent chromosomal fragility and repeat expansion at the FMR1 locus upon replication fork stalling at CGG repeats, and the possible involvement of Human DNA helicase B (HDHB) at unwinding CGG repeat structures to aid replication fork progression. We present data that show HDHB localization to fragile sites specifically during S-phase even in unperturbed cells, suggesting that HDHB may help prevent or repair replication fork stalling. Furthermore, we discuss other possible DNA and RNA helicases that are capable of unwinding secondary CGG repeat structures.
DNA Helicases Involved in Resolution of Secondary Repeat Structures During Replication Fork Progression
Despite the threats to genome stability posed by the secondary structures adopted by repeats, replication most often proceeds through repeat regions without the expression of chromosomal fragility. This suggests the presence of molecular mechanisms that help replication fork progression through repeat regions. At the heart of these mechanisms are DNA helicase(s) responsible for unwinding the secondary structures adopted by repeats. Helicases separate strands of a DNA double helix or a self-annealed RNA molecule using the energy from ATP hydrolysis, a process characterized by the breaking of hydrogen bonds between annealed nucleotide bases. DNA helicases translocate on double-stranded DNA (dsDNA) or single-stranded DNA (ssDNA) to unwind dsDNA into ssDNA or remove proteins bound to DNA. There is a plethora of helicases encoded by the genome, each performing a specialized function dictated by their enzymatic properties as well as their interaction partners.
Conserved among vertebrates, DNA helicase B (DHB) contains seven helicase motifs of superfamily 1 with sequence similarity to homologous recombination proteins prokaryotic RecD and bacteriophage T4 dda. It works with 5’ to 3’ polarity. The C-terminus of HDHB contains a phosphorylation-dependent subcellular localization domain (PSLD; Figure 1A). PSLD is responsible for nuclear localization in G1-phase and phosphorylation-dependent nuclear export of HDHB at the G1/S transition (Gu et al., 2004). HDHB was shown to associate with pre-replication complex components Cdc45 and TopBP1 (Taneja et al., 2002; Gerhardt et al., 2015). HDHB also interacts with Cyclin E and A (Gu et al., 2004) and ssDNA-binding protein RPA (Guler et al., 2012). It was reported that its helicase activity is necessary for replication initiation (Taneja et al., 2002). At G1/S, majority of HDHB is exported from the nucleus. However, a low level of HDHB is retained on bulk chromatin during S-phase and this fraction is increased in cells exposed to agents that stall fork progression. Furthermore, DNA damage leads to HDHB accumulation on chromatin particularly during S-phase (Guler et al., 2012). Single stranded DNA-bound RPA at stalled replication forks recruit HDHB (Figure 1A) by a direct protein interaction between HDHB and RPA.
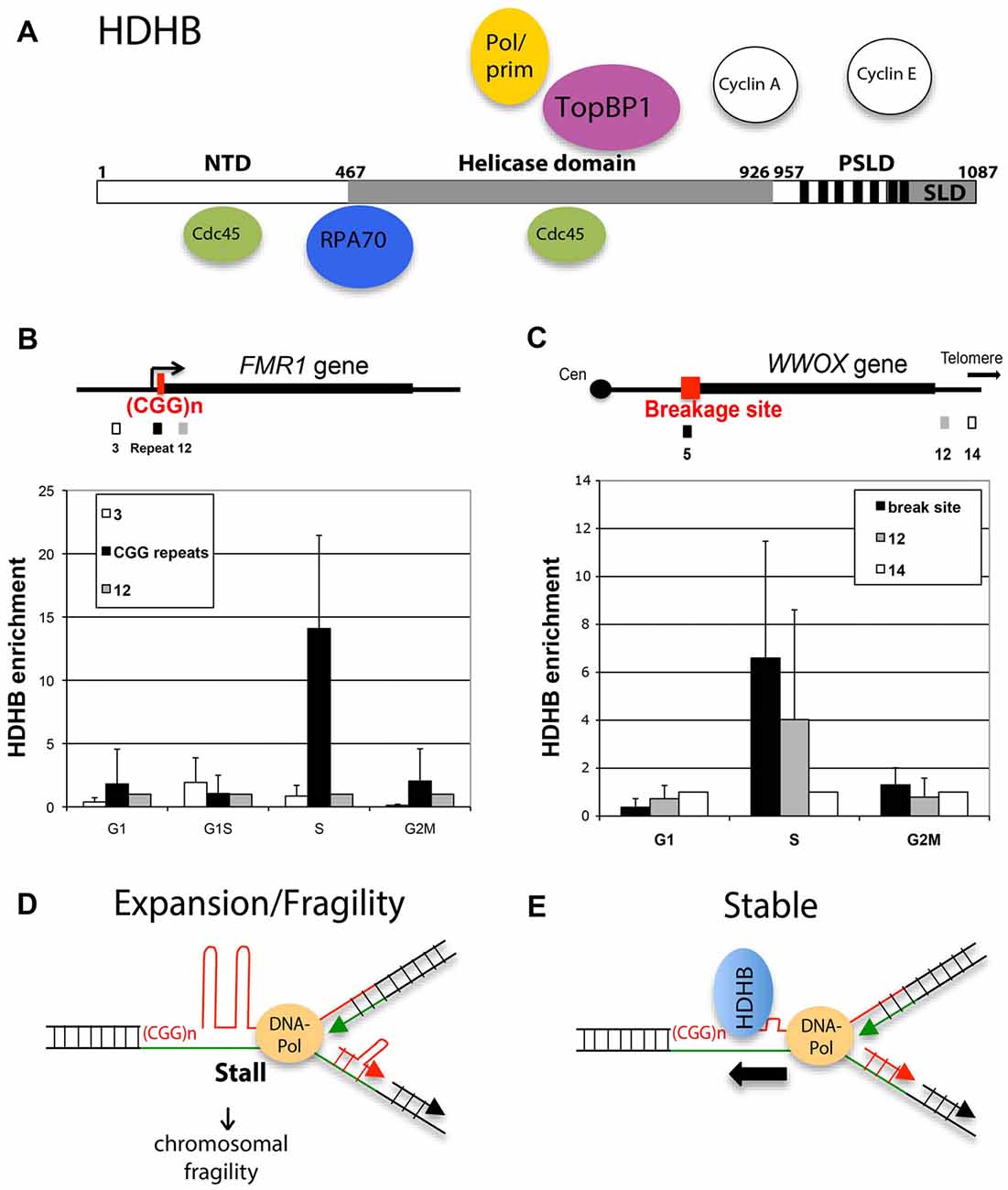
Figure 1. Human DNA helicase B (HDHB) resides at fragile sites in particular at the CGG repeats at human fragile X mental retardation (FMR1) gene locus. (A) Functional regions of HDHB. An N-terminal domain (NTD) is uncharacterized. The central domain (gray; residues 467–926) contains the seven superfamily I helicase motifs. The C-terminal domain contains consensus CDK phosphorylation sites (vertical black bars) and a subcellular localization domain (SLD), which together constitute a phosphorylation-dependent SLD (PSLD). Replication proteins, which are binding to HDHB are indicated. (B) Maps showing FMR1 gene locus and three primer sets and (C) the common fragile sites (CFS) FRA16D and three primer sets. ChIP experiments were performed using U2OS cells synchronized in G1, S and G2/M cells and affinity-purified polyclonal HDHB antibody or rabbit IgG as control as described before (Gerhardt et al., 2006, 2015). HDHB enrichment at each site in each of three independent experiments is plotted on the Y-axis. Horizontal bars show the average HDHB enrichment at each time point and error bars are indicated. (D,E) Model for repeat expansion and fragility after fork stalling at secondary CGG repeat structures. HDHB could prevent genome instability by resolving the non-canonical repeat structures.
In addition to HDHB, there may be other helicases to help manage replication stress induced by repeat structures. Particularly, helicases capable of resolving G-quadruplexes adopted by guanine-rich stretches of DNA such as CGG repeats are of interest. G-quadruplexes may require specialized machinery to unwind them so that replication fork can progress through. One of the helicases capable of resolving G-quadruplexes is another superfamily 1 member with sequence similarity to RecD, called Pif1 helicase. Closely related to S. cerevisiae Rrm3 helicase, Pif1 was shown to promote replication fork progression through genomic regions that contain G-quadruplex sites (Paeschke et al., 2011). Furthermore, Pif1 functions to prevent G-quadruplex-associated DNA damage (Paeschke et al., 2013). Another helicase implicated at resolving G-quadruplexes is Dna2 nuclease-helicase, which also has roles in telomere maintenance in addition to Okazaki fragments-processing while traveling with the fork (Lin et al., 2013). Moreover, RecQ helicase members Bloom’s syndrome helicase (BLM), Werner’s syndrome helicase (WRN), and Fanconi anemia group J (FANCJ) helicase are all capable of resolving G-quadruplexes and accumulate at stalled replication forks (London et al., 2008). Of particular note, WRN was previously reported to unwind CGG repeats in vitro (Fry and Loeb, 1999). Even though none of these helicases were yet shown to associate with the FMR1 gene, it remains to be investigated whether these helicases can help replication forks progress through the CGG repeats at the FMR1 locus.
HDHB Is Recruited to the Repeats at the FMR1 Gene and at Common Fragile Sites
Replication forks may stall naturally without any additional perturbation at hard to replicate regions such as repetitive regions. Replication fork stalling initially results in long stretches of ssDNA that get coated with RPA. To facilitate replication fork recovery, ssDNA-bound RPA recruits DNA damage response proteins, including helicases like HDHB (Guler et al., 2012).
To investigate whether HDHB associates with replication forks stalled at repetitive DNA sequences, in particular to the CGG repeats at FMR1 locus and AT-rich repeats at FRA16D, we performed chromatin immunoprecipitation (ChIP) experiments. U2OS cells were blocked in G2/M-phase and released for 5 h (G1), or synchronized in G1/S-phase and released for 6 (S) or 9 h (G2/M), followed by formaldehyde treatment to crosslink chromatin. Chromatin was isolated, sheared and immunoprecipitated using purified polyclonal HDHB antibody or non-immune control antibody (Gerhardt et al., 2015). HDHB enrichment in immunoprecipitated chromatin was measured by quantitative real-time PCR using one primer pair amplifying DNA segment containing the repeat region/break site (Figures 1B,C) and two primer pairs in a distal region (FMR1 gene: primer pair 3 and 12 (Gray et al., 2007); FRA16D/WWOX gene primer pair 12 and 14). In S-phase cells, HDHB was significantly enriched on chromatin in the repeat region of fragile sites relative to the distal regions. HDHB enrichment in both repeat regions was reduced in G1- and G2/M-phase. We found similar results at a second CFS, FRA3B (data not shown). These results show that HDHB is enriched in S-phase at the repetitive DNA sequences and suggest that HDHB is recruited to stalled replication forks at repetitive regions such as CGG repeats at the FMR1 gene.
Upon recruitment to the fork, HDHB can unwind the secondary repeat structures formed by CGG repeats ahead of DNA-polymerase (Figures 1D,E). Repeat expansion following DNA polymerase slippage at stalled replication forks can be so prevented (Figure 1E) as well as chromosomal fragility, which could result from DNA breaks induced by replication fork stalling. It would be interesting to determine whether FXS and premutation patients have a decreased HDHB protein level or HDHB helicase activity, and whether such differences can affect replication fork stalling, chromosomal fragility and CGG repeat expansion.
RNA Helicases Involved in Prevention of Secondary CGG Repeat RNA Structures
The presence of FMR1 mRNA in intranuclear inclusions (Tassone et al., 2004) in premutation patients, as observed in brain tissues from FXTAS patients (Galloway and Nelson, 2009), and increased FMR1 mRNA level in PM carriers (Tassone et al., 2000) led to the suggestion that a toxic RNA gain-of-function mechanism might be responsible for FXTAS development. An increased FMR1 mRNA level was also noticed in ovarian granulosa cells of female carriers with a PM (Elizur et al., 2014). Since RNAs containing CGG repeats can adopt secondary structures in vitro (Napierala et al., 2005), it is likely that FMR1 mRNA assumes non-canonical RNA structures in vivo as well.
It was previously shown that the expansion to FM is reduced by AGG interruptions within the premutated allele (Yrigollen et al., 2012; Nolin et al., 2014). AGG interruptions are located at the 5’ end of the CGG repeat sequence in FMR1 gene (Kunst and Warren, 1994; Kunst et al., 1996, 1997). These interruptions were proposed to stabilize the repeats (Nelson et al., 2013), potentially by preventing the formation of secondary repeat DNA structures within the cell. AGG interruptions could also prevent formation of secondary structures in FMR1 mRNA. Indeed, we recently found that an increase in number of AGG interruptions, from none or one to two, is associated with lower risk of FXDOR in patients carrying a PM (Lekovich et al., 2017). We propose a model where AGG interruptions lowers the probability of secondary repeat structure formation in FMR1 mRNA, and hence pathogenesis, in the ovaries of women carrying a PM.
Other than cis-acting elements like AGG interruptions described above, RNA helicases could also prevent repeat-containing RNA to form non-canonical secondary structures. One candidate for unwinding rCGG repeat structures is Rm62, Drosophila ortholog of p68/DDX5 RNA helicase. p68 RNA helicase is a prototypical DEAD-box RNA helicase that has been implicated in transcriptional regulation, pre-mRNA splicing, and nucleocytoplasmic shuttling. Rm62 overexpression rescued neurodegeneration in flies expressing 90 CGG repeats (Qurashi et al., 2011). Additionally, RNA helicases p68/DDX5 and DDX6 were reported to unwind expanded CUG repeats in myotonic dystrophy (Laurent et al., 2012; Pettersson et al., 2014), making these helicases of therapeutic interest. Another helicase able to unwind G-quadruplexes and R-loop structures is RNA helicase A (RHA, also called DHX9, and nuclear DNA helicase II). RHA unwinds DNA–DNA, RNA–RNA and DNA–RNA duplexes with 3’ to 5’ direction. It acts preferentially on RNA substrates.
Additional proteins, involved in preventing the formation of secondary RNA structures, are heterogeneous nuclear ribonucleoproteins A2/B1 (hnRNP A2/B1). HnRNP A2/B1 have a role in packaging nascent mRNA, alternative splicing and cytoplasmic RNA trafficking, translation and stabilization. Found in intranuclear inclusions of FXTAS patients (Iwahashi et al., 2006), hnRNP A2/B1 were described to act as an RNA chaperone destabilizing RNA structures formed by CGG repeats (Khateb et al., 2004; Ofer et al., 2009). HnRNP A2/B1 overexpression rescues neurodegeneration in drosophila expressing 90 CGG repeats (Jin et al., 2007; Sofola et al., 2007). It remains to be determined if these helicases or proteins unwinding secondary structures are deregulated in patients and whether helicase deficiency promotes the pathogenicity observed in patients with a premutation.
Conclusion
Repeats present a challenge for the replication machinery and other cellular processes involving repetitive DNA and RNA. Therefore, mechanisms to resolve secondary structures that repeats might adopt are needed. Specialized helicases are able to unwind secondary DNA and RNA structures, which can otherwise lead to replication fork stalling or toxic RNA, respectively. Replication fork stalling at endogenous repeats is reported in cells derived from FXS and FRDA patients (Gerhardt et al., 2014, 2016). This can lead to genomic instability by DNA polymerase slippage-induced repeat expansion or chromosomal fragility when occurred at RFS and CFS (Madireddy et al., 2016). Toxic RNAs, on the other hand, may disrupt cellular processes particularly by sequestering proteins important for RNA function. Therefore, resolution of secondary structures, which DNA or RNA repeats adopt, by DNA/RNA helicases is a crucial mechanism that could help prevent repeat-induced diseases. Consequently, helicases that resolve such secondary structures adopted by DNA or RNA repeats may constitute a crucial toolbox cells employ to help prevent repeat-induced diseases such as fragile X syndrome.
Author Contributions
GG and JG performed the experiments. GG, JG and ZR wrote and edited the manuscript.
Funding
This work was supported by the Starr Foundation and Perelman research recruitment gift (Weill Cornell Medicine) and NIH/NINDS R03 NS106216 (JG).
Conflict of Interest Statement
GG is an employee of Celgene.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Barlow, J. H., Faryabi, R. B., Callén, E., Wong, N., Malhowski, A., Chen, H. T., et al. (2013). Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632. doi: 10.1016/j.cell.2013.01.006
Durkin, S. G., and Glover, T. W. (2007). Chromosome fragile sites. Annu. Rev. Genet. 41, 169–192. doi: 10.1146/annurev.genet.41.042007.165900
Elizur, S. E., Lebovitz, O., Derech-Haim, S., Dratviman-Storobinsky, O., Feldman, B., Dor, J., et al. (2014). Elevated levels of FMR1 mRNA in granulosa cells are associated with low ovarian reserve in FMR1 premutation carriers. PLoS One 9:e105121. doi: 10.1371/journal.pone.0105121
Fry, M., and Loeb, L. A. (1994). The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. U S A 91, 4950–4954. doi: 10.1073/pnas.91.11.4950
Fry, M., and Loeb, L. A. (1999). Human werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. J. Biol. Chem. 274, 12797–12802. doi: 10.1074/jbc.274.18.12797
Gacy, A. M., Goellner, G., Juranić, N., Macura, S., and McMurray, C. T. (1995). Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 81, 533–540. doi: 10.1016/0092-8674(95)90074-8
Gacy, A. M., and McMurray, C. T. (1998). Influence of hairpins on template reannealing at trinucleotide repeat duplexes: a model for slipped DNA. Biochemistry 37, 9426–9434. doi: 10.1021/bi980157s
Galloway, J. N., and Nelson, D. L. (2009). Evidence for RNA-mediated toxicity in the fragile X-associated tremor/ataxia syndrome. Future Neurol. 4:785. doi: 10.2217/fnl.09.44
Gerhardt, J., Bhalla, A. D., Butler, J. S., Puckett, J. W., Dervan, P. B., Rosenwaks, Z., et al. (2016). Stalled DNA replication forks at the endogenous GAA repeats drive repeat expansion in friedreich’s ataxia cells. Cell Rep. 16, 1218–1227. doi: 10.1016/j.celrep.2016.06.075
Gerhardt, J., Guler, G. D., and Fanning, E. (2015). Human DNA helicase B interacts with the replication initiation protein Cdc45 and facilitates Cdc45 binding onto chromatin. Exp. Cell Res. 334, 283–293. doi: 10.1016/j.yexcr.2015.04.014
Gerhardt, J., Jafar, S., Spindler, M. P., Ott, E., and Schepers, A. (2006). Identification of new human origins of DNA replication by an origin-trapping assay. Mol. Cell. Biol. 26, 7731–7746. doi: 10.1128/MCB.01392-06
Gerhardt, J., Tomishima, M. J., Zaninovic, N., Colak, D., Yan, Z., Zhan, Q., et al. (2014). The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol. Cell 53, 19–31. doi: 10.1016/j.molcel.2013.10.029
Glover, T. W., and Stein, C. K. (1987). Induction of sister chromatid exchanges at common fragile sites. Am. J. Hum. Genet. 41, 882–890.
Glover, T. W., and Stein, C. K. (1988). Chromosome breakage and recombination at fragile sites. Am. J. Hum. Genet. 43, 265–273.
Gray, S. J., Gerhardt, J., Doerfler, W., Small, L. E., and Fanning, E. (2007). An origin of DNA replication in the promoter region of the human fragile X mental retardation (FMR1) gene. Mol. Cell. Biol. 27, 426–437. doi: 10.1128/MCB.01382-06
Groh, M., Lufino, M. M., Wade-Martins, R., and Gromak, N. (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 10:e1004318. doi: 10.1371/journal.pgen.1004318
Gu, J., Xia, X., Yan, P., Liu, H., Podust, V. N., Reynolds, A. B., et al. (2004). Cell cycle-dependent regulation of a human DNA helicase that localizes in DNA damage foci. Mol. Biol. Cell 15, 3320–3332. doi: 10.1091/mbc.e04-03-0227
Guler, G. D., Liu, H., Vaithiyalingam, S., Arnett, D. R., Kremmer, E., Chazin, W. J., et al. (2012). Human DNA helicase B (HDHB) binds to replication protein A and facilitates cellular recovery from replication stress. J. Biol. Chem. 287, 6469–6481. doi: 10.1074/jbc.M111.324582
Hagerman, R., and Hagerman, P. (2013). Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 12, 786–798. doi: 10.1016/s1474-4422(13)70125-x
Hagerman, R. J., Leehey, M., Heinrichs, W., Tassone, F., Wilson, R., Hills, J., et al. (2001). Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57, 127–130. doi: 10.1212/WNL.57.1.127
Hansen, R. S., Canfield, T. K., Lamb, M. M., Gartler, S. M., and Laird, C. D. (1993). Association of fragile X syndrome with delayed replication of the FMR1 gene. Cell 73, 1403–1409. doi: 10.1016/0092-8674(93)90365-w
Iwahashi, C. K., Yasui, D. H., An, H. J., Greco, C. M., Tassone, F., Nannen, K., et al. (2006). Protein composition of the intranuclear inclusions of FXTAS. Brain 129, 256–271. doi: 10.1093/brain/awh650
Jin, P., Duan, R., Qurashi, A., Qin, Y., Tian, D., Rosser, T. C., et al. (2007). Pur α binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55, 556–564. doi: 10.1016/j.neuron.2007.07.020
Khateb, S., Weisman-Shomer, P., Hershco, I., Loeb, L. A., and Fry, M. (2004). Destabilization of tetraplex structures of the fragile X repeat sequence (CGG)n is mediated by homolog-conserved domains in three members of the hnRNP family. Nucleic Acids Res. 32, 4145–4154. doi: 10.1093/nar/gkh745
Kronquist, K. E., Sherman, S. L., and Spector, E. B. (2008). Clinical significance of tri-nucleotide repeats in Fragile X testing: a clarification of American College of Medical Genetics guidelines. Genet. Med. 10, 845–847. doi: 10.1097/GIM.0b013e31818b0c8a
Kunst, C. B., Leeflang, E. P., Iber, J. C., Arnheim, N., and Warren, S. T. (1997). The effect of FMR1 CGG repeat interruptions on mutation frequency as measured by sperm typing. J. Med. Genet. 34, 627–631. doi: 10.1136/jmg.34.8.627
Kunst, C. B., and Warren, S. T. (1994). Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell 77, 853–861. doi: 10.1016/0092-8674(94)90134-1
Kunst, C. B., Zerylnick, C., Karickhoff, L., Eichler, E., Bullard, J., Chalifoux, M., et al. (1996). FMR1 in global populations. Am. J. Hum. Genet. 58, 513–522.
Laurent, F. X., Sureau, A., Klein, A. F., Trouslard, F., Gasnier, E., Furling, D., et al. (2012). New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic Acids Res. 40, 3159–3171. doi: 10.1093/nar/gkr1228
Lekovich, J., Man, L., Xu, K., Canon, C., Lilienthal, D., Stewart, J. D., et al. (2017). CGG repeat length and AGG interruptions as indicators of fragile X-associated diminished ovarian reserve. Genet. Med. . doi: 10.1038/gim.2017.220 [Epub ahead of print]
Lin, W., Sampathi, S., Dai, H., Liu, C., Zhou, M., Hu, J., et al. (2013). Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 32, 1425–1439. doi: 10.1038/emboj.2013.88
London, T. B., Barber, L. J., Mosedale, G., Kelly, G. P., Balasubramanian, S., Hickson, I. D., et al. (2008). FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J. Biol. Chem. 283, 36132–36139. doi: 10.1074/jbc.M808152200
Loomis, E. W., Sanz, L. A., ChéDin, F., and Hagerman, P. J. (2014). Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 10:e1004294. doi: 10.1371/journal.pgen.1004294
Madireddy, A., and Gerhardt, J. (2017). Replication through repetitive DNA elements and their role in human diseases. Adv. Exp. Med. Biol. 1042, 549–581. doi: 10.1007/978-981-10-6955-0_23
Madireddy, A., Kosiyatrakul, S. T., Boisvert, R. A., Herrera-Moyano, E., García-Rubio, M. L., Gerhardt, J., et al. (2016). FANCD2 facilitates replication through common fragile sites. Mol. Cell 64, 388–404. doi: 10.1016/j.molcel.2016.09.017
Man, L., Lekovich, J., Rosenwaks, Z., and Gerhardt, J. (2017). Fragile X-associated diminished ovarian reserve and primary ovarian insufficiency from molecular mechanisms to clinical manifestations. Front. Mol. Neurosci. 10:290. doi: 10.3389/fnmol.2017.00290
Napierala, M., Michalowski, D., de Mezer, M., and Krzyzosiak, W. J. (2005). Facile FMR1 mRNA structure regulation by interruptions in CGG repeats. Nucleic Acids Res. 33, 451–463. doi: 10.1093/nar/gki186
Nelson, D. L., Orr, H. T., and Warren, S. T. (2013). The unstable repeats—three evolving faces of neurological disease. Neuron 77, 825–843. doi: 10.1016/j.neuron.2013.02.022
Nolin, S. L., Glicksman, A., Ersalesi, N., Dobkin, C., Brown, W. T., Cao, R., et al. (2014). Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 17, 358–364. doi: 10.1038/gim.2014.106
Ofer, N., Weisman-Shomer, P., Shklover, J., and Fry, M. (2009). The quadruplex r(CGG)n destabilizing cationic porphyrin TMPyP4 cooperates with hnRNPs to increase the translation efficiency of fragile X premutation mRNA. Nucleic Acids Res. 37, 2712–2722. doi: 10.1093/nar/gkp130
Paeschke, K., Bochman, M. L., Garcia, P. D., Cejka, P., Friedman, K. L., Kowalczykowski, S. C., et al. (2013). Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 497, 458–462. doi: 10.1038/nature12149
Paeschke, K., Capra, J. A., and Zakian, V. A. (2011). DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145, 678–691. doi: 10.1016/j.cell.2011.04.015
Pelletier, R., Krasilnikova, M. M., Samadashwily, G. M., Lahue, R., and Mirkin, S. M. (2003). Replication and expansion of trinucleotide repeats in yeast. Mol. Cell. Biol. 23, 1349–1357. doi: 10.1128/mcb.23.4.1349-1357.2003
Pettersson, O. J., Aagaard, L., Andrejeva, D., Thomsen, R., Jensen, T. G., and Damgaard, C. K. (2014). DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1. Nucleic Acids Res. 42, 7186–7200. doi: 10.1093/nar/gku352
Qurashi, A., Li, W., Zhou, J. Y., Peng, J., and Jin, P. (2011). Nuclear accumulation of stress response mRNAs contributes to the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS Genet. 7:e1002102. doi: 10.1371/journal.pgen.1002102
Samadashwily, G. M., Raca, G., and Mirkin, S. M. (1997). Trinucleotide repeats affect DNA replication in vivo. Nat. Genet. 17, 298–304. doi: 10.1038/ng1197-298
Sherman, S. L. (2000). Premature ovarian failure in the fragile X syndrome. Am. J. Med. Genet. 97, 189–194. doi: 10.1002/1096-8628(200023)97:3<189::aid-ajmg1036>3.0.co;2-j
Sofola, O. A., Jin, P., Qin, Y., Duan, R., Liu, H., de Haro, M., et al. (2007). RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55, 565–571. doi: 10.1016/j.neuron.2007.07.021
Subramanian, P. S., Nelson, D. L., and Chinault, A. C. (1996). Large domains of apparent delayed replication timing associated with triplet repeat expansion at FRAXA and FRAXE. Am. J. Hum. Genet. 59, 407–416.
Taneja, P., Gu, J., Peng, R., Carrick, R., Uchiumi, F., Ott, R. D., et al. (2002). A dominant-negative mutant of human DNA helicase B blocks the onset of chromosomal DNA replication. J. Biol. Chem. 277, 40853–40861. doi: 10.1074/jbc.M208067200
Tassone, F., Hagerman, R. J., Taylor, A. K., Gane, L. W., Godfrey, T. E., and Hagerman, P. J. (2000). Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 66, 6–15. doi: 10.1086/302720
Tassone, F., Iwahashi, C., and Hagerman, P. J. (2004). FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol. 1, 103–105. doi: 10.4161/rna.1.2.1035
Todd, P. K., Oh, S. Y., Krans, A., He, F., Sellier, C., Frazer, M., et al. (2013). CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78, 440–455. doi: 10.3410/f.718009175.793476334
Voineagu, I., Surka, C. F., Shishkin, A. A., Krasilnikova, M. M., and Mirkin, S. M. (2009). Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 16, 226–228. doi: 10.1038/nsmb.1527
Wang, L., Paradee, W., Mullins, C., Shridhar, R., Rosati, R., Wilke, C. M., et al. (1997). Aphidicolin-induced FRA3B breakpoints cluster in two distinct regions. Genomics 41, 485–488. doi: 10.1006/geno.1997.4690
Keywords: replication, helicases, fragile X syndrome, fragile sites, secondary structure, repeats
Citation: Guler GD, Rosenwaks Z and Gerhardt J (2018) Human DNA Helicase B as a Candidate for Unwinding Secondary CGG Repeat Structures at the Fragile X Mental Retardation Gene. Front. Mol. Neurosci. 11:138. doi: 10.3389/fnmol.2018.00138
Received: 19 February 2018; Accepted: 04 April 2018;
Published: 30 April 2018.
Edited by:
Regina Dahlhaus, Friedrich-Alexander-Universität Erlangen-Nürnberg, GermanyReviewed by:
Corrado Romano, Associazione Oasi Maria SS. Onlus (IRCCS), ItalyChang-hui Shen, College of Staten Island, United States
Copyright © 2018 Guler, Rosenwaks and Gerhardt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeannine Gerhardt, jeg2039@med.cornell.edu