
Comprehensive Protein Interactome Analysis of a Key RNA Helicase: Detection of Novel Stress Granule Proteins


Abstract
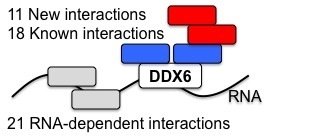
:
1. Introduction
2. Results
2.1. Identification of DDX6-Interacting Proteins



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name | SG | TL | PB | DC | Status | SF |
|---|---|---|---|---|---|---|---|
| ATXN2 | ataxin 2 | X | X | X | BG | X | |
| ATXN2L | ataxin 2-like | X | X | BG | X | ||
| C1QBP | complement component 1, q subcomponent binding protein | New | X | ||||
| DCP1B | DCP1 decapping enzyme homolog B | X | X | BG | |||
| DDX1 | DEAD (Asp-Glu-Ala-Asp) box helicase 1 | X | X | Inf | X | ||
| DDX17 | DEAD (Asp-Glu-Ala-Asp) box helicase 17 | New | X | ||||
| EDC3 | enhancer of mRNA decapping 3 homolog | X | X | BG | |||
| EDC4 | enhancer of mRNA decapping 4 | X | X | BG | |||
| EIF4ENIF1 | eukaryotic translation initiation factor 4E nuclear import factor 1 | BG | |||||
| ERH | enhancer of rudimentary homolog (Drosophila) | New | |||||
| FMR1 | fragile X mental retardation 1 | X | X | New | |||
| FXR2 | fragile X mental retardation, autosomal homolog 2 | X | New | ||||
| G3BP2 | GTPase activating protein (SH3 domain) binding protein 2 | X | BG | ||||
| GRAN1/ FAM195A | family with sequence similarity 195, member A | X*** | BG | ||||
| GRAN2/ FAM195B | family with sequence similarity 195, member B | X*** | BG | ||||
| HNRNPC | heterogeneous nuclear ribonucleoprotein C (C1/C2) | New | X | ||||
| HNRNPM | heterogeneous nuclear ribonucleoprotein M | New | X | ||||
| LARP4 | La ribonucleoprotein domain family, member 4 | New | |||||
| LSM12 | LSM12 homolog | BG | |||||
| LSM14A | LSM14A, SCD6 homolog A | X | X | X | BG | ||
| LSM14B | LSM14B, SCD6 homolog B | X | BG | X | |||
| NUFIP2 | nuclear fragile X mental retardation protein interacting protein 2 | X*** | BG | ||||
| PABPC1 | poly(A) binding protein, cytoplasmic 1 | X | X | Inf | X | ||
| PABPC3 | poly(A) binding protein, cytoplasmic 3 | X | Inf | ||||
| PABPC4 | poly(A) binding protein, cytoplasmic 4 (inducible form) | X | X | Inf | X | ||
| PATL1 | protein associated with topoisomerase II homolog 1 | X | X | BG | |||
| RTCB | RNA 2',3'-cyclic phosphate and 5'-OH ligase | New | X | ||||
| THRAP3 | thyroid hormone receptor associated protein 3 | New | X | ||||
| TIF1B | tripartite motif containing 28 | New |
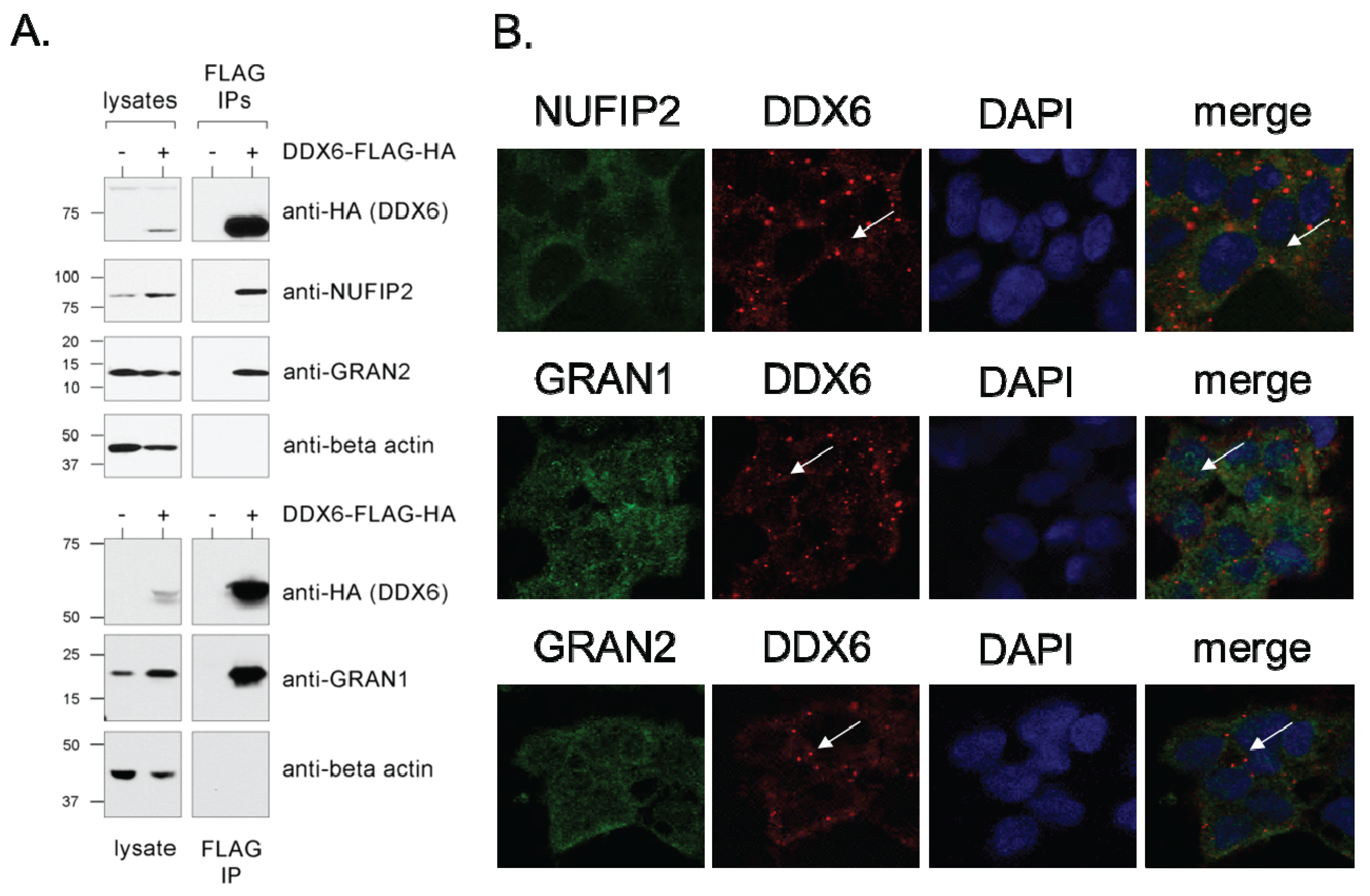
2.2. Validation and Characterization of Direct Interactions with NUFIP2, GRAN1 (FAM195A), and GRAN2 (FAM195B)


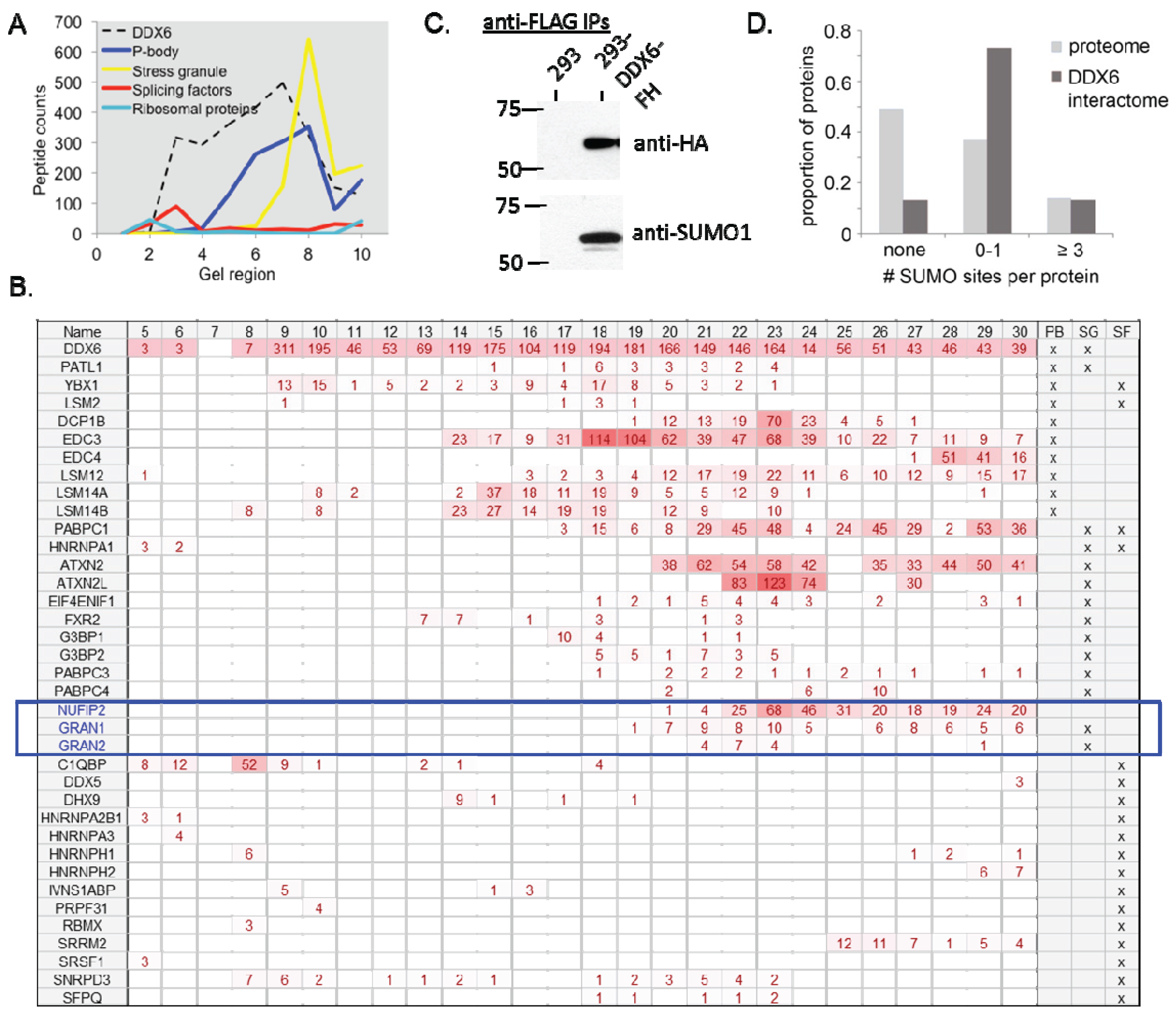
2.3. Further Functional Characterization of the DDX6 Interactome

3. Discussion
4. Experimental Section
4.1. Generation of Cell Lines
4.2. Cell Culture
4.3. Immunoprecipitation
4.4. Western Blot
4.5. Blue Native Polyacrylamide Gel Electrophoresis
4.6. Sample Preparation for Mass Spectrometry
4.7. Mass Spectrometry
4.8. Immunofluorescence Imaging
4.9. Bioinformatics and Data Analysis
4.10. Mass Spectrometry Data Analysis
4.11. Sumoylation Bioinformatics Analysis
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgements
Author Contributions
Conflicts of Interest
References
- De Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Guell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Baltz, A.G.; Munschauer, M.; Schwanhausser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell. 2012, 46, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.C.; Yi, H.; Eichelbaum, K.; Fohr, S.; Fischer, B.; You, K.T.; Castello, A.; Krijgsveld, J.; Hentze, M.W.; Kim, V.N. The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Ostareck, D.H.; Naarmann-de Vries, I.S.; Ostareck-Lederer, A. DDX6 and its orthologs as modulators of cellular and viral RNA expression. Wiley Interdiscip. Rev. RNA 2014, 5, 659–678. [Google Scholar] [CrossRef] [PubMed]
- Iio, A.; Takagi, T.; Miki, K.; Naoe, T.; Nakayama, A.; Akao, Y. DDX6 post-transcriptionally down-regulates miR-143/145 expression through host gene NCR143/145 in cancer cells. Biochim. Biophys. Acta 2013, 1829, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Matsumoto, K.; Ohguchi, K.; Nakagawa, Y.; Yoshida, H. Human DEAD-box/RNA unwindase Rck/p54 contributes to maintenance of cell growth by affecting cell cycle in cultured cells. Int. J. Oncol. 2006, 29, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vilaro, G.; Fernandez-Carrillo, C.; Mensa, L.; Miquel, R.; Sanjuan, X.; Forns, X.; Pérez-del-Pulgar, S.; Díez, J. Hepatitis C virus infection inhibits P-body granule formation in human livers. J. Hepatol. 2015, 62, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Scheller, N.; Mina, L.B.; Galao, R.P.; Chari, A.; Gimenez-Barcons, M.; Noueiry, A.; Fischer, U.; Meyerhans, A.; Díez, J. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc. Natl. Acad. Sci. USA 2009, 106, 13517–13522. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, J.; Boccaccio, G.L. Translation and silencing in RNA granules: A tale of sand grains. Front. Mol. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Coller, J.M.; Tucker, M.; Sheth, U.; Valencia-Sanchez, M.A.; Parker, R. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA 2001, 7, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Weis, K. The DEAD box protein Dhh1 stimulates the decapping enzyme Dcp1. EMBO J. 2002, 21, 2788–2797. [Google Scholar] [CrossRef] [PubMed]
- Coller, J.; Parker, R. General translational repression by activators of mRNA decapping. Cell 2005, 122, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Sweet, T.; Kovalak, C.; Coller, J. The DEAD-box protein Dhh1 promotes decapping by slowing ribosome movement. PLoS Biol. 2012, 10, e1001342. [Google Scholar] [CrossRef] [PubMed]
- Nissan, T.; Rajyaguru, P.; She, M.; Song, H.; Parker, R. Decapping activators in Saccharomyces cerevisiae act by multiple mechanisms. Mol. Cell 2010, 39, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Meng, S.; Lu, Y.; Trombly, M.I.; Chen, J.; Lin, C.; Turk, A.; Wang, X. Mammalian hyperplastic discs homolog EDD regulates miRNA-mediated gene silencing. Mol. Cell. 2011, 43, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Rouya, C.; Siddiqui, N.; Morita, M.; Duchaine, T.F.; Fabian, M.R.; Sonenberg, N. Human DDX6 effects miRNA-mediated gene silencing via direct binding to CNOT1. RNA 2014, 20, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Seto, M.; Yamamoto, K.; Iida, S.; Nakazawa, S.; Inazawa, J.; Abe, T.; Takahashi, T.; Ueda, R. The RCK gene associated with t(11;14) translocation is distinct from the MLL/ALL-1 gene with t(4;11) and t(11;19) translocations. Cancer Res. 1992, 52, 6083–6087. [Google Scholar] [PubMed]
- Lin, F.; Wang, R.; Shen, J.J.; Wang, X.; Gao, P.; Dong, K.; Zhang, H.Z. Knockdown of RCK/p54 expression by RNAi inhibits proliferation of human colorectal cancer cells in vitro and in vivo. Cancer Biol. Ther. 2008, 7, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Morikawa, H.; Hirata, I.; Shiozaki, M.; Matsumoto, A.; Maemura, K.; Nishikawa, T.; Niki, M.; Tanigawa, N.; Ikegami, M.; et al. Overexpression of Rck/p54, a DEAD box protein, in human colorectal tumours. Br. J. Cancer 1999, 80, 914–917. [Google Scholar] [CrossRef] [PubMed]
- Stary, S.; Vinatzer, U.; Mullauer, L.; Raderer, M.; Birner, P.; Streubel, B. t(11;14)(q23;q32) involving IGH and DDX6 in nodal marginal zone lymphoma. Genes Chromosomes Cancer 2013, 52, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Chahar, H.S.; Chen, S.; Manjunath, N. P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 2013, 436, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jangra, R.K.; Yi, M.; Lemon, S.M. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J. Virol. 2010, 84, 6810–6824. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Molter, B.; Geary, C.D.; McNevin, J.; McElrath, J.; Giri, S.; Klein, K.C.; Lingappa, J.R. HIV-1 Gag co-opts a cellular complex containing DDX6, a helicase that facilitates capsid assembly. J. Cell. Biol. 2012, 198, 439–456. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; Lujan, P.; Jackson, D.L.; Emerman, M.; Linial, M.L. The DEAD-box RNA helicase DDX6 is required for efficient encapsidation of a retroviral genome. PLoS Pathog. 2011, 7, e1002303. [Google Scholar] [CrossRef] [PubMed]
- Strahl-Bolsinger, S.; Tanner, W. A yeast gene encoding a putative RNA helicase of the “DEAD”-box family. Yeast 1993, 9, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, H.; Nakagawa, T.; Uno, Y.; Kitamura, K.; Shimoda, C. The Ste13+ gene encoding a putative RNA helicase is essential for nitrogen starvation-induced G1 arrest and initiation of sexual development in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Gene 1994, 244, 456–464. [Google Scholar] [CrossRef]
- Navarro, R.E.; Shim, E.Y.; Kohara, Y.; Singson, A.; Blackwell, T.K. Cgh-1, a conserved predicted RNA helicase required for gametogenesis and protection from physiological germline apoptosis in C. elegans. Development 2001, 128, 3221–3232. [Google Scholar] [PubMed]
- De Valoir, T.; Tucker, M.A.; Belikoff, E.J.; Camp, L.A.; Bolduc, C.; Beckingham, K. A second maternally expressed Drosophila gene encodes a putative RNA helicase of the “DEAD box” family. Proc. Natl. Acad. Sci. USA 1991, 88, 2113–2117. [Google Scholar] [CrossRef] [PubMed]
- Ladomery, M.; Wade, E.; Sommerville, J. Xp54, the Xenopus homologue of human RNA helicase p54, is an integral component of stored mRNP particles in oocytes. Nucleic Acids Res. 1997, 25, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Yunis, J.J. Cloning, expression and localization of an RNA helicase gene from a human lymphoid cell line with chromosomal breakpoint 11q23.3. Nucleic Acids Res. 1992, 20, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Cougot, N.; Babajko, S.; Seraphin, B. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell. Biol. 2004, 165, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.J.; Fritzler, M.J. Relationship of other cytoplasmic ribonucleoprotein bodies (cRNPB) to GW/P bodies. Adv. Exp. Med. Biol. 2013, 768, 213–242. [Google Scholar] [PubMed]
- Nonhoff, U.; Ralser, M.; Welzel, F.; Piccini, I.; Balzereit, D.; Yaspo, M.L.; Lehrach, H.; Krobitsch, S. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol. Biol. Cell. 2007, 18, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Wilczynska, A.; Aigueperse, C.; Kress, M.; Dautry, F.; Weil, D. The translational regulator CPEB1 provides a link between dcp1 bodies and stress granules. J. Cell. Sci. 2005, 118, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Presnyak, V.; Coller, J. The DHH1/RCKp54 family of helicases: An ancient family of proteins that promote translational silencing. Biochim. Biophys. Acta. 2013, 1829, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Fenger-Gron, M.; Fillman, C.; Norrild, B.; Lykke-Andersen, J. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol. Cell. 2005, 20, 905–915. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.; Naarmann-de Vries, I.S.; Urlaub, H.; Lue, H.; Bernhagen, J.; Ostareck, D.H.; Ostareck-Lederer, A. Identification of DEAD-box RNA helicase 6 (DDX6) as a cellular modulator of vascular endothelial growth factor expression under hypoxia. J. Biol. Chem. 2013, 288, 5815–5827. [Google Scholar] [CrossRef] [PubMed]
- Minshall, N.; Thom, G.; Standart, N. A conserved role of a DEAD box helicase in mRNA masking. RNA 2001, 7, 1728–1742. [Google Scholar] [CrossRef] [PubMed]
- Stoecklin, G.; Kedersha, N. Relationship of GW/P-bodies with stress granules. Adv. Exp. Med. Biol. 2013, 768, 197–211. [Google Scholar] [PubMed]
- Chu, C.Y.; Rana, T.M. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006, 4, e210. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, T.; Zekri, L.; Braun, J.E.; Izaurralde, E. miRISC recruits decapping factors to miRNA targets to enhance their degradation. Nucleic Acids Res. 2013, 41, 8692–8705. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, S.; Okawa, S.; Hillje, A.L.; Gonzalez-Cano, L.; del Sol, A.; Schwamborn, J.C. The RNA helicase DDX6 regulates cell-fate specification in neural stem cells via miRNAs. Nucleic Acids Res. 2015, 43, 2638–2654. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Boland, A.; Kuzuoglu-Ozturk, D.; Bawankar, P.; Loh, B.; Chang, C.T.; Weichenrieder, O.; Izaurralde, E. A DDX6-CNOT1 complex and W-binding pockets in CNOT9 reveal direct links between miRNA target recognition and silencing. Mol. Cell 2014, 54, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Ozgur, S.; Chekulaeva, M.; Stoecklin, G. Human Pat1b connects deadenylation with mRNA decapping and controls the assembly of processing bodies. Mol. Cell. Biol. 2010, 30, 4308–4323. [Google Scholar] [CrossRef] [PubMed]
- Kaehler, C.; Isensee, J.; Nonhoff, U.; Terrey, M.; Hucho, T.; Lehrach, H.; Krobitsch, S. Ataxin-2-like is a regulator of stress granules and processing bodies. PLoS ONE 2012, 7, e50134. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Sheth, U. P bodies and the control of mRNA translation and degradation. Mol. Cell 2007, 25, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Phalora, P.K.; Sherer, N.M.; Wolinsky, S.M.; Swanson, C.M.; Malim, M.H. HIV-1 replication and APOBEC3 antiviral activity are not regulated by P bodies. J. Virol. 2012, 86, 11712–11724. [Google Scholar] [CrossRef] [PubMed]
- Andrei, M.A.; Ingelfinger, D.; Heintzmann, R.; Achsel, T.; Rivera-Pomar, R.; Lührmann, R. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA 2005, 11, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, L.; Fu, B.; Berman, M.A.; Diallo, A.; Dorf, M.E. TRIM65 regulates microRNA activity by ubiquitination of TNRC6. Proc. Natl. Acad. Sci. USA 2014, 111, 6970–6975. [Google Scholar] [CrossRef] [PubMed]
- Rolland, T.; Tasan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Bardoni, B.; Castets, M.; Huot, M.E.; Schenck, A.; Adinolfi, S.; Corbin, F.; Pastore, A.; Khandjian, E.W.; Mandel, J.L. 82-FIP, a novel FMRP (fragile X mental retardation protein) interacting protein, shows a cell cycle-dependent intracellular localization. Hum. Mol. Genet. 2003, 12, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Hollingworth, D.; Adinolfi, S.; Castets, M.; Kelly, G.; Frenkiel, T.A.; Bardoni, B.; Pastore, A. The structure of the N-terminal domain of the fragile X mental retardation protein: A platform for protein-protein interaction. Structure 2006, 14, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.; Mistry, J.; Tate, J.; Hotz, H.; Ceric, G.; Forslund, K.; Eddy, S.R.; Sonnhammer, E.L.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Fridell, R.A.; Benson, R.E.; Hua, J.; Cullen, B.R. Protein sequence requirements for function of the human T-cell leukemia virus type 1 Rex nuclear export signal delineated by a novel in vivo randomization-selection assay. Mol. Cell. Biol. 1996, 16, 4207–4214. [Google Scholar] [PubMed]
- La Cour, T.; Kiemer, L.; Molgaard, A.; Gupta, R.; Skriver, K.; Brunak, S. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 2004, 17, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Reijns, M.A.; Alexander, R.D.; Spiller, M.P.; Beggs, J.D. A role for Q/N-rich aggregation-prone regions in P-body localization. J. Cell. Sci. 2008, 121, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; Cramer, W.A.; von Jagow, G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 1994, 217, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; von Jagow, G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991, 199, 223–231. [Google Scholar] [CrossRef]
- Wittig, I.; Braun, H.P.; Schagger, H. Blue native PAGE. Nat. Protoc. 2006, 1, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Petersen-Mahrt, S.K.; Estmer, C.; Ohrmalm, C.; Matthews, D.A.; Russell, W.C.; Russell, W.C.; Akusjärvi, G. The splicing factor-associated protein, p32, regulates RNA splicing by inhibiting ASF/SF2 RNA binding and phosphorylation. EMBO J. 1999, 18, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Deng, H.; Li, X.; Wu, X.; Tang, Q.; Chang, T.H.; Peng, H.; Rauscher, F.J., 3rd; Ozato, K.; Zhu, F. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 2011, 187, 4754–4763. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.M. Identification of SUMOylated proteins in neuroblastoma cells after treatment with hydrogen peroxide or ascorbate. BMB Rep. 2010, 43, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Blomster, H.A.; Hietakangas, V.; Wu, J.; Kouvonen, P.; Hautaniemi, S.; Sistonen, L. Novel proteomics strategy brings insight into the prevalence of SUMO-2 target sites. Mol. Cell. Proteomics 2009, 8, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Golebiowski, F.; Matic, I.; Tatham, M.H.; Cole, C.; Yin, Y.; Nakamura, A.; Cox, J.; Barton, G.J.; Mann, M.; Hay, R.T. System-wide changes to SUMO modifications in response to heat shock. Sci. Signal. 2009. [Google Scholar] [CrossRef] [PubMed]
- Lamoliatte, F.; Bonneil, E.; Durette, C.; Caron-Lizotte, O.; Wildemann, D.; Zerweck, J.; Wenshuk, H.; Thibault, P. Targeted identification of SUMOylation sites in human proteins using affinity enrichment and paralog-specific reporter ions. Mol. Cell. Proteomics 2013, 12, 2536–2550. [Google Scholar] [CrossRef] [PubMed]
- Manza, L.L.; Codreanu, S.G.; Stamer, S.L.; Smith, D.L.; Wells, K.S.; Roberts, R.L.; Liebler, D.C. Global shifts in protein sumoylation in response to electrophile and oxidative stress. Chem. Res. Toxicol. 2004, 17, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Barrett-Wilt, G.A.; Hua, Z.; Vierstra, R.D. Proteomic analyses identify a diverse array of nuclear processes affected by small ubiquitin-like modifier conjugation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2010, 107, 16512–16517. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Scalf, M.; Rytz, T.C.; Hubler, S.L.; Smith, L.M.; Vierstra, R.D. Quantitative proteomics reveals factors regulating RNA biology as dynamic targets of stress-induced SUMOylation in Arabidopsis. Mol. Cell. Proteomics 2013, 12, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Vertegaal, A.C.; Andersen, J.S.; Ogg, S.C.; Hay, R.T.; Mann, M.; Lamond, A.I. Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteins revealed by quantitative proteomics. Mol. Cell. Proteomics 2006, 5, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Westman, B.J.; Lamond, A.I. A role for SUMOylation in snoRNP biogenesis revealed by quantitative proteomics. Nucleus 2011, 2, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; D’Souza, R.C.; Yang, B.; Verlaan-de Vries, M.; Mann, M.; Vertegaal, A.C. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014, 21, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Blomster, H.A.; Imanishi, S.Y.; Siimes, J.; Kastu, J.; Morrice, N.A.; Eriksson, J.E.; Sistonen, L. In vivo identification of sumoylation sites by a signature tag and cysteine-targeted affinity purification. J. Biol. Chem. 2010, 285, 19324–19329. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.; Tatham, M.H.; Plechanovova, A.; Matic, I.; Garg, A.K.; Hay, R.T. Purification and identification of endogenous polySUMO conjugates. EMBO Rep. 2011, 12, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Galisson, F.; Mahrouche, L.; Courcelles, M.; Bonneil, E.; Meloche, S.; Chelbi-Alix, M.K.; Thibaultc, P. A novel proteomics approach to identify SUMOylated proteins and their modification sites in human cells. Mol. Cell. Proteomics 2011, 10, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Matic, I.; Schimmel, J.; Hendriks, I.A.; van Santen, M.A.; van de Rijke, F.; van Dam, H.; Gnad, F.; Mann, M.; Vertegaal, A.C. Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Mol. Cell 2010, 39, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Acosta, G.; Russell, W.K.; Deyrieux, A.; Russell, D.H.; Wilson, V.G. A universal strategy for proteomic studies of SUMO and other ubiquitin-like modifiers. Mol. Cell. Proteomics 2005, 4, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, J.; Larsen, K.M.; Matic, I.; van Hagen, M.; Cox, J.; Mann, M.; Andersen, J.S.; Vertegaal, A.C. The ubiquitin-proteasome system is a key component of the SUMO-2/3 cycle. Mol. Cell. Proteomics 2008, 7, 2107–2122. [Google Scholar] [CrossRef] [PubMed]
- Tatham, M.H.; Matic, I.; Mann, M.; Hay, R.T. Comparative proteomic analysis identifies a role for SUMO in protein quality control. Sci. Signal. 2011, 4, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Vertegaal, A.C.; Ogg, S.C.; Jaffray, E.; Rodriguez, M.S.; Hay, R.T.; Andersen, J.S.; Mann, M.; Lamond, A.I. A proteomic study of SUMO-2 target proteins. J. Biol. Chem. 2004, 279, 33791–33798. [Google Scholar] [CrossRef] [PubMed]
- Weston, A.; Sommerville, J. Xp54 and related (DDX6-like) RNA helicases: Roles in messenger RNP assembly, translation regulation and RNA degradation. Nucleic Acids Res. 2006, 34, 3082–3094. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Genes Dev. 2013, 27, 2628–2641. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Anderson, P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007, 431, 61–81. [Google Scholar] [PubMed]
- Chang, L.C.; Lee, F.J. The RNA helicase Dhh1p cooperates with Rbp1p to promote porin mRNA decay via its non-conserved C-terminal domain. Nucleic Acids Res. 2012, 40, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Ayache, J.; Benard, M.; Ernoult-Lange, M.; Minshall, N.; Standart, N.; Kress, M.; Weil, D. P-body assembly requires DDX6 repression complexes rather than decay or Ataxin2/2L complexes. Mol. Biol. Cell 2015. [Google Scholar] [CrossRef] [PubMed]
- Monzo, K.; Papoulas, O.; Cantin, G.T.; Wang, Y.; Yates, J.R., 3rd; Sisson, J.C. Fragile X mental retardation protein controls trailer hitch expression and cleavage furrow formation in Drosophila embryos. Proc. Natl. Acad. Sci. USA 2006, 103, 18160–18165. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, G.; King, A.; Rappsilber, J.; Calvio, C.; Watson, M.; Sleeman, J.; Lamond, A.; Mann, M. Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat. Genet. 1998, 20, 46–50. [Google Scholar] [PubMed]
- Sharma, S.; Kohlstaedt, L.A.; Damianov, A.; Rio, D.C.; Black, D.L. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat. Struct. Mol. Biol. 2008, 15, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Stevens, S.W.; Ryan, D.E.; Ge, H.Y.; Moore, R.E.; Young, M.K.; Lee, T.D.; Abelson, J. Composition and functional characterization of the yeast spliceosomal penta-snRNP. Mol. Cell 2002, 9, 31–44. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed]
- O’Geen, H.; Farnham, P.J. TRIM28: The transcription factor encyclopedia. Genome Biol. 2012. [Google Scholar] [CrossRef]
- Smillie, D.A.; Sommerville, J. RNA helicase p54 (DDX6) is a shuttling protein involved in nuclear assembly of stored mRNP particles. J. Cell. Sci. 2002, 115, 395–407. [Google Scholar] [PubMed]
- Marblestone, J.G.; Edavettal, S.C.; Lim, Y.; Lim, P.; Zuo, X.; Tauseef, R.B. Comparison of SUMO fusion technology with traditional gene fusion systems: Enhanced expression and solubility with SUMO. Protein Sci. 2006, 15, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell. Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, M.; Taylor, J.P.; Parker, R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 2013, 154, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed]
- Antonov, A.V. BioProfiling.de: Analytical web portal for high-throughput cell biology. Nucleic Acids Res. 2011, 39, W323–W327. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Fenyo, D.; Eriksson, J.; Beavis, R. Mass spectrometric protein identification using the global proteome machine. Methods Mol. Biol. 2010, 673, 189–202. [Google Scholar] [PubMed]
- Consortium, U.P. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2012, 41, D190–D195. [Google Scholar]
- Powell, D.W.; Weaver, C.M.; Jennings, J.L.; McAfee, K.J.; He, Y.; Weil, P.A.; Link, A.J. Cluster analysis of mass spectrometry data reveals a novel component of SAGA. Mol. Cell. Biol. 2004, 24, 7249–7259. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Cote, R.G.; Csordas, A.; Dianes, J.A.; Fabregat, A.; Foster, J.M.; Griss, J.; Alpi, E.; Birim, M.; Contell, J.; et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: Status in 2013. Nucleic Acids Res. 2013, 41, D1063–D1069. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Gao, X.; Jin, C.; Zhu, M.; Wang, X.; Shaw, A.; Wen, L.; Yao, X.; Yu, X. Systematic study of protein sumoylation: Development of a site-specific predictor of SUMOsp 2.0. Proteomics 2009, 9, 3409–3412. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bish, R.; Cuevas-Polo, N.; Cheng, Z.; Hambardzumyan, D.; Munschauer, M.; Landthaler, M.; Vogel, C. Comprehensive Protein Interactome Analysis of a Key RNA Helicase: Detection of Novel Stress Granule Proteins. Biomolecules 2015, 5, 1441-1466. https://doi.org/10.3390/biom5031441
Bish R, Cuevas-Polo N, Cheng Z, Hambardzumyan D, Munschauer M, Landthaler M, Vogel C. Comprehensive Protein Interactome Analysis of a Key RNA Helicase: Detection of Novel Stress Granule Proteins. Biomolecules. 2015; 5(3):1441-1466. https://doi.org/10.3390/biom5031441
Chicago/Turabian StyleBish, Rebecca, Nerea Cuevas-Polo, Zhe Cheng, Dolores Hambardzumyan, Mathias Munschauer, Markus Landthaler, and Christine Vogel. 2015. "Comprehensive Protein Interactome Analysis of a Key RNA Helicase: Detection of Novel Stress Granule Proteins" Biomolecules 5, no. 3: 1441-1466. https://doi.org/10.3390/biom5031441