Mutated Rnf43 Aggravates Helicobacter Pylori-Induced Gastric Pathology


1
Institut für Medizinische Mikrobiologie, Immunologie und Hygiene, Technische Universität München, 81675 Munich, Germany
2
Institut für Pathologie, Klinikum Bayreuth, 95445 Bayreuth, Germany
*
Authors to whom correspondence should be addressed.
Cancers 2019, 11(3), 372; https://doi.org/10.3390/cancers11030372
Submission received: 14 February 2019
/
Revised: 10 March 2019
/
Accepted: 13 March 2019
/
Published: 16 March 2019
(This article belongs to the Special Issue Helicobacter pylori Associated Cancer)
Abstract
:The E3 ubiquitin ligase ring finger protein 43 (RNF43) is frequently mutated in gastric tumors and loss of RNF43 expression was suggested to be one of the key events during the transition from adenoma to gastric carcinoma. Functional studies on RNF43 have shown that it acts as a tumor suppressor by negatively regulating Wnt signaling. Interestingly, we observed that RNF43H292R/H295R mice bearing two point mutations in the ring domain displayed thickening of the mucosa at early age but did not develop neoplasia. In this study, we infected these mice for 6 months with Helicobacter pylori, which has been described as one of the major risk factors for gastric cancer. Mice bearing mutant RNF43H292R/H295R showed higher gastritis scores upon H. pylori infection compared to wild-type mice, accompanied by increased lymphocyte infiltration and Ifng levels. Furthermore, infected Rnf43 mutant mice developed atrophy, hyperplasia and MUC2 expressing metaplasia and displayed higher levels of the gastric stem cell marker CD44 and canonical NF-κB signaling. In summary, our results show that transactivating mutations in the tumor suppressor Rnf43 can worsen H. pylori induced pathology.
1. Introduction
Gastric cancer has been described as a multifactorial disease with high interindividual and intraindividual heterogeneity [1]. In recent years, much effort has been applied to define molecular signatures that may be useful in classifying and developing targeted therapeutic approaches for gastric cancer patients. To this end, analysis of the mutational landscape of gastric cancer has allowed generation of better molecular classifications and the definition of important genes driving gastric carcinogenesis. In this context, the E3 ubiquitin ligase ring finger protein 43 (RNF43) was found to be frequently mutated in gastric tumors [2,3,4] highlighting an important role for RNF43 in gastric carcinogenesis. RNF43 expression was shown to be lost in poorly differentiated gastric cancers [5], while mutations were concentrated in gastric carcinomas adjacent to adenomas [6], indicating an important role for RNF43 in the transition from adenoma to carcinoma. In addition, RNF43 downregulation in gastric tumors was associated with metastasis, TNM staging and poor survival [7], suggesting that loss of RNF43 function is one of the key events in gastric carcinogenesis. Our recent results using mice carrying a 57 bp deletion in exon 8 of Rnf43 (Rnf43ΔEx8), confirmed an important role for RNF43 in gastric homeostasis, since mutant mice showed gastric hyperplasia, although lesions did not develop to neoplasia [8].
The tumor suppressor function of RNF43 is based on its ability to inhibit Wnt signaling at the level of Frizzled (FZD) receptors [9,10] as well as its capacity to sequester TCF4 to the nuclear envelope thereby disrupting transcription of Wnt target genes [11]. Interestingly, introduction of two point mutations (RNF43H292R/H295R) not only abolished its inhibitory effect but rather transactivated Wnt signaling [10,11], suggesting that certain mutations can have a further deleterious effect by enhancing Wnt signaling. Indeed, we identified in patients some mutations having this Wnt-transactivating effect in colon cells [11]. Furthermore, in the murine stomach, the introduction of the two point mutations leading to the amino acid substitutions H292R/H295R in the ring domain of RNF43 led to marked thickening of the gastric mucosa, hyperplasia and cellular atypia at early ages [8]. Whether these pathological changes depend on alterations of Wnt signaling could not be determined. Therefore, the identification of other signaling pathways or targeted genes needs to be further addressed.
Apart from mutations in important genes, one of the main risk factors for gastric cancer development is chronic infection with H. pylori, which is classified as a class I carcinogen [12]. H. pylori induces strong Th1 and Th17 inflammatory responses, but at the same time favors the expansion of regulatory T cells, which create an immunosuppressive environment, possibly contributing to gastric cancer development [13,14,15]. H. pylori-induced chronic gastritis may progress to gastric atrophy, metaplasia and eventually to gastric cancer over years, in a process defined as the Correa pathway [16]. During this process, H. pylori de-regulates several signaling pathways, including the canonical and non-canonical NF-κB pathway [17,18] and the EGFR signaling pathway [19,20], and also promotes a mutation-prone milieu through ROS production and subsequent induction of DNA damage [21]. In addition, H. pylori can target the gastric stem cell compartment, leading to aberrant epithelial cell proliferation, metaplasia, and altered differentiation. Specifically, H. pylori was reported to accelerate the proliferation and expansion of LGR5+ cells [22] as well as CD44+ cells [23], which constitute important populations of stem cells in the stomach. Notably, expression of RNF43 was exclusively found in LGR5 intestinal crypt stem cells [10], while overexpression of RNF43 in gastric cells led to decreased protein levels of LGR5 [7]. In addition, xenografts derived from gastric RNF43 knockdown cells showed enhanced expression of the stem cell markers SOX2 and CD44, correlating with enhanced tumor growth [8]. Although these observations suggest an important role for RNF43 in the control of gastrointestinal stem cell homeostasis, it is still unknown whether RNF43 is also expressed in stomach stem cells or to what extent it controls the expression of other gastric stem cell markers in vivo.
Considering the important role of RNF43 in gastric homeostasis and the high frequency of mutations observed in gastric tumors, as well as the carcinogenic potential of H. pylori infection, in the present study we sought to investigate how chronic H. pylori infection would affect the onset and development of gastric pathology in mice carrying mutated Rnf43. Our results show that H. pylori infection worsens gastric pathology of RNF43H292R/H295R mice and suggest that RNF43 mutations in combination with sustained chronic inflammation contribute to the development of gastric malignancies.
2. Results
2.1. RNF43H292R/H295R Mice Show Enhanced H. pylori-Induced Gastritis Inflammation
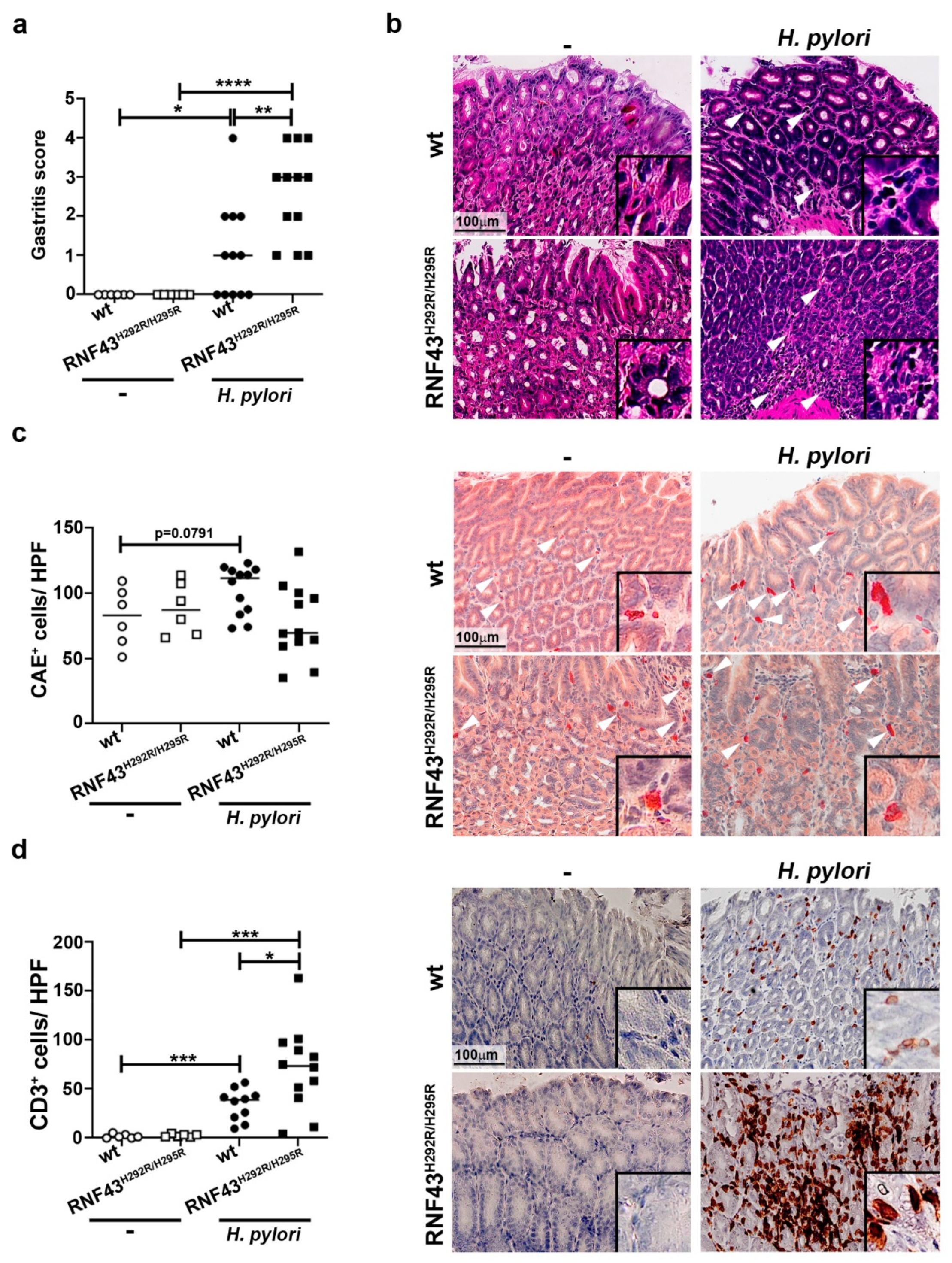
To analyze how the presence of RNF43 transactivating mutations affects the inflammatory response to H. pylori chronic infection, we infected RNF43H292R/H295R mice with the pathogenic H. pylori strain PMSS1 for six months. No significant differences in bacterial colonization were detected between wild-type and RNF43H292R/H295R mice (Figure S1); however, RNF43H292R/H295R mice showed higher inflammation scores in the stomach, evaluated according to the updated Sydney system for gastritis classification [24] (Figure 1a,b). Notably, no differences in neutrophil infiltration were detected between infected wild-type and RNF43H292R/H295R mice at this point of chronic infection (Figure 1c). In contrast, lymphocytic infiltration, as detected by immunohistochemical staining of CD3+ cells, was higher in infected RNF43H292R/H295R compared to infected wild-type mice (Figure 1d).
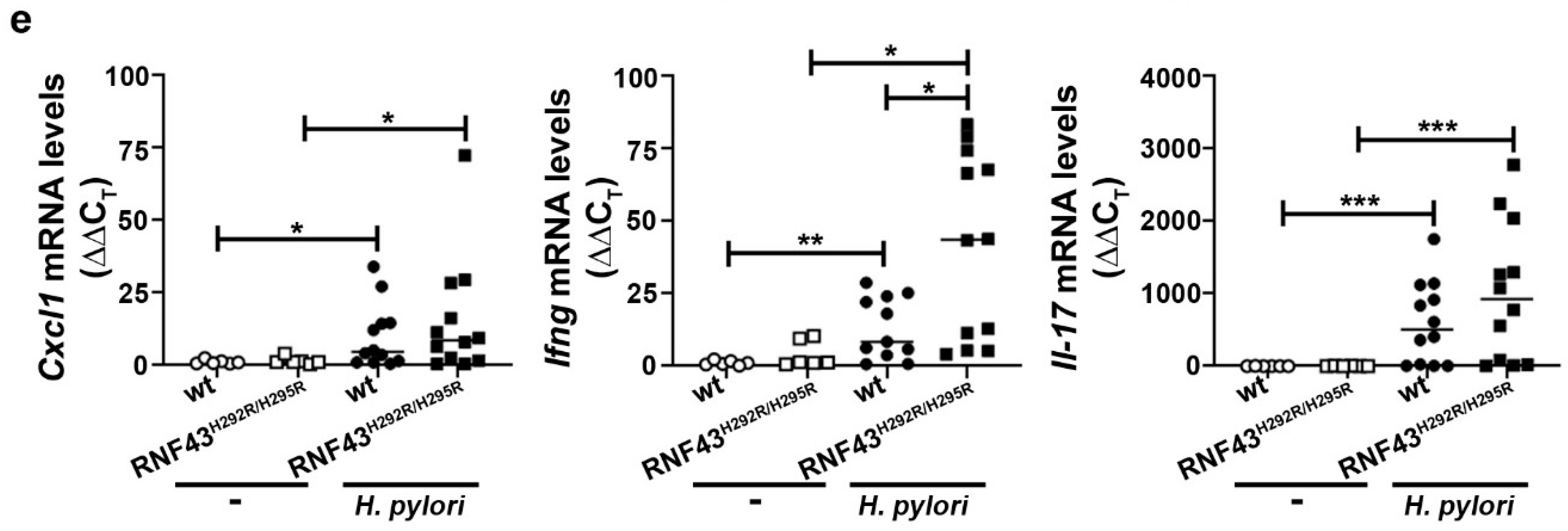
To further characterize the immune response of RNF43H292R/H295R mice compared to wild-type mice towards H. pylori infection, we analyzed the expression of different cytokines typically induced by H. pylori infection: Cxcl1 (murine homologue of IL-8), as it relates to innate immunity, Ifng as a marker of Th1 responses, and Il-17 as a marker of Th17 responses. H. pylori-infected wild-type mice showed increased expression levels of Cxcl1 compared to uninfected mice (Figure 1e). Similar results were observed when comparing infected RNF43H292R/H295R to uninfected RNF43H292R/H295R mice, while no differences were detected between wild-type and RNF43H292R/H295R mice upon infection (Figure 1e). As expected, H. pylori induced Infg expression in the stomachs of infected mice (Figure 1e). Interestingly, this expression was increased in infected RNF43H292R/H295R mice, which showed higher levels of Ifng mRNA compared to infected wild-type mice (Figure 1e). H. pylori infection also induced IL-17 responses, as detected by high levels of Il-17 expression upon infection (Figure 1e). However, no significant differences between wild-type and RNF43H292R/H295R mice were observed (Figure 1e).
Together these results suggest that transactivating mutations of RNF43 aggravate gastric inflammation in response to H. pylori.
2.2. H. pylori Increases Gastric Pathology of RNF43H292R/H295R Mice
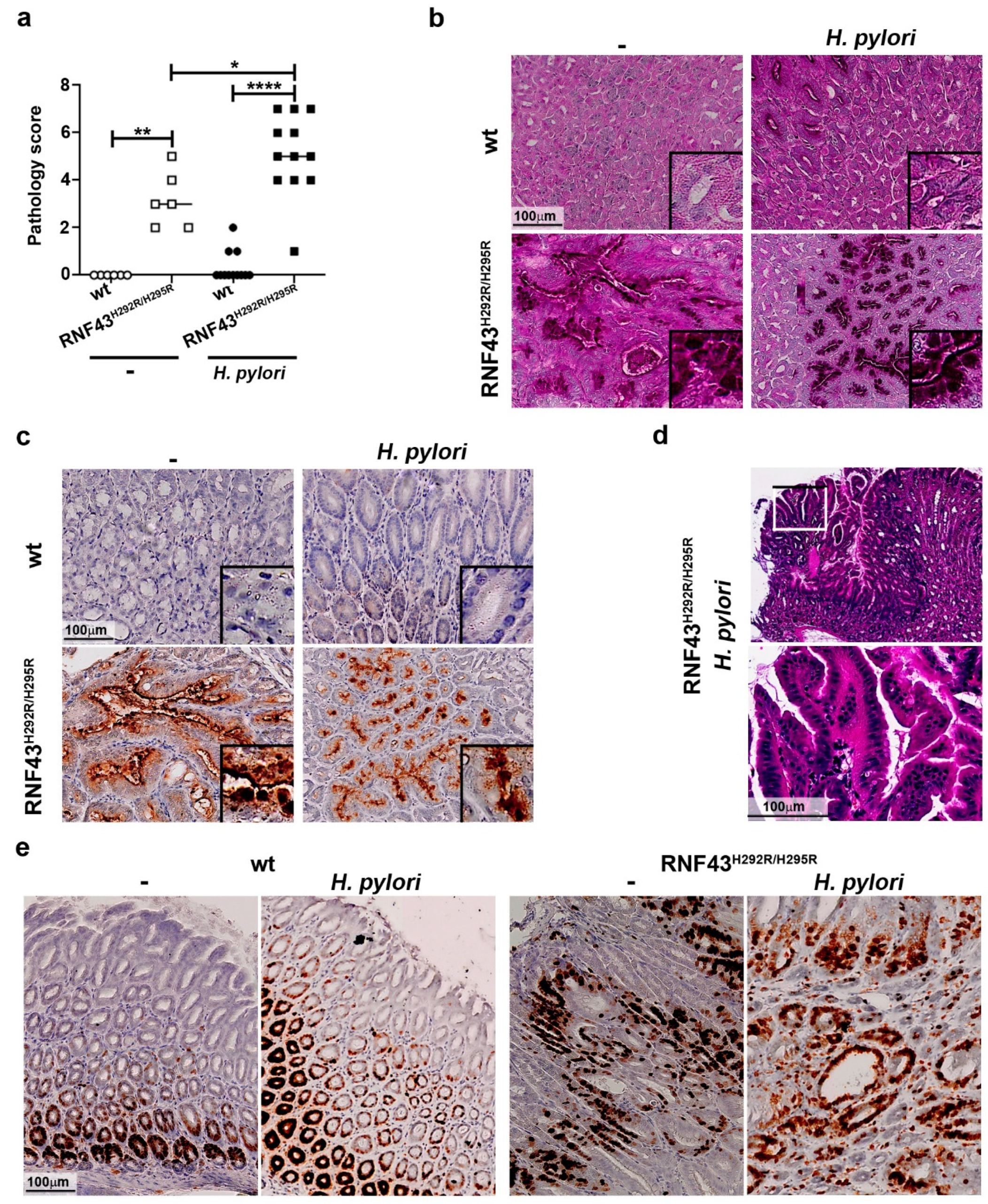
We have recently reported that RNF43H292R/H295R mice show gastric hyperproliferation [8]. To explore whether H. pylori infection may influence gastric pathology in mice carrying a transactivating mutation of Rnf43, we analyzed the gastric mucosa of mice infected for six months. Macroscopically, we could detect lesions in the stomachs of Rnf43 mutant mice, especially in the corpus, which were aggravated upon infection (Figure S2a). We established a pathology score based on the presence of atrophy, metaplasia, hyperplasia and reactive changes. Chronic H. pylori infection increased gastric pathology of RNF43H292R/H295R mice (Figure 2a). Atrophy and metaplasia were detected only in the corpus of RNF43H292R/H295R mice, under basal conditions as well as after H. pylori infection (Figure 2b). Metaplasia was observed to be multifocal in uninfected RNF43H292R/H295R mice. Upon infection, metaplasia could be detected in most of the fields analyzed for some mice. Metaplasia was characterized by the expression of high levels of MUC2 (Figure 2c). Notably, no signs of atrophy or metaplasia were observed in the antrums of the mice (Figure S2b). Gastric pathology progressed to hyperplasia in 9 out of 12 (75%) of the infected RNF43H292R/H295R mice, while 2 out 4 (50%) of RNF43H292R/H295R mice showed hyperplasia without infection (Figure 2d). Hyperplasia was observed only in 2 out of 12 (17%) of infected wild-type mice. Reactive changes (nuclear enlargement, nuclear hyperchromasia, prominent nuclei and nuclear atypia) were detected in 25% of the uninfected RNF43H292R/H295R mice. Upon H. pylori infection, 100% of the RNF43H292R/H295R mice showed such reactive changes, while those were present in only 25% of the wild-type mice upon H. pylori infection. The lesions observed in Rnf43 mutant mice were highly proliferative, as detected by Ki67 staining (Figure 2e). These results indicate that gastric pathology of RNF43H292R/H295R mice is worsened by H. pylori infection.
2.3. RNF43 Transactivating Mutations Favor the Expression of Genes Related to Gastric Carcinogenesis
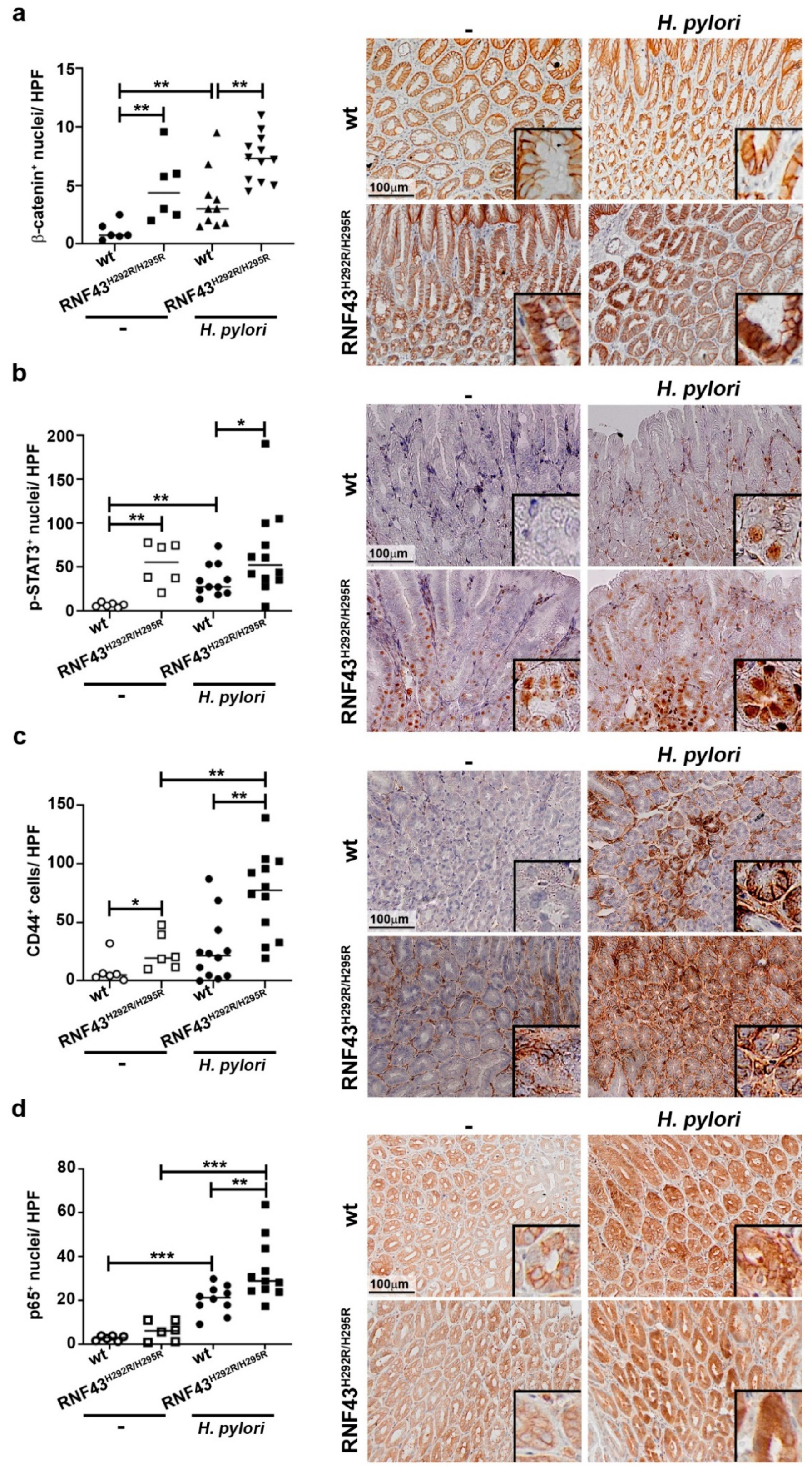
We analyzed the expression of genes related to the development of gastric tumors. We first studied the activation status of Wnt signaling by analyzing nuclear β-catenin. RNF43H292R/H295R mice showed enhanced activation of the pathway compared to wild-type mice (Figure 3a). Notably, H. pylori infection led to increased nuclear translocation of β-catenin in wild-type as well as in RNF43H292R/H295R mice (Figure 3a).
Next, we analyzed the expression of Sox2, as previous studies have shown SOX2 to be deregulated during gastric carcinogenesis [25]. RNF43H292R/H295R mice showed more expression of SOX2 in the stomach than wild-type mice (Figure S3a,b), although the difference was not significant. H. pylori infection slightly decreased SOX2 expression in wild-type mice, while infected RNF43H292R/H295R mice showed markedly lower levels of SOX2 upon infection when compared to uninfected mutant mice (Figure S3a).
STAT3 signaling has been linked to the development of gastric tumors in mice [26]. H. pylori infection induced phosphorylation of STAT3 in wild-type mice (Figure 3b). Under basal conditions, RNF43H292R/H295R mice showed a higher number of p-STAT3+ cells compared to wild-type mice. Interestingly, H. pylori infection did not alter the phosphorylation of STAT3 in RNF43H292R/H295R mice (Figure 3b), but RNF43H292R/H295R mice presented a higher number of p-STAT3+ cells compared to infected wild-type mice (Figure 3b). These results indicate that changes in STAT3 signaling may contribute to the gastric pathology observed in RNF43H292R/H295R mice, but not to the increased pathology driven by H. pylori infection.
We also analyzed the expression of the stem cell marker CD44, which has been associated with initiation and progression of gastric cancer and previous studies have shown that CD44+ cells are targeted during H. pylori infection [27]. We observed a higher number of CD44+ cells in the stomach of uninfected RNF43H292R/H295R mice compared to wild-type control mice (Figure 3c). H. pylori infection led to increased numbers of CD44+ cells in wild-type mice, although the differences were not statistically significant (Figure 3c). Notably, infected RNF43H292R/H295R mice displayed much higher numbers of CD44+ cells in the gastric mucosa upon H. pylori infection compared to uninfected RNF43H292R/H295R mice as well as infected wild-type mice (Figure 3c), suggesting H. pylori-targeted expansion of CD44+ gastric stem cells is enhanced in the presence of RNF43 transactivating mutations.
Finally, given the importance of NF-ĸB signaling during H. pylori infection and gastric cancer development, we analyzed activation of NF-ĸB in the stomach by assessing the number of cells presenting nuclear expression of p65. We observed that upon infection with H. pylori, NF-ĸB was activated in wild-type mice, as expected (Figure 3d). RNF43H292R/H295R mice showed no basal activation of the pathway. Upon infection, RNF43H292R/H295R mice showed enhanced p65 nuclear translocation in gastric cells compared to uninfected RNF43H292R/H295R and infected wild-type mice. These observations suggest that NF-ĸB might be involved in the enhanced pathology observed in RNF43H292R/H295R mice in response to H. pylori infection. In addition, we analyzed the expression of the canonical and non-canonical NF-ĸB target genes Cxcl10 and Cxcl13, respectively. Cxcl10 mRNA levels were higher in infected wild-type mice compared to uninfected mice (Figure S3c). Infected RNF43H292R/H295R mice showed higher levels of Cxcl10 compared to uninfected RNF43H292R/H295R mice as well as to infected wild-type mice, confirming increased activation of canonical NF-ĸB. Although H. pylori induced the expression of Cxcl13 in the stomach (Figure S3d), as we have previously reported [18], we could not detect differences between infected wild-type and RNF43H292R/H295R mice, excluding the involvement of non-canonical NF-ĸB in the phenotype observed for infected RNF43H292R/H295R mice.
Together, our results indicate that increased pathology induced by H. pylori in the presence of RNF43 transactivating mutations may be supported by increased expression of CD44 and hyperactivation of canonical NF-ĸB.
3. Discussion
Gastric cancer is the fifth most frequently diagnosed cancer and the third leading cause of cancer related deaths worldwide [28]. Whole exome and whole genome sequencing studies reported RNF43 to be frequently mutated in gastric tumors. RNF43 mutations are more often found in microsatellite instable tumors [2,3,4] and truncating mutations are the most common type of mutations [3]. Using a model of CRISPR/Cas9 engineered mice, we found that the introduction of two point mutations in the ring domain of murine Rnf43 did not impact intestinal homeostasis, but induced relevant changes in the stomach architecture of RNF43H292R/H295R mutant mice [8]. Nevertheless, pathology did not progress to neoplasia, indicating that mutations in RNF43 alone are not sufficient to drive malignant transformation. In the present study, we combined the presence of mutated Rnf43 with chronic H. pylori infection, since the latter is a major risk factor for the development of gastric cancer. We observed that RNF43H292R/H295R mice showed increased inflammation characterized by high lymphocytic infiltration and IFNγ production, and worsened pathology compared to uninfected RNF43H292R/H295R and infected wild-type mice. This suggests that mutations in RNF43 render the gastric mucosa more susceptible to H. pylori infection.
When exploring possible mechanisms driving pathology in Rnf43 mutant mice, we analyzed phosphorylation of STAT3, since this signaling pathway has been previously related to gastric carcinogenesis [26]. RNF43H292R/H295R mice presented increased number of p-STAT3+ nuclei in the gastric mucosa compared to wild-type mice. However, H. pylori infection did not further enhance STAT3 activation, suggesting that STAT3 signaling may be involved in the pathology observed in Rnf43 mutant mice already under basal conditions, and may favor enhanced pathology upon infection by crosstalk with other signaling pathways.
We further observed that mice carrying mutated Rnf43 showed enhanced nuclear accumulation of β-catenin in the stomach. Several components of the Wnt signaling pathway, including activators as well as suppressors, have been described to be de-regulated during gastric carcinogenesis (reviewed in [29]). The Wnt pathway plays a critical role in stem cell proliferation, but it also supports the development and renewal of cancer stem cells. Although the origin of gastric cancer stem cells is still unclear, it is suggested that they are responsible for the formation, maintenance, and continuous growth of the tumors [30,31,32]. Notably, gastric cancer stem cells are characterized by the expression of CD44 [33,34], which is a target gene of the Wnt signaling pathway [33]. We observed that RNF43H292R/H295R mice showed higher levels of CD44 than wild-type mice under basal conditions. This could be related to a de-regulated Wnt pathway as a result of Rnf43 mutation. Interestingly, H. pylori infection led to an even higher number of CD44+ cells in the stomach. H. pylori has been previously shown to target and expand the CD44+ gastric stem cell compartment [23], while inhibition of CD44 blocked proliferation and cancer progression in H. pylori-infected gerbils [27]. Further, experiments using CD44−/− mice showed that CD44 is crucial for the development of mucous metaplasia during H. pylori infection [35]. In humans, progression of precancerous gastric lesions was associated with increased levels of CD44 in H. pylori-infected subjects [35]. In line with these observations, the expression of CD44 in the stomach of RNF43H292R/H295R mice was related to the presence of metaplasia and progression of the gastric lesions, and both pathology and CD44 expression were associated with H. pylori infection. The expansion of CD44+ gastric cells in mice was suggested to be induced by a cooperative effect of PGE2-mediated inflammation and Wnt signaling, resulting in the development of gastric tumors [36]. In addition, Wnt-1 was related to CD44 expression [37], while inhibition of the Wnt/β-catenin pathway suppressed gastric cancer stem cells [37]. These observations confirmed an important role for Wnt signaling in the control of CD44+ gastric stem cells. However, H. pylori has been described to enhance Wnt signaling by inducing nuclear β-catenin accumulation independently of upstream components of the Wnt pathway [38,39,40]. Therefore, H. pylori-enhanced expansion of CD44+ cells observed in RNF43H292R/H295R mice may be further regulated by an additional mechanism independent of Wnt. In fact, the expression of CD44 in different cancer cells has been reported to be regulated by NF-ĸB [41,42,43], a signaling pathway that is rapidly activated in gastric epithelial cells upon H. pylori infection [44]. Thus, NF-ĸB signaling is a major contributor to Helicobacter-induced gastric pathology and epithelial malignant transformation [45,46]. In light of our results, it is tempting to speculate that activation of NF-ĸB by the infection further up-regulates the expression of CD44, thereby inducing the progression of gastric lesions to more severe pathology. Nevertheless, it is still unclear why activation of NF-ĸB is higher in RNF43H292R/H295R mice than in wild-type mice upon infection; thus, this deserves further investigation.
Although mutations in RNF43 seem to lead to a more severe pathology in response to H. pylori infection, it has not been addressed whether H. pylori may induce mutations in RNF43. To date, no correlations between H. pylori infection and RNF43 mutation status in patients have been established. However, analysis of RNF43 mutations during progression of tumors from low grade to high grade dysplasia and early gastric cancer revealed that mutations in RNF43 occurred at the stage of high grade dysplasia and early gastric tumors [6]. This indicates that H. pylori-induced preneoplastic changes precede RNF43 mutations. Considering the high prevalence of H. pylori infection and the increased incidence of RNF43 mutations detected in gastric tumors, further studies should be conducted to understand how both events interrelate.
Together, our results using a novel mouse model provide evidence that a chronic inflammatory milieu contributes to progression of gastric lesions when RNF43 is mutated in gastric cells. This finding opens the pathway to further investigations in order to address whether H. pylori infection aggravates cancer progression in subjects carrying mutations in RNF43, as well as the molecular mechanisms engaged.
4. Materials and Methods
4.1. Helicobacter Pylori Infection
RNF43H292R/H295R mice were generated by introducing two point mutations in the RING domain of Rnf43 through homology directed repair, as described in [8]. 6–8-week old mice were infected twice with 2 × 108 H. pylori strain PMSS1 [47] diluted in 200 µL brain-heart-infusion (BHI) containing 20% FCS by oral gavage and sacrificed after 6 months.
To this end, PMSS1 was cultured on Wilkins-Chalgren (WC) Dent agar plates in a microaerophilic atmosphere (5% O2, 10% CO2). For infection, bacteria were collected in BHI 20% FCS and density was measured at OD600 (OD600 1 = 2 × 108 bacteria/mL).
All animal experiments were conducted in compliance with European guidelines for the care and use of laboratory animals and were approved by the Bavarian Government (Regierung von Oberbayern, AZ.55.2-1-54-2532-196-2016).
4.2. Quantitative PCR
Murine stomach tissue pieces were homogenized with a Precellys lysing Kit and RNA was extracted using a Maxwell 16 LEV simply RNA Tissue Kit and a Maxwell 16 MDx Instrument (Promega, Madison, WI, USA) RNA was retrotranscribed using Moloney Murine Leukemia Virus Reverse Transcriptase RNase H- Point Mutant (Promega). GoTaq qPCR Mastermix (Promega) and a CFX384 system (Bio-Rad, Hercules, CA, USA) were used to assess transcript abundance with the recommended standard quantitative PCR cycling program. The PCR conditions used were 40 cycles of amplification with 15 s denaturation at 95 °C and 1 min annealing and amplification at 60 °C. Normalization to GAPDH and the comparative ΔΔCT method was used to analyze target gene expression. Primers used in this study are summarized in Table 1.
4.3. Immunohistochemistry
Mice were dissected and tissue was fixed in 4% formaldehyde and subsequently embedded in paraffin. For histologic evaluation, hematoxylin and eosin, chloroacetate esterase staining and periodic acid Schiff (PAS) staining were performed. For immunohistochemistry, 10 mM sodium citrate (pH 6) or 1 mM EDTA (pH 8) (p-STAT3) was used for heat induced antigen retrieval. Primary antibodies were applied overnight or for 1 h (MUC2) at room temperature (Table 2) and detected by horseradish peroxidase coupled secondary antibodies. Signal was detected using diaminobenzidine (DAB), and slides were scanned and analyzed using an Olympus Virtual Slide Imaging system (Olympus, Shinjuku, Japan).
To assess the pathology observed in the stomach a pathology score was established by assessing the degree of atrophy (0–3), the presence of metaplasia (0-absent, 1-multifocal, 2-in most fields, 3-widespread in all fields), the presence of hyperplasia (0-absent, 1-slight, 2-moderate, 3-severe), and reactive changes (0-absent, 1-present).
5. Conclusions
Mutation of Rnf43 worsens H. pylori-induced pathology in the stomach. Rnf43 mutations increase the levels of nuclear β-catenin and of phosphorylated STAT3 in the gastric mucosa, while H. pylori in addition enhances activation of NF-κB signaling and expression of the stem cell marker CD44. Thus, the activation of these different signaling pathways in concert seem to aggravate pathology in the stomach. This data provides functional hints for a novel role of RNF43 mutations in gastric carcinogenesis.
Supplementary Materials
The following are available online at https://www.mdpi.com/2072-6694/11/3/372/s1, Figure S1: Colony forming units in the stomach of H. pylori-infected mice, Figure S2: Gastric pathology in RNF43H292R/H295R mice is mainly observed in the corpus, Figure S3: Representative images and quantification of SOX2 expression (a) and mRNA expression levels of Sox2 (b), Cxcl10 (c) and Cxcl13 (d) in the stomach of control and infected mice.
Author Contributions
Methodology, V.N.; formal analysis, V.N., M.V. and R.M.-L.; writing—original draft preparation, V.N. and R.M.-L.; writing—review and editing, V.N., M.V., M.G. and R.M.-L.; supervision, R.M.-L.
Funding
This research was funded by the German Research Foundation (DFG), grant number GE2042 12-1, and the DFG and the Technical University of Munich (TUM) in the framework of the Open Access Publishing Program.
Acknowledgments
Authors thank Monika Schön, Andreas Wanisch and Marta Garrido for excellent technical support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aichler, M.; Luber, B.; Lordick, F.; Walch, A. Proteomic and metabolic prediction of response to therapy in gastric cancer. World J. Gastroenterol. 2014, 20, 13648–13657. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nature Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Maruvka, Y.E.; Mouw, K.W.; Karlic, R.; Parasuraman, P.; Kamburov, A.; Polak, P.; Haradhvala, N.J.; Hess, J.M.; Rheinbay, E.; Brody, Y.; et al. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nature Biotechnol. 2017, 35, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Qin, H.Z.; Xi, H.Q.; Wei, B.; Xia, S.Y.; Chen, L. RNF43 inhibits cancer cell proliferation and could be a potential prognostic factor for human gastric carcinoma. Cell. Physiol. Biochem. 2015, 36, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Min, B.H.; Hwang, J.; Kim, N.K.; Park, G.; Kang, S.Y.; Ahn, S.; Ahn, S.; Ha, S.Y.; Lee, Y.K.; Kushima, R.; et al. Dysregulated WNT signalling and recurrent mutations of the tumour suppressor RNF43 in early gastric carcinogenesis. J. Pathol. 2016, 10. [Google Scholar] [CrossRef]
- Gao, Y.; Cai, A.; Xi, H.; Li, J.; Xu, W.; Zhang, Y.; Zhang, K.; Cui, J.; Wu, X.; Wei, B.; et al. Ring finger protein 43 associates with gastric cancer progression and attenuates the stemness of gastric cancer stem-like cells via the Wnt-beta/catenin signaling pathway. Stem Cell Res. Ther. 2017, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Neumeyer, V.; Grandl, M.; Dietl, A.; Brutau-Abia, A.; Allgauer, M.; Kalali, B.; Zhang, Y.; Pan, K.F.; Steiger, K.; Vieth, M.; et al. Loss of endogenous RNF43 function enhances proliferation and tumour growth of intestinal and gastric cells. Carcinogenesis 2018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Lu, B.; Hsieh, M.H.; Bagdasarian, L.; Meyer, R.; et al. Inactivating mutations of RNF43 confer WNT dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12649–12654. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of WNT receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef]
- Loregger, A.; Grandl, M.; Mejias-Luque, R.; Allgauer, M.; Degenhart, K.; Haselmann, V.; Oikonomou, C.; Hatzis, P.; Janssen, K.P.; Nitsche, U.; et al. The E3 ligase RNF43 inhibits WNT signaling downstream of mutated beta-catenin by sequestering TCF4 to the nuclear membrane. Sci. Signal. 2015, 8, ra90. [Google Scholar] [CrossRef]
- Anonymous. Infection with Helicobacter pylori. IARC Monogr. Eval. Carcinog. Risks. Hum. 1994, 61, 177–240. [Google Scholar]
- Mejias-Luque, R.; Gerhard, M. Immune evasion strategies and persistence of Helicobacter pylori. Curr. Top. Microbiol. Immunol. 2017, 400, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Kandulski, A.; Malfertheiner, P.; Wex, T. Role of regulatory T-cells in H. pylori-induced gastritis and gastric cancer. Anticancer Res. 2010, 30, 1093–1103. [Google Scholar] [PubMed]
- Capitani, N.; Codolo, G.; Vallese, F.; Minervini, G.; Grassi, A.; Cianchi, F.; Troilo, A.; Fischer, W.; Zanotti, G.; Baldari, C.T.; et al. The lipoprotein HP1454 of Helicobacter pylori regulates T-cell response by shaping T-cell receptor signalling. Cell. Microbiol. 2019, e13006. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society award lecture on cancer epidemiology and prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Lamb, A.; Chen, L.F. Role of the Helicobacter pylori-induced inflammatory response in the development of gastric cancer. J. Cell. Biochem. 2013, 114, 491–497. [Google Scholar] [CrossRef]
- Mejias-Luque, R.; Zoller, J.; Anderl, F.; Loew-Gil, E.; Vieth, M.; Adler, T.; Engler, D.B.; Urban, S.; Browning, J.L.; Muller, A.; et al. Lymphotoxin beta receptor signalling executes Helicobacter pylori-driven gastric inflammation in a T4SS-dependent manner. Gut 2017, 66, 1369–1381. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Asim, M.; Piazuelo, M.B.; Yan, F.; Barry, D.P.; Sierra, J.C.; Delgado, A.G.; Hill, S.; Casero, R.A., Jr.; Bravo, L.E.; et al. Activation of EGFR and ERBB2 by Helicobacter pylori results in survival of gastric epithelial cells with DNA damage. Gastroenterology 2014, 146, 1739–1751. [Google Scholar] [CrossRef]
- Sierra, J.C.; Asim, M.; Verriere, T.G.; Piazuelo, M.B.; Suarez, G.; Romero-Gallo, J.; Delgado, A.G.; Wroblewski, L.E.; Barry, D.P.; Peek, R.M., Jr.; et al. Epidermal growth factor receptor inhibition downregulates Helicobacter pylori-induced epithelial inflammatory responses, DNA damage and gastric carcinogenesis. Gut 2017. [Google Scholar] [CrossRef]
- Wen, J.; Wang, Y.; Gao, C.; Zhang, G.; You, Q.; Zhang, W.; Zhang, Z.; Wang, S.; Peng, G.; Shen, L. Helicobacter pylori infection promotes Aquaporin 3 expression via the ROS-HIF-1alpha-AQP3-ROS loop in stomach mucosa: A potential novel mechanism for cancer pathogenesis. Oncogene 2018, 37, 3549–3561. [Google Scholar] [CrossRef] [PubMed]
- Sigal, M.; Rothenberg, M.E.; Logan, C.Y.; Lee, J.Y.; Honaker, R.W.; Cooper, R.L.; Passarelli, B.; Camorlinga, M.; Bouley, D.M.; Alvarez, G.; et al. Helicobacter pylori activates and expands Lgr5(+) stem cells through direct colonization of the gastric glands. Gastroenterology 2015, 148, 1392–1404.e21. [Google Scholar] [CrossRef]
- Khurana, S.S.; Riehl, T.E.; Moore, B.D.; Fassan, M.; Rugge, M.; Romero-Gallo, J.; Noto, J.; Peek, R.M., Jr.; Stenson, W.F.; Mills, J.C. The hyaluronic acid receptor CD44 coordinates normal and metaplastic gastric epithelial progenitor cell proliferation. J. Biological Chem. 2013, 288, 16085–16097. [Google Scholar] [CrossRef]
- Dixon, M.F.; Genta, R.M.; Yardley, J.H.; Correa, P. Classification and grading of gastritis. The updated Sydney system. International workshop on the histopathology of gastritis, Houston 1994. Am. J. Surg. Pathol. 1996, 20, 1161–1181. [Google Scholar] [CrossRef]
- Carrasco-Garcia, E.; Santos, J.C.; Garcia, I.; Brianti, M.; Garcia-Puga, M.; Pedrazzoli, J., Jr.; Matheu, A.; Ribeiro, M.L. Paradoxical role of SOX2 in gastric cancer. Am. J. Cancer Res. 2016, 6, 701–713. [Google Scholar]
- Jenkins, B.J.; Grail, D.; Nheu, T.; Najdovska, M.; Wang, B.; Waring, P.; Inglese, M.; McLoughlin, R.M.; Jones, S.A.; Topley, N.; et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat. Med. 2005, 11, 845–852. [Google Scholar] [CrossRef]
- Bertaux-Skeirik, N.; Feng, R.; Schumacher, M.A.; Li, J.; Mahe, M.M.; Engevik, A.C.; Javier, J.E.; Peek, R.M., Jr.; Ottemann, K.; Orian-Rousseau, V.; et al. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog. 2015, 11, e1004663. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Chiurillo, M.A. Role of the WNT/beta-catenin pathway in gastric cancer: An in-depth literature review. World J. Exp. Med. 2015, 5, 84–102. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.L.; Xiang, D.; Shigdar, S.; Macdonald, J.; Li, Y.; Wang, T.; Pu, C.; Wang, Z.; Qiao, L.; Duan, W. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014, 344, 180–187. [Google Scholar] [CrossRef]
- Brungs, D.; Aghmesheh, M.; Vine, K.L.; Becker, T.M.; Carolan, M.G.; Ranson, M. Gastric cancer stem cells: Evidence, potential markers, and clinical implications. J. Gastroenterol. 2016, 51, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.; El-Rayes, B. Identifying and targeting cancer stem cells in the treatment of gastric cancer. Cancer 2017, 123, 1303–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, C.; He, F.; Cai, Y.; Yang, H. Identification of CD44+CD24+ gastric cancer stem cells. J. Cancer Res. Clin. Oncol. 2011, 137, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Garay, J.; Piazuelo, M.B.; Majumdar, S.; Li, L.; Trillo-Tinoco, J.; Del Valle, L.; Schneider, B.G.; Delgado, A.G.; Wilson, K.T.; Correa, P.; et al. The homing receptor CD44 is involved in the progression of precancerous gastric lesions in patients infected with Helicobacter pylori and in development of mucous metaplasia in mice. Cancer Lett. 2016, 371, 90–98. [Google Scholar] [CrossRef]
- Ishimoto, T.; Oshima, H.; Oshima, M.; Kai, K.; Torii, R.; Masuko, T.; Baba, H.; Saya, H.; Nagano, O. CD44+ slow-cycling tumor cell expansion is triggered by cooperative actions of WNT and prostaglandin E2 in gastric tumorigenesis. Cancer Sci. 2010, 101, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Fan, S.; Ma, W.; Fan, P.; Wang, B.; Zhang, J.; Wang, H.; Tang, B.; Zhang, Q.; Yu, X.; et al. Roles of WNT/beta-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 2014, 5, e1039. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Israel, D.A.; Washington, M.K.; Krishna, U.; Fox, J.G.; Rogers, A.B.; Neish, A.S.; Collier-Hyams, L.; Perez-Perez, G.I.; Hatakeyama, M.; et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2005, 102, 10646–10651. [Google Scholar] [CrossRef] [PubMed]
- Nagy, T.A.; Wroblewski, L.E.; Wang, D.; Piazuelo, M.B.; Delgado, A.; Romero-Gallo, J.; Noto, J.; Israel, D.A.; Ogden, S.R.; Correa, P.; et al. beta-Catenin and p120 mediate PPARdelta-dependent proliferation induced by Helicobacter pylori in human and rodent epithelia. Gastroenterology 2011, 141, 553–564. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Piazuelo, M.B.; Chaturvedi, R.; Schumacher, M.; Aihara, E.; Feng, R.; Noto, J.M.; Delgado, A.; Israel, D.A.; Zavros, Y.; et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut 2015, 64, 720–730. [Google Scholar] [CrossRef]
- Smith, S.M.; Cai, L. Cell specific CD44 expression in breast cancer requires the interaction of AP-1 and NFkappaB with a novel cis-element. PLoS ONE 2012, 7, e50867. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Lyu, Y.L.; Cai, L. NF-kappaB affects proliferation and invasiveness of breast cancer cells by regulating CD44 expression. PLoS ONE 2014, 9, e106966. [Google Scholar] [CrossRef] [PubMed]
- Haria, D.; Trinh, B.Q.; Ko, S.Y.; Barengo, N.; Liu, J.; Naora, H. The homeoprotein DLX4 stimulates NF-kappaB activation and CD44-mediated tumor-mesothelial cell interactions in ovarian cancer. Am. J. Pathol. 2015, 185, 2298–2308. [Google Scholar] [CrossRef]
- Keates, S.; Hitti, Y.S.; Upton, M.; Kelly, C.P. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology 1997, 113, 1099–1109. [Google Scholar] [CrossRef]
- Shibata, W.; Takaishi, S.; Muthupalani, S.; Pritchard, D.M.; Whary, M.T.; Rogers, A.B.; Fox, J.G.; Betz, K.S.; Kaestner, K.H.; Karin, M.; et al. Conditional deletion of IkappaB-kinase-beta accelerates helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenterology 2010, 138, 1022–1034.e1. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, M.D.; Williams, J.M.; Duckworth, C.A.; O’Hara, A.; Hanedi, A.; Varro, A.; Caamano, J.H.; Pritchard, D.M. Signaling mediated by the NF-kappaB sub-units NF-kappaB1, NF-kappaB2 and c-Rel differentially regulate Helicobacter felis-induced gastric carcinogenesis in C57BL/6 mice. Oncogene 2013, 32, 5563–5573. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; O’Rourke, J.; Ungria, M.C.d.; Robertson, B.; Daskalopoulos, G.; Dixon, M.F. A standardized mouse model of Helicobacter pylori infection: Introducing the Sydney strain. Gastroenterology 1997, 112, 1386–1397. [Google Scholar] [CrossRef]
- Carlstedt, I.; Herrmann, A.; Hovenberg, H.; Lindell, G.; Nordman, H.; Wickstrom, C.; Davies, J.R. ‘Soluble’ and ‘insoluble’ mucins—Identification of distinct populations. Biochem. Soc. Trans. 1995, 23, 845–851. [Google Scholar] [CrossRef]
Figure 1.
H. pylori infection worsens gastric inflammation of RNF43H292R/H295R mice. (a) Gastritis score. Gastric inflammation was assessed according to the updated Sydney system for gastritis classification after 6 months of infection with the H. pylori strain PMSS1. (b) Representative images of H&E stained gastric sections showing infiltration of inflammatory cells (arrows) in the corpus of control and PMSS1 infected mice. (c) Quantification (positive cells per high power field (20× objective)) and representative images of chloracetate esterase (CAE) stained tissue samples. (d) Quantification of CD3+ cells per high power field and representative images of stained tissue samples. (e) mRNA levels of Cxcl1, Ifng and Il-17 detected in gastric homogenates. Values were normalized to Gapdh and differences expressed as ΔΔCT to control uninfected mice. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001. Mann-Whitney Test (pairwise comparisons).
Figure 1.
H. pylori infection worsens gastric inflammation of RNF43H292R/H295R mice. (a) Gastritis score. Gastric inflammation was assessed according to the updated Sydney system for gastritis classification after 6 months of infection with the H. pylori strain PMSS1. (b) Representative images of H&E stained gastric sections showing infiltration of inflammatory cells (arrows) in the corpus of control and PMSS1 infected mice. (c) Quantification (positive cells per high power field (20× objective)) and representative images of chloracetate esterase (CAE) stained tissue samples. (d) Quantification of CD3+ cells per high power field and representative images of stained tissue samples. (e) mRNA levels of Cxcl1, Ifng and Il-17 detected in gastric homogenates. Values were normalized to Gapdh and differences expressed as ΔΔCT to control uninfected mice. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001. Mann-Whitney Test (pairwise comparisons).


Figure 2.
H. pylori enhances gastric pathology of RNF43H292R/H295R mice. (a) Pathology score. The presence of atrophy, metaplasia, hyperplasia and reactive changes was assessed in antrum and corpus after 6-month H. pylori infection. (b) Representative images of periodic acid Schiff (PAS) stained gastric sections. (c) MUC2 expression detected in gastric tissue sections by immunohistochemistry. (d) Representative picture and higher magnification image of hyperplasia in the stomach of an infected RNF43H292R/H295R mouse. * p ≤ 0.05; ** p ≤ 0.01; **** p ≤ 0.0001. Mann-Whitney Test (pairwise comparisons). (e) Ki67 staining denoting cellular proliferation in the stomach.
Figure 2.
H. pylori enhances gastric pathology of RNF43H292R/H295R mice. (a) Pathology score. The presence of atrophy, metaplasia, hyperplasia and reactive changes was assessed in antrum and corpus after 6-month H. pylori infection. (b) Representative images of periodic acid Schiff (PAS) stained gastric sections. (c) MUC2 expression detected in gastric tissue sections by immunohistochemistry. (d) Representative picture and higher magnification image of hyperplasia in the stomach of an infected RNF43H292R/H295R mouse. * p ≤ 0.05; ** p ≤ 0.01; **** p ≤ 0.0001. Mann-Whitney Test (pairwise comparisons). (e) Ki67 staining denoting cellular proliferation in the stomach.

Figure 3.
H. pylori enhances CD44 expression and NF-ĸB activation in the stomach of RNF43H292R/H295R. Number of β-catenin (a), p-STAT3 (b), CD44 (c) and nuclear p65+ cells (d) in the stomach of wild-type and RNF43H292R/H295R mice under basal conditions and upon 6-month H. pylori infection. Representative images are shown. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. Mann-Whitney Test (pairwise comparisons).
Figure 3.
H. pylori enhances CD44 expression and NF-ĸB activation in the stomach of RNF43H292R/H295R. Number of β-catenin (a), p-STAT3 (b), CD44 (c) and nuclear p65+ cells (d) in the stomach of wild-type and RNF43H292R/H295R mice under basal conditions and upon 6-month H. pylori infection. Representative images are shown. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. Mann-Whitney Test (pairwise comparisons).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Sequence of the primers used in this study.
| Gene Name | Forward | Reverse |
|---|---|---|
| Cxcl10 | AAGTGCTGCCGTCATTTTCT | CCTATGGCCCTCATTCTCAC |
| Cxcl13 | ATATGTGTGAATCCTCGTGCCA | GGGAGTTGAAGACAGACTTTTGC |
| Gapdh | GCCTTCTCCATGGTGGTGAA | GCACAGTCAAGGCCGAGAAT |
| Ifng | TCAAGTGGCATAGATGTGGAAGAA | TGGCTCTGCAGGATTTTCATG |
| Il-17 | GCTCCAGAAGGCCCTCAGA | AGCTTTCCCTCCGCATTGA |
| Cxcl1 | TGCACCCAAACCGAAGTCAT | TTGTCAGAAGCCAGCGTTCAC |
| Sox2 | CATGGGCTCTGTGGTCAAGT | CGGGGAGGTACATGCTGATC |
Table 2.
Antibodies used in this study.
| Target | Clone | Company |
|---|---|---|
| CD3 | SP7 | Thermo Fisher |
| CD44 | E7K2Y | Cell Signaling |
| MUC2 | LUM 2.3 | [48] |
| p-STAT3 | D3A7 | Cell Signaling |
| p65 | D14E12 | Cell Signaling |
| SOX2 | C70B1 | Cell Signaling |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Neumeyer, V.; Vieth, M.; Gerhard, M.; Mejías-Luque, R. Mutated Rnf43 Aggravates Helicobacter Pylori-Induced Gastric Pathology. Cancers 2019, 11, 372. https://doi.org/10.3390/cancers11030372
AMA Style
Neumeyer V, Vieth M, Gerhard M, Mejías-Luque R. Mutated Rnf43 Aggravates Helicobacter Pylori-Induced Gastric Pathology. Cancers. 2019; 11(3):372. https://doi.org/10.3390/cancers11030372
Chicago/Turabian StyleNeumeyer, Victoria, Michael Vieth, Markus Gerhard, and Raquel Mejías-Luque. 2019. "Mutated Rnf43 Aggravates Helicobacter Pylori-Induced Gastric Pathology" Cancers 11, no. 3: 372. https://doi.org/10.3390/cancers11030372
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.