The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma


1
Institute for Human Health & Disease Intervention, Department of Chemistry & Biochemistry, and the Center for Molecular Biology & Biotechnology, Florida Atlantic University, Jupiter, FL 33458, USA
2
Department of Chemistry, The Scripps Research Institute/Scripps Florida, Jupiter, FL 33458, USA
Cells 2019, 8(9), 984; https://doi.org/10.3390/cells8090984
Submission received: 2 August 2019
/
Revised: 22 August 2019
/
Accepted: 26 August 2019
/
Published: 27 August 2019
(This article belongs to the Special Issue Matrix Metalloproteinases: From Structure to Function)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The pursuit of matrix metalloproteinase (MMP) inhibitors began in earnest over three decades ago. Initial clinical trials were disappointing, resulting in a negative view of MMPs as therapeutic targets. As a better understanding of MMP biology and inhibitor pharmacokinetic properties emerged, it became clear that initial MMP inhibitor clinical trials were held prematurely. Further complicating matters were problematic conclusions drawn from animal model studies. The most recent generation of MMP inhibitors have desirable selectivities and improved pharmacokinetics, resulting in improved toxicity profiles. Application of selective MMP inhibitors led to the conclusion that MMP-2, MMP-9, MMP-13, and MT1-MMP are not involved in musculoskeletal syndrome, a common side effect observed with broad spectrum MMP inhibitors. Specific activities within a single MMP can now be inhibited. Better definition of the roles of MMPs in immunological responses and inflammation will help inform clinic trials, and multiple studies indicate that modulating MMP activity can improve immunotherapy. There is a U.S. Food and Drug Administration (FDA)-approved MMP inhibitor for periodontal disease, and several MMP inhibitors are in clinic trials, targeting a variety of maladies including gastric cancer, diabetic foot ulcers, and multiple sclerosis. It is clearly time to move on from the dogma of viewing MMP inhibition as intractable.
1. Introduction
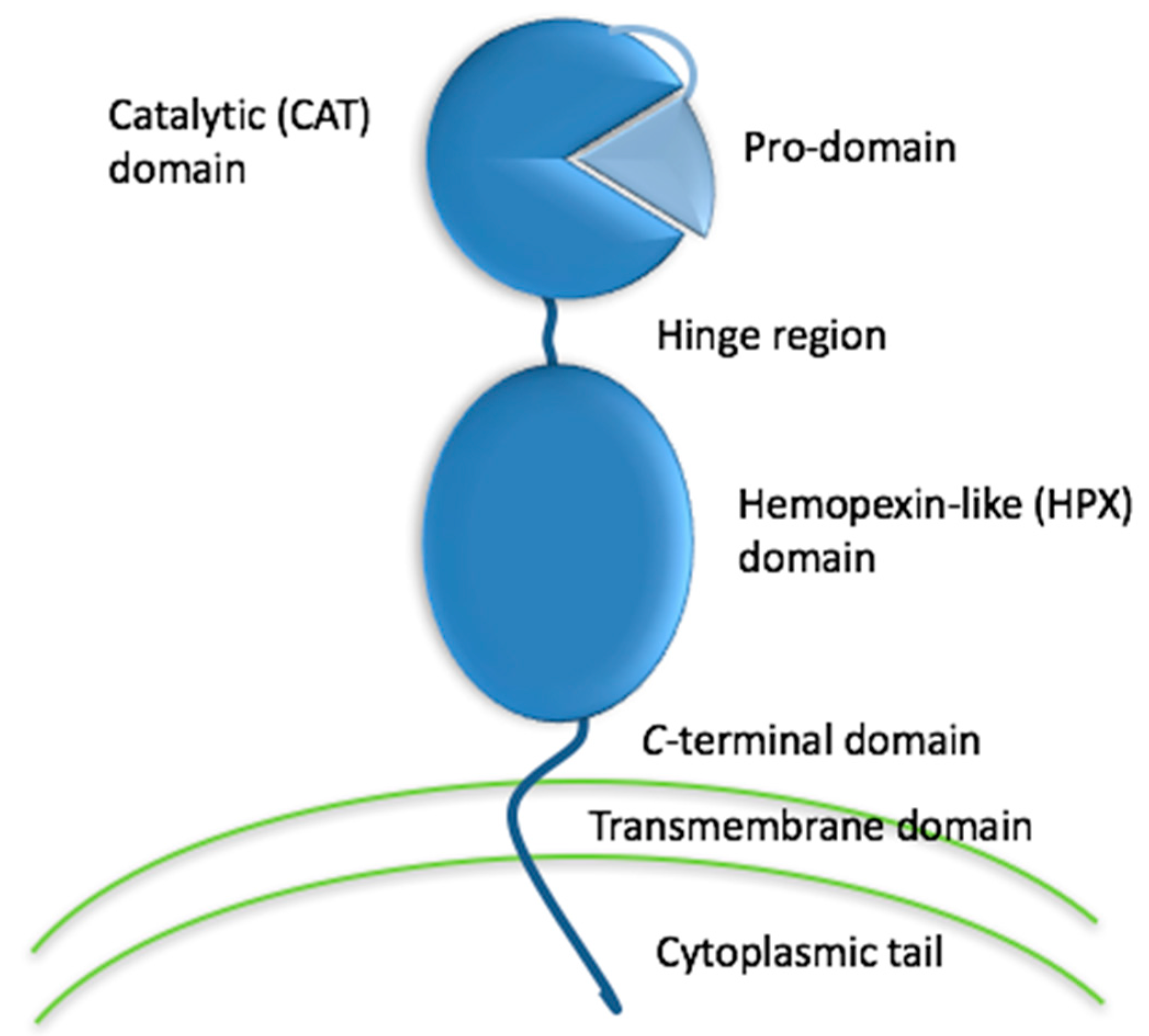
Activities of the matrix metalloproteinase (MMP) family (Figure 1) have long been correlated to disease initiation and progression [1,2,3,4,5,6]. For example, collagenolytic activity had been observed in tumor tissues, cells, and extracts since the late 1960s [7,8,9,10,11,12]. Initially, MMPs were considered as potential targets in cancer and osteoarthritis [13,14,15,16]. Subsequently, roles for MMPs emerged in neurodegenerative, infectious, and cardiovascular diseases [17]. MMPs can be present in three forms, latent (zymogen), activated, and activated but in complex with natural inhibitors such as tissue inhibitor of metalloproteinases (TIMPs). During conditions where active MMPs are overproduced, the regulation by TIMPs may be overwhelmed [18]. Developing inhibitors of the transcription, activation, or activity of MMPs thus represents an attractive therapeutic prospect [19].
Members of the MMP family are also essential for the maintenance of healthy processes, such as wound healing (e.g., MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-10, MMP-13, and MT1-MMP), inflammatory response (e.g., MMP-2, MMP-3, MMP-7, MMP-8, MMP-9, MMP-10, MMP-12, MT1-MMP, MMP-19, and MMP-28), and angiogenesis (e.g., MMP-2, MMP-9, and MT1-MMP) [5,20,21,22,23]. Thus, inhibiting all MMP family members at the same time would be detrimental, as indicated by early clinical trials conducted with broad spectrum MMP inhibitors [24,25]. In addition to problems of selectivity, the low bioavailability and poor metabolic profile of broad spectrum MMP inhibitors led to limited beneficial effect and thus did not justify the further pursuit of clinical trials [18]. Overall, the previous poor performance of MMP inhibitors in clinical trials has been attributed to (a) inhibition of other metalloenzymes, (b) lack of specificity within the MMP family, (c) poor pharmacokinetics, (d) dose-limiting side effects/toxicity, (e) in vivo instability, and (f) low oral availability/inability to assess inhibition efficacy [26,27]. New classes of MMP inhibitors need to address these factors. In addition, as the role of MMPs may change from detrimental to beneficial, or vice versa, during the course of disease, the mechanism and/or substrate profile of MMPs needs to be clearly identified when designing clinical trial protocols. Finally, there needs to be a precise understanding as to which MMPs are relevant in specific diseases, and the forms (active versus inactive) that these MMPs may be present in.
2. Problems Associated with MMP Inhibition, Real and Imagined
2.1. MSS Induced by Broad Spectrum Inhibitors
Initial strategies for designing MMP inhibitors focused on the necessity of Zn2+ in the catalytic mechanism [14,28]. Small molecules such as hydroxylamine, which has the property of chelating Zn2+, were utilized as templates to create inhibitors with strong Zn2+ chelating properties [13,14]. Unfortunately, clinical trials using broad spectrum inhibitors often led to severe side effects and had to be discontinued [18,29,30]. More specifically, the poor selectivity of broad spectrum inhibitors resulted in musculoskeletal triad (arthralgia, myalgia, tendinitis)/musculoskeletal syndrome (MSS) and gastrointestinal disorders [31]. Development of roughly thirty MMP inhibitor-based anti-arthritic drugs were discontinued in clinical trials due to the occurrence of MSS [32,33,34].
MSS does not appear to be caused by the off-target inhibition of a single enzyme, but rather inhibition of a combination of several MMPs and/or possibly other related enzymes [35]. Among enzymes initially proposed to play role in MSS development were MMP-1 [35], MMP-2 [36], MMP-9 [37], MT1-MMP [38], and ADAM family members [35]. MSS was also attributed to combined inhibition of MMP-1 and ADAM17 [39] or ADAM10 and ADAM17 [30]. A pyrimidine-2,4,6-trione derivative that inhibited MT1-MMP, MMP-2, and MMP-9 was not associated with MSS [40], and no MSS was observed in animals treated with the MMP-13 selective inhibitor ALS 1-0635, even at an ALS 1-0635 concentration 200-fold greater than that of marimastat known to induce this condition [41]. Thus, MMP-2, MMP-9, MMP-13, and MT1-MMP are no longer considered to be involved in MSS. MSS has been evaluated in rat models, which may have certain caveats (as rodents have MMP-1a as a homolog to human MMP-1 [42]), but broad spectrum MMP inhibition results in fibroproliferation affecting the patellar tendon and other intra- and periarticular connective tissues in rats, mimicking MSS.
2.2. Off-Target Interactions of Hydroxamic Acid-Based Inhibitors
Hydroxamic acid-based inhibitors designed for MMPs, such as BB-94, were found to inhibit ADAM family members, such as ADAM17, more potently then MMPs [43]. Hydroxamic acids have poor selectivity for Zn2+ over other divalent first row transition metals [44]. Activity-based protein profiling probes [45] were designed based on GM6001, a hydroxamic acid that chelates Zn2+ and was thought to selectively inhibit MMPs. When analyzing the metalloproteinases increased in invasive melanoma, the enzymes that were affinity-purified were neprilysin (an integral membrane metalloproteinase), leucine aminopeptidase, and dipeptidylpeptidase III [45]. These results suggest that nonspecific inhibition of metalloproteinases (not limited to the MMP family) may explain the toxicity observed with MMP inhibitors in clinical trials [33,46,47].
2.3. Metabolic Instability of Hydroxamic Acid-Based Inhibitors
In addition to a lack of selectivity, hydroxamic acids are problematic compounds due to their susceptibility to a variety of metabolic events. Hydroxamic acids can be hydrolyzed to carboxylic acids [50,51], which are typically less active than the parent compound. In addition, hydrolysis results in the production of hydroxylamine, a toxic byproduct [52]. Hydroxamic acids may also be reduced to amides [53], or O-glucuronylated or O-sulfated [51,54,55]. Hydroxamic acid stability in plasma is dependent upon esterase activity [56], specifically arylesterases and carboxylesterases [57]. The stability of hydroxamic acid-containing compounds is dependent upon (a) the accessibility of the esterase to the hydroxamic acid, (b) favorable or unfavorable interactions of the esterase with other constituents present in the compound, (c) compound hydrophilicity (logP), and (d) number of hydrogen bond donors in the compound [57]. Significant differences have also been noted in hydroxamic acid stability in rat versus human plasma [57].
2.4. Animal Models
Genetically modified (knockout) mice have been utilized to delineate the roles MMPs in normal physiological function and pathological states. Unfortunately, the identification of passenger mutations that accompanied MMP knockouts raised serious concerns as to the interpretation of results from disease models in which MMPs were implicated [58]. For example, the protection of Mmp7, Mmp8, or Mmp13 null mice from lipopolysaccharide lethality [59,60,61] may well be due to a Casp11-inactivating passenger mutation [58,62].
The passenger mutation effect may lead to questions about knockout animal results when, in fact, the results have been interpreted correctly. For example, MMP-8 knockout mice were used to determine the role of MMP-8 in wound healing [63]. MMP-8 was found to contribute to the resolution of inflammation and promote re-epithelization, aiding wound repair [63]. Transplantation of bone marrow from wild-type animals restored normal wound healing in MMP-8 knockout mice [63]. As indicated above, interpretation of activity alterations in Mmp8 null mice may be complicated due to Casp11 inactivation. However, the positive contribution of MMP-8 to diabetic wound healing was confirmed by chemical inhibition approaches [20,64].
High levels of MMP-8 have been correlated with fatal outcomes in sepsis [65,66]. Subsequently, MMP-8 knockout mice demonstrated increased survival compared to wild-type animals when subjected to the cecal ligation and puncture (CLP) model of sepsis [66]. Survival was also improved upon the application of an MMP inhibitor ((3R)-(+)-[2-(4-methoxybenzenesulfonyl)-1,2,3,4-tetrahydroisoquinoline-3-hydroxamate] or GlyΨ{PO2H-CH2}Ile-His-Lys-Gln THPI) to wild-type mice subjected to CLP [66,67], and transplantation of bone marrow from wild-type animals into MMP-8 knockout mice subjected to CLP compared with transplantation of bone marrow from MMP-8 knockout mice into wild-type animals subjected to CLP [68]. In the case of inhibition, the inhibitory compounds may not have been selective for just MMP-8. The effect in sepsis may be due to a combination of, for example, MMP-8 and MMP-13 inhibition. Along these lines, a bispecific nanobody that inhibited MMP-8 and tumor necrosis factor receptor 1 offered complete protection in mice subjected to endotoxemia and CLP [69].
Comparison of MMP-9 knockout and wild-type mice found 34 plasma glycoproteins significantly different between the two, including Ecm1, periostin, and fibronectin [70]. The differing proteome background between the MMP-9 knockout and wild-type mice suggested that disease models utilizing pharmacological inhibition versus knockout of target enzymes may have different downstream results [70]. Indeed, it was found that CD36 (a phagocytic marker in macrophages) was reduced post-myocardial infarction in animals treated with an MMP-9 inhibitor but increased post-myocardial infarction in MMP-9 knockout mice [70]. It is important to note that in the knockout mice, MMP-9 was completely removed, while the MMP inhibitor reduced MMP activity by 30% [70]. Such differences in relative MMP-9 inhibition, as well as the aforementioned differences in proteome background, can produce different outcomes in the two animal model systems.
An additional concern with the application of MMP knockout mice for disease models are compensatory effects on other MMPs. For example, in the aforementioned study of the role of MMP-8 in wound healing, MMP-8 knockout mice were found to have increased levels of MMP-9 in the wound area compared with wild-type mice [63]. In contrast, the expression pattern of MMP-13 had a more restricted distribution in the MMP-8 knockout mice compared with the wild-type mice [63]. MMP-13 knockout mice exhibited enhanced expression of MMP-8 in wound areas compared with wild-type mice [71]. These compensatory effects complicate interpretation of results in which inhibitor-treated wild-type animals are compared with knockout animals.
Beyond knockout studies, there are examples of where initial interpretation of animal models has provided incorrect results for evaluating the roles of MMPs in disease. Based on animal studies, MMP-9 was viewed as an appropriate target for modulating colitis [72]. Clinical trials of a monoclonal antibody that inhibits MMP-9 (GS-5745/andecaliximab) for ulcerative colitis (UC) were subsequently undertaken [73,74]. A later study utilizing three different mouse models of colitis indicated that MMP-9 upregulation was a consequence, rather than a cause, of intestinal inflammation [75]. Clinical remission or response was not observed in the UC trial [74]. In addition to the initial colitis animal model results being misleading, the lack of success of GS-5745/andecaliximab in the UC clinical trial may also have been due to the antibody having higher affinity for proMMP-9 (KD = 0.008–0.043 nM) compared with active MMP-9 (KD = 2.0–6.6 nM) [76], and thus much of the applied inhibitor may not have been bound to the active form of the enzyme.
2.5. The Complexity of the Protease Web
MMPs operate in linear pathways (direct substrates), amplification cascades, and regulatory circuits, resulting in a complex “protease web” (Figure 2) [33]. Therapeutic intervention should restore a normal MMP protease web, but is daunting in terms of possible side effects due to MMP inhibition [77]. It has been recommended that a systems biology approach be utilized to predict consequences of MMP inhibition [77]. Although many substrates have been identified and validated for MMPs [17,78], the kinetics for substrate turnover are often not reported. In addition, in vitro activity towards a substrate does not automatically indicate the processing of the substrate occurs in vivo. As has been noted, although MMPs can process chemokines, lack of proteolysis of a specific chemokine has not been observed in MMP knockout mice [79]. Since chemokine substrates of MMPs are often cleaved by multiple proteases, proteolytic redundancy may explain why chemokines are still processed in MMP knockout mice.
MT1-MMP has a significant role in angiogenesis, whereby it can exhibit both pro-angiogenic and anti-angiogenic behaviors [80,81,82,83,84,85,86,87,88]. These contrasting behaviors point to the importance of spatial and temporal expression of MT1-MMP. Similar concerns have arisen for MMP inhibition following ischemic stroke, where MMPs can disrupt the neurovascular system and blood–brain barrier, leading to cell death and cerebral edema, but can also contribute to neurorepair via angio-vasculogenesis, gliogenesis, and neurogenesis [89]. The same is true in neurological infectious diseases and multiple sclerosis, where inhibition during acute inflammation can be beneficial while inhibition during repair processes can be detrimental [6].
The complexity of the protease web is not necessarily a deterrent for inhibitor development. Angiotensin-converting enzyme (ACE) is well recognized for converting angiotensin I to angiotensin II and degrading bradykinin. ACE has an extensive substrate profile [90], including degrading acetyl-Pro-Gly-Pro. Acetyl-Pro-Gly-Pro serves to reduce neutrophilic inflammation and matrix degradation in chronic obstructive pulmonary disease (COPD) [91]. ACE also converts β-amyloid protein 42 (Aβ42, which is neurotoxic) to Aβ40 (which is neuroprotective) [92]. Despite a complex protease web for ACE, clinical applications of ACE inhibitors have existed for decades. A recent concern has arisen based on ACE inhibition resulting in increased brain amyloid deposition and apoptotic neurons in mice [93]. Whether this translates to human disease remains to be seen.
3. Development of Improved MMP Inhibitors
3.1. Creating Selective MMP Inhibitors
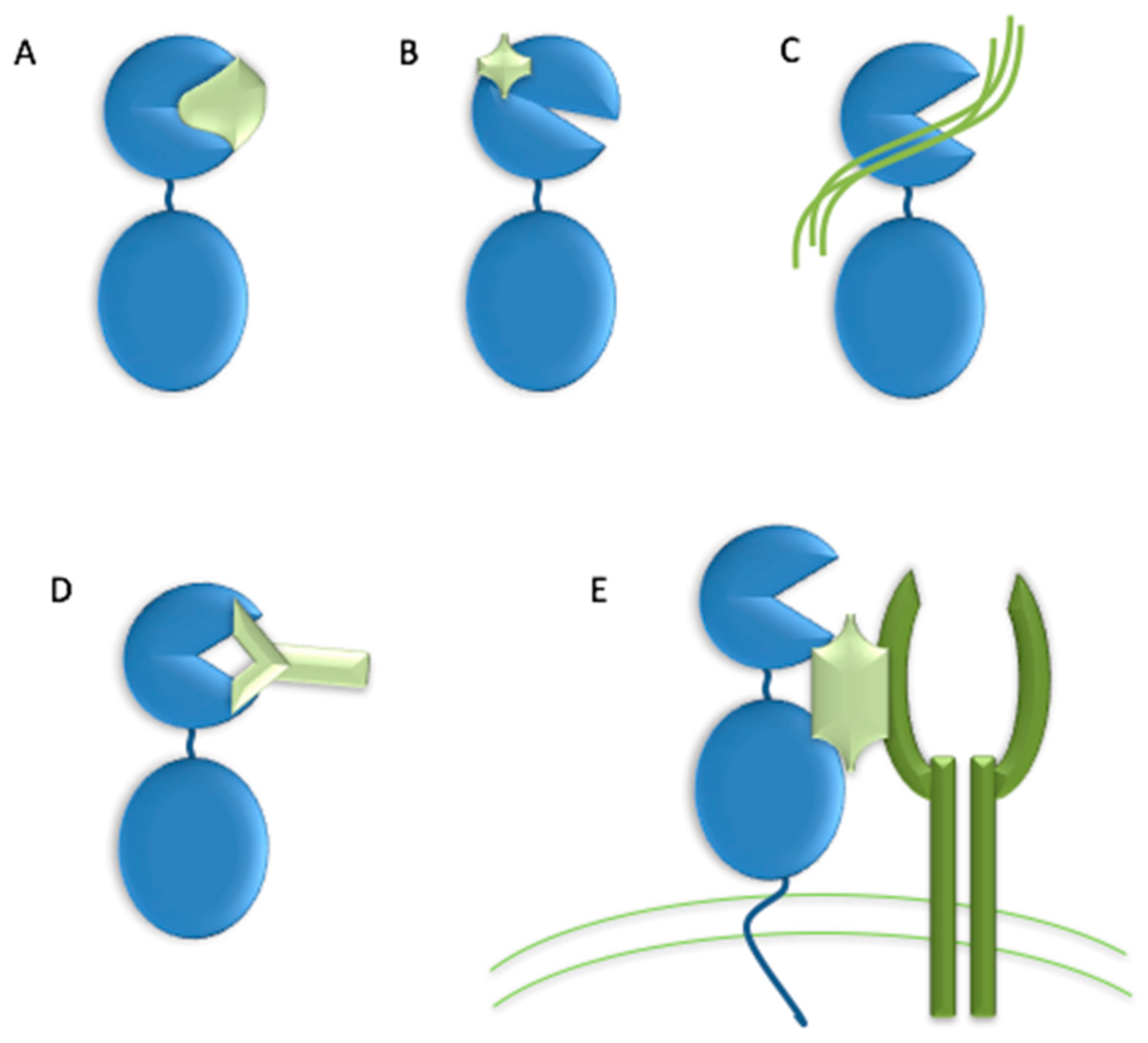
The high homology within the MMP family has made the development of selective inhibitors difficult. Ideally, an MMP inhibitor should exhibit a ~3 orders of magnitude difference in Ki between the target MMP and other MMPs [32,48]. Numerous strategies have been suggested for creating selective MMP inhibitors, including (a) application of endogenous-like inhibitors (TIMP analogs), (b) exosite targeting (i.e., HPX domain or MMP-2/MMP-9 fibronectin type II inserts), (c) combination of exosite binding and metal chelation, (d) function blocking antibodies, and (e) disrupting MMP interactions with cell surface binding partners (Figure 3) [94].
Modification of TIMPs has been explored in an attempt to design selective MMP inhibitors [95]. It was initially observed that mutation of Thr2 to Ala in TIMP-1 resulted in an inhibitor that was much less effective towards MMP-1 compared with MMP-2 and MMP-3 [96] Additional mutations of Thr2 furthered the concept that TIMPs could be selectively targeted [97]. Mutation of residues 2, 4, and/or 68 produced a TIMP-1 variant (Thr2Arg/Val4Ile) that effectively inhibited MMP-2 and MMP-3, but did not inhibit MMP-1 [98]. In turn, mutation of Thr98 to Leu converted TIMP-1 to an effective inhibitor of MT1-MMP [99], although multiple substitutions were required to fully explain why wild-type TIMP-1 was a poor inhibitor of MT1-MMP [99,100]. The TIMP-1 triple mutant Val4Ala/Pro6Val/Thr98Leu, a very effective inhibitor of MT1-MMP (Ki = 1.66 nM) [99], was fused to a glycosyl-phosphatidyl inositol anchor for targeting to the cell membrane [101]. Mutation of Thr2 to Gly resulted in a TIMP-1 variant with virtually complete selectivity between MMP-9 (Ki = 2.1 nM) and MMP-2 (Ki > 40 μM), while mutation of Thr2 to Arg and replacement of the TIMP-1 AB loop with the TIMP-2 AB loop produced a TIMP-1 variant with selectivity for MMP-2 and MMP-9 [100]. The TIMP-2 mutant Ser2Asp/Ser4Ala was effective against MMP-1 (Ki = 34 nM), while poorly inhibiting MMP-3, MMP-7, and MT1-MMP [102]. The TIMP-2 mutant Ser4Asp/Ile35Leu/Asn38Ser/Ser68Asp/Val71Ser/His97Ser/Thr99Phe had greatly enhanced specificity for MT1-MMP compared with MMP-9, while the TIMP-2 mutant Ser4Pro/Ile35Pro/Asn38Trp/Ser68Asn/His97Lys/Thr99Lys offered the opposite specificity [103]. Computational and mutational analysis of TIMP-2 interactions with multiple MMPs indicated that TIMP-2 was not optimized to bind to any specific MMP, and thus many affinity-enhancing mutations were possible [104]. Five mutations in TIMP-2, designed to optimize interactions with MT1-MMP (Ile35Met/Asn38Asp/Ser68Asn/Val71Gly/His97Arg), resulted in an inhibitor with Ki = 0.9 pM and selectivity over MMP-2 and MMP-10 [105]. While much work has focused on mutations in the N-terminal domain of TIMPs, the C-terminal domain also impacts MMP selectivity. Replacement of the TIMP-1 C-terminal domain with the TIMP-2 C-terminal produced a chimera (T1:T2) that was a much more effective inhibitor of MT1-MMP and MMP-19 than the wild-type TIMP-1 [106]. Subsequent studies led to the identification of residues in both the N- and C-termini of TIMP-1 that impacted binding to MMP-3 [107]. The importance of cooperativity between TIMP-1 N- and C-terminal domains was clearly demonstrated, and set the stage for additional considerations in the design of mutant TIMPs as selective metalloproteinase inhibitors. TIMP-2 mutagenesis has been used to engineer a bi-functional protein that binds to the αvβ3 integrin with KD = 130 nM and inhibited MT1-MMP with Ki = 77 pM [108]. Concerns exist about the short half-life of TIMPs in vivo, but conjugation of 20 kDa mPEG chains to TIMP-1 Lys residues dramatically extended the half-life in mouse plasma (from 1.1 to 28 h), yet did not significantly impact inhibitory activities either in vitro or in vivo [109]. An additional concern raised about TIMP-based inhibitors is in their ability to penetrate cartilage [110], as needed for osteoarthritis applications.
There are multiple examples of MMP-13 small molecule inhibitors that do not interact with the active site Zn2+, but rather bind via so called secondary binding/exosites or allosteric sites [78,111]. Many of these inhibitors take advantage of the MMP-13 P1’ subsite. However, distinct drawbacks to these inhibitors have been reported. For inhibitors presented as organic anions, binding to human organic anion transporter 3 resulted in nephrotoxicity [112]. Inhibitors possessing carboxylic acids may generate reactive metabolites through protein conjugation of the resulting acyl glucuronide [112,113]. Pyrimidine-2-carboxamide-4-one-based inhibitors have exhibited poor bioavailability, low volume of distribution, poor metabolic stability, and/or P450 3A4 inhibition [114]. Obtaining appropriate kinetic solubilities for MMP-13 inhibitors has proven challenging [115,116].
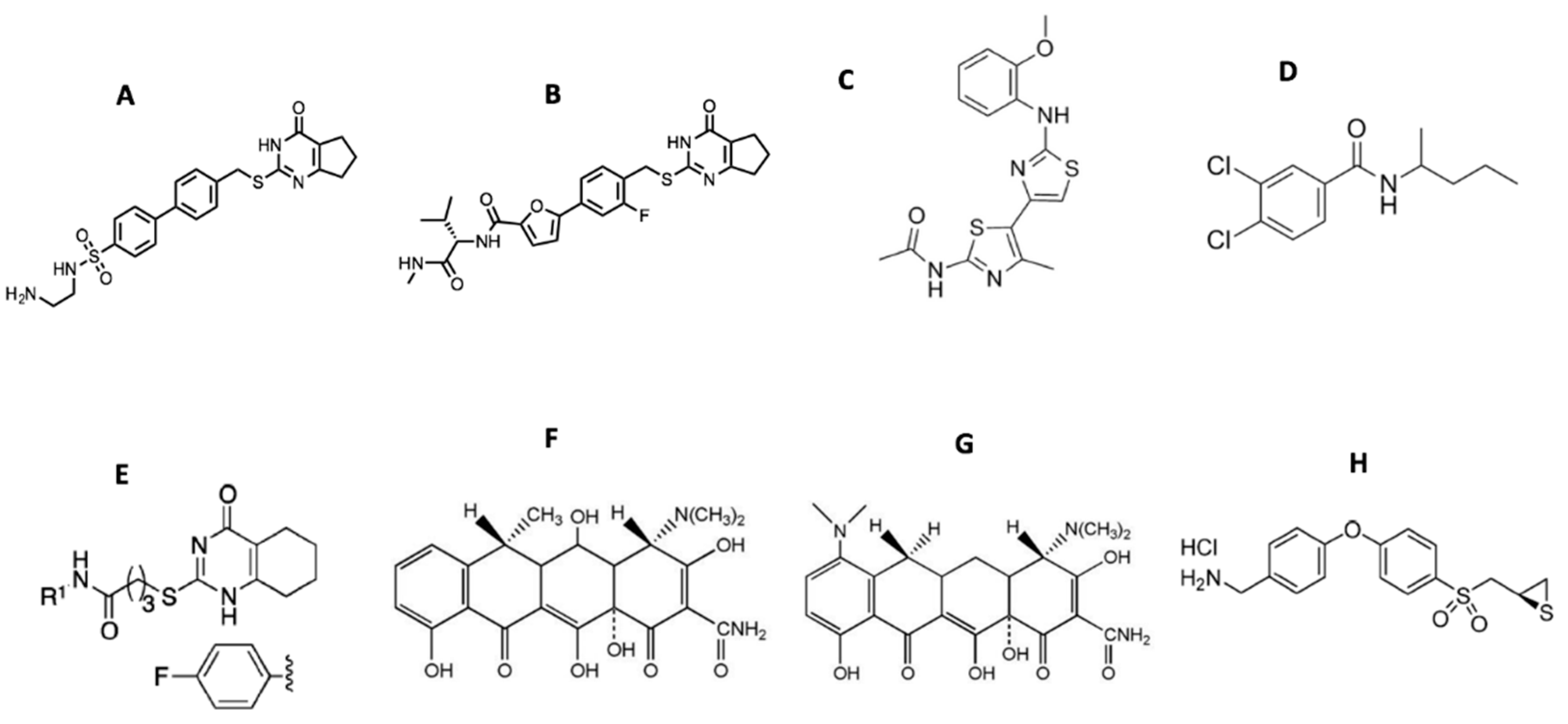
Starting from the 2-(arylmethylthio)-cyclopentapyrimidin-4-one scaffold identified in a high throughput screen [115,117,118], we synthesized more than 130 compounds with 10d and (S)-17b (as numbered when initially published) emerging as the two most promising inhibitors (Figure 4) [119,120]. Both compounds inhibited MMP-13 with low nanomolar IC50 values and exhibited an excellent selectivity profile within the MMP family (>170-fold and >1000-fold selectivity, respectively, compared to MMP-1, MMP-2, MMP-9, and MT1-MMP) [119]. Compound (S)-17b had a long plasma half-life (T1/2 = 2.93 h) and a low clearance rate (Cl = 0.18 mL/min/kg) after IV administration in rats [120]. Due to design considerations based on activity profiles and prior data, 10d and (S)-17b avoid many of the aforementioned pitfalls of MMP-13 inhibitors, particularly poor solubility and metabolic stability, as well as the potential for nephrotoxicity and generation of reactive metabolites.
Compound JNJ0966 [N-(2-((2-methoxyphenyl)amino)-4’-methyl-[4,5’-bithiazol]-2’-yl)acetamide] (Figure 4) inhibited the activation of proMMP-9 and the migration of HT1080 cells [121]. Intratumoral injections of compound NSC405020 [3,4-dichloro-N-(1-methylbutyl)benzamide] (Figure 4), an MT1-MMP HPX domain binder, reduced MCF7-β3/MT tumor xenograft size significantly [122]. Screening of oligomer libraries comprised of peptoid and peptide tertiary acid units led to the identification of chemically diverse MT1-MMP inhibitors [123]. One drawback is that these aforementioned compounds typically possess only modest affinities.
As discussed earlier, hydroxylamine was utilized as a template to create MMP inhibitors possessing hydroxamic acid moieties. Hydroxamic acids bind Zn2+ in a bidentate fashion [44]. Carboxylic acids bind Zn2+ in a monodentate fashion, and thus are more weakly bound to the MMP active site Zn2+ than hydroxamic acids [44]. The weaker binding to Zn2+ could reduce nonspecific inhibition. Phosphinic acids bind Zn2+ in a monodentate fashion, as one phosphinic oxygen binds the MMP active site Zn2+ while the second phosphinic oxygen hydrogen bonds to the active site Glu [44]. In addition to a possible advantage due to weaker Zn2+ binding, phosphinic acids may have improved metabolic stability compared with hydroxamic acids [48].
A variety of phosphorus-based peptides have been developed as MMP inhibitors, including phosphonates/phosphonic acids [124,125,126,127,128,129,130,131], phosphoramidates [132,133,134], phosphonamidates [133,135,136], and phosphinates/phosphinic peptides [136,137,138,139,140,141,142,143,144,145,146,147,148]. Phosphinates/phosphinic peptides contain a hydrolytically stable, tetrahedral phosphinic pseudo-dipeptide moiety which mimics the tetrahedral intermediate formed during Zn2+ metalloproteinase-catalyzed amide bond hydrolysis. Phosphinic peptides offer the possibility of interacting with both the primed and unprimed sides of the MMP active site cleft [43]. We have reported that phosphinic triple-helical peptides (THPs) behave as effective transition state analog inhibitors of collagenolytic MMPs [67,149,150,151,152]. These triple-helical peptide inhibitors (THPIs) have proven effective in mouse models of multiple sclerosis, sepsis, and myocardial infarction [67,153]. THPs have been found to be reasonably stable to general proteolysis, as observed in vitro in mouse, rat, and human serum and/or plasma and in vivo in mice and rats [154,155,156,157,158,159]. THPs were found to exhibit high stability (72%) when administered orally to rats [160].
Function blocking metallobodies (SDS3 and SDS4) were generated by immunization directed at the catalytic Zn2+ and enzyme surface epitopes in activated MMP-9 [161]. SDS3 was shown, in both prophylactic and therapeutic applications, to protect mice from dextran sodium sulfate-induced colitis [161].
Numerous MMP inhibitory antibodies have been developed [23,26,95,162]. Mouse mAb REGA-3G12, a selective inhibitor of MMP-9 [163], prevented interleukin-8-induced mobilization of hematopoietic progenitor cells in rhesus monkeys [164]. Two monoclonal anti-MMP-9 antibodies, AB0041 and AB0046, were shown to inhibit tumor growth and metastasis in a model of colorectal carcinoma [72]. A humanized version of AB0041, GS-5745/andecaliximab, was generated for use in clinical trials [72]. As mentioned earlier, GS-5745 inhibited proMMP-9 activation and non-competitively inhibited MMP-9 activity [76].
DX-2400, a selective, fully human MT1-MMP antibody inhibited metastasis in a breast cancer xenograft mouse model [165]. DX-2400 also enhanced tumor response to radiation therapy [166]. Two humanized scFv antibodies, CHA and CHL, generated against the MT1-MMP HPX domain inhibited HT1080 invasion of type I collagen [167]. Human scFv-Fc antibody E3 bound to the MT1-MMP CAT domain and inhibited type I collagen binding [168]. A second generation E3 clone (E2_C6) inhibited tumor growth and metastasis [168]. Human antibody Fab libraries were synthesized, where the Peptide G sequence (Phe-Ser-Ile-Ala-His-Glu) [169] was incorporated into complementarity-determining region (CDR)-H3 [170]. The resulting Fab 1F8 inhibited MT1-MMP CAT domain activity [170]. Screening of a phage displayed synthetic humanized Fab library led to the identification of Fab 3369, which inhibited the activity of the MT1-MMP CAT domain [171]. IgG 3369 treatment of MDA-MB-231 mammary orthotopic xenograft mice reduced lung metastases, collagen processing, and tumor density of CD31+ blood vessels [171].
mAb 9E8 inhibited MT1-MMP activation of proMMP-2, but not other MT1-MMP catalytic activities [172]. Antibody LOOPAb also inhibited MT1-MMP activation of proMMP-2 but not MT1-MMP collagenolysis [173]. These antibodies represent approaches in which a specific activity of an MMP is inhibited, which would prove especially beneficial in cases where maintaining certain activities of the MMP are desirable to support normal physiological functions.
The LEM-2/15 antibody was generated using a cyclic peptide mimicking the MT1-MMP CAT domain V-B loop (residues 218-233) [174]. A minimized Fab fragment of LEM-2/15 [175] significantly increased the ability of virally infected mice to fight off secondary Streptococcus pneumoniae bacterial infection [176].
It has been noted that antibody antigen binding sites are not complementary to the concave shape of catalytic clefts, as antigen binding sites are planar or concave [95]. To overcome this, the convex-shaped paratope of camelid antibodies was incorporated into the human antibody scaffold [177]. The resulting Fab 3A2 was a competitive MT1-MMP inhibitor which reduced metastasis in a melanoma mouse model [178].
There are several inhibitors that act by disrupting interactions of MMPs with cell surface binding partners. The MMP-9 HPX domain binds to B chronic lymphocytic leukemia (B-CLL) cells via the α4β1 integrin [179]. A variant of the MMP-9 HPX domain containing blades III and IV (B3B4) interacted with the α4β1 integrin, and a peptide derived from this region (peptide P3, residues 654-674, PFPGVPLDTHDVFQYREKAYFC) inhibited cell adhesion. A truncated version of peptide P3 (P3a, FPGVPLDTHDVFQYREK) inhibited B-CLL and MEC-1 cell adhesion to proMMP-9 and inhibited B-CLL cell transendothelial migration and intracellular survival signals. The compound N-(4-fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanamide (Figure 4) prevented association of proMMP-9 with the α4β1 integrin and CD44, resulting in the dissociation of epidermal growth factor receptor (EGFR) from the β1 integrin subunit and CD44 [180].
3.2. Timing of MMP Inhibition
Often overlooked is that the timing of MMP inhibitor application may be critical to achieve the desired therapeutic effect. Application of marimastat in combination with gemcitabine significantly improved survival in pancreatic cancer patients with disease confined to the pancreas [182]. Presurgical treatment with the oral MMP inhibitor SD-7300 (N-hydroxy-1-(2-methoxyethyl)-4-[4-[4-(trifluoromethoxy)phenoxy]piperidin-1-yl]sulfonylpiperidine-4-carboxamide, with high inhibitory activity towards MMP-2, MMP-9, and MMP-13) improved survival from 67 to 92% in a mouse breast cancer model and significantly decreased the risk of recurrence [183]. The “window of opportunity” for MMP inhibitor application in cancer is often in premetastatic disease [30,184,185,186]. In the future, MMP inhibitors may need to be considered in metastasis prevention trials.
MMP-9 is a key contributor to the “angiogenic switch” during carcinogenesis of pancreatic islets [187]. However, MMP-9 deficiency in pancreatic ductal adenocarcinoma (PDAC) mouse models resulted in more invasive tumors and an increase in desmoplastic stroma [188]. The absence of MMP-9 led to increased interleukin 6 levels in the bone marrow, which activated tumor cell STAT3 signaling and promoted PDAC invasion and metastasis [188]. Thus, MMP-9 represents an anti-target in the later stage of pancreatic cancer.
3.3. Side Effects of Selective MMP Inhibitors
The monoclonal anti-MMP-9 antibody GS-5745/andecaliximab has been evaluated under several clinical trials. The outcome of a randomized placebo controlled phase 1b single and multiple ascending dose-ranging clinical trial on 72 patients diagnosed with moderately to severely active UC showed that GS-5745/andecaliximab was safe and well tolerated [73]. A phase 2/3 UC study with 165 patients treated over 8 weeks further indicated that GS-5745/andecaliximab was well tolerated [74]. A phase 1b trial investigating the safety, pharmacokinetics, and disease-related outcomes for 15 rheumatoid arthritis patients demonstrated that GS-5745/andecaliximab was safe, with adverse events that were only grade 1 or 2 in severity and no indication of MSS [189]. A phase 1 GS-5745/andecaliximab dose-finding trial of 13 patients with advanced solid tumors found no dose-limiting toxicity at any concentration (200, 600, or 1800 mg IV every 2 weeks), with adverse events that were only grade 1 or 2 in severity [190]. These clinical trials provide further evidence that selective MMP inhibitors can be developed without inducing MSS.
4. The Outlook for Current MMP Inhibitors
The lack of success in early MMP clinical trials has created a strong negative bias toward MMP inhibition as a clinical strategy. In actuality, MMP clinical trials were undertaken prematurely [191,192], due to insufficient knowledge of MMP biology and the choice of hydroxamic acid as the active component of the inhibitors. Unique modes of action have been used to develop the next generation of MMP inhibitors. Clinical trials utilizing antibodies have provided evidence that selective MMP inhibitors do not induce MSS. The therapeutic potential of anti-MMP antibodies has yet to be realized, with one concern being limited extravascular distribution [189,193]. Antibodies are also subject to proteolysis, may be removed from circulation rapidly, and are costly. Nonetheless, antibodies have provided truly selective, high affinity MMP inhibitors. In addition, selective, high affinity inhibitors can be developed for MMPs based on triple-helical structure. Triple-helical peptide inhibitors (THPIs) have excellent pharmacokinetic properties compared with other peptide-based therapeutics.
4.1. MMP Inhibition and the Immune System
It has been proposed that the major role of MMPs is in modulation of inflammatory and immune processes [17,79]. MMP processing can either activate or inhibit a variety of chemokines and cytokines [79,194]. For example, MMP-9 cleaves interleukin-8 and increases the activity of the chemokine 10-fold for neutrophil activation and chemotaxis [195]. In turn, neutrophils release a TIMP-free form of MMP-9 which promotes angiogenesis [196]. Overall, MMPs can cleave virtually all human chemokines [197]. A better understanding of MMPs in the immune system and in inflammatory diseases may be necessary before proceeding with cancer-based clinical trials [191].
Insight into the roles of MMPs in the immune system has revealed potential applications of MMP inhibitors to improve immunotherapy. Monoclonal MMP-9 antibody AB0046, which inhibited tumor growth and metastasis in a surgical orthotopic xenograft model of colorectal carcinoma [72], improved immune responses to tumors, as the inhibition of MMP-9 reversed MMP-9 inactivation of T-cell chemoattractant CXCR3 ligands (CXCL9, CXCL10, and CXCL11) [198]. MT1-MMP shed tumor cell MHC class I chain-related molecule A (MICA) [199]. Engagement of MICA to NKG2D stimulated natural killer (NK) and T-cell antitumor activity [199]. Protection of MICA stimulated antitumor immunity and reduced metastasis in a humanized melanoma mouse model [200].
Examination of tumor cryosections in an MT1-MMP antibody treated MDA-MB-231 triple-negative breast cancer xenograft mouse model revealed an increased density of iNOS+ cells (a marker of anti-tumor M1 tumor-associated macrophages) and Granzyme B+ cells [171]. Application of an MT1-MMP antibody to a 4T1 triple-negative breast cancer mouse model shifted macrophages towards the antitumor M1-like phenotype and reduced activated TGFβ (an immunosuppressive cytokine) [166].
4.2. MMP Inhibitors in the Clinic and in Clinical Trials
There is an established history of clinically successful MMP inhibitors. Periostat, which is doxycycline (Figure 4) formulated at a sub-antimicrobial dose, is a U.S. Food and Drug Administration (FDA)-approved MMP inhibitor for periodontal disease treatment [201,202,203,204]. Doxycycline is a broad-spectrum MMP inhibitor with IC50 values in the range of 2–50 μM [18]. Tetracyclines have shown promise in other MMP indications. Treatment of 16 relapsing–remitting multiple sclerosis patients with a doxycycline/interferon combination for 4 months (ClinicalTrials.gov Identifier NCT00246324) reduced brain lesions and MMP-9 serum levels and improved Expanded Disability Status Scale values [205]. The treatment was considered effective, safe, and well tolerated [205,206]. In other trials for relapsing–remitting multiple sclerosis, minocycline (Figure 4) treatment resulted in no new active lesions after 1 month with detection up to 6 months for 10 patients [207] while conversion from a first demyelinating event (clinically isolated syndrome) to multiple sclerosis was lowered by 18.5% over 6 months but was not lowered over 24 months for 72 patients [208]. Combination treatment of minocycline with glatiramer acetate, compared with glatiramer alone, in 44 patients showed the combination therapy reduced the number of lesions by 63–65% and lowered the risk of relapse after 8–9 months [209]. The treatment was considered safe and well tolerated [209]. Additional clinical trials using minocycline for multiple sclerosis treatment are reportedly upcoming [6]. In similar fashion to doxycycline, minocycline is a broad-spectrum MMP inhibitor, with IC50 values in the 100–300 μM range [18].
Minocycline has also been examined for improving neurological outcomes following stroke [210,211]. MMP-9 levels are increased following cerebral ischemia, and are further enhanced by tissue plasminogen activator (tPA) stroke treatment [212,213,214]. Administered within 6 h of stroke symptom onset in 60 patients, minocyline was well tolerated up to 10 mg/kg intravenous administration and had a half-life of 24 h [210]. Minocycline decreased MMP-9 levels at 24 h and to baseline in 72 h in 36 patients treated with tPA [211]. Incyclinide (a.k.a. Metastat, COL-3), a chemically modified tetracycline with no antibiotic activity, showed a significant regression of AIDS-related Kaposi’s sarcoma with a significant reduction of MMP-2 and MMP-9 plasma levels [215].
Clinical trials using anti-MMP antibodies have moved forward. GS5745/andecaliximab in combination with mFOLFOX6 (oxaliplatin, leucovorin, and 5-fluorouracil) was utilized in a phase I clinical trial of 40 patients with HER2-negative gastric and gastroesophageal junction adenocarcinoma [190]. The overall response rate for all patients was 47.5% (7.5% complete response and 40% partial response), with no observed MSS [190]. As described earlier, one concern about GS5745/andecaliximab is the much higher affinity for proMMP-9 compared with active MMP-9 [76].
Topical application of MMP inhibitors may also prove to be efficacious. In human diabetic foot ulcers, levels of active MMP-8 and MMP-9 are increased [216]. MMP-9 has been identified as a limiting factor in healing, while MMP-8 facilitates wound repair [20,64,216]. The compound (R)-ND-336 (Figure 4) selectively inhibits MMP-2, MMP-9, and MT1-MMP with Ki < 100 nM, while weakly inhibiting MMP-8 [216]. Thus, (R)-ND-336 has the desired inhibitory profile for application to diabetic foot ulcers. Additionally, (R)-ND-336 is a slow binding inhibitor of MMP-2, MMP-9, and MT1-MMP with a long residence time [216]. (R)-ND-336 is more efficacious than becaplermin in animal models of wound healing, and thus represents a promising topical treatment for diabetic foot ulcers [64,216]. Clinical trials for (R)-ND-336 utility are anticipated (https://ideacenter.nd.edu/news-events/news/new-hope-for-the-treatment-of-diabetic-foot-ulcers/).
A related, promising therapeutic approach is the use of MMPs to activate prodrugs and imaging agents or facilitate drug delivery [27,217,218,219,220,221]. A concern with these approaches is the “selectivity” of the peptide sequences used for activation [217,218,219,222]. ICT2588 is a vascular disrupting agent designed to be activated by MT1-MMP, containing an Arg-Ser-Cit-Gly~Hof-Tyr-Leu-Tyr sequence (where Cit is citrulline and Hof is homophenylalanine) with azademethylcolchicine at the C-terminus and fluorescein (FITC) at the N-terminus [223,224]. Azademethylcolchicine functions as the vascular disrupting agent. Following hydrolysis at the Gly~Hof bond by MT1-MMP, exopeptidase activity removes the remaining Hof-Tyr-Leu-Tyr sequence to liberate azademethylcolchicine [223]. ICT2588 produced a 90% decrease in functional tumor vasculature in HT1080 tumor-bearing mice, and co-administration of ICT2588 and doxorubicin was significantly more effective in reducing tumor volume compared with either ICT2588 or doxorubicin alone [223]. Conjugation of a variant of ICT2588 (FITC-βAla-Cys-Arg-Ser-Cit-Gly~Hof-Tyr-Leu-Tyr-azademethylcolchicine) with a cross-linked iron oxide (CLIO) nanocarrier resulted in a theranostic that allowed for magnetic resonance imaging of drug delivery and accumulation in tumors [220,225]. Co-administration of the CLIO-ICT with temozolomide resulted in decreased tumor size and increased survival in glioblastoma xenograft mice [226]. ICT2588 was scheduled to begin Phase 1 clinical trials in 2018 (http://www.incanthera.com/what-we-do/technology-pipeline/).
5. Conclusions
There is considerable promise in the recent generation of MMP inhibitors. Valuable lessons were learned from prior clinical trial failures, including the need for selective and metabolically stable inhibitors. Proteomics approaches have better defined the impact of MMP activity on biological systems. Short-term indications may be needed when anti-target activities are of concern. While there is great reliance on mouse models for examining MMP in vivo behaviors, it has been recognized that animal models may not clearly reflect human conditions and mechanism of disease. Consideration of MMP secondary binding sites (exosites) offers the best opportunity for development of selective inhibitors. The FDA approval of an MMP inhibitor and the advancement of numerous MMP inhibitors to clinical trials clearly indicates that it is time to depart from the dogma of viewing MMP inhibition as intractable.
Author Contributions
Writing—original draft preparation, review, and editing, G.B.F.
Funding
MMP inhibitor studies in the author’s laboratory have been supported by the National Institutes of Health (CA098799, CA239214, AR063795, MH078948, and NHLBI Contract 268201000036C-0-0-1), the James and Esther King Biomedical Research Program (8JK01), the US–Israel Binational Science Foundation (2015180), the Multiple Sclerosis National Research Institute, the Center for Molecular Biology & Biotechnology at Florida Atlantic University, and the State of Florida, Executive Office of the Governor’s Department of Economic Opportunity.
Acknowledgments
Conflicts of Interest
The author is Vice President of MMP Biopharma, Inc.
References
- Fingleton, B. Matrix metalloproteinases as valid clinical targets. Curr. Pharm. Design 2007, 13, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Van den Steen, P.E.; Sang, Q.-X.A.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Nagase, H. Progress in matrix metalloproteinase research. Mol. Aspects Med. 2008, 29, 290–308. [Google Scholar] [CrossRef] [Green Version]
- Candelario-Jalil, E.; Yang, Y.; Rosenberg, G.A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 2009, 158, 983–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Muri, L.; Leppert, D.; Grandgirard, D.; Leib, S.L. MMPs and ADAMs in neurological infectious diseases and multiple sclerosis. Cell Mol. Life Sci. 2019, 76, 3097–3116. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.M.; Williams, D.C. In Vitro Evidence of Neutral Collagenase Activity in an Invasive Mammalian Tumour. Nature 1969, 221, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.C.; Levy, B.M.; Simpson, J.W. Collagenolytic Activity of Sarcoma Tissues in Culture. Nature 1970, 228, 366–367. [Google Scholar] [CrossRef]
- Dresden, M.H.; Heilman, S.A.; Schmidt, J.D. Collagenolytic Enzymes in Human Neoplasms. Cancer Res. 1972, 32, 993–996. [Google Scholar]
- Labrosse, K.R.; Liener, I.E.; Hargrave, P.A. A sensitive assay for collagenolytic activity using tritiated collagen. Anal. Biochem. 1976, 70, 218–223. [Google Scholar] [CrossRef]
- Kuettner, K.E.; Soble, L.; Croxen, R.L.; Marczynslea, B.; Hiti, J.; Harper, E. Tumor cell collagenase and its inhibition by a cartilage-derived protease inhibitor. Science 1977, 196, 653–654. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; DeClerck, Y.A. Destruction of Extracellular Matrices Containing Glycoproteins, Elastin, and Collagen by Metastatic Human Tumor Cells. Cancer Res. 1980, 40, 3222–3227. [Google Scholar] [PubMed]
- Beckett, R.P.; Whittaker, M. Matrix metalloproteinase inhibitors 1998. Exp. Opin. Ther. Patents 1998, 8, 259–282. [Google Scholar] [CrossRef]
- Whittaker, M.; Floyd, C.D.; Brown, P.; Gearing, A.J.H. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999, 99, 2735–2776. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Welsch, D.J.; Pelletier, J.-P. Metalloproteases and inhibitors in arthritic diseases. Best Practice Res. Clin. Rheumatol. 2001, 15, 805–829. [Google Scholar] [CrossRef] [PubMed]
- Burrage, P.S.; Brinckerhoff, C.E. Molecular Targets in Osteoarthritis: Metalloproteinases and Their Inhibitors. Curr. Drug Targets 2007, 8, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Overall, C.M.; Dufour, A. Matrix metalloproteinases in the CNS: Interferons get nervous. Cell Mol. Life Sci. 2019, 76, 3083–3095. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.C.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M.; Lopez-Otin, C. Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef]
- Gao, M.; Nguyen, T.T.; Suckow, M.A.; Wolter, W.R.; Gooyit, M.; Mobashery, S.; Chang, M. Acceleration of diabetic wound healing using a novel protease-anti-protease combination therapy. Proc. Natl. Acad. Sci. USA 2015, 112, 15226–15231. [Google Scholar] [CrossRef]
- Caley, M.P.; Martins, V.L.; O’Toole, E.A. Metalloproteinases and Wound Healing. Adv. Wound Care (New Rochelle) 2015, 4, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fingleton, B. Matrix metalloproteinases as regulators of inflammatory processes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2036–2042. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. Mechanisms of action of novel drugs targeting angiogenesis-promoting matrix metalloproteinases. Front. Immunol. 2019, 10, 1278. [Google Scholar] [CrossRef] [PubMed]
- Hadler-Olsen, E.; Winberg, J.O.; Uhlin-Hansen, L. Matrix metalloproteinases in cancer: Their value as diagnostic and prognostic markers and therapeutic targets. Tumour Biol. 2013, 34, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.S.; Overall, C.M. Updated biological roles for matrix metalloproteinases and new "intracellular" substrates revealed by degradomics. Biochemistry 2009, 48, 10830–10845. [Google Scholar] [CrossRef]
- Sela-Passwell, N.; Rosenblum, G.; Shoham, T.; Sagi, I. Structural and functional bases for allosteric control of MMP activities: Can it pave the path for selective inhibition? Biochim. Biophys. Acta 2010, 1803, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Tauro, M.; McGuire, J.; Lynch, C.C. New approaches to selectively target cancer-associated matrix metalloproteinase activity. Cancer Metastasis Rev. 2014, 33, 1043–1057. [Google Scholar] [CrossRef]
- Beckett, R.P.; Davidson, A.H.; Drummond, A.H.; Huxley, P.; Whittaker, M. Recent advances in matrix metalloproteinase inhibitor research. Drug Design Today 1996, 1, 16–26. [Google Scholar] [CrossRef]
- Radisky, E.S.; Raeeszadeh-Sarmazdeh, M.; Radisky, D.C. Therapeutic Potential of Matrix Metalloproteinase Inhibition in Breast Cancer. J. Cell. Biochem. 2017, 118, 3531–3548. [Google Scholar] [CrossRef] [Green Version]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Rudek, M.A.; Venitz, J.; Figg, W.D. Matrix Metalloproteinase Inhibitors: Do They Have A Place in Anticancer Therapy? Pharmacotherapy 2002, 22, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M.; Kleifeld, O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br. J. Cancer 2006, 94, 941–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overall, C.M.; Kleifeld, O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Fingleton, B. MMPs as therapeutic targets--still a viable option? Semin. Cell Dev. Biol. 2008, 19, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.T. The importance of estimating the therapeutic index in the development of matrix metalloproteinase inhibitors. Cardiovasc. Res. 2006, 69, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Martignetti, J.A.; Aqeel, A.A.; Sewairi, W.A.; Boumah, C.E.; Kambouris, M.; Mayouf, S.A.; Sheth, K.V.; Eid, W.A.; Dowling, O.; Harris, J.; et al. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat. Genet. 2001, 28, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Shipley, J.M.; Bergers, G.; Berger, J.E.; Helms, J.A.; Hanahan, D.; Shapiro, S.D.; Senior, R.M.; Werb, Z. MMP-9/Gelatinase B Is a Key Regulator of Growth Plate Angiogenesis and Apoptosis of Hypertrophic Chondrocytes. Cell 1998, 93, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Holmbeck, K.; Bianco, P.; Caterina, J.; Yamada, S.; Kromer, M.; Kuznetsov, S.A.; Mankani, M.; Robey, P.G.; Poole, A.R.; Pidoux, I.; et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 1999, 99, 81–92. [Google Scholar] [CrossRef]
- Becker, D.P.; Barta, T.E.; Bedell, L.J.; Boehm, T.L.; Bond, B.R.; Carroll, J.; Carron, C.P.; DeCrescenzo, G.A.; Easton, A.M.; Freskos, J.N.; et al. Orally active MMP-1 sparing α-tetrahydropyranyl and α-piperidinyl sulfone matrix metalloproteinase (MMP) inhibitors with efficacy in cancer, arthritis, and cardiovascular disease. J. Med. Chem. 2010, 53, 6653–6680. [Google Scholar] [CrossRef]
- Devy, L.; Dransfield, D.T. New strategies for the next generation of matrix-metalloproteinase inhibitors: Selectively targeting membrane-anchored MMPs with therapeutic antibodies. Biochem. Res. Int. 2011, 2011, 191670. [Google Scholar] [CrossRef]
- Baragi, V.M.; Becher, G.; Bendele, A.M.; Biesinger, R.; Bluhm, H.; Boer, J.; Deng, H.; Dodd, R.; Essers, M.; Feuerstein, T.; et al. A new class of potent matrix metalloproteinase 13 inhibitors for potential treatment of osteoarthritis: Evidence of histologic and clinical efficacy without musculoskeletal toxicity in rat models. Arthritis Rheum. 2009, 60, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.J.; Luo, C.; O’Callaghan, K.; Hinds, P.W.; Covic, L.; Kuliopulos, A. Matrix metalloproteinase-1a promotes tumorigenesis and metastasis. J. Biol. Chem. 2012, 287, 24330–24338. [Google Scholar] [CrossRef] [PubMed]
- Cuniasse, P.; Devel, L.; Makaritis, A.; Beau, F.; Georgiadis, D.; Matziari, M.; Yiotakis, A.; Dive, V. Future challenges facing the development of specific active-site-directed synthetic inhibitors of MMPs. Biochimie 2005, 87, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Puerta, D.T.; Cohen, S.M. A bioinorganic perspective on matrix metalloproteinase inhibition. Curr. Topics Med. Chem. 2004, 4, 1551–1573. [Google Scholar] [CrossRef]
- Saghatelian, A.; Jessani, N.; Joseph, A.; Humphrey, M.; Cravatt, B.F. Activity-based probes for the proteomic profiling of metalloproteases. Proc. Natl. Acad. Sci. USA 2004, 101, 10000–10005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vihinen, P.; Ala-aho, R.; Kahari, V.-M. Matrix metalloproteinases as therapeutic targets in cancer. Curr. Cancer Drug Targets 2005, 5, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Fingleton, B. Matrix metalloproteinases: Roles in cancer and metastasis. Front. Biosci. 2006, 11, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.A.; Major Jourden, J.L.; Miller, M.T.; Cohen, S.M. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta 2010, 1803, 72–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, A.; Romero-Perez, D.; Jacobsen, J.A.; Villarreal, F.J.; Cohen, S.M. Zinc-binding groups modulate selective inhibition of MMPs. ChemMedChem 2008, 3, 812–820. [Google Scholar] [CrossRef]
- Sanderson, L.; Taylor, G.W.; Aboagye, E.O.; Alao, J.P.; Latigo, J.R.; Coombes, R.C.; Vigushin, D.M. Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin a after intraperitoneal administration to mice. Drug Metab. Dispos. 2004, 32, 1132–1138. [Google Scholar] [CrossRef]
- Du, L.; Musson, D.G.; Wang, A.Q. Stability studies of vorinostat and its two metabolites in human plasma, serum and urine. J. Pharm. Biomed. Anal. 2006, 42, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Weisburger, J.H.; Weisburger, E.K. Biochemical formation and pharmacological, toxicological, and pathological properties of hydroxylamines and hydroxamic acids. Pharmacol. Rev. 1973, 25, 1–66. [Google Scholar] [PubMed]
- Kitamura, S.; Sugihara, K.; Tatsumi, K. Reductase activity of aldehyde oxidase toward the carcinogen N-hydroxy-2-acetylaminofluorene and the related hydroxamic acids. Biochem. Mol. Biol. Int. 1994, 34, 1197–1203. [Google Scholar] [PubMed]
- Mulder, G.J.; Meerman, J.H. Sulfation and glucuronidation as competing pathways in the metabolism of hydroxamic acids: The role of N,O-sulfonation in chemical carcinogenesis of aromatic amines. Environ. Health Perspect. 1983, 49, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Lee, C.; Kook, Y.J.; Oh, S.J.; Kang, J.S.; Kim, H.J.; Han, G. Improving potency and metabolic stability by introducing an alkenyl linker to pyridine-based histone deacetylase inhibitors for orally available RUNX3 modulators. Eur. J. Med. Chem. 2017, 126, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Flipo, M.; Charton, J.; Hocine, A.; Dassonneville, S.; Deprez, B.; Deprez-Poulain, R. Hydroxamates: Relationships between structure and plasma stability. J. Med. Chem. 2009, 52, 6790–6802. [Google Scholar] [CrossRef]
- Hermant, P.; Bosc, D.; Piveteau, C.; Gealageas, R.; Lam, B.; Ronco, C.; Roignant, M.; Tolojanahary, H.; Jean, L.; Renard, P.Y.; et al. Controlling Plasma Stability of Hydroxamic Acids: A MedChem Toolbox. J. Med. Chem. 2017, 60, 9067–9089. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Hulpiau, P.; Martens, L.; Vandenbroucke, R.E.; Van Wonterghem, E.; Perry, S.W.; Bruggeman, I.; Divert, T.; Choi, S.M.; Vuylsteke, M.; et al. Passenger Mutations Confound Interpretation of All Genetically Modified Congenic Mice. Immunity 2015, 43, 200–209. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroucke, R.E.; Dejonckheere, E.; Van Lint, P.; Demeestere, D.; Van Wonterghem, E.; Vanlaere, I.; Puimège, L.; Van Hauwermeiren, F.; De Rycke, R.; Mc Guire, C.; et al. Matrix metalloprotease 8-dependent extracellular matrix cleavage at the blood-CSF barrier contributes to lethality during systemic inflammatory diseases. J. Neurosci. 2012, 32, 9805–9816. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Dejonckheere, E.; Van Hauwermeiren, F.; Lodens, S.; De Rycke, R.; Van Wonterghem, E.; Staes, A.; Gevaert, K.; López-Otin, C.; Libert, C. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol. Med. 2013, 5, 1000–1016. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroucke, R.E.; Vanlaere, I.; Van Hauwermeiren, F.; Van Wonterghem, E.; Wilson, C.; Libert, C. Pro-inflammatory effects of matrix metalloproteinase 7 in acute inflammation. Mucosal Immunol. 2014, 7, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Miura, M.; Jung, Y.K.; Zhu, H.; Li, E.; Yuan, J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 1998, 92, 501–509. [Google Scholar] [CrossRef]
- Gutiérrez-Fernández, A.; Inada, M.; Balbín, M.; Fueyo, A.; Pitiot, A.S.; Astudillo, A.; Hirose, K.; Hirata, M.; Shapiro, S.D.; Noël, A.; et al. Increased inflammation delays wound healing in mice deficient in collagenase-2 (MMP-8). FASEB J. 2007, 21, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.I.; Nguyen, T.T.; Peng, Z.; Chang, M. Targeting MMP-9 in Diabetic Foot Ulcers. Pharmaceuticals 2019, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Lauhio, A.; Hästbacka, J.; Pettilä, V.; Tervahartiala, T.; Karlsson, S.; Varpula, T.; Varpula, M.; Ruokonen, E.; Sorsa, T.; Kolho, E. Serum MMP-8, -9 and TIMP-1 in sepsis: High serum levels of MMP-8 and TIMP-1 are associated with fatal outcome in a multicentre, prospective cohort study. Hypothetical impact of tetracyclines. Pharmacol. Res. 2011, 64, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Solan, P.D.; Dunsmore, K.E.; Denenberg, A.G.; Odoms, K.; Zingarelli, B.; Wong, H.R. A novel role for matrix metalloproteinase-8 in sepsis. Crit. Care Med. 2012, 40, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, M.; Tokmina-Roszyk, D.; Onwuha-Ekpete, L.; Harmon, K.; Robichaud, T.; Fuerst, R.; Stawikowska, R.; Steffensen, B.; Roush, W.R.; Wong, H.; et al. Second Generation Triple-Helical Peptide Transition State Analog Matrix Metalloproteinase Inhibitors. J. Med. Chem. 2017, 60, 3814–3827. [Google Scholar] [CrossRef]
- Atkinson, S.J.; Nolan, M.; Klingbeil, L.; Harmon, K.; Lahni, P.; Zingarelli, B.; Wong, H.R. Intestine-Derived Matrix Metalloproteinase-8 Is a Critical Mediator of Polymicrobial Peritonitis. Crit. Care Med. 2016, 44, e200–e206. [Google Scholar] [CrossRef]
- Steeland, S.; Van Ryckeghem, S.; Vandewalle, J.; Ballegeer, M.; Van Wonterghem, E.; Eggermont, M.; Decruyenaere, J.; De Bus, L.; Libert, C.; Vandenbroucke, R.E. Simultaneous Inhibition of Tumor Necrosis Factor Receptor 1 and Matrix Metalloproteinase 8 Completely Protects Against Acute Inflammation and Sepsis. Crit. Care Med. 2018, 46, e67–e75. [Google Scholar] [CrossRef]
- Iyer, R.P.; de Castro Brás, L.E.; Patterson, N.L.; Bhowmick, M.; Flynn, E.R.; Asher, M.; Cannon, P.L.; Deleon-Pennell, K.Y.; Fields, G.B.; Lindsey, M.L. Early matrix metalloproteinase-9 inhibition post-myocardial infarction worsens cardiac dysfunction by delaying inflammation resolution. J. Mol. Cell Cardiol. 2016, 100, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Hartenstein, B.; Dittrich, B.T.; Stickens, D.; Heyer, B.; Vu, T.H.; Teurich, S.; Schorpp-Kistner, M.; Werb, Z.; Angel, P. Epidermal development and wound healing in matrix metalloproteinase 13-deficient mice. J. Investig. Dermatol. 2006, 126, 486–496. [Google Scholar] [CrossRef]
- Marshall, D.C.; Lyman, S.K.; McCauley, S.; Kovalenko, M.; Spangler, R.; Liu, C.; Lee, M.; O’Sullivan, C.; Barry-Hamilton, V.; Ghermazien, H.; et al. Selective Allosteric Inhibition of MMP9 Is Efficacious in Preclinical Models of Ulcerative Colitis and Colorectal Cancer. PLoS ONE 2015, 10, e0127063. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Bhandari, B.R.; Fogel, R.; Onken, J.; Yen, E.; Zhao, X.; Jiang, Z.; Ge, D.; Xin, Y.; Ye, Z.; et al. Randomised clinical trial: A phase 1, dose-ranging study of the anti-matrix metalloproteinase-9 monoclonal antibody GS-5745 versus placebo for ulcerative colitis. Aliment. Pharmacol. Ther. 2016, 44, 157–169. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Bhandari, B.R.; Randall, C.; Younes, Z.H.; Romanczyk, T.; Xin, Y.; Wendt, E.; Chai, H.; McKevitt, M.; Zhao, S.; et al. Andecaliximab [Anti-matrix Metalloproteinase-9] Induction Therapy for Ulcerative Colitis: A Randomised, Double-Blind, Placebo-Controlled, Phase 2/3 Study in Patients With Moderate to Severe Disease. J. Crohns Colitis 2018, 12, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- de Bruyn, M.; Breynaert, C.; Arijs, I.; De Hertogh, G.; Geboes, K.; Thijs, G.; Matteoli, G.; Hu, J.; Van Damme, J.; Arnold, B.; et al. Inhibition of gelatinase B/MMP-9 does not attenuate colitis in murine models of inflammatory bowel disease. Nat. Commun. 2017, 8, 15384. [Google Scholar] [CrossRef]
- Appleby, T.C.; Greenstein, A.E.; Hung, M.; Liclican, A.; Velasquez, M.; Villaseñor, A.G.; Wang, R.; Wong, M.H.; Liu, X.; Papalia, G.A.; et al. Biochemical characterization and structure determination of a potent, selective antibody inhibitor of human MMP9. J. Biol. Chem. 2017, 292, 6810–6820. [Google Scholar] [CrossRef] [Green Version]
- Butler, G.S.; Overall, C.M. Proteomic validation of protease drug targets: Pharmacoproteomics of matrix metalloproteinase inhibitor drugs using isotope-coded affinity tag labelling and tandem mass spectrometry. Curr. Pharm. Design 2007, 13, 263–270. [Google Scholar] [CrossRef]
- Amar, S.; Minond, D.; Fields, G.B. Clinical implications of compounds designed to inhibit ECM-modifying metalloproteinases. Proteomics 2017, 17, 1600389. [Google Scholar] [CrossRef]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix Metalloproteinases As Modulators Of Inflammation And Innate Immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef]
- Zhou, Z.; Apte, S.S.; Soininen, R.; Cao, R.; Baaklini, G.Y.; Rauser, R.W.; Wang, J.; Cao, Y.; Tryggvason, K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase 1. Proc. Natl. Acad. Sci. USA 2000, 97, 4052–4057. [Google Scholar] [CrossRef]
- Koike, T.; Vernon, R.B.; Hamner, M.A.; Sadoun, E.; Reed, M.J. MT1-MMP, but not secreted MMPs, influences the migration of human microvascular endothelial cells in 3-dimensional collagen gels. J. Cell. Biochem. 2002, 86, 748–758. [Google Scholar] [CrossRef]
- Chun, T.-H.; Sabeh, F.; Ota, I.; Murphy, H.; McDonagh, K.T.; Holmbeck, K.; Birkedal-Hansen, H.; Allen, E.D.; Weiss, S.J. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J. Cell Biol. 2004, 167, 757–767. [Google Scholar] [CrossRef]
- Saunders, W.B.; Bohnsack, B.L.; Faske, J.B.; Anthis, N.J.; Bayless, K.J.; Hirschi, K.K.; Davis, G.E. Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J. Cell Biol. 2006, 175, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Genís, l.; Gálvez, B.G.; Gonzalo, P.; Arroyo, A.G. MT1-MMP: Universal or particular player in angiogenesis? Cancer Metastasis Rev. 2006, 25, 77–86. [Google Scholar] [CrossRef]
- Stratman, A.N.; Saunders, W.B.; Sacharidou, A.; Koh, W.; Fisher, K.E.; Zawieja, D.C.; Davis, M.J.; Davis, G.E. Endothelial cell lumen and vascular guidance tunnel formatinon requires MT1-MMP-dependent proteolysis in 3-dimensional collagen matrices. Blood 2009, 114, 237–247. [Google Scholar] [CrossRef]
- Onimaru, M.; Yonemitsu, Y.; Suzuki, H.; Fujii, T.; Sueishi, K. An autocrine linkage between matrix metalloproteinase-14 and Tie-2 via ectodomain shedding modulates angiopoietin-1-dependent function in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 818–826. [Google Scholar] [CrossRef]
- Sacharidou, A.; Koh, W.; Stratman, A.N.; Mayo, A.M.; Fisher, K.E.; Davis, G.E. Endothelial lumen signaling complexes control 3D matrix-specific tubulogenesis through interdependent Cdc42- and MT1-MMP-mediated events. Blood 2010, 115, 5259–5269. [Google Scholar] [CrossRef]
- Sounni, N.E.; Paye, A.; Host, L.; Noël, A. MT-MMPs as Regulators of Vessel Stability Associated with Angiogenesis. Front. Pharmacol. 2011, 2, 111. [Google Scholar] [CrossRef]
- Montaner, J.; Ramiro, L.; Simats, A.; Hernández-Guillamon, M.; Delgado, P.; Bustamante, A.; Rosell, A. Matrix metalloproteinases and ADAMs in stroke. Cell Mol. Life Sci. 2019, 76, 3117–3140. [Google Scholar] [CrossRef]
- Bernstein, K.E.; Ong, F.S.; Blackwell, W.L.; Shah, K.H.; Giani, J.F.; Gonzalez-Villalobos, R.A.; Shen, X.Z.; Fuchs, S.; Touyz, R.M. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol. Rev. 2012, 65, 1–46. [Google Scholar] [CrossRef]
- O’Reilly, P.J.; Ding, Q.; Akthar, S.; Cai, G.; Genschmer, K.R.; Patel, D.F.; Jackson, P.L.; Viera, L.; Roda, M.; Locy, M.L.; et al. Angiotensin-converting enzyme defines matrikine-regulated inflammation and fibrosis. JCI Insight 2017, 2, e91923. [Google Scholar] [CrossRef]
- Zou, K.; Yamaguchi, H.; Akatsu, H.; Sakamoto, T.; Ko, M.; Mizoguchi, K.; Gong, J.S.; Yu, W.; Yamamoto, T.; Kosaka, K.; et al. Angiotensin-converting enzyme converts amyloid beta-protein 1-42 (Abeta(1-42)) to Abeta(1-40), and its inhibition enhances brain Abeta deposition. J. Neurosci. 2007, 27, 8628–8635. [Google Scholar] [CrossRef]
- Liu, S.; Ando, F.; Fujita, Y.; Liu, J.; Maeda, T.; Shen, X.; Kikuchi, K.; Matsumoto, A.; Yokomori, M.; Tanabe-Fujimura, C.; et al. A clinical dose of angiotensin-converting enzyme (ACE) inhibitor and heterozygous ACE deletion exacerbate Alzheimer’s disease pathology in mice. J. Biol. Chem. 2019, 294, 9760–9770. [Google Scholar] [CrossRef]
- Sela-Passwell, N.; Trahtenherts, A.; Krüger, A.; Sagi, I. New opportunities in drug design of metalloproteinase inhibitors: Combination between structure-function experimental approaches and systems biology. Expert Opin. Drug Discov. 2011, 6, 527–542. [Google Scholar] [CrossRef]
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors - Novel strategies bring new prospects. Biochim. Biophys. Acta 2017, 1864, 1927–1939. [Google Scholar] [CrossRef]
- Huang, W.; Meng, Q.; Suzuki, K.; Nagase, H.; Brew, K. Mutational study of the amino-termina domain of human tissue inhibitor of metalloproteinases 1 (TIMP-1) locates an inhibitory region for matrix metalloproteinases. J. Biol. Chem. 1997, 272, 22086–22091. [Google Scholar] [CrossRef]
- Meng, Q.; Malinovskii, V.A.; Huang, W.; Hu, Y.; Chung, L.; Nagase, H.; Bode, W.; Maskos, K.; Brew, K. Residue 2 of TIMP-1 is a major determinant of affinity and spcificity for MMPs but effects of substitutions do not correlate with those of corresponding P1’ residue of substrate. J. Biol. Chem. 1999, 274, 10184–10189. [Google Scholar] [CrossRef]
- Wei, S.; Chen, Y.; Chung, L.; Nagase, H.; Brew, K. Protein engineering of the tissue inhibitor of metalloproteinase 1 (TIMP-1) inhibitory domain. J. Biol. Chem. 2003, 278, 9831–9834. [Google Scholar] [CrossRef]
- Lee, M.-H.; Rapti, M.; Murphy, G. Unveiling the surface epitopes that render tissue inhibitor of metalloproteinase-1 inactive against membrane type 1-matrix metalloproteinase. J. Biol. Chem. 2003, 278, 40224–40230. [Google Scholar] [CrossRef]
- Hamze, A.B.; Wei, S.; Bahudhanapati, H.; Kota, S.; Acharya, K.R.; Brew, K. Constraining specificity in the N-domain of tissue inhibitor of metalloproteinases-1; gelatinase-selective inhibitors. Protein Sci. 2007, 16, 1905–1913. [Google Scholar] [CrossRef]
- Jiang, B.; Liu, J.; Lee, M.H. Targeting a Designer TIMP-1 to the Cell Surface for Effective MT1-MMP Inhibition: A Potential Role for the Prion Protein in Renal Carcinoma Therapy. Molecules 2019, 24, 255. [Google Scholar] [CrossRef]
- Bahudhanapati, H.; Zhang, Y.; Sidhu, S.S.; Brew, K. Phage display of tissue inhibitor of metalloproteinases-2 (TIMP-2): Identification of selective inhibitors of collagenase-1 (metalloproteinase 1 (MMP-1)). J. Biol. Chem. 2011, 286, 31761–31770. [Google Scholar] [CrossRef]
- Arkadash, V.; Radisky, E.S.; Papo, N. Combinatorial engineering of N-TIMP2 variants that selectively inhibit MMP9 and MMP14 function in the cell. Oncotarget 2018, 9, 32036–32053. [Google Scholar] [CrossRef]
- Sharabi, O.; Shirian, J.; Grossman, M.; Lebendiker, M.; Sagi, I.; Shifman, J. Affinity- and specificity-enhancing mutations are frequent in multispecific interactions between TIMP2 and MMPs. PLoS ONE 2014, 9, e93712. [Google Scholar] [CrossRef]
- Arkadash, V.; Yosef, G.; Shirian, J.; Cohen, I.; Horev, Y.; Grossman, M.; Sagi, I.; Radisky, E.S.; Shifman, J.M.; Papo, N. Development of High Affinity and High Specificity Inhibitors of Matrix Metalloproteinase 14 through Computational Design and Directed Evolution. J. Biol. Chem. 2017, 292, 3481–3495. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.X.; Rapti, M.; Tsigkou, A.; Lee, M.H. Expanding the Activity of Tissue Inhibitors of Metalloproteinase (TIMP)-1 against Surface-Anchored Metalloproteinases by the Replacement of Its C-Terminal Domain: Implications for Anti-Cancer Effects. PLoS ONE 2015, 10, e0136384. [Google Scholar] [CrossRef]
- Raeeszadeh-Sarmazdeh, M.; Greene, K.A.; Sankaran, B.; Downey, G.P.; Radisky, D.C.; Radisky, E.S. Directed evolution of the metalloproteinase inhibitor TIMP-1 reveals that its N- and C-terminal domains cooperate in matrix metalloproteinase recognition. J. Biol. Chem. 2019, 294, 9476–9488. [Google Scholar] [CrossRef]
- Yosef, G.; Arkadash, V.; Papo, N. Targeting the MMP-14/MMP-2/integrin αvβ3 axis with multispecific N-TIMP2-based antagonists for cancer therapy. J. Biol. Chem. 2018, 293, 13310–13326. [Google Scholar] [CrossRef]
- Batra, J.; Robinson, J.; Mehner, C.; Hockla, A.; Miller, E.; Radisky, D.C.; Radisky, E.S. PEGylation extends circulation half-life while preserving in vitro and in vivo activity of tissue inhibitor of metalloproteinases-1 (TIMP-1). PLoS ONE 2012, 7, e50028. [Google Scholar] [CrossRef]
- Li, N.G.; Shib, Z.H.; Tang, Y.P.; Duan, J.A. Selective matrix metalloproteinase inhibitors for cancer. Curr. Med. Chem. 2009, 16, 3805–3827. [Google Scholar] [CrossRef]
- Xie, X.-W.; Wan, R.-Z.; Liu, Z.-P. Recent research advances in selective matrix metalloproteinase-13 inhibitors as anti-osteoarthritis agents. ChemMedChem 2017, 12, 1157–1168. [Google Scholar] [CrossRef]
- Ruminski, P.G.; Massa, M.; Strohbach, J.; Hanau, C.E.; Schmidt, M.; Scholten, J.A.; Fletcher, T.R.; Hamper, B.C.; Carroll, J.N.; Shieh, H.S.; et al. Discovery of N-(4-Fluoro-3-methoxybenzyl)-6-(2-(((2S,5R)-5-(hydroxymethyl)-1,4-dioxan-2-yl)methyl)-2H-tetrazol-5-yl)-2-methylpyrimidine-4-carboxamide. A Highly Selective and Orally Bioavailable Matrix Metalloproteinase-13 Inhibitor for the Potential Treatment of Osteoarthritis. J. Med. Chem. 2016, 59, 313–327. [Google Scholar]
- Sallusti, B.C.; Sabordo, L.; Evans, A.M.; Nation, R.L. Hepatic disposition of electrophilic acyl glucuronide conjugates. Curr. Drug Metab. 2000, 1, 163–180. [Google Scholar] [CrossRef]
- Nara, H.; Sato, K.; Kaieda, A.; Oki, H.; Kuno, H.; Santou, T.; Kanzaki, N.; Terauchi, J.; Uchikawa, O.; Kori, M. Design, synthesis, and biological activity of novel, potent, and highly selective fused pyrimidine-2-carboxamide-4-one-based matrix metalloproteinase (MMP)-13 zinc-binding inhibitors. Bioorg. Med. Chem. 2016, 24, 6149–6165. [Google Scholar] [CrossRef]
- Spicer, T.P.; Jiang, J.; Taylor, A.B.; Hart, P.J.; Roush, W.R.; Fields, G.B.; Hodder, P.S.; Minond, D. Characterization of Selective Exosite-Binding Inhibitors of Matrix Metalloproteinase 13 That Prevent Articular Cartilage Degradation In Vitro. J. Med. Chem. 2014, 57, 9598–9611. [Google Scholar] [CrossRef]
- Nara, H.; Sato, K.; Naito, T.; Mototani, H.; Oki, H.; Yamamoto, Y.; Kuno, H.; Santou, T.; Kanzaki, N.; Terauchi, J.; et al. Thieno[2,3-d]pyrimidine-2-carboxamides bearing a carboxybenzene group at 5-position: Highly potent, selective, and orally available MMP-13 inhibitors interacting with the S1″ binding site. Bioorg. Med. Chem. 2014, 22, 5487–5505. [Google Scholar] [CrossRef]
- Lauer-Fields, J.L.; Minond, D.; Chase, P.S.; Baillargeon, P.E.; Saldanha, S.A.; Stawikowska, R.; Hodder, P.; Fields, G.B. High throughput screening of potentially selective MMP-13 exosite inhibitors utilizing a triple-helical FRET substrate. Bioorg. Med. Chem. 2009, 17, 990–1005. [Google Scholar] [CrossRef] [Green Version]
- Roth, J.; Minond, D.; Darout, E.; Liu, Q.; Lauer, J.; Hodder, P.; Fields, G.B.; Roush, W.R. Identification of novel, exosite-binding matrix metalloproteinase-13 inhibitor scaffolds. Bioorg. Med. Chem. Lett. 2011, 21, 7180–7184. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Fuerst, R.; Knapinska, A.M.; Taylor, A.; Smith, L.; Cao, X.; Hart, P.J.; Fields, G.B.; Roush, W.R. Structure-based design and synthesis of potent and selective matrix metalloproteinase 13 inhibitors. J. Med. Chem. 2017, 60, 5816–5825. [Google Scholar] [CrossRef]
- Fuerst, R.; Choi, J.Y.; Knapinska, A.M.; Smith, L.; Cameron, M.D.; Ruiz, C.H.; Fields, G.B.; Roush, W.R. Development of matrix metalloproteinase 13 inhibitors - A structure-activity/structure-property relationship study. Bioorg. Med. Chem. 2018, 26, 4984–4995. [Google Scholar] [CrossRef]
- Scannevin, R.H.; Alexander, R.; Haarlander, T.M.; Burke, S.L.; Singer, M.; Huo, C.; Zhang, Y.M.; Maguire, D.; Spurlino, J.; Deckman, I.; et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 2017, 292, 17963–17974. [Google Scholar] [CrossRef] [Green Version]
- Remacle, A.G.; Golubkov, V.S.; Shiryaev, S.A.; Dahl, R.; Stebbins, J.L.; Chernov, A.V.; Cheltsov, A.V.; Pellecchia, M.; Strongin, A.Y. Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res. 2012, 72, 2339–2349. [Google Scholar] [CrossRef]
- Gao, Y.; Amar, S.; Pahwa, S.; Fields, G.B.; Kodadek, T. Rapid Lead Discovery Through Iterative Screening of One Bead One Compound Libraries. ACS Comb. Sci. 2015, 17, 49–59. [Google Scholar] [CrossRef]
- Hunter, D.J.; Bird, J.; Cassidy, F.; De Mello, R.C.; Harper, G.P.; Karran, E.H.; Markwell, R.E.; Miles-Williams, A.J.; Ward, R.W. Aminophosphonic acid containing inhibitors of human collagenase: Modification of the P1 residue. Bioorg. Med. Chem. Lett. 1994, 4, 2833–2836. [Google Scholar] [CrossRef]
- Morphy, J.R.; Beeley, N.R.A.; Boyce, B.A.; Leonard, J.; Mason, B.; Millican, A.; Millar, K.; O’Connel, J.P.; Porter, J. Potent and selective inhibitors of gelatinase-A 2. carboxylic and phosphonic acid derivatives. Bioorg. Med. Chem. Lett. 1994, 4, 2747–2752. [Google Scholar] [CrossRef]
- Gavuzzo, E.; Pochetti, G.; Mazza, F.; Gallina, C.; Gorini, B.; D’Alessio, S.; Pieper, M.; Tschesche, H.; Tucker, P.A. Two crystal structures of human neutrophil collagenase, one complexed with a primed- and the other with an unprimed-side inhibitor: Implications for drug design. J. Med. Chem. 2000, 43, 3377–3385. [Google Scholar] [CrossRef]
- Breuer, E.; Salomon, C.J.; Katz, Y.; Chen, W.; Lu, S.; Röschenthaler, G.V.; Hadar, R.; Reich, R. Carbamoylphosphonates, a new class of in vivo active matrix metalloproteinase inhibitors. 1. Alkyl- and cycloalkylcarbamoylphosphonic acids. J. Med. Chem. 2004, 47, 2826–2832. [Google Scholar] [CrossRef]
- Breuer, E.; Katz, Y.; Hadar, R.; Reich, R. Carbamoylphosphonate MMP inhibitors. Part 4: The influence of chirality and geometrical isomerism on the potency and selectivity of inhibition Tetrahedron: Asymmetry 2004, 15, 2415–2420. [Google Scholar]
- Pochetti, G.; Gavuzzo, E.; Campestre, C.; Agamennone, M.; Tortorella, P.; Consalvi, V.; Gallina, C.; Hiller, O.; Tschesche, H.; Tucker, P.A.; et al. Structural insight into the stereoselective inhibition of MMP-8 by enantiomeric sulfonamide phosphonates. J. Med. Chem. 2006, 49, 923–931. [Google Scholar] [CrossRef]
- Hoffman, A.; Qadri, B.; Frant, J.; Katz, Y.; Bhusare, S.R.; Breuer, E.; Hadar, R.; Reich, R. Carbamoylphosphonate matrix metalloproteinase inhibitors 6: Cis-2-aminocyclohexylcarbamoylphosphonic acid, a novel orally active antimetastatic matrix metalloproteinase-2 selective inhibitor--synthesis and pharmacodynamic and pharmacokinetic analysis. J. Med. Chem. 2008, 51, 1406–1414. [Google Scholar] [CrossRef]
- Frant, J.; Veerendhar, A.; Chernilovsky, T.; Nedvetzki, S.; Vaksman, O.; Hoffman, A.; Breuer, E.; Reich, R. Orally active, antimetastatic, nontoxic diphenyl ether-derived carbamoylphosphonate matrix metalloproteinase inhibitors. ChemMedChem 2011, 6, 1471–1477. [Google Scholar] [CrossRef]
- Kortylewicz, Z.P.; Galardy, R.E. Phosphoramidate peptide inhibitors of human skin fibroblast collagenase. J. Med. Chem. 1990, 33, 263–273. [Google Scholar] [CrossRef]
- Galardy, R.E.; Grobelny, D.; Kortylewicz, Z.P.; Poncz, L. Inhibition of human skin fibroblast collagenase by phosphorus-containing peptides. Matrix Suppl. 1992, 1, 259–262. [Google Scholar]
- Mendes, D.E.; Wong-On-Wing, A.; Berkman, C.E. Phosphoramidate-based peptidomimetic inhibitors of membrane type-1 matrix metalloproteinase. J. Enzyme Inhib. Med. Chem. 2016, 31, 167–171. [Google Scholar] [CrossRef]
- Izquierdo-Martin, M.; Stein, R.L. Mechanistic studies on the inhibition of stromelysin by a peptide phosphonamidate. Bioorg. Med. Chem. 1993, 1, 19–26. [Google Scholar] [CrossRef]
- Caldwell, C.G.; Sahoo, S.P.; Polo, S.A.; Eversole, R.R.; Lanza, T.J.; Mills, S.C.; Niedzwiecki, L.M.; Izquierdo-Martin, M.; Chang, B.C.; Harrison, R.K.; et al. Phosphinic acid inhibitors of matrix metalloproteinases. Bioorg. Med. Chem. Lett. 1996, 6, 323–328. [Google Scholar] [CrossRef]
- Goulet, J.L.; Kinneary, J.F.; Durette, P.L.; Stein, R.L.; Harrison, R.K.; Izquierdo-Martin, M.; Kuo, D.W.; Lin, T.-Y.; Hagmann, W.K. Inhibition of stromelysin-1 (MMP-3) by peptidyl phosphinic acids. Bioorg. Med. Chem. Lett. 1994, 4, 1221–1224. [Google Scholar] [CrossRef]
- Vassiliou, S.; Mucha, A.; Cuniasse, P.; Georgiadis, D.; Lucet-Levannier, K.; Beau, F.; Kannan, R.; Murphy, G.; Knauper, V.; Rio, M.C.; et al. Phosphinic pseudo-tripeptides as potent inhibitors of matrix metalloproteinases: A structure-activity study. J. Med. Chem. 1999, 42, 2610–2620. [Google Scholar] [CrossRef]
- Buchardt, J.; Ferreras, M.; Krog-Jensen, C.; Delaisse, J.-M.; Foged, N.T.; Meldal, M. Phosphinic peptide matrix metalloproteinase-9 inhibitors by solid-phase synthesis using a building block approach. Chem. Eur. J. 1999, 5, 2877–2884. [Google Scholar] [CrossRef]
- Reiter, L.A.; Rizzi, J.P.; Pandit, J.; Lasut, M.J.; McGahee, S.M.; Parikh, V.D.; Blake, J.F.; Danley, D.E.; Laird, E.R.; Lopez-Anaya, A.; et al. Inhibition of MMP-1 and MMP-13 with phosphinic acids that exploit binding in the S2 pocket. Bioorg. Med. Chem. Lett. 1999, 9, 127–132. [Google Scholar] [CrossRef]
- Buchardt, J.; Schiodt, C.B.; Krog-Jensen, C.; Delaissé, J.-M.; Foged, N.T.; Meldal, M. Solid phase combinatorial library of phosphinic peptides for discovery of matrix metalloproteinase inhibitors. J. Comb. Chem. 2000, 2, 624–638. [Google Scholar] [CrossRef]
- Schiodt, C.B.; Buchardt, J.; Terp, G.E.; Christensen, U.; Brink, M.; Larsen, Y.B.; Meldal, M.; Foged, N.T. Phosphinic peptide inhibitors of macrophage metalloelastase (MMP-12): Selectivity and mechanism of binding. Current Med. Chem. 2001, 8, 967–976. [Google Scholar] [CrossRef]
- Gall, A.L.; Ruff, M.; Kannan, R.; Cuniasse, P.; Yiotakis, A.; Dive, V.; Rio, M.C.; Basset, P.; Moras, D. Crystal structure of the stromelysin-3 (MMP-11) catalytic domain complexed with a phosphinic inhibitor mimicking the transition-state. J. Mol. Biol. 2001, 307, 577–586. [Google Scholar] [CrossRef]
- Reiter, L.A.; Mitchell, P.G.; Martinelli, G.J.; Lopresti-Morrow, L.L.; Yocum, S.A.; Eskra, J.D. Phosphinic acid-based MMP-13 inhibitors that spare MMP-1 and MMP-3. Bioorg. Med. Chem. Lett. 2003, 13, 2331–2336. [Google Scholar] [CrossRef]
- Makaritis, A.; Georgiadis, D.; Dive, V.; Yiotakis, A. Diastereoselective solution and multipin-based combinatorial array synthesis of a novel class of potent phosphinic metalloprotease inhibitors. Chem. Eur. J. 2003, 9, 2079–2094. [Google Scholar] [CrossRef]
- Bianchini, G.; Aschi, M.; Cavicchio, G.; Crucianelli, M.; Preziuso, S.; Gallina, C.; Nastari, A.; Gavuzzo, E.; Mazza, F. Design, modelling, synthesis and biological evaluation of peptidomimetic phosphinates as inhibitors of matrix metalloproteinases MMP-2 and MMP-8. Bioorg. Med. Chem. 2005, 13, 4740–4749. [Google Scholar] [CrossRef]
- Devel, L.; Rogakos, V.; David, A.; Makaritis, A.; Beau, F.; Cuniasse, P.; Yiotakis, A.; Dive, V. Development of selective inhibitors and substrate of matrix metalloproteinase-12. J. Biol. Chem. 2006, 281, 11152–11160. [Google Scholar] [CrossRef]
- Tochowicz, A.; Maskos, K.; Huber, R.; Oltenfreiter, R.; Dive, V.; Yiotakis, A.; Zanda, M.; Bode, W.; Goettig, P. Crystal structure of MMP-9 complexes with five inhibitors: Contribution of the flexible Arg424 side-chain to selectivity. J. Mol. Biol. 2007, 371, 989–1006. [Google Scholar] [CrossRef]
- Lauer-Fields, J.L.; Brew, K.; Whitehead, J.K.; Li, S.; Hammer, R.P.; Fields, G.B. Triple-helical transition-state analogs: A new class of selective matrix metalloproteinase inhibitors. J. Am. Chem. Soc. 2007, 129, 10408–10417. [Google Scholar] [CrossRef]
- Lauer-Fields, J.L.; Whitehead, J.K.; Li, S.; Hammer, R.P.; Brew, K.; Fields, G.B. Selective modulation of matrix metalloproteinase 9 (MMP-9) functions via exosite inhibition. J. Biol. Chem. 2008, 283, 20087–20095. [Google Scholar] [CrossRef]
- Lauer-Fields, J.L.; Chalmers, M.J.; Busby, S.A.; Minond, D.; Griffin, P.R.; Fields, G.B. Identification of Specific Hemopexin-like Domain Residues That Facilitate Matrix Metalloproteinase Collagenolytic Activity. J. Biol. Chem. 2009, 284, 24017–24024. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, M.; Stawikowska, R.; Tokmina-Roszyk, D.; Fields, G.B. Matrix Metalloproteinase Inhibition by Heterotrimeric Triple-Helical Peptide Transition State Analogs. ChemBioChem 2015, 16, 1084–1092. [Google Scholar] [CrossRef]
- de Castro Brás, L.E.; Cates, C.A.; DeLeon-Pennell, K.Y.; Ma, Y.; Iyer, R.P.; Halade, G.V.; Yabluchanskiy, A.; Fields, G.B.; Weintraub, S.T.; Lindsey, M.L. Citrate Synthase is a Novel In Vivo Matrix Metalloproteinase-9 Substrate that Regulates Mitochondrial Function in the Post-Myocardial Infarction Left Ventricle. Antioxid. Redox Signal. 2014, 21, 1974–1985. [Google Scholar] [CrossRef]
- Fan, C.-Y.; Huang, C.-C.; Chiu, W.-C.; Lai, C.-C.; Liou, G.-G.; Li, H.-C.; Chou, M.-Y. Production of multivalent protein binders using a self-trimerizing collagen-like peptide scaffold. FASEB J. 2008, 22, 3795–3804. [Google Scholar] [CrossRef]
- Ndinguri, M.W.; Zheleznyak, A.; Lauer, J.L.; Anderson, C.J.; Fields, G.B. Application of collagen-model triple-helical peptide-amphiphiles for CD44 targeted drug delivery systems. J. Drug Delivery 2012, 2012, 592602. [Google Scholar] [CrossRef]
- Yasui, H.; Yamazaki, C.M.; Nose, H.; Awada, C.; Takao, T.; Koide, T. Potential of collagen-like triple helical peptides as drug carriers: Their In vivo distribution, metabolism, and excretion profiles in rodents. Pept. Sci. 2013, 100, 705–713. [Google Scholar] [CrossRef]
- Yamazaki, C.M.; Nakase, I.; Endo, H.; Kishimoto, S.; Mashiyama, Y.; Masuda, R.; Futaki, S.; Koide, T. Collagen-like cell-penetrating peptides. Angew. Chem. Int. Ed Engl. 2013, 52, 5497–5500. [Google Scholar] [CrossRef]
- Shinde, A.; Feher, K.M.; Hu, C.; Slowinska, K. Peptide internalization enabled by folding: Triple-helical cell-penetrating peptides. J. Pept. Sci. 2015, 21, 77–84. [Google Scholar] [CrossRef]
- Masuda, R.; Hayashi, R.; Nose, H.; Taguchi, A.; Hayashi, Y.; Yasui, H.; Koide, T. Development of a carboplatin derivative conjugated with a collagen-like triple-helical peptide. Future Med. Chem. 2018, 10, 619–629. [Google Scholar] [CrossRef]
- Koide, T.; Yamamoto, N.; Taira, K.B.; Yasui, H. Fecal Excretion of Orally Administered Collagen-Like Peptides in Rats: Contribution of the Triple-Helical Conformation to Their Stability. Biol. Pharm. Bull. 2016, 39, 135–137. [Google Scholar] [CrossRef] [Green Version]
- Sela-Passwell, N.; Kikkeri, R.; Dym, O.; Rozenberg, H.; Margalit, R.; Arad-Yellin, R.; Eisenstein, M.; Brenner, O.; Shoham, T.; Danon, T.; et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 2011, 18, 143–147. [Google Scholar] [CrossRef]
- Santamaria, S.; de Groot, R. Monoclonal antibodies against metzincin targets. Br. J. Pharmacol. 2019, 176, 52–66. [Google Scholar] [CrossRef]
- Paemen, L.; Martens, E.; Masure, S.; Opdenakker, G. Monoclonal antibodies specific for natural human neutrophil gelatinase B used for affinity purification, quantitation by two-site ELISA and inhibition of enzymatic activity. Eur. J. Biochem. 1995, 234, 759–765. [Google Scholar] [CrossRef]
- Pruijt, J.F.; Fibbe, W.E.; Laterveer, L.; Pieters, R.A.; Lindley, I.J.; Paemen, L.; Masure, S.; Willemze, R.; Opdenakker, G. Prevention of interleukin-8-induced mobilization of hematopoietic progenitor cells in rhesus monkeys by inhibitory antibodies against the metalloproteinase gelatinase B (MMP-9). Proc. Natl. Acad. Sci. USA 1999, 96, 10863–10868. [Google Scholar] [CrossRef]
- Devy, L.; Huang, L.; Naa, L.; Yanamandra, N.; Pieters, H.; Frans, N.; Chang, E.; Tao, Q.; Vanhove, M.; Lejeune, A.; et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009, 69, 1517–1526. [Google Scholar] [CrossRef]
- Ager, E.I.; Kozin, S.V.; Kirkpatrick, N.D.; Seano, G.; Kodack, D.P.; Askoxylakis, V.; Huang, Y.; Goel, S.; Snuderl, M.; Muzikansky, A.; et al. Blockade of MMP14 activity in murine breast carcinomas: Implications for macrophages, vessels, and radiotherapy. J. Natl. Cancer Inst. 2015, 107, djv017. [Google Scholar] [CrossRef]
- Basu, B.; Correa de Sampaio, P.; Mohammed, H.; Fogarasi, M.; Corrie, P.; Watkins, N.A.; Smethurst, P.A.; English, W.R.; Ouwehand, W.H.; Murphy, G. Inhibition of MT1-MMP activity using functional antibody fragments selected against its hemopexin domain. Int. J. Biochem. Cell Biol. 2012, 44, 393–403. [Google Scholar] [CrossRef]
- Botkjaer, K.A.; Kwok, H.F.; Terp, M.G.; Karatt-Vellatt, A.; Santamaria, S.; McCafferty, J.; Andreasen, P.A.; Itoh, Y.; Ditzel, H.J.; Murphy, G. Development of a specific affinity-matured exosite inhibitor to MT1-MMP that efficiently inhibits tumor cell invasion in vitro and metastasis in vivo. Oncotarget 2016, 7, 16773–16792. [Google Scholar] [CrossRef]
- Suojanen, J.; Salo, T.; Koivunen, E.; Sorsa, T.; Pirilä, E. A novel and selective membrane type-1 matrix metalloproteinase (MT1-MMP) inhibitor reduces cancer cell motility and tumor growth. Cancer Biol. Ther. 2009, 8, 2362–2370. [Google Scholar] [CrossRef] [Green Version]
- Nam, D.H.; Rodriguez, C.; Remacle, A.G.; Strongin, A.Y.; Ge, X. Active-site MMP-selective antibody inhibitors discovered from convex paratope synthetic libraries. Proc. Natl. Acad. Sci. USA 2016, 113, 14970–14975. [Google Scholar] [CrossRef] [Green Version]
- Ling, B.; Watt, K.; Banerjee, S.; Newsted, D.; Truesdell, P.; Adams, J.; Sidhu, S.S.; Craig, A.W. A novel immunotherapy targeting MMP-14 limits hypoxia, immune suppression and metastasis in triple-negative breast cancer models. Oncotarget 2017, 8, 58372–58385. [Google Scholar] [CrossRef]
- Ingvarsen, S.; Porse, A.; Erpicum, C.; Maertens, L.; Jürgensen, H.J.; Madsen, D.H.; Melander, M.C.; Gårdsvoll, H.; Høyer-Hansen, G.; Noel, A.; et al. Targeting a single function of the multifunctional matrix metalloproteinase MT1-MMP: Impact on lymphangiogenesis. J. Biol. Chem. 2013, 288, 10195–10204. [Google Scholar] [CrossRef]
- Woskowicz, A.M.; Weaver, S.A.; Shitomi, Y.; Ito, N.; Itoh, Y. MT-LOOP-dependent localization of membrane type I matrix metalloproteinase (MT1-MMP) to the cell adhesion complexes promotes cancer cell invasion. J. Biol. Chem. 2013, 288, 35126–35137. [Google Scholar] [CrossRef]
- Gálvez, B.G.; Matías-Román, S.; Albar, J.P.; Sánchez-Madrid, F.; Arroyo, A.G. Membrane type 1-matrix metalloproteinase is activated during migration of human endothelial cells and modulates endothelial motility and matrix remodeling. J. Biol. Chem. 2001, 276, 37491–37500. [Google Scholar] [CrossRef]
- Udi, Y.; Grossman, M.; Solomonov, I.; Dym, O.; Rozenberg, H.; Moreno, V.; Cuniasse, P.; Dive, V.; Arroyo, A.G.; Sagi, I. Inhibition mechanism of membrane metalloprotease by an exosite-swiveling conformational antibody. Structure 2015, 23, 104–115. [Google Scholar] [CrossRef]
- Talmi-Frank, D.; Altboum, Z.; Solomonov, I.; Udi, Y.; Jaitin, D.A.; Klepfish, M.; David, E.; Zhuravlev, A.; Keren-Shaul, H.; Winter, D.R.; et al. Extracellular Matrix Proteolysis by MT1-MMP Contributes to Influenza-Related Tissue Damage and Mortality. Cell Host Microbe 2016, 20, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Nam, D.H.; Fang, K.; Rodriguez, C.; Lopez, T.; Ge, X. Generation of inhibitory monoclonal antibodies targeting matrix metalloproteinase-14 by motif grafting and CDR optimization. Protein Eng. Des. Sel. 2017, 30, 113–118. [Google Scholar] [CrossRef]
- Remacle, A.G.; Cieplak, P.; Nam, D.H.; Shiryaev, S.A.; Ge, X.; Strongin, A.Y. Selective function-blocking monoclonal human antibody highlights the important role of membrane type-1 matrix metalloproteinase (MT1-MMP) in metastasis. Oncotarget 2017, 8, 2781–2799. [Google Scholar] [CrossRef]
- Ugarte-Berzal, E.; Bailón, E.; Amigo-Jiménez, I.; Vituri, C.L.; del Cerro, M.H.; Terol, M.J.; Albar, J.P.; Rivas, G.; García-Marco, J.A.; García-Pardo, A. A 17-residue sequence from the matrix metalloproteinase-9 (MMP-9) hemopexin domain binds α4β1 integrin and inhibits MMP-9-induced functions in chronic lymphocytic leukemia B cells. J. Biol. Chem. 2012, 287, 27601–27613. [Google Scholar] [CrossRef]
- Alford, V.M.; Kamath, A.; Ren, X.; Kumar, K.; Gan, Q.; Awwa, M.; Tong, M.; Seeliger, M.A.; Cao, J.; Ojima, I.; et al. Targeting the Hemopexin-like Domain of Latent Matrix Metalloproteinase-9 (proMMP-9) with a Small Molecule Inhibitor Prevents the Formation of Focal Adhesion Junctions. ACS Chem. Biol. 2017, 12, 2788–2803. [Google Scholar] [CrossRef]
- Cao, J.; Kozarekar, P.; Pavlaki, M.; Chiarelli, C.; Bahou, W.F.; Zucker, S. Distinct roles for the catalytic and hemopexin domains of membrane type 1-matrix metalloproteinase in substrate degradation and cell migration. J. Biol. Chem. 2004, 279, 14129–14139. [Google Scholar] [CrossRef]
- Bramhall, S.R.; Schulz, J.; Nemunaitis, J.; Brown, P.D.; Baillet, M.; Buckels, J.A. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br. J. Cancer 2002, 87, 161–167. [Google Scholar] [CrossRef]
- Winer, A.; Janosky, M.; Harrison, B.; Zhong, J.; Moussai, D.; Siyah, P.; Schatz-Siemers, N.; Zeng, J.; Adams, S.; Mignatti, P. Inhibition of Breast Cancer Metastasis by Presurgical Treatment with an Oral Matrix Metalloproteinase Inhibitor: A Preclinical Proof-of-Principle Study. Mol. Cancer Ther. 2016, 15, 2370–2377. [Google Scholar] [CrossRef]
- Rasmussen, H.S.; McCann, P.P. Matrix metalloproteinase inhibition as a novel anticancer strategy: A review with special focus on batimastat and marimastat. Pharmacol. Ther. 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Bloomston, M.; Zervos, E.E.; Rosemurgy, A.S., II. Matrix metalloproteinases and their role in pancreatic cancer: A review of preclinical studies and clinical trials. Ann. Surg. Oncol. 2002, 9, 668–674. [Google Scholar] [CrossRef]
- Kim, H.; Chung, H.; Kim, J.; Choi, D.H.; Shin, Y.; Kang, Y.G.; Kim, B.M.; Seo, S.U.; Chung, S.; Seok, S.H. Macrophages-Triggered Sequential Remodeling of Endothelium-Interstitial Matrix to Form Pre-Metastatic Niche in Microfluidic Tumor Microenvironment. Adv. Sci. 2019, 6, 1900195. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.A.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Grünwald, B.; Vandooren, J.; Gerg, M.; Ahomaa, K.; Hunger, A.; Berchtold, S.; Akbareian, S.; Schaten, S.; Knolle, P.; Edwards, D.R.; et al. Systemic Ablation of MMP-9 Triggers Invasive Growth and Metastasis of Pancreatic Cancer via Deregulation of IL6 Expression in the Bone Marrow. Mol. Cancer Res. 2016, 14, 1147–1158. [Google Scholar] [CrossRef]
- Gossage, D.L.; Cieslarová, B.; Ap, S.; Zheng, H.; Xin, Y.; Lal, P.; Chen, G.; Smith, V.; Sundy, J.S. Phase 1b Study of the Safety, Pharmacokinetics, and Disease-Related Outcomes of the Matrix Metalloproteinase-9 Inhibitor Andecaliximab in Patients With Rheumatoid Arthritis. Clin. Ther. 2018, 40, 156–165.e155. [Google Scholar] [CrossRef]
- Shah, M.A.; Starodub, A.; Sharma, S.; Berlin, J.; Patel, M.; Wainberg, Z.A.; Chaves, J.; Gordon, M.; Windsor, K.; Brachmann, C.B.; et al. Andecaliximab/GS-5745 Alone and Combined with mFOLFOX6 in Advanced Gastric and Gastroesophageal Junction Adenocarcinoma: Results from a Phase I Study. Clin. Cancer Res. 2018, 24, 3829–3837. [Google Scholar] [CrossRef] [Green Version]
- Vandooren, J.; Van den Steen, P.E.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 222–272. [Google Scholar] [CrossRef]
- Murphy, G. Riding the metalloproteinase roller coaster. J. Biol. Chem. 2017, 292, 7708–7718. [Google Scholar] [CrossRef]
- Amar, S.; Fields, G.B. Potential clinical implications of recent MMP inhibitor design strategies. Exp. Rev. Proteomics 2015, 12, 445–447. [Google Scholar] [CrossRef]
- Cauwe, B.; Van den Steen, P.E.; Opdenakker, G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 113–185. [Google Scholar] [CrossRef]
- Van den Steen, P.E.; Proost, P.; Wuyts, A.; Van Damme, J.; Opdenakker, G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood 2000, 96, 2673–2681. [Google Scholar]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.H.; Overall, C.M. Cytokine substrates: MMP regulation of inflammatory signaling molecules. In The Cancer Degradome; Edwards, D., Hoyer-Hansen, G., Blasi, F., Sloane, B.F., Eds.; Springer: New York, NY, USA, 2008; pp. 519–539. [Google Scholar]
- Juric, V.; O’Sullivan, C.; Stefanutti, E.; Kovalenko, M.; Greenstein, A.; Barry-Hamilton, V.; Mikaelian, I.; Degenhardt, J.; Yue, P.; Smith, V.; et al. MMP-9 inhibition promotes anti-tumor immunity through disruption of biochemical and physical barriers to T-cell trafficking to tumors. PLoS ONE 2018, 13, e0207255. [Google Scholar] [CrossRef]
- Liu, G.; Atteridge, C.L.; Wang, X.; Lundgren, A.D.; Wu, J.D. The membrane type matrix metalloproteinase MMP14 mediates constitutive shedding of MHC class I chain-related molecule A independent of A disintegrin and metalloproteinases. J. Immunol. 2010, 184, 3346–3350. [Google Scholar] [CrossRef]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef]
- Golub, L.M.; Lee, H.M.; Ryan, M.E.; Giannobile, W.V.; Payne, J.; Sorsa, T. Tetracyclines inhibit connective tissue breakdown by multiple non-antimicrobial mechanisms. Adv. Dent. Res. 1998, 12, 12–26. [Google Scholar] [CrossRef]
- Ashley, R.A. Clinical trials of a matrix metalloproteinase inhibitor in human periodontal disease: SDD Clinical Research Team. Ann. N. Y. Acad. Sci. 1999, 878, 335–346. [Google Scholar] [CrossRef]
- Preshaw, P.M.; Hefti, A.F.; Novak, M.J.; Michalowicz, B.S.; Pihlstrom, B.L.; Schoor, R.; Trummel, C.L.; Dean, J.; Van Dyke, T.E.; Walker, C.B.; et al. Subantimicrobial dose doxycycline enhances the efficacy of scaling and root planing in chronic periodontitis: A multicenter trial. J. Periodontol. 2004, 75, 1068–1076. [Google Scholar] [CrossRef]
- Boelen, G.J.; Boute, L.; d’Hoop, J.; EzEldeen, M.; Lambrichts, I.; Opdenakker, G. Matrix metalloproteinases and inhibitors in dentistry. Clin. Oral Investig. 2019, 23, 2823–2835. [Google Scholar] [CrossRef]
- Minagar, A.; Alexander, J.S.; Schwendimann, R.N.; Kelley, R.E.; Gonzalez-Toledo, E.; Jimenez, J.J.; Mauro, L.; Jy, W.; Smith, S.J. Combination therapy with interferon beta-1a and doxycycline in multiple sclerosis: An open-label trial. Arch. Neurol. 2008, 65, 199–204. [Google Scholar] [CrossRef]
- Rempe, R.G.; Hartz, A.M.; Bauer, B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef]
- Metz, L.M.; Zhang, Y.; Yeung, M.; Patry, D.G.; Bell, R.B.; Stoian, C.A.; Yong, V.W.; Patten, S.B.; Duquette, P.; Antel, J.P.; et al. Minocycline reduces gadolinium-enhancing magnetic resonance imaging lesions in multiple sclerosis. Ann. Neurol. 2004, 55, 756. [Google Scholar] [CrossRef]
- Metz, L.M.; Li, D.K.B.; Traboulsee, A.L.; Duquette, P.; Eliasziw, M.; Cerchiaro, G.; Greenfield, J.; Riddehough, A.; Yeung, M.; Kremenchutzky, M.; et al. Trial of Minocycline in a Clinically Isolated Syndrome of Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 2122–2133. [Google Scholar] [CrossRef] [Green Version]
- Metz, L.M.; Li, D.; Traboulsee, A.; Myles, M.L.; Duquette, P.; Godin, J.; Constantin, M.; Yong, V.W.; GA/minocycline Study Investigators. Glatiramer acetate in combination with minocycline in patients with relapsing--remitting multiple sclerosis: Results of a Canadian, multicenter, double-blind, placebo-controlled trial. Mult. Scler. 2009, 15, 1183–1194. [Google Scholar] [CrossRef]
- Fagan, S.C.; Waller, J.L.; Nichols, F.T.; Edwards, D.J.; Pettigrew, L.C.; Clark, W.M.; Hall, C.E.; Switzer, J.A.; Ergul, A.; Hess, D.C. Minocycline to improve neurologic outcome in stroke (MINOS): A dose-finding study. Stroke 2010, 41, 2283–2287. [Google Scholar] [CrossRef]
- Switzer, J.A.; Hess, D.C.; Ergul, A.; Waller, J.L.; Machado, L.S.; Portik-Dobos, V.; Pettigrew, L.C.; Clark, W.M.; Fagan, S.C. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke 2011, 42, 2633–2635. [Google Scholar] [CrossRef]
- Romanic, A.M.; White, R.F.; Arleth, A.J.; Ohlstein, E.H.; Barone, F.C. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: Inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 1998, 29, 1020–1030. [Google Scholar] [CrossRef]
- Wang, X.; Lee, S.R.; Arai, K.; Lee, S.R.; Tsuji, K.; Rebeck, G.W.; Lo, E.H. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat. Med. 2003, 9, 1313–1317. [Google Scholar] [CrossRef]
- Ning, M.; Furie, K.L.; Koroshetz, W.J.; Lee, H.; Barron, M.; Lederer, M.; Wang, X.; Zhu, M.; Sorensen, A.G.; Lo, E.H.; et al. Association between tPA therapy and raised early matrix metalloproteinase-9 in acute stroke. Neurology 2006, 66, 1550–1555. [Google Scholar] [CrossRef]
- Dezube, B.J.; Krown, S.E.; Lee, J.Y.; Bauer, K.S.; Aboulafia, D.M. Randomized phase II trial of matrix metalloproteinase inhibitor COL-3 in AIDS-related Kaposi’s sarcoma: An AIDS Malignancy Consortium Study. J. Clin. Oncol. 2006, 24, 1389–1394. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ding, D.; Wolter, W.R.; Pérez, R.L.; Champion, M.M.; Mahasenan, K.V.; Hesek, D.; Lee, M.; Schroeder, V.A.; Jones, J.I.; et al. Validation of Matrix Metalloproteinase-9 (MMP-9) as a Novel Target for Treatment of Diabetic Foot Ulcers in Humans and Discovery of a Potent and Selective Small-Molecule MMP-9 Inhibitor That Accelerates Healing. J. Med. Chem. 2018, 61, 8825–8837. [Google Scholar] [CrossRef]
- Vartak, D.G.; Gemeinhart, R.A. Matrix metalloproteinases: Underutilized targets for drug delivery. J. Drug. Targeting 2007, 15, 1–20. [Google Scholar] [CrossRef]
- Fields, G.B. Protease-activated delivery and imaging systems. In The Cancer Degradome—Proteases in Cancer Biology; Edwards, D., Hoyer-Hansen, G., Blasi, F., Sloane, B., Eds.; Springer: New York, NY, USA, 2008; pp. 827–851. [Google Scholar]
- Atkinson, J.M.; Siller, C.S.; Gill, J.H. Tumour endoproteases: The cutting edge of cancer drug delivery? Br. J. Pharmacol. 2008, 153, 1344–1352. [Google Scholar] [CrossRef]
- Vandooren, J.; Opdenakker, G.; Loadman, P.M.; Edwards, D.R. Proteases in cancer drug delivery. Adv. Drug Deliv. Rev. 2016, 97, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Daldrup-Link, H.E. Rethinking Brain Cancer Therapy: Tumor Enzyme Activatable Theranostic Nanoparticles. Mol. Imaging 2017, 16, 1–4. [Google Scholar] [CrossRef]
- Jain, M.; Harburn, J.J.; Gill, J.H.; Loadman, P.M.; Falconer, R.A.; Mooney, C.A.; Cobb, S.L.; Berry, D.J. Rationalized Computer-Aided Design of Matrix-Metalloprotease-Selective Prodrugs. J. Med. Chem. 2017, 60, 4496–4502. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, J.M.; Falconer, R.A.; Edwards, D.R.; Pennington, C.J.; Siller, C.S.; Shnyder, S.D.; Bibby, M.C.; Patterson, L.H.; Loadman, P.M.; Gill, J.H. Development of a novel tumor-targeted vascular disrupting agent activated by membrane-type matrix metalloproteinases. Cancer Res. 2010, 70, 6902–6912. [Google Scholar] [CrossRef]
- Gill, J.H.; Loadman, P.M.; Shnyder, S.D.; Cooper, P.; Atkinson, J.M.; Ribeiro Morais, G.; Patterson, L.H.; Falconer, R.A. Tumor-targeted prodrug ICT2588 demonstrates therapeutic activity against solid tumors and reduced potential for cardiovascular toxicity. Mol. Pharm. 2014, 11, 1294–1300. [Google Scholar] [CrossRef]
- Ansari, C.; Tikhomirov, G.A.; Hong, S.H.; Falconer, R.A.; Loadman, P.M.; Gill, J.H.; Castaneda, R.; Hazard, F.K.; Tong, L.; Lenkov, O.D.; et al. Development of novel tumor-targeted theranostic nanoparticles activated by membrane-type matrix metalloproteinases for combined cancer magnetic resonance imaging and therapy. Small 2014, 10, 566–575. [Google Scholar] [CrossRef]
- Mohanty, S.; Chen, Z.; Li, K.; Morais, G.R.; Klockow, J.; Yerneni, K.; Pisani, L.; Chin, F.T.; Mitra, S.; Cheshier, S.; et al. A Novel Theranostic Strategy for MMP-14-Expressing Glioblastomas Impacts Survival. Mol. Cancer Ther. 2017, 16, 1909–1921. [Google Scholar] [CrossRef]
Figure 1.
Schematic depiction of the domains of a representative MMP family member, membrane type 1 matrix metalloproteinase (MT1-MMP).
Figure 1.
Schematic depiction of the domains of a representative MMP family member, membrane type 1 matrix metalloproteinase (MT1-MMP).

Figure 2.
The “protease web”, in which proteases act in linear pathways, amplification cascades (with a one-way flow of information), or in circuits (with information feedback) [33]. Figure reproduced with permission of the Nature Publishing Group.
Figure 2.
The “protease web”, in which proteases act in linear pathways, amplification cascades (with a one-way flow of information), or in circuits (with information feedback) [33]. Figure reproduced with permission of the Nature Publishing Group.

Figure 3.
Strategies for creating selective MMP inhibitors. (A) endogenous-like inhibitors, (B) exosite targeting inhibitors, (C) combination of exosite binding and metal chelating inhibitor, (D) function blocking antibodies, and (E) inhibitor disrupting MMP interactions with cell surface binding partners.
Figure 3.
Strategies for creating selective MMP inhibitors. (A) endogenous-like inhibitors, (B) exosite targeting inhibitors, (C) combination of exosite binding and metal chelating inhibitor, (D) function blocking antibodies, and (E) inhibitor disrupting MMP interactions with cell surface binding partners.

Figure 4.
Structures of small molecule MMP inhibitors (A) 10d, (B) (S)-17b, (C) JNJ0966 [N-(2-((2-methoxyphenyl)amino)-4’-methyl-[4,5’-bithiazol]-2’-yl)acetamide], (D) NSC405020 [3,4-dichloro-N-(1-methylbutyl)benzamide], (E) N-(4-fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanamide, (F) doxycycline, (G) minocycline, and (H) (R)-ND-336.
Figure 4.
Structures of small molecule MMP inhibitors (A) 10d, (B) (S)-17b, (C) JNJ0966 [N-(2-((2-methoxyphenyl)amino)-4’-methyl-[4,5’-bithiazol]-2’-yl)acetamide], (D) NSC405020 [3,4-dichloro-N-(1-methylbutyl)benzamide], (E) N-(4-fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanamide, (F) doxycycline, (G) minocycline, and (H) (R)-ND-336.

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. https://doi.org/10.3390/cells8090984
AMA Style
Fields GB. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells. 2019; 8(9):984. https://doi.org/10.3390/cells8090984
Chicago/Turabian StyleFields, Gregg B. 2019. "The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma" Cells 8, no. 9: 984. https://doi.org/10.3390/cells8090984
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.