Electrochemical Characterization of Escherichia coli Adaptive Response Protein AidB

Abstract
:1. Introduction
2. Results
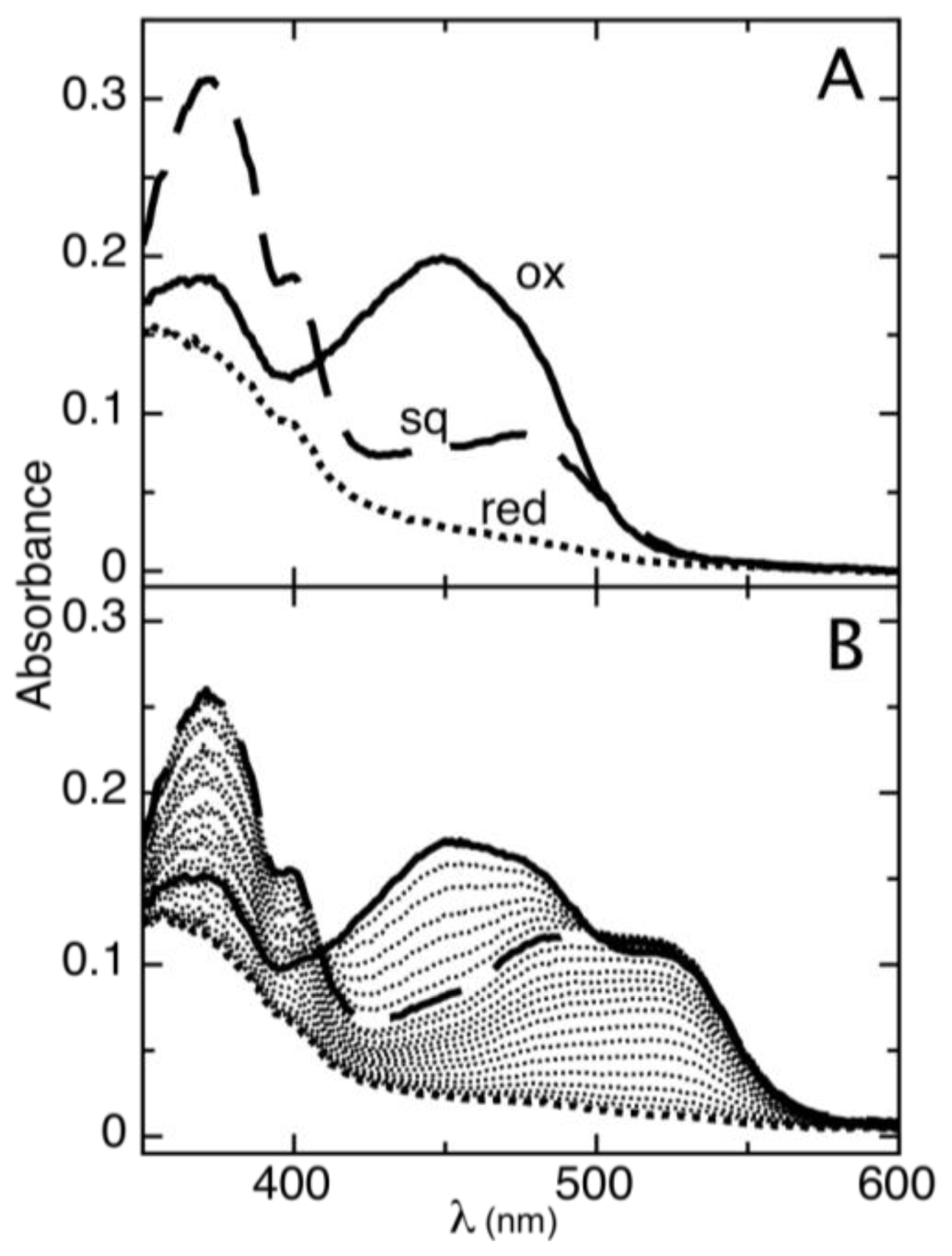
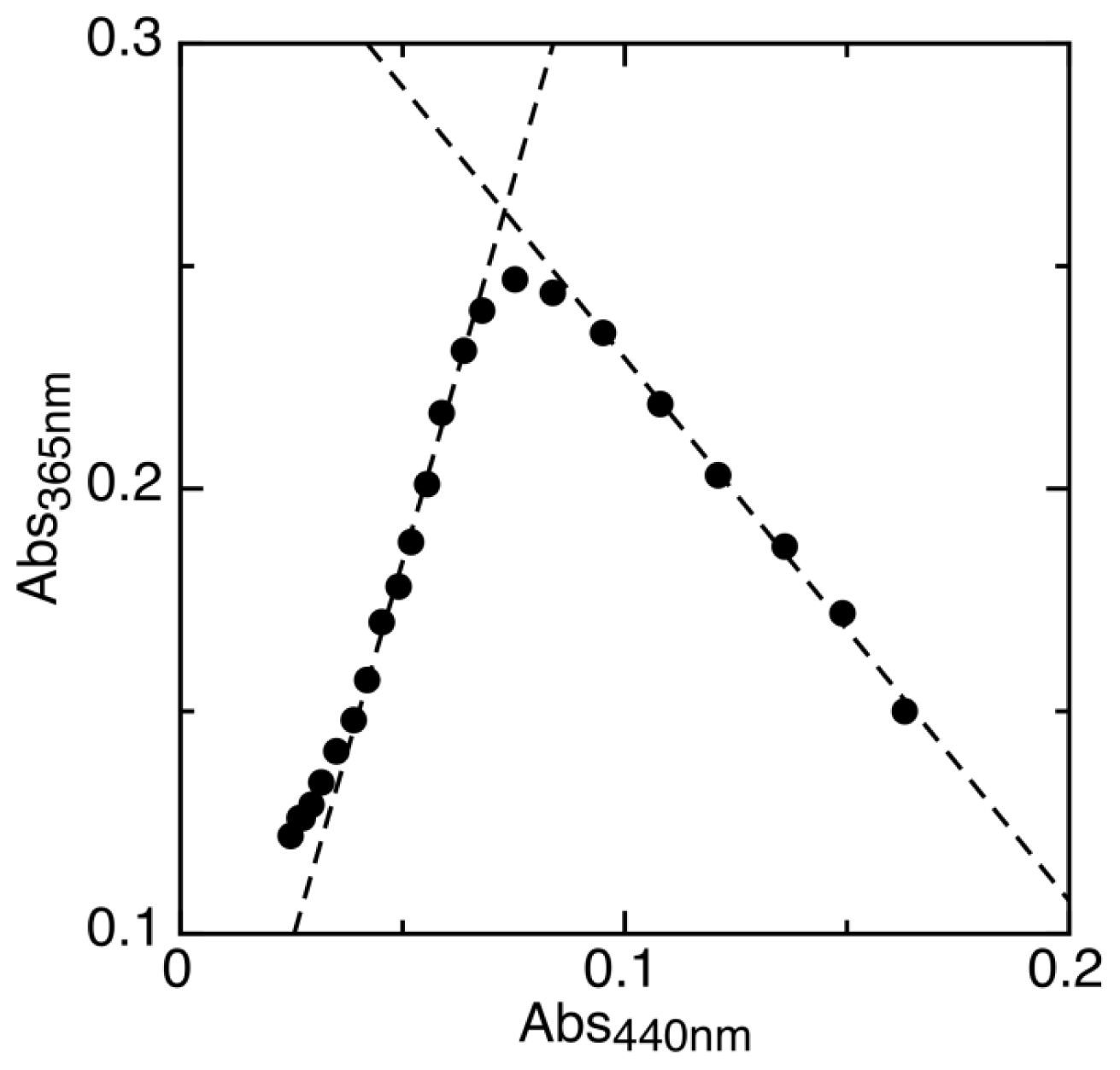
2.1. Determination of Reduction Potentials for AidB
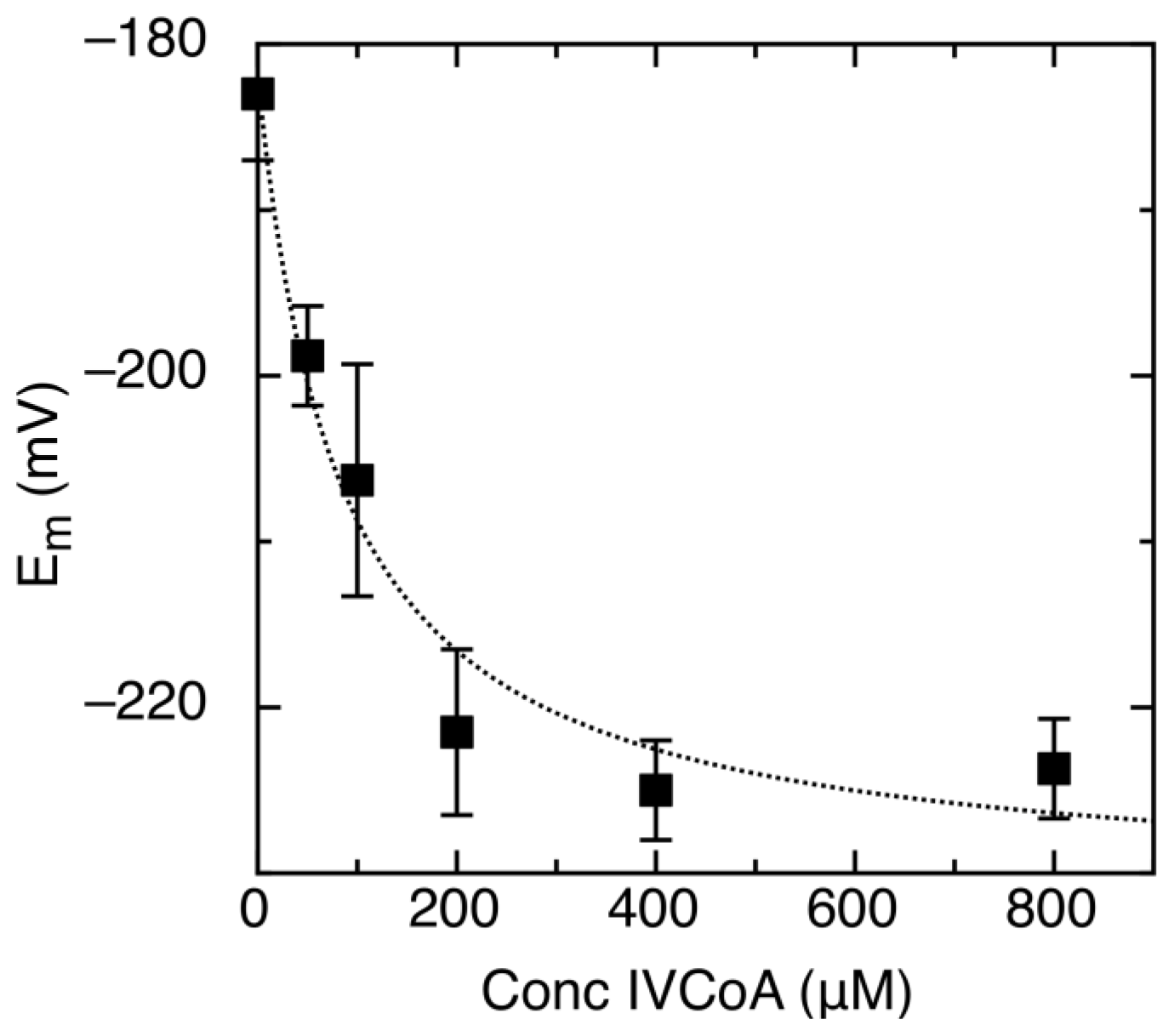
2.2. Effect of IVCoA on Reduction Potential
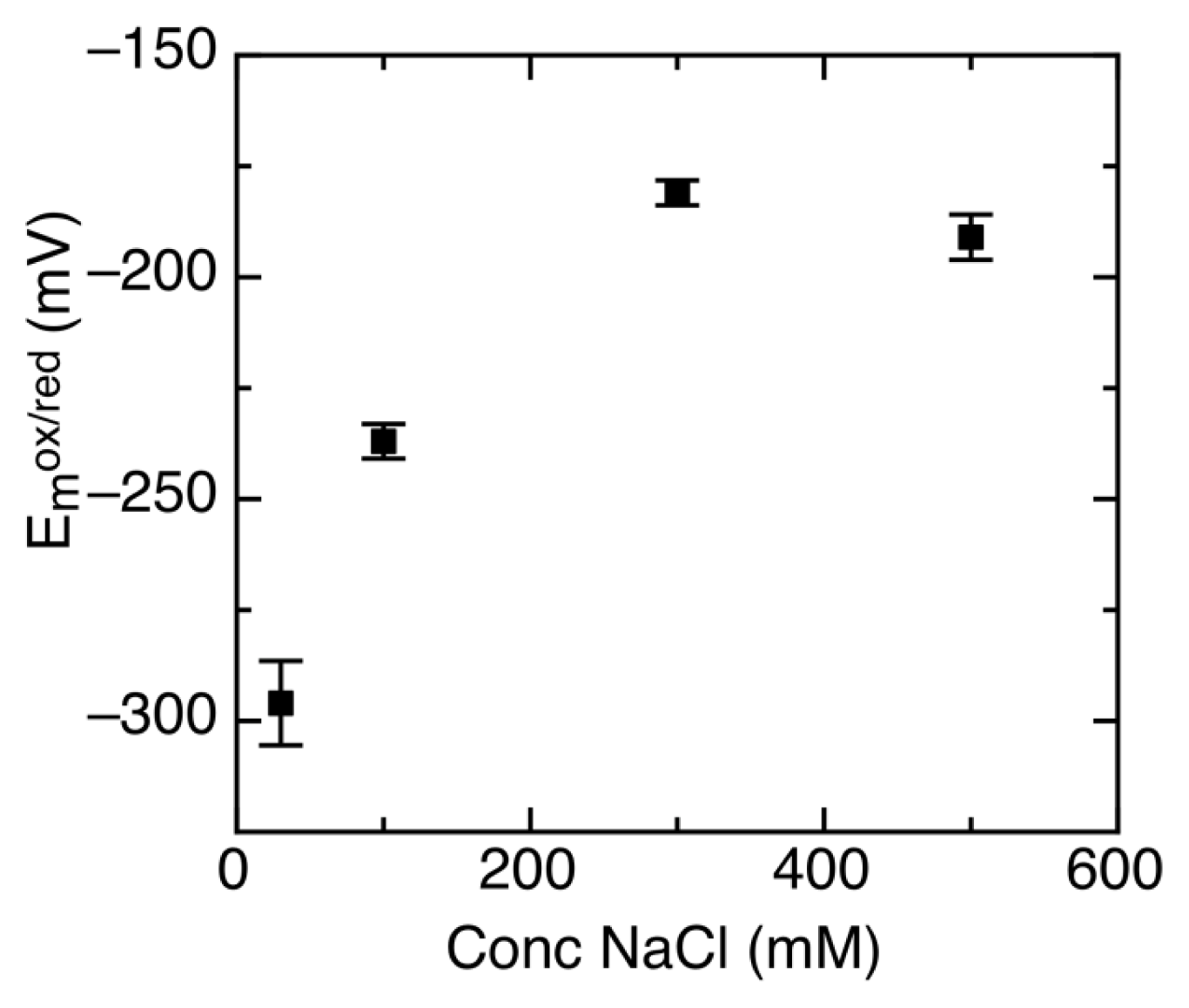
2.3. Ionic Strength Dependent Reduction Potentials
2.4. Effect of DNA on Reduction Potential
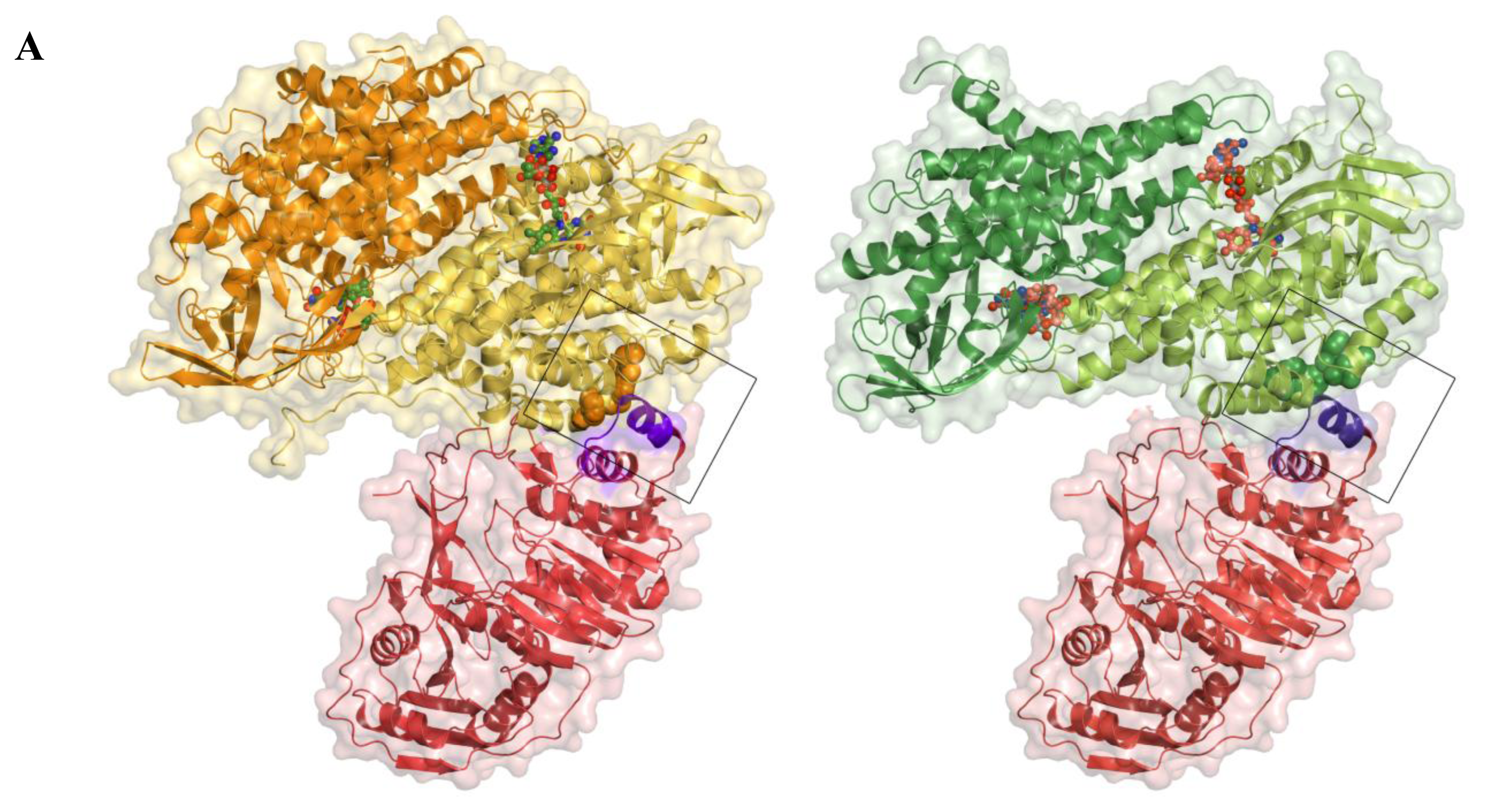
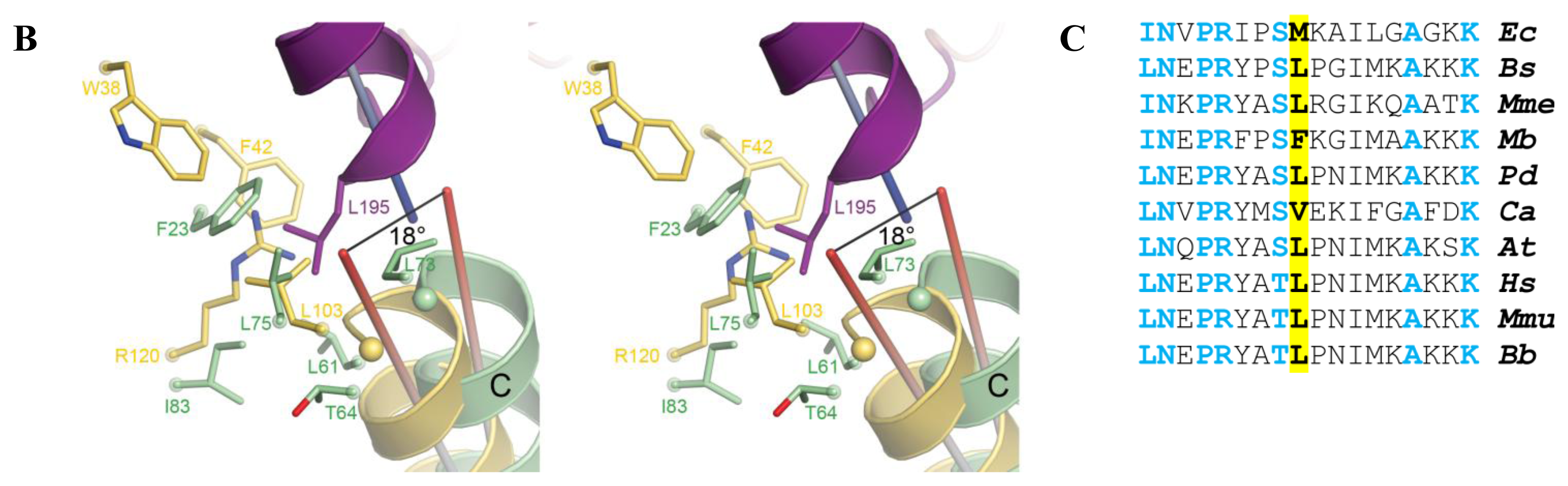
2.5. AidB-ETF Docking Model
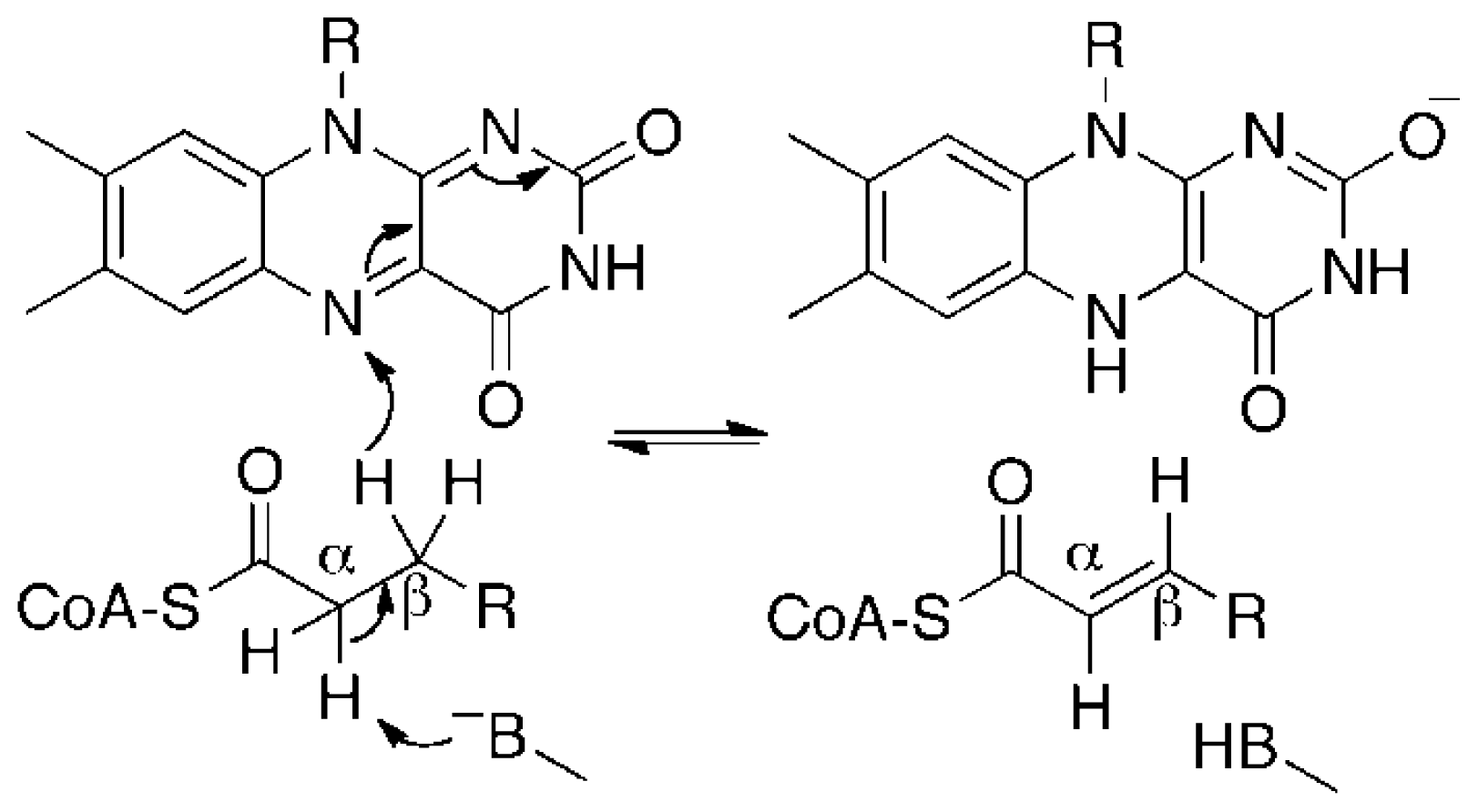
3. Discussion
4. Experimental Section
4.1. Redox Potentiometry
4.2. Structural Modeling of Electron Transfer Flavoprotein Docking
5. Conclusions
Acknowledgments
- Conflict of interestThe authors declare no conflict of interest.
References
- Volkert, M.R. Adaptive response of Escherichia coli to alkylation damage. Environ. Mol. Mutagen 1988, 11, 241–255. [Google Scholar]
- Volkert, M.R.; Nguyen, D.C. Induction of specific Escherichia coli genes by sublethal treatments with alkylating agents. Proc. Natl. Acad. Sci. USA 1984, 81, 4110–4114. [Google Scholar]
- Sedgwick, B. Repairing DNA-methylation damage. Nat. Rev. Mol. Cell Biol 2004, 5, 148–157. [Google Scholar]
- Samson, L.; Cairns, J. A new pathway for DNA repair in Escherichia coli. Nature 1977, 267, 281–283. [Google Scholar]
- Sedgwick, B.; Robins, P.; Totty, N.; Lindahl, T. Functional domains and methyl acceptor sites of the Escherichia coli ada protein. J. Biol. Chem 1988, 263, 4430–4433. [Google Scholar]
- Demple, B.; Sedgwick, B.; Robins, P.; Totty, N.; Waterfield, M.D.; Lindahl, T. Active site and complete sequence of the suicidal methyltransferase that counters alkylation mutagenesis. Proc. Natl. Acad. Sci. USA 1985, 82, 2688–2692. [Google Scholar]
- Landini, P.; Volkert, M.R. Regulatory responses of the adaptive response to alkylation damage: A simple regulon with complex regulatory features. J. Bacteriol 2000, 182, 6543–6549. [Google Scholar]
- Sedgwick, B.; Lindahl, T. Recent progress on the Ada response for inducible repair of DNA alkylation damage. Oncogene 2002, 21, 8886–8894. [Google Scholar]
- Evensen, G.; Seeberg, E. Adaptation to alkylation resistance involves the induction of a DNA glycosylase. Nature 1982, 296, 773–775. [Google Scholar]
- Karran, P.; Hjelmgren, T.; Lindahl, T. Induction of a DNA glycosylase for N-methylated purines is part of the adaptive response to alkylating agents. Nature 1982, 296, 770–773. [Google Scholar]
- Trewick, S.C.; Henshaw, T.F.; Hausinger, R.P.; Lindahl, T.; Sedgwick, B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 2002, 419, 174–178. [Google Scholar]
- Falnes, P.O.; Johansen, R.F.; Seeberg, E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature 2002, 419, 178–182. [Google Scholar]
- Falnes, P.O. Repair of 3-methylthymine and 1-methylguanine lesions by bacterial and human AlkB proteins. Nucleic Acids Res 2004, 32, 6260–6267. [Google Scholar]
- Delaney, J.C.; Smeester, L.; Wong, C.; Frick, L.E.; Taghizadeh, K.; Wishnok, J.S.; Drennan, C.L.; Samson, L.D.; Essigmann, J.M. AlkB reverses etheno DNA lesions caused by lipid oxidation in vitro and in vivo. Nat. Struct. Mol. Biol 2005, 12, 855–860. [Google Scholar]
- Frick, L.E.; Delaney, J.C.; Wong, C.; Drennan, C.L.; Essigmann, J.M. Alleviation of 1,N6-ethanoadenine genotoxicity by the Escherichia coli adaptive response protein AlkB. Proc. Natl. Acad. Sci. USA 2007, 104, 755–760. [Google Scholar]
- Landini, P.; Hajec, L.I.; Volkert, M.R. Structure and transcriptional regulation of the Escherichia coli adaptive response gene aidB. J. Bacteriol 1994, 176, 6583–6589. [Google Scholar]
- Rohankhedkar, M.S.; Mulrooney, S.B.; Wedemeyer, W.J.; Hausinger, R.P. The AidB component of the Escherichia coli adaptive response to alkylating agents is a flavin-containing, DNA-binding protein. J. Bacteriol 2006, 188, 223–230. [Google Scholar]
- Bowles, T.; Metz, A.H.; O’Quin, J.; Wawrzak, Z.; Eichman, B.F. Structure and DNA binding of alkylation response protein AidB. Proc. Natl. Acad. Sci. USA 2008, 105, 15299–15304. [Google Scholar]
- Rippa, V.; Duilio, A.; di Pasquale, P.; Amoresano, A.; Landini, P.; Volkert, M.R. Preferential DNA damage prevention by the E. coli AidB gene: A new mechanism for the protection of specific genes. DNA Repair 2011, 10, 934–941. [Google Scholar]
- Mulrooney, S.B.; Howard, M.J.; Hausinger, R.P. The Escherichia coli alkylation response protein AidB is a redox partner of flavodoxin and binds RNA and acyl carrier protein. Arch. Biochem. Biophys 2011, 513, 81–86. [Google Scholar]
- Hamill, M.J.; Jost, M.; Wong, C.; Elliott, S.J.; Drennan, C.L. Flavin-induced oligomerization in Escherichia coli adaptive response protein AidB. Biochemistry 2011, 50, 10159–10169. [Google Scholar]
- Massey, V. The chemical and biological versatility of riboflavin. Biochem. Soc. Trans 2000, 28, 283–296. [Google Scholar]
- Massey, V.; Palmer, G. On the existence of spectrally distinct classes of flavoprotein semiquinones. A new method for the quantitative production of flavoprotein semiquinones. Biochemistry 1966, 5, 3181–3189. [Google Scholar]
- Clark, W.M. Oxidation-Reduction Potentials of Organic Systems; The Williams & Wilkins Company: Baltimore, MD, USA; p. 1960.
- Toogood, H.S.; Leys, D.; Scrutton, N.S. Dynamics driving function: New insights from electron transferring flavoproteins and partner complexes. FEBS J 2007, 274, 5481–5504. [Google Scholar]
- Roberts, D.L.; Salazar, D.; Fulmer, J.P.; Frerman, F.E.; Kim, J.J. Crystal structure of Paracoccus denitrificans electron transfer flavoprotein: Structural and electrostatic analysis of a conserved flavin binding domain. Biochemistry 1999, 38, 1977–1989. [Google Scholar]
- Roberts, D.L.; Frerman, F.E.; Kim, J.J. Three-dimensional structure of human electron transfer flavoprotein to 2.1-A resolution. Proc. Natl. Acad. Sci. USA 1996, 93, 14355–14360. [Google Scholar]
- Leys, D.; Basran, J.; Talfournier, F.; Sutcliffe, M.J.; Scrutton, N.S. Extensive conformational sampling in a ternary electron transfer complex. Nat. Struct. Biol 2003, 10, 219–225. [Google Scholar]
- Toogood, H.S.; van Thiel, A.; Basran, J.; Sutcliffe, M.J.; Scrutton, N.S.; Leys, D. Extensive domain motion and electron transfer in the human electron transferring flavoprotein.medium chain Acyl-CoA dehydrogenase complex. J. Biol. Chem 2004, 279, 32904–32912. [Google Scholar]
- Chohan, K.K.; Jones, M.; Grossmann, J.G.; Frerman, F.E.; Scrutton, N.S.; Sutcliffe, M.J. Protein dynamics enhance electronic coupling in electron transfer complexes. J. Biol. Chem 2001, 276, 34142–34147. [Google Scholar]
- Swanson, M.A.; Kathirvelu, V.; Majtan, T.; Frerman, F.E.; Eaton, G.R.; Eaton, S.S. Electron transfer flavoprotein domain II orientation monitored using double electron-electron resonance between an enzymatically reduced, native FAD cofactor, and spin labels. Protein Sci 2011, 20, 610–620. [Google Scholar]
- Kim, J.J.; Wang, M.; Paschke, R. Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate. Proc. Natl. Acad. Sci. USA 1993, 90, 7523–7527. [Google Scholar]
- Ghisla, S.; Thorpe, C. Acyl-CoA dehydrogenases. A mechanistic overview. Eur. J. Biochem 2004, 271, 494–508. [Google Scholar]
- Lenn, N.D.; Stankovich, M.T.; Liu, H.W. Regulation of the redox potential of general acyl-CoA dehydrogenase by substrate binding. Biochemistry 1990, 29, 3709–3715. [Google Scholar]
- Fink, C.W.; Stankovich, M.T.; Soltysik, S. Oxidation-reduction potentials of butyryl-CoA dehydrogenase. Biochemistry 1986, 25, 6637–6643. [Google Scholar]
- Pace, C.P.; Stankovich, M.T. Oxidation-reduction properties of short-chain acyl-CoA dehydrogenase: Effects of substrate analogs. Arch. Biochem. Biophys 1994, 313, 261–266. [Google Scholar]
- Stankovich, M.T.; Soltysik, S. Regulation of the butyryl-CoA dehydrogenase by substrate and product binding. Biochemistry 1987, 26, 2627–2632. [Google Scholar]
- Johnson, B.D.; Stankovich, M.T. Influence of two substrate analogues on thermodynamic properties of medium-chain acyl-CoA dehydrogenase. Biochemistry 1993, 32, 10779–10785. [Google Scholar]
- Byron, C.M.; Stankovich, M.T.; Husain, M. Spectral and electrochemical properties of glutaryl-CoA dehydrogenase from Paracoccus denitrificans. Biochemistry 1990, 29, 3691–3700. [Google Scholar]
- Saenger, A.K.; Nguyen, T.V.; Vockley, J.; Stankovich, M.T. Thermodynamic regulation of human short-chain acyl-CoA dehydrogenase by substrate and product binding. Biochemistry 2005, 44, 16043–16053. [Google Scholar]
- Becker, D.F.; Fuchs, J.A.; Stankovich, M.T. Product binding modulates the thermodynamic properties of a Megasphaera elsdenii short-chain acyl-CoA dehydrogenase active-site mutant. Biochemistry 1994, 33, 7082–7087. [Google Scholar]
- He, M.; Burghardt, T.P.; Vockley, J. A novel approach to the characterization of substrate specificity in short/branched chain Acyl-CoA dehydrogenase. J. Biol. Chem. 2003, 278, 37974–37986. [Google Scholar]
- Frerman, F.E.; Miziorko, H.M.; Beckmann, J.D. Enzyme-activated inhibitors, alternate substrates, and a dead end inhibitor of the general Acyl-Coa dehydrogenase. J. Biol. Chem 1980, 255, 1192–1198. [Google Scholar]
- Maier, E.M.; Gersting, S.W.; Kemter, K.F.; Jank, J.M.; Reindl, M.; Messing, D.D.; Truger, M.S.; Sommerhoff, C.P.; Muntau, A.C. Protein misfolding is the molecular mechanism underlying MCADD identified in newborn screening. Hum. Mol. Genet 2009, 18, 1612–1623. [Google Scholar]
- Husain, M.; Steenkamp, D.J. Partial purification and characterization of glutaryl-coenzyme A dehydrogenase, electron transfer flavoprotein, and electron transfer flavoprotein-Q oxidoreductase from Paracoccus denitrificans. J. Bacteriol 1985, 163, 709–715. [Google Scholar]
- Vetter, H., Jr; Knappe, J. Flavodoxin and ferredoxin of Escherichia coli. Hoppe. Seylers Z. Physiol. Chem. 1971, 352, 433–446. [Google Scholar]
- Massey, V.A. Simple Method for the Determination of Redox Potentials. In Flavins and Flavoproteins; Curti, B., Rochi, S., Zanetti, G., Eds.; Water DeGruyter & Co.: Berlin, Germany, 1991; pp. 59–66. [Google Scholar]
- Stiehler, R.D.; Chen, T.T.; Clark, W.M. Studies on oxidation-reduction. XVIII. simple Safranines. J. Am. Chem. Soc 1933, 55, 891–907. [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.5.0.4; LLC: Schrödinger. Available online: http://www.pymol.org/ accessed on 8 December 2012.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Emox/sq | Emsq/red | Emox/red | % sq | pH | Radical | Ref. |
|---|---|---|---|---|---|---|---|
| AidB (300 mM NaCl) | −103 | −258 | −181 | 92 | 8.0 | anionic | - |
| AidB + IVCoA (300 mM NaCl) | −148 | −297 | −222 | 91 | 8.0 | anionic | - |
| AidB (100 mM NaCl) | −160 | −315 | −237 | 92 | 8.0 | anionic | - |
| AidB + pUC19 (100 mM NaCl) | −121 | −274 | −197 | 92 | 8.0 | anionic | - |
| AidB + 28-mer (100 mM NaCl) | −129 | −282 | −205 | 92 | 8.0 | anionic | - |
| MeSCAD | - | - | −79 | 5 | 7.0 | neutral | [35] |
| MeSCAD + S/P | - | - | −19 | 0 | 7.0 | - | [37] |
| MeSCAD + Butyl-CoA | - | - | −79 | 0 | 6.9 | - | [36] |
| MeSCAD + Acetoacetyl-CoA | - | - | −180 | 0 | 7.0 | - | [35] |
| PdGCD | - | - | −85 | 0 | 6.4 | - | [39] |
| PdGCD + S/P | - | - | 30 | 5 | 6.4 | anionic | [39] |
| PdGCD + Acetoacetyl-CoA | −154 † | −104 † | −129 | 15 | 6.4 | neutral | [39] |
| pMCAD | −166 | −129 | −136 | 20 | 7.6 | neutral | [38] |
| pMCAD + S/P | - | - | −26 | 0 | 7.6 | - | [34] |
| pMCAD + Butyl-CoA | −189 † | −155 † | −172 | 20 | 7.6 | neutral | [38] |
| hSCAD * | - | - | −141 | ≤5 | 7.6 | - | [40] |
| hSCAD * + Butyryl-CoA | - | - | −103 | ≤5 | 7.6 | - | [40] |
| hSCAD * + Octanoyl-CoA | - | - | −161 | ≤5 | 7.6 | - | [40] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hamill, M.J.; Jost, M.; Wong, C.; Bene, N.C.; Drennan, C.L.; Elliott, S.J. Electrochemical Characterization of Escherichia coli Adaptive Response Protein AidB. Int. J. Mol. Sci. 2012, 13, 16899-16915. https://doi.org/10.3390/ijms131216899
Hamill MJ, Jost M, Wong C, Bene NC, Drennan CL, Elliott SJ. Electrochemical Characterization of Escherichia coli Adaptive Response Protein AidB. International Journal of Molecular Sciences. 2012; 13(12):16899-16915. https://doi.org/10.3390/ijms131216899
Chicago/Turabian StyleHamill, Michael J., Marco Jost, Cintyu Wong, Nicholas C. Bene, Catherine L. Drennan, and Sean J. Elliott. 2012. "Electrochemical Characterization of Escherichia coli Adaptive Response Protein AidB" International Journal of Molecular Sciences 13, no. 12: 16899-16915. https://doi.org/10.3390/ijms131216899