
Diabetic Microvascular Disease and Pulmonary Fibrosis: The Contribution of Platelets and Systemic Inflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
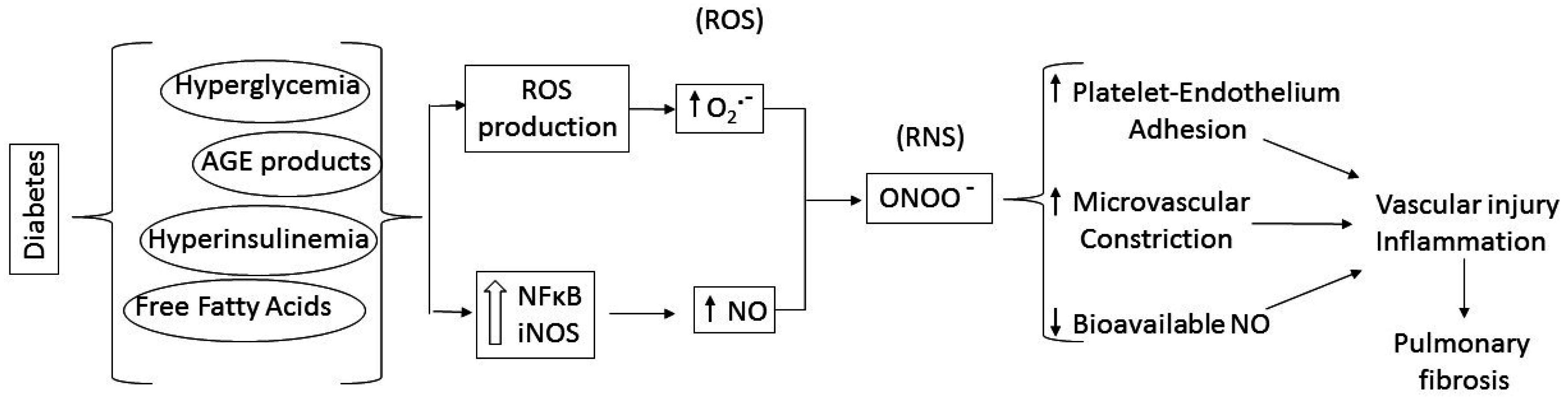
:1. Diabetic Lung Fibrosis
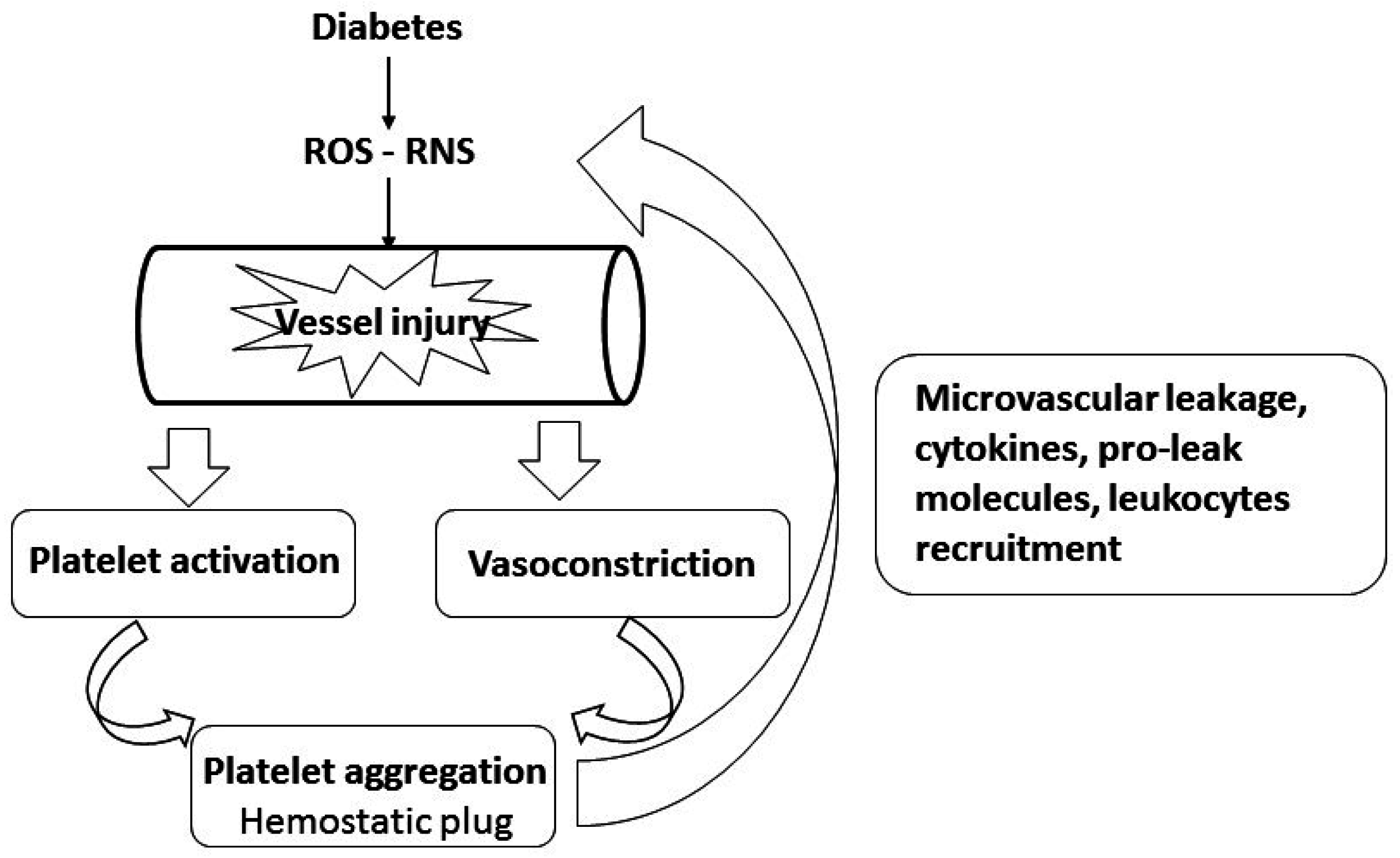
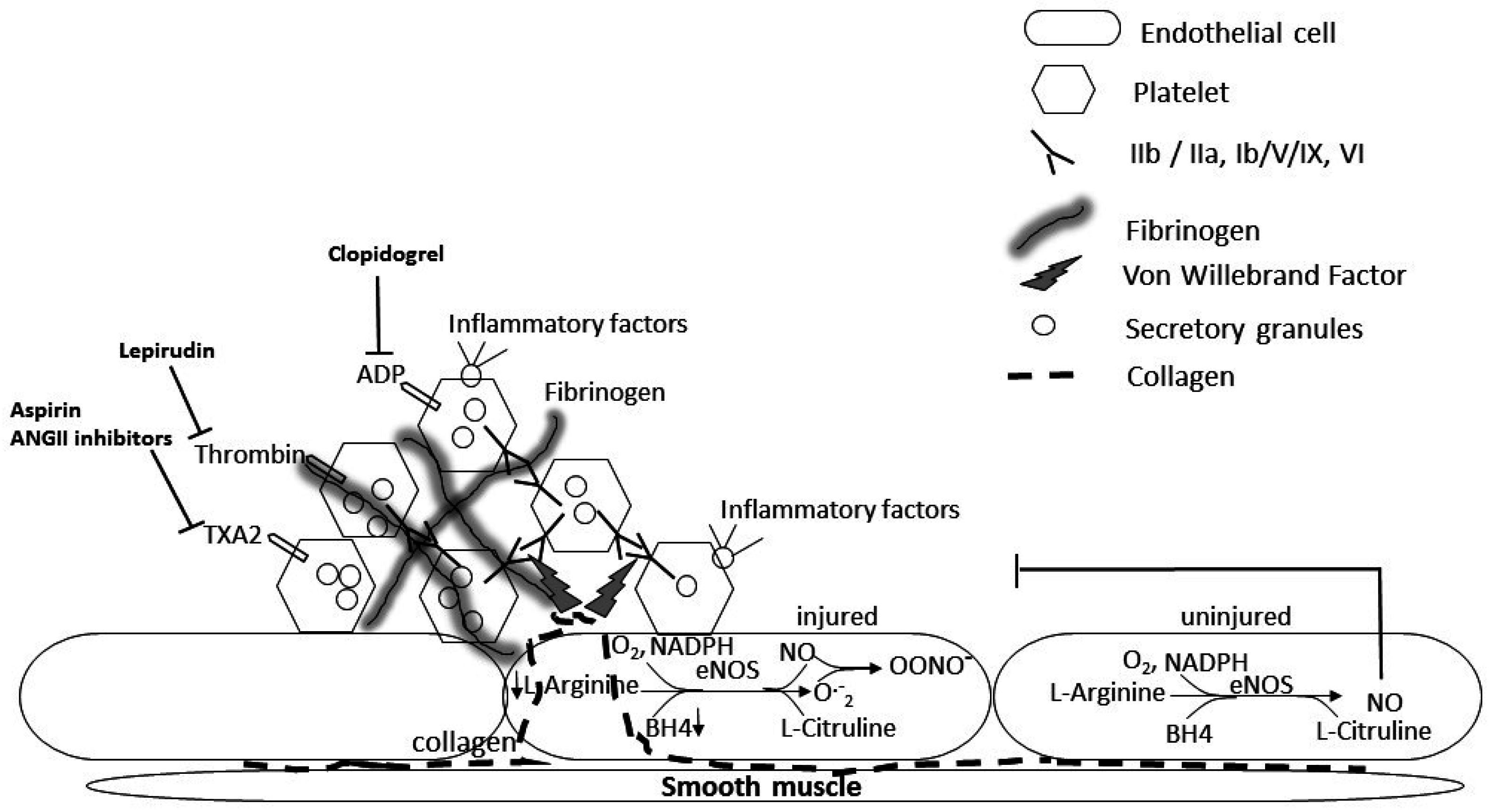
2. A Role for Platelets in the Pathology of Diabetes-Induced Microvascular and Lung Disease
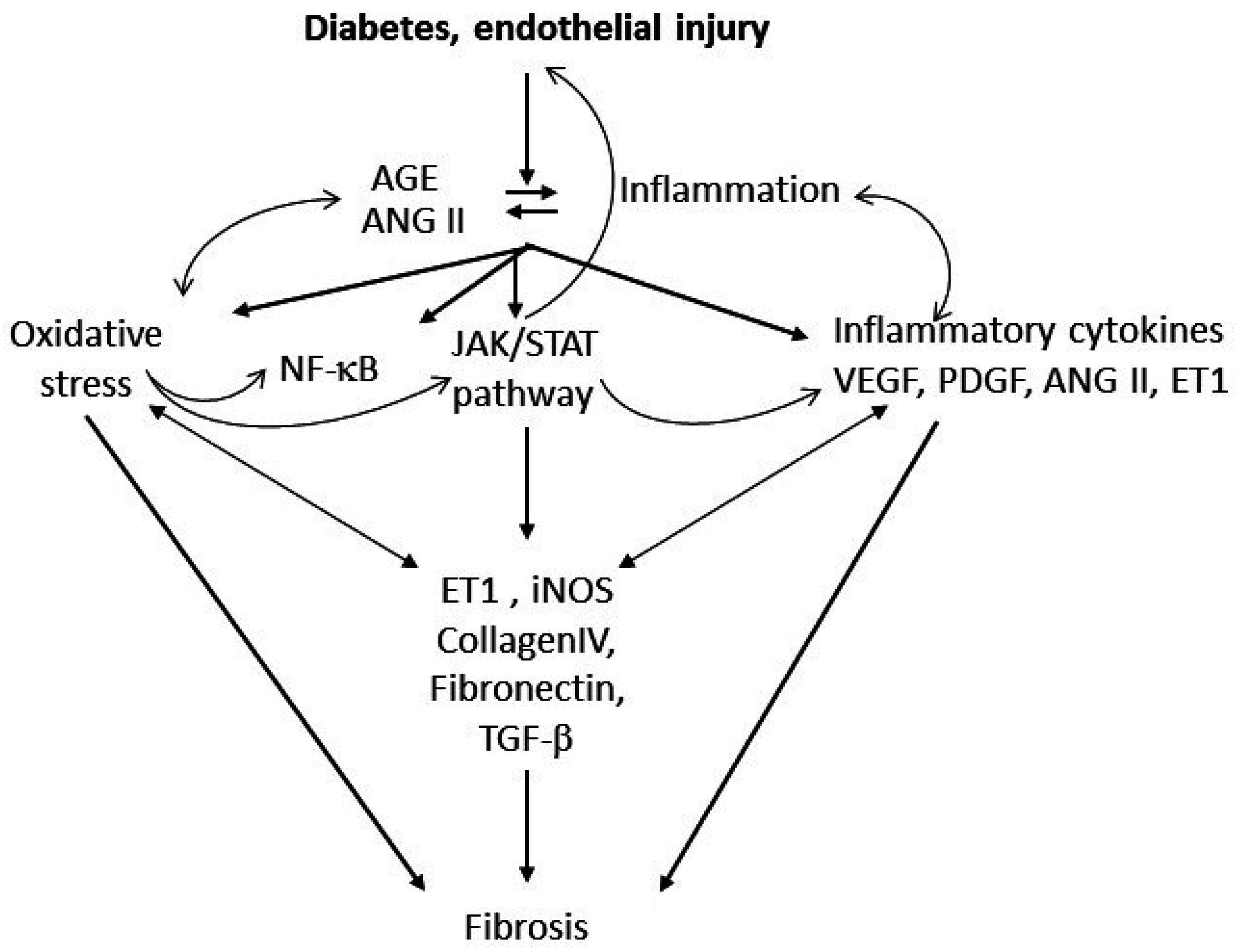
3. Putative Signaling Mediating Diabetes-Induced Microvascular Disease and Lung Fibrosis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Centers for Disease Control and Prevention (CDC). 2014 National Diabetes Statistics Report; CDC: Atlanta, GA, USA, 2014. Available online: http://www.cdc.gov/diabetes/data/statistics/2014statisticsreport.html (accessed on 3 November 2016).
- Zhang, Z.; Wang, S.; Zhou, S.; Yan, X.; Wang, Y.; Chen, J.; Mellen, N.; Kong, M.; Gu, J.; Tan, Y.; et al. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of LKB1/AMPK pathway. J. Mol. Cell. Cardiol. 2014, 77, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Kang, Y.J. Oxidative stress and diabetic cardiomyopathy. Cardiovasc. Toxicol. 2001, 1, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, S.F.; Quesenberry, C.P., Jr.; van den Eeden, S.K.; Shan, J.; Ferrara, A. Patients diagnosed with diabetes are at increased risk for asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, and pneumonia but not lung cancer. Diabetes Care 2010, 33, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Weynand, B.; Jonckheere, A.; Frans, A.; Rahier, J. Diabetes mellitus induces a thickening of the pulmonary basal lamina. Respiration 1999, 66, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tan, Y.; Zhao, F.; Ma, Z.; Wang, Y.; Zheng, S.; Epstein, P.N.; Yu, J.; Yin, X.; Zheng, Y.; et al. Angiotensin II plays a critical role in diabetic pulmonary fibrosis most likely via activation of NADPH oxidase-mediated nitrosative damage. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E132–E144. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xue, Q.; Miao, L.; Cai, L. Pulmonary fibrosis: A possible diabetic complication. Diabetes Metab. Res. Rev. 2011, 27, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Uz-Zaman, S.; Banerjee, J.; Singhamahapatra, A.; Dey, P.K.; Roy, A.; Roy, K.; Roy Basu, K. Assessment of lung function by spirometry and diffusion study and effect of glycemic control on pulmonary function in type 2 diabetes mellitus patients of the eastern India. J. Clin. Diagn. Res. 2014, 8, BC01–BC04. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ma, Z.; Guo, Z.; Zhao, F.; Wang, Y.; Cai, L.; Yang, J. Type 1 diabetes mellitus is an independent risk factor for pulmonary fibrosis. Cell Biochem. Biophys. 2014, 70, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.O. Pulmonary complications in diabetes mellitus. Mymensingh Med. J. 2014, 23, 603–605. [Google Scholar] [PubMed]
- Cai, L. Diabetic cardiomyopathy and its prevention by metallothionein: Experimental evidence, possible mechanisms and clinical implications. Curr. Med. Chem. 2007, 14, 2193–2203. [Google Scholar] [CrossRef] [PubMed]
- Cai, L. Suppression of nitrative damage by metallothionein in diabetic heart contributes to the prevention of cardiomyopathy. Free Radic. Biol. Med. 2006, 41, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Bernardo, B.C.; McMullen, J.R.; Ritchie, R.H. Diabetic cardiomyopathy: Mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol. Ther. 2014, 142, 375–415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cheng, Y.; Gu, J.; Wang, S.; Zhou, S.; Wang, Y.; Tan, Y.; Feng, W.; Fu, Y.; Mellen, N.; et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin. Sci. 2016, 130, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Fouty, B. Diabetes and the pulmonary circulation. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L725–L726. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.D. Lung dysfunction in diabetes. Diabetes Care 2003, 26, 1915–1918. [Google Scholar] [CrossRef] [PubMed]
- Mirrakhimov, A.E. Chronic Obstructive pulmonary disease and glucose metabolism: A bitter sweet symphony. Cardiovasc. Diabetol. 2012, 11, 132–158. [Google Scholar] [CrossRef] [PubMed]
- Kroll, M.H.; Afshar-Kharghan, V. Platelets in pulmonary vascular physiology and pathology. Pulm. Circ. 2012, 2, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Klein, O.L.; Jones, M.; Lee, M.; Collard, H.R.; Smith, L.J. Reduced lung diffusion capacity in type 2 diabetes is independent of heart failure. Diabetes Res. Clin. Pract. 2012, 96, e73–e75. [Google Scholar] [CrossRef] [PubMed]
- Baldi, J.C.; Hofman, P.L. Does careful glycemic control improve aerobic capacity in subjects with type 1 diabets? Exerc. Sport Sci. Rev. 2010, 38, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Wheatly, C.M.; Baldi, J.C.; Cassuto, N.A.; Foxx-Lupo, W.T.; Synder, E.M. Glycemic control influences lung membrane diffusion and oxygen saturation in exercise-trained subjects with type 1 diabetes. Alveolar-capillary membrane conductance in type 1 diabetes. Eur. J. Appl. Physiol. 2011, 111, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, E.I.; Demidov, I. Gas exchange function of the lungs in patients with type 1 diabetes mellitus (in Russian). Ter. Arkh. 2008, 80, 63–66. [Google Scholar] [PubMed]
- Sandler, M.; Bunn, A.E.; Stewart, R.I. Cross-section study of pulmonary function in patients with insulin-dependent diabetes mellitus. Am. Rev. Respir. Dis. 1987, 135, 223–229. [Google Scholar] [PubMed]
- Banga, J.D.; Sixma, J.J. Diabetes mellitus, vascular disease and thrombosis. Clin. Haematol. 1986, 15, 465–492. [Google Scholar] [PubMed]
- Colwell, J.A. Antiplatelet drugs and prevention of microvascular disease in diabetes mellitus. Metabolism 1992, 41, 7–10. [Google Scholar] [CrossRef]
- Kuziemski, K.; Pienkowska, J.; Slominski, W.; Jassem, E.; Studniarek, M. Pulmonary capillary permeability and pulmonary microangiopathy in diabetes mellitus. Diabetes Res. Clin. Pract. 2015, 108, e56–e59. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.M.; Ovechkin, A.V.; Mowbray, J.G.; Robinson, T.W.; Lominadze, D. Effects of pulmonary ischemia-reperfusion on platelet adhesion in subpleural arterioles in rabbits. Microvasc. Res. 2004, 67, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Imamura, T.; Okamura, T. Alteration of nitric oxide-mediated blood flow regulationin diabetes mellitus. Pharmacol. Ther. 2010, 127, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.M.; Tentolouris, C.; Papageaorgiu, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Naseem, K.M.; Roberts, W. Nitric oxide at a glance. Platelets 2011, 22, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Piknova, B.; Huang, P.L.; Nogushi, C.T.; Schechter, A.N. Effect of Blood Nitrite and Nitrate Levels on Murine Platelet Function. PLoS ONE 2013, 8, e55699. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Umeda, K.; Sugihara, N.; Yoshio, H.; Ino, H.; Takeda, R.; Okada, Y.; Nakanishi, I. Collagen remodelling in myocardia of patients with diabetes. J. Clin. Pathol. 1993, 46, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Asbun, J.; Villarreal, F.J. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 47, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Black, M.J.; D’Amore, A.; Auden, A.; Stamp, L.; Osicka, T.; Panagiotopoulos, S.; Jerums, G. Chronic type 1 diabetes in spontaneously hypertensive rats leads to exacerbated cardiac fibrosis. Cardiovasc. Pathol. 2010, 19, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Kiriazis, H.; Du, X.J.; Love, J.E.; Jandeleit-Dahm, K.A.; Forbes, J.M.; McMullen, J.R.; Ritchie, R.H. Coenzyme q10 attenuates diastolic dysfunction, cardiomyocyte hypertrophy and cardiac fibrosis in the db/db mouse model of type 2 diabetes. Diabetologia 2012, 55, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Jesel, L.; Abbas, M.; Morel, N. Prothrombotic changes in diabetes mellitus. Semin. Thromb. Hemost. 2013, 39, 477–488. [Google Scholar] [PubMed]
- Dixon, J.T.; Gozal, E.; Roberts, A.M. Platelet-mediated vascular dysfunction during acute lung injury. Arch. Physiol. Biochem. 2012, 118, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, A.S.; Zimmerman, G.A. Platelets in lung biology. Annu. Rev. Physiol. 2013, 75, 569–591. [Google Scholar] [CrossRef] [PubMed]
- Ho-Tin-Noe, B.; Demers, M.; Wagner, D.D. How platelets safeguard vascular integrity. J. Thromb. Haemost. 2011, 9 (Suppl. S1), 56–65. [Google Scholar] [CrossRef] [PubMed]
- Ovechkin, A.V.; Lominadze, D.; Sedoris, K.C.; Gozal, E.; Robinson, T.W.; Roberts, A.M. Inhibition of inducible nitric oxide synthase attenuates platelet adhesion in subpleural arterioles caused by lung ischemia-reperfusion in rabbits. J. Appl. Physiol. 2005, 99, 2423–2432. [Google Scholar] [CrossRef] [PubMed]
- Ovechkin, A.V.; Lominadze, D.; Sedoris, K.C.; Robinson, T.W.; Tyagi, S.C.; Roberts, A.M. Lung ischemia-reperfusion injury: Implications of oxidative stress and platelet-arteriolar wall interactions. Arch. Physiol. Biochem. 2007, 113, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bozza, F.A.; Shah, A.M.; Weyrich, A.S.; Zimmerman, G.A. Amicus or adversary: Platelets in lung biology, acute injury, and inflammation. Am. J. Respir. Cell Mol. Biol. 2009, 40, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Zucker-Franklin, D.; Philipp, C.S. Platelet production in the pulmonary capillary bed: New ultrastructural evidence for an old concept. Am. J. Pathol. 2000, 157, 69–74. [Google Scholar] [CrossRef]
- Wagner, D.D.; Fernette, P.S. The vessel wall and its interactions. Blood 2008, 111, 5271–5281. [Google Scholar] [CrossRef] [PubMed]
- Bridges, J.M.; Dalby, A.M.; Millar, J.H.D.; Weaver, J.A. An effect of d-glucose on platelet stickiness. Lancet 1965, 1, 75–77. [Google Scholar] [CrossRef]
- Kakouros, N.; Rade, J.J.; Kourliouros, A.; Resar, J.R. Platelet function in patients with diabetes mellitus from a theoretical to practical perspective. Int. J. Endocrinol. 2011, 2011, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Watala, C.; Boncer, M.; Golanski, J.; Koziolkiewicz, W.; Trojanowski, Z.; Walkowiak, B. Platelet membrane lipid fluidity and intraplatelet calcium mobilization in type 2 diabetes mellitus. Eur. J. Haematol. 1998, 61, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, G.; Wascher, T.C.; Kostner, G.M.; Graier, W.F. Alterations in platelet Ca2+ signalling in diabetic patients is due to increased formation of superoxide anions and reduced nitric oxide production. Diabetologia 1999, 42, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Keating, F.K.; Sobelm, B.E.; Schneider, D.J. Effect of increased concentrations of glucose on platelet reactivity in healthy subjects and in patients with and without diabetes. Am. J. Cardiol. 2003, 92, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.J. Factors contributing to increased platelet reactivity in people with diabetes. Diabetes Care 2009, 32, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, J.L.; Gomez-Hospital, J.A.; Angiolillo, D.J. Platelet abnormalities in diabetes mellitus. Diabetes Vasc. Dis. Res. 2010, 7, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, B.; Tschoepe, D. Anti-thrombotic therapy in diabetic patients: Revisited. Expert Rev. Cardiovasc. Ther. 2011, 9, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Ang, L.; Palakodeki, V.; Khalid, A.; Tsimikas, S.; Idrees, Z.; Tran, P.; Clopton, P.; Zafar, N.; Bromberg, G.; Keramati, S.; et al. Elevated plasma fibrinogen and diabetes mellitus are associated with lower inhibition of platelet reactivity with Clopidogrel. J. Am. Coll. Cardiol. 2008, 52, 1052–1089. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Simeone, P.; Lani, R.; Davi, G. Platelets and diabetes mellitus. Prostaglandins Other Lipid Mediat. 2015, 120, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Mercer, P.F.; Chambers, R.C. Coagulation and coagulation signaling in fibrosis. Biochem. Biophys. Acta 2013, 1832, 1018–1027. [Google Scholar] [PubMed]
- Sen, U.; Tyagi, N.; Patibandla, P.K.; Dean, W.L.; Tyagi, S.C.; Roberts, A.M.; Lominadze, D. Fibrinogen-induced production of endothelin-1 from endothelial cells. Am. J. Physiol. Cell Physiol. 2009, 296, C840–C847. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lang, S.; Zhai, Z.; Ling, L.; Kahr, W.H.A.; Chen, P.; Brkic, J.; Spring, C.M.; Flick, M.J.; Degen, J.L.; et al. Fibrinogen is required for maintenance of platelet intracellular and cell-surface P-selectin expression. Blood 2009, 114, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Dees, C.; Akhmestshina, A.; Zerr, P.; Reich, N.; Palumbo, K.; Horn, A.; Jungel, A.; Beyer, C.; Kronke, G.; Zwerina, J.; et al. Platelet-derived serotonin links vascular disease and tissue fibrosis. J. Exp. Med. 2011, 208, 961–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijs, T.; Deckmyn, H.; Broos, K. Model systems of genetically modified platelets. Blood 2012, 119, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Gumieniczek, A.; Krzywdzinska, M.; Nowak, M. Modulation of nitrosative/oxidative stress in the lung of hyperglycemic rabbits by two antidiabetics, pioglitazone and repaglinide. Exp. Lung Res. 2009, 35, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E. Oxidative stress and platelets. Arterioscler. Thromb. Vasc. Biol. 2008, 28, s11–s16. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Redondo, P.C.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Endogenously generated reactive oxygen species reduce PMCA activity in platelets from patients with non-insulin-dependent diabetes mellitus. Platelets 2006, 17, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, J.G.; Maral-Sanz, J.; Frazziano, G.; Gomez-Villalobos, M.J.; Flores-Hernandez, J.; Monjaraz, E.; Gogolludo, A.; Pezer-Vizcaino, F. Diabetes induces pulmonary artery endothelial dysfunction by NADPH oxidase induction. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L727–L732. [Google Scholar] [CrossRef] [PubMed]
- Sacan, O.; Turkyilmaz, I.B.; Bayrak, B.B.; Mutlu, O.; Akev, N.; Yanardag, R. Zinc supplementation ameliorates glycoprotein components and oxidative stress changes in the lung of streptozotocin diabetic rats. Biometals 2016, 29, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Brosius, F.C., III. New insights into the mechanisms of fibrosis and sclerosis in diabetic nephropathy. Rev. Endocr. Metab. Disord. 2008, 9, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Kaneki, M.; Shimizu, N.; Yamada, D.; Chang, K. Nitrosative stress and pathology of insulin resistance. Antioxid. Redox Signal. 2007, 9, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Soskic, S.S.; Dobutovic, B.D.; Sudar, E.M.; Obradovic, M.M.; Niklolic, D.M.; Djordjevic, J.D.; Radak, D.J.; Mikjailidis, D.P. Regulation of inducible nitric oxide synthase (iNOS) and its potential role in insulin resistance, diabetes, and heart failure. Open Cardiovasc. Med. J. 2011, 5, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.H.; Hart, J.L.; Woodman, O.L. Impairment of both nitric oxide-mediated and EDHF-type relaxation in small mesenteric arteries from rats with streptozotocin-induced diabetes. Br. J. Pharmacol. 2011, 162, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, S.S.; Vanhoutte, P.M.; Leung, S.W.S.; Suppian, R.; Yusof, M.I.; Ghulam Rasool, A.H. Reduced nitric-oxide-mediated relaxation and endothelial nitric oxide synthase expression in the tail arteries of streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2016, 773, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Fetterman, J.L.; Holbrook, M.; Flint, N.; Feng, B.; Breton-Romero, R.; Linder, E.A.; Berk, B.D.; Dues, M.A.; Farb, M.G.; Gokce, N.; et al. Restoration of autophagy in endothelial cells from patients with diabetes mellitus improves nitric oxide signaling. Atherosclerosis 2016, 247, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and the ugly. Am. J. Physiol. 1996, 271, C1424–C1427. [Google Scholar] [PubMed]
- Dias-Junior, C.A.; Bruno de Assis Cau, S.; Tanus-Santos, J.E. Role of nitric oxide in the control of the pulmonary circulation: Physiologycal, pathophysiological and therapeutic implications. J. Bras. Pneumol. 2008, 34, 412–419. [Google Scholar] [PubMed]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Duplain, H.; Burcelin, R.; Sartori, C.; Cook, S.; Egli, M.; Lepori, M.; Vollenweider, P.; Pedrazzini, T.; Nicod, P.; Thorens, B.; et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 2001, 104, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Perreault, M.; Marette, A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat. Med. 2001, 7, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Filho, M.A.; Ueno, M.; Hirabara, S.M.; Seabra, A.B.; Carvalheira, J.B.; de Oliveira, M.G.; Velloso, L.A.; Curi, R.; Saad, M.J. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: A novel mechanism of insulin resistance. Diabetes 2005, 54, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.; Vollenweider, P.; Menard, B.; Thalmann, S.; Sartori, C.; Perrin, C.; Nicod, P.; Scherrer, U. Increase eNO and pulmonary iNOS expression in eNOS null mice. Eur. Respir. J. 2003, 21, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Razzuk, S.J.; Zellers, T.M. Balloon dilation of porcine pulmonary arteries decreases endothelium-dependent relaxation and increases vasoconstriction to aggregating platelets. Circulation 1995, 91, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.M.; Lominadze, D.; Dassanayaka, S.; Sachleben, L.R., Jr.; Juniel, C.L.; Gozal, E. Pulmonary Microvascular Constriction and Oxidative Stress in the Intact-ventilated Mouse Lung during Acute Inhibition of Nitric Oxide Synthase. FASEB J. 2011, 25, 1102.8. [Google Scholar]
- Sedoris, K.C.; Gozal, E.; Ovechkin, A.V.; Theile, A.R.; Roberts, A.R. Interplay of endothelial and inducible nitric oxide synthases modulates the vascular response to ischaemia-reperfusion in the rabbit lung. Acta Physiol. 2012, 204, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Tannous, M.; Rabini, R.A.; Vignini, A.; Moretti, N.; Fumelli, P.; Mazzanti, L.; Mutus, B. Evidence for iNOS-dependent peroxinitrite production in diabetic platelets. Diabetologia 1999, 42, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, S.; Ji, H.; Zhang, Z.; Chen, J.; Tan, Y.; Wintergerst, K.; Zheng, Y.; Sun, J.; Cai, L. Broccoli sprout extract prevents diabetic cardiomyopathy via Nrf2 activation in db/db T2DM mice. Sci. Rep. 2016, 6, 30252. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Luob, L.; Zhang, Z.; Gu, J.; Chen, J.; McClung Payne, K.; Tan, Y.; Wang, Y.; Yin, X.; Zhang, X.; et al. Zinc deficiency exacerbates while zinc supplement attenuates cardiac hypertrophy in high-fat diet-induced obese mice through modulating p38 MAPK-dependent signaling. Toxicol. Lett. 2016, 258, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Liu, S.; Miao, L.; Cai, L. Prevention of diabetic complications by activation of Nrf2: Diabetic cardiomyopathy and nephropathy. Exp. Diabetes Res. 2012, 2012, 216512. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Miao, X.; Zhang, L.; Epstein, P.N.; Mellen, N.; Zheng, Y.; Wang, Y.; Cai, L. Zinc rescue of Akt2 gene deletion-linked murine cardiac dysfunction and pathological changes is metallothionein-dependent. J. Mol. Cell. Cardiol. 2014, 74, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.; Zhao, T.; Zhu, Z.; Huang, B.; Ma, W.; Wang, Y.; Tan, Y.; Chakrabarti, S.; Li, X.; Jin, L.; et al. Metallothionein prevents cardiac pathological changes in diabetes by modulating nitration and inactivation of cardiac ATP synthase. J. Nutr. Biochem. 2014, 25, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Wang, Y.; Sun, J.; Tan, Y.; Cai, L.; Zheng, Y.; Su, G.; Liu, Q.; Wang, Y. Zinc protects against diabetes-induced pathogenic changes in the aorta: Roles of metallothionein and nuclear factor (eythroid-derived 2)-like 2. Cardiovasc. Diabetol. 2013, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.S.; Manea, A.; Heltianu, C. Inhibition of JAK/STAT signaling pathway prevents high-glucose-induces increase in endothelin-1 synthesis in human endothelial cells. Cell Tissue Res. 2010, 340, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Banes-Berceli, A.K.L.; Ketsawatsomkron, P.; Ogbi, S.; Patel, B.; Pollock, D.M.; Marrero, M.B. Angiotensin II and endothelin-1 augment the vascular complications of diabetes via JAK2 activation. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Banes, A.K.L.; Shaw, S.M.; Tawfik, A.; Patel, B.P.; Ogbi, S.; Fulton, D.; Marrero, M.B. Activation of the JAK/STAT pathway in vascular smooth muscle by serotonin. Am. J. Physiol. Cell Physiol. 2005, 288, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Prele, C.M.; Yao, E.; O’Donoghu, R.J.J.; Mutsaers, S.E.; Knight, D.A. STAT3: A central mediator of pulmonary fibrosis? Proc. Am. Thorac. Soc. 2012, 9, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Gurzov, E.N.; Stanley, W.J.; Pappas, E.G.; Thomas, H.E.; Gough, D.J. The JAK/STAT pathway in obesity and diabetes. FEBS J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gao, O.; Wolfgang, M.J.; Neschen, S.; Morino, K.; Horvath, T.L.; Shulman, G.I.; Fu, X.Y. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility and thermal dysregulation. Proc. Nat. Acad. Sci. USA 2004, 101, 4661–4666. [Google Scholar] [CrossRef] [PubMed]
- Heitmeier, M.R.; Scarim, A.L.; Corbett, J.A. Prolonged STAT1 activation is associated with interferon-gamma priming for interleukin-1-induced inducible nitric-oxide synthase expression by islets of Langerhans. J. Biol. Chem. 1999, 274, 29266–29273. [Google Scholar] [CrossRef] [PubMed]
- Moore, F.; Naamane, N.; Colli, M.L.; Bouckenooghe, T.; Ortis, F.; Gurzov, E.N.; Igoillo-Esteve, M.; Mathieu, C.; Bontempi, G.; Thykjaer, T.; et al. STAT1 is a master regulator of pancreatic β-cell apoptosis and islet inflammation. J. Biol. Chem. 2011, 286, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, Y.; Dong, L.; Li, M.; Cai, W. Anti-inflammatory effect of resveratrol through the suppression of NF-κB and JAK/STAT signaling pathways. Acta Biochim. Biophys. Sin. 2015, 47, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yang, L.; Luo, J.; Zhang, C.; Xia, Y.; Ma, T.; Kong, L. Sophoraflavanone G from Sophora alopecuroides inhibits lipopolysaccharides-induced inflammation in RAW264.7 cells by targeting PI3K/Akt, JAK/STAT and Nrf2/HO-1 pathways. Int. Immunopharmacol. 2016, 38, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zeng, K.; Ma, X.; Song, F.; Jiang, Y.; Tu, P.; Wang, X. Resokaempferol-mediated anti-inflammatory effects on activated macrophages via the inhibition of JAK2/STAT3, NF-κB and JNK/p38 MAPK signaling pathways. Int. Immunopharmacol. 2016, 38, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Al-Rasheed, N.M.; Al-Rasheed, N.M.; Hasan, I.H.; Al-Amin, M.A.; Al-Ajmi, H.N.; Mahmoud, A.M. Sitagliptin attenuates cardiomyopathy by modulating the JAK/STAT Signaling pathway in experimental diabetic rats. Drug Des. Dev. Ther. 2016, 10, 2095–2107. [Google Scholar]
- Walters, D.M.; Antao-Menezes, A.; Ingram, J.L.; Rice, A.B.; Nyska, A.; Tani, Y.; Kleeberger, S.R.; Bonne, J.C. Susceptibility of signal transducer and activator of transcription-1-deficient mice to pulmonary fibrosis. Am. J. Pathol. 2005, 167, 1221–1229. [Google Scholar] [CrossRef]
- Jeong, W.I.; Park, O.; Radaeva, S.; Gao, B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cells proliferationand stimulating NK cell toxicity. Hepatology 2006, 44, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Liao, B.; Zeng, M.; Zhu, C.; Fan, X.M. The effects of aerosolized STA1 antisense oligonucleotides on rat pulmonary fibrosis. Cell. Mol. Immunol. 2009, 6, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Yokoyama, A.; Onari, Y.; Shoda, H.; Haruta, Y.; Hattori, N.; Naka, T.; Kohno, N. Suppressor of cytokine signaling 1 inhibits pulmonary inflammation and fibrosis. J. Allergy Clin. Immunol. 2008, 121, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Cetkovic-Cvrlje, M.; Dragt, A.L.; Vassilev, A.; Liu, X.P.; Uckun, F.M. Targetting JAK3 with JANEX-1 for prevention of autoimmune type 1 diabetes in NOD mice. Clin. Immunol. 2003, 106, 213–225. [Google Scholar] [CrossRef]
- Lv, N.; Kim, E.K.; Song, M.Y.; Choi, H.N.; Moon, W.S.; Park, S.J.; Park, J.W.; Kwon, K.B.; Park, B.H. JANEX-1, a JAK3 inhibitor, protects pancreatic islets from cytokine toxicity through downregulation of NF-κB activation and the JAK/STAT pathway. Exp. Cell Res. 2009, 315, 2064–2071. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Lin, K.C.; Huang, S.Y.; Thomas, P.A.; Wu, Y.H.; Wu, H.C.; Lin, K.H.; Sheu, J.R. Role of a Janus kinase 2-dependent signaling pathway in platelet activation. Thromb. Res. 2014, 133, 1088–1096. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jagadapillai, R.; Rane, M.J.; Lin, X.; Roberts, A.M.; Hoyle, G.W.; Cai, L.; Gozal, E. Diabetic Microvascular Disease and Pulmonary Fibrosis: The Contribution of Platelets and Systemic Inflammation. Int. J. Mol. Sci. 2016, 17, 1853. https://doi.org/10.3390/ijms17111853
Jagadapillai R, Rane MJ, Lin X, Roberts AM, Hoyle GW, Cai L, Gozal E. Diabetic Microvascular Disease and Pulmonary Fibrosis: The Contribution of Platelets and Systemic Inflammation. International Journal of Molecular Sciences. 2016; 17(11):1853. https://doi.org/10.3390/ijms17111853
Chicago/Turabian StyleJagadapillai, Rekha, Madhavi J. Rane, Xingyu Lin, Andrew M. Roberts, Gary W. Hoyle, Lu Cai, and Evelyne Gozal. 2016. "Diabetic Microvascular Disease and Pulmonary Fibrosis: The Contribution of Platelets and Systemic Inflammation" International Journal of Molecular Sciences 17, no. 11: 1853. https://doi.org/10.3390/ijms17111853