2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents

 , ,
, ,
Abstract
:1. Introduction
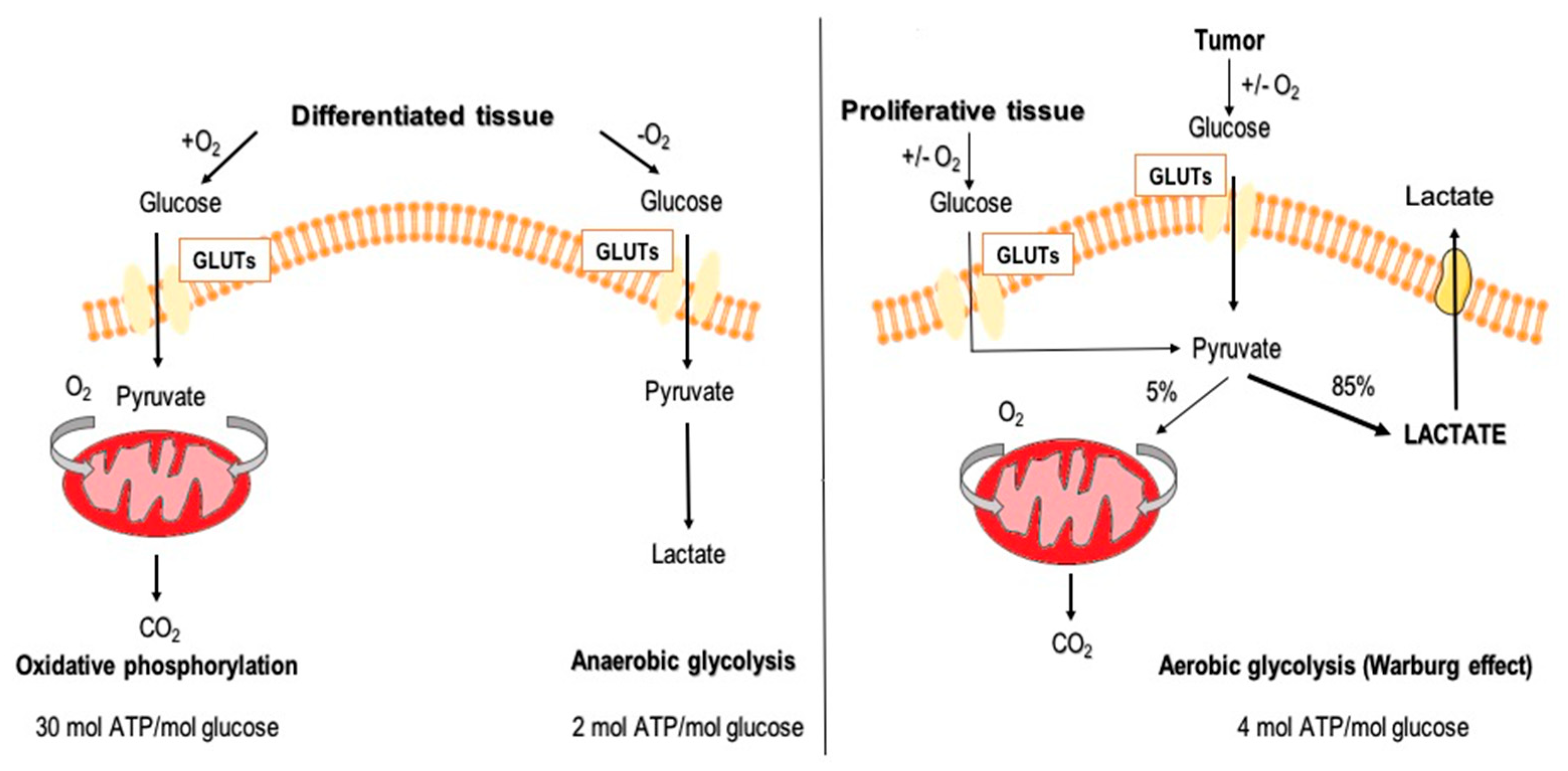
1.1. Glucose Metabolism
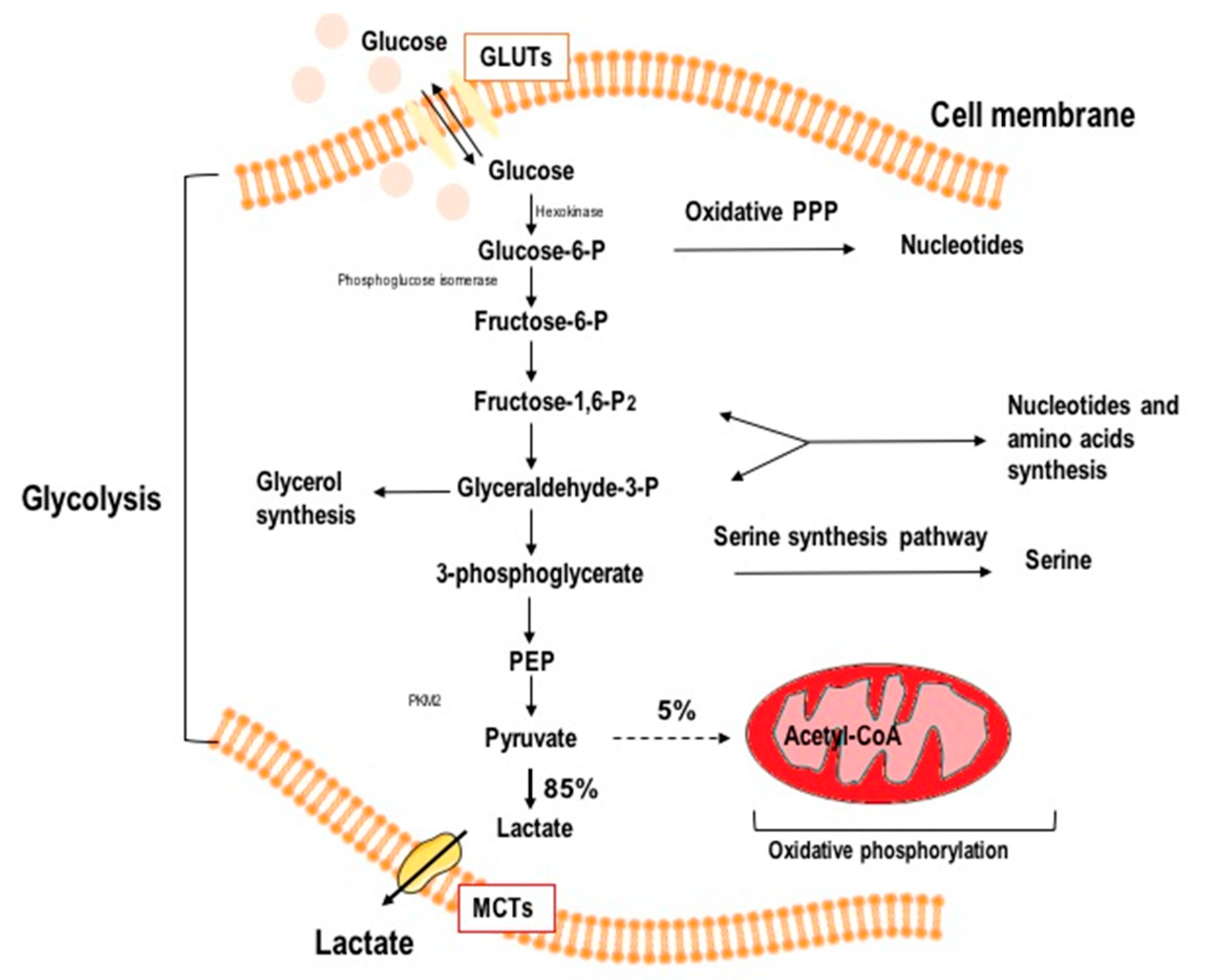
1.2. Aerobic Glycolysis in Cancer Cells
2. Glycolysis as a Target in Anticancer Therapy
3. 2-DG in Cancer Diagnostics
4. Biological Activity of 2-DG in Cancer Cells
4.1. Glycolysis Inhibition
4.2. Autophagy Induction
4.3. Apoptosis Induction
4.4. Protein N-Glycosylation
5. Preclinical and Clinical Studies of 2-DG in Anticancer Therapy
5.1. 2-DG and Cytotoxic Chemotherapeutics
5.2. 2-DG and Radiotherapy Co-Treatment
6. Novel 2-DG Analogs
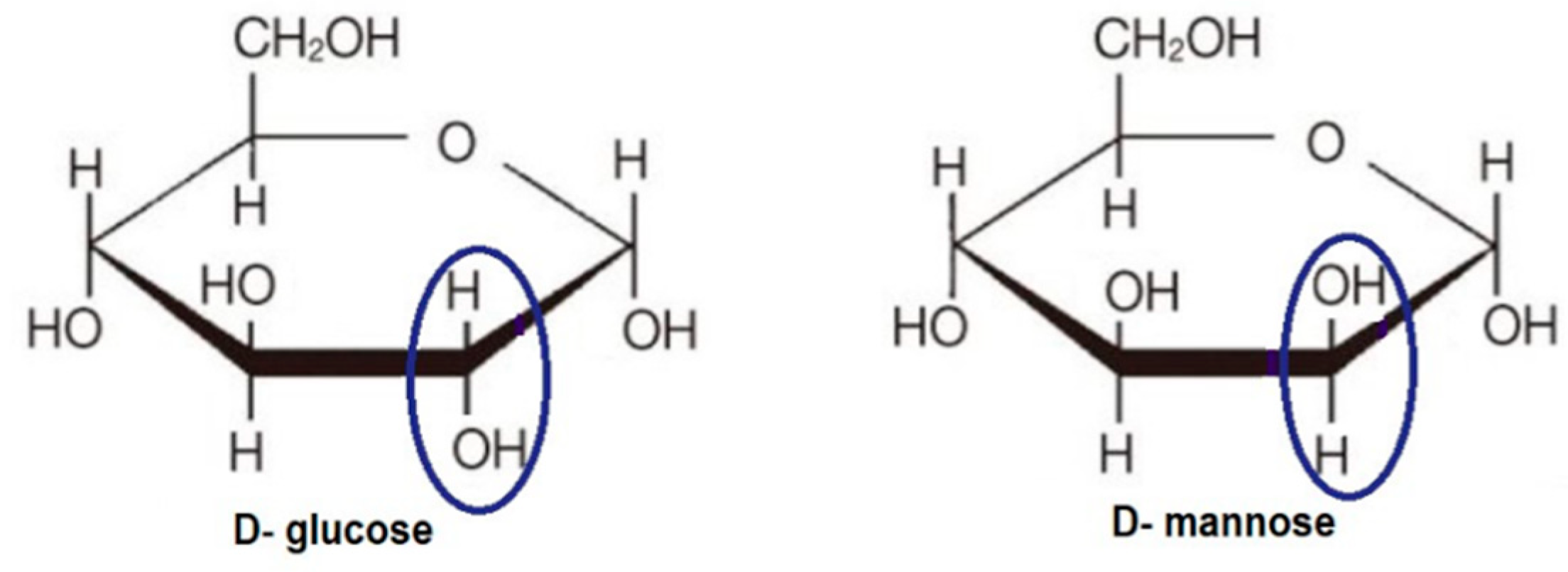
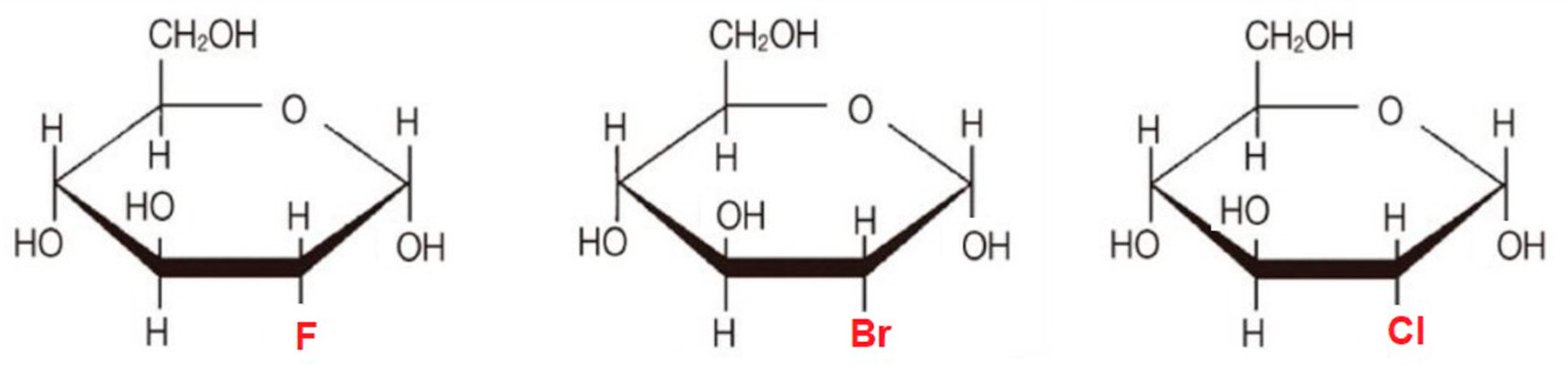
6.1. 2-Halogen Substituted d-Glucose
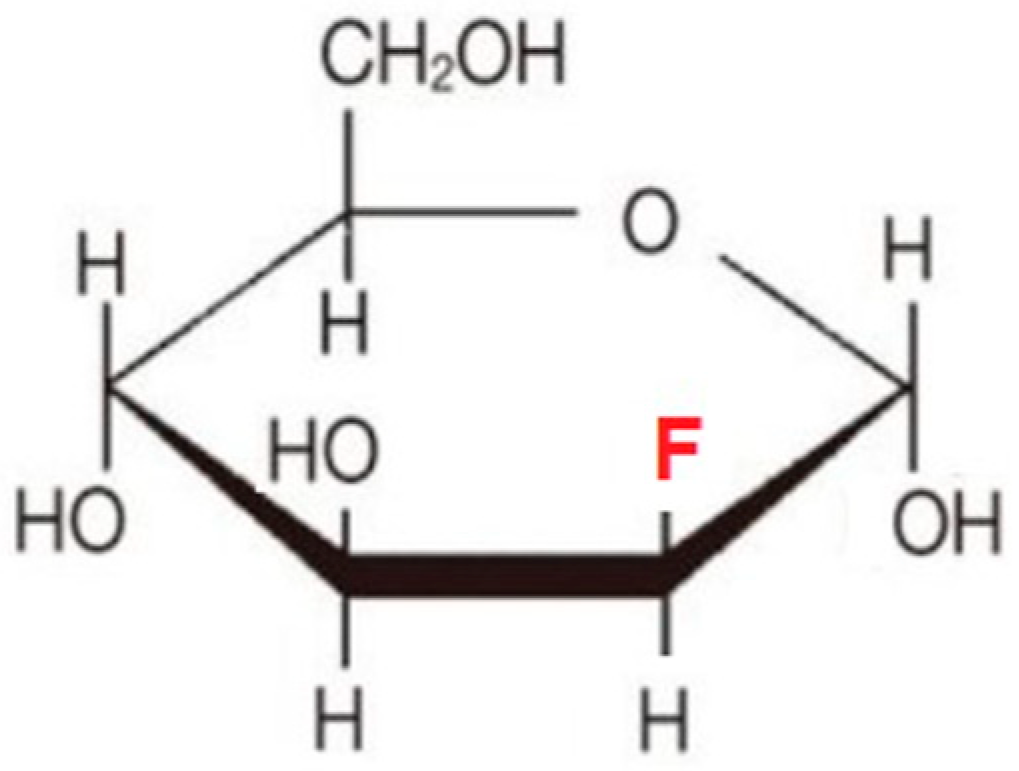
6.2. Fluoro-Hexose Compounds
6.3. Acetates of 2-Deoxy Monosaccharides as Prodrugs
7. Perspectives
Funding
Conflicts of Interest
Abbreviations
| 2-DG | 2-deoxy-d-glucose |
| GLUTs | glucose transporter proteins |
| ROS | reactive oxygen species |
| F-6-P | fructose-6-P |
| PGI | phosphoglucose-isomerase |
| PFK | phosphofructokinase |
| PEP | phosphophenol pyruvate |
| PK | pyruvate kinase |
| GBM | glioblastoma multiforme |
| BBB | blood brain barrier |
| 18F-DG | F-18 2-fluoro-2-deoxy-d-glucose |
| 3-BrPA | 3-bromopuryvate |
| 2-DG-6-P | 2-deoxy-d-glucose-6-phosphate |
| F-1 6-BP | F-6-P to fructose-1,6-biphosphate |
| AMPK | AMP-activated protein kinase |
| UPR | unfolded protein response |
| ER | endoplasmic reticulum |
| DHEA | dehydroepiandrosterone |
| 5-FU | 5-fluorouracil |
| IR | ionizing radiation |
| 2-FG | 2-fluoro-2-deoxy-d-glucose |
| 2-CG | 2-chloro-2-deoxy-d-glucose |
| 2-BG | 2-bromo-2-deoxy-d-glucose |
| HK1 | hexokinase 1 |
| 2-FM | 2-fluoro-2-deoxy-d-mannose |
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. Versuche an uberlebendem carcinomgewebe. Klin. Wochenschr. 1923, 2, 776–777. [Google Scholar] [CrossRef]
- Muz, B.; De la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Tirone, T.A.; Brunicardi, F.C. Overview of glucose regulation. World J. Surg. 2001, 25, 461–467. [Google Scholar] [CrossRef]
- Wood, I.S.; Trayhurn, P. Glucose transporters (GLUT and SGLT): Expanded families of sugar transport proteins. Br. J. Nutr. 2003, 89, 3–9. [Google Scholar] [CrossRef]
- Herling, A.; König, M.; Bulik, S.; Holzhütter, H.-G. Enzymatic features of the glucose metabolism in tumor cells. FEBS J. 2011, 278, 2436–2459. [Google Scholar] [CrossRef]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef]
- Wilson, D.F. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide Association of Hypoxia-inducible Factor (HIF)-1α and HIF-2α DNA Binding with Expression Profiling of Hypoxia-inducible Transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Pouyssegur, J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Targeting glucose metabolism for cancer therapy. J. Exp. Med. 2012, 209, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-d-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Paolicchi, E.; Gemignani, F.; Krstic-Demonacos, M.; Dedhar, S.; Mutti, L.; Landi, S. Targeting hypoxic response for cancer therapy. Oncotarget 2016, 7, 13464. [Google Scholar] [CrossRef] [Green Version]
- Blum, R.; Kloog, Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014, 5, e1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Batra, S.; Adekola, K.U.A.; Rosen, S.T.; Shanmugam, M. Cancer metabolism as a therapeutic target. Oncology 2013, 27, 460–467. [Google Scholar]
- Gong, L.; Wei, Y.; Yu, X.; Peng, J.; Leng, X. 3-Bromopyruvic acid, a hexokinase II inhibitor, is an effective antitumor agent on hepatoma cells: In vitro and in vivo findings. Anticancer Agents Med. Chem. 2014, 14, 771–776. [Google Scholar] [CrossRef]
- Ralph, S.J. Arsenic-based antineoplastic drugs and their mechanisms of action. Met. Based Drugs 2008, 2008, 260146. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, H.; Lu, W.; Huang, P. Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochim. Biophys. Acta 2009, 1787, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Aft, R.L.; Zhang, F.W.; Gius, D. Evaluation of 2-deoxy-d-glucose as a chemotherapeutic agent: Mechanism of cell death. Br. J. Cancer 2002, 87, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, J.C.; Krishan, A.; Lampidis, T.J. Greater cell cycle inhibition and cytotoxicity induced by 2-deoxy-d-glucose in tumor cells treated under hypoxic vs. aerobic conditions. Cancer Chemother. Pharmacol. 2004, 53, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Kunjithapatham, R.; Geschwind, J.-F.H.; Rao, P.P.; Boronina, T.N.; Cole, R.N.; Ganapathy-Kanniappan, S. Systemic administration of 3-bromopyruvate reveals its interaction with serum proteins in a rat model. BMC Res. Notes 2013, 6, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navale, A.M.; Paranjape, A.N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 2016, 8, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Cura, A.J.; Carruthers, A. Role of monosaccharide transport proteins in carbohydrate assimilation, distribution, metabolism, and homeostasis. Compr. Physiol. 2012, 2, 863–914. [Google Scholar]
- Jin, T.; Mehrens, H.; Wang, P.; Kim, S.-G. Glucose metabolism-weighted imaging with chemical exchange-sensitive MRI of 2-deoxyglucose (2DG) in brain: Sensitivity and biological sources. Neuroimage 2016, 143, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Mees, G.; Dierckx, R.; Vangestel, C.; Laukens, D.; Damme, N.V.; De Wiele, C.V. Pharmacologic Activation of Tumor Hypoxia: A Means to Increase Tumor 2-Deoxy-2-[F-18]Fluoro-d-Glucose Uptake? Mol. Imaging 2013, 12, 49–58. [Google Scholar]
- Zhou, H.; Luby-Phelps, K.; Mickey, B.E.; Habib, A.A.; Mason, R.P.; Zhao, D. Dynamic near-infrared optical imaging of 2-deoxyglucose uptake by intracranial glioma of athymic mice. PLoS ONE 2009, 4, e8051. [Google Scholar] [CrossRef] [Green Version]
- Sokoloff, L. Relation between physiological function and energy metabolism in the central nervous system. J. Neurochem. 1977, 29, 13–26. [Google Scholar] [CrossRef]
- Schmidt, K.C.; Lucignani, G.; Sokoloff, L. Fluorine-18-fluorodeoxyglucose PET to determine regional cerebral glucose utilization: A re-examination. J. Nucl. Med. 1996, 37, 394–399. [Google Scholar]
- Xi, H.; Kurtoglu, M.; Lamipidis, T.J. The wonders of 2-deoxy-d-glucose. IUBMB J. 2014, 66, 110–121. [Google Scholar] [CrossRef]
- Lai, M.; Vassallo, I.; Lanz, B.; Poitry-Yamate, C.; Hamou, M.-F.; Cudalbu, C.; Gruetter, R.; Hegi, M.E. In vivo characterization of brain metabolism by 1H MRS, 13C MRS and 18FDG PET reveals significant glucose oxidation of invasively growing glioma cells. Int. J. Cancer 2018, 143, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, C.; Faria, S.; Devine, C.; Patnana, M.; Sagebiel, T.; Iyer, R.B.; Bhosale, P.R. [18F]-2-Fluoro-2-Deoxy-d-glucose-PET Assessment of Cervical Cancer. PET Clin. 2018, 13, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, F.; Wang, J.; Luo, H.; Wang, Z. 2-DG-regulated RIP and cFLIP effect on liver cancer cells apoptosis induced by TRAIL. Med. Sci. Monit. 2015, 10, 3442–3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magier, Z.; Jarzyna, R. The role of glucose transporters in human metabolic regulation. Postepy Biochem. 2013, 59, 70–82. [Google Scholar] [PubMed]
- Bost, F.; Decoux-Poullot, A.-G.; Tanti, J.F.; Clavel, S. Energy disruptors: Rising stars in anticancer therapy? Oncogenesis 2016, 5, e188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, H.; Kurtoglu, M.; Liu, H.; Wangpaitchitr, M.; You, M.; Liu, X.; Savaraj, N.; Lampidis, T.J. 2-Deoxy-d-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 2011, 67, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Lin, S.C. AMP-activated protein kinase—Not just an energy sensor. F1000 Res. 2017, 6, 1724. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. Physiological Functions of Autophagy. In Autophagy in Infection and Immunity; Levine, B., Yoshimori, T., Deretic, V., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 71–84. ISBN 978-3-642-00302-8. [Google Scholar]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Cheong, H. Integrating autophagy and metabolism in cancer. Arch. Pharm. Res. 2015, 38, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pflaum, J.; Schlosser, S.; Müller, M. p53 Family and Cellular Stress Responses in Cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef]
- Kulkarni, Y.M.; Kaushik, V.; Azad, N.; Wright, C.; Rojanasakul, Y.; O’Doherty, G.; Iyer, A.K.V. Autophagy-Induced Apoptosis in Lung Cancer Cells by a Novel Digitoxin Analog. J. Cell. Physiol. 2016, 231, 817–828. [Google Scholar] [CrossRef] [Green Version]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Shutt, D.C.; O’Dorisio, M.S.; Aykin-Burns, N.; Spitz, D.R. 2-deoxy-d-glucose induces oxidative stress and cell killing in human neuroblastoma cells. Cancer. Biol. Ther. 2010, 9, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Debnath, J.; Baehrecke, E.H.; Kroemer, G. Does autophagy contribute to cell death? Autophagy 2005, 1, 66–74. [Google Scholar] [CrossRef]
- Muñoz-Pinedo, C.; Ruiz-Ruiz, C.; De Almodóvar, C.R.; Palacios, C.; López-Rivas, A. Inhibition of Glucose Metabolism Sensitizes Tumor Cells to Death Receptor-triggered Apoptosis through Enhancement of Death-inducing Signaling Complex Formation and Apical Procaspase-8 Processing. J. Biol. Chem. 2003, 278, 12759–12768. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.-H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar]
- Wang, Q.; Liang, B.; Shirwany, N.A.; Zou, M.-H. 2-Deoxy-d-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS ONE 2011, 6, e17234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutros, J.; Almasan, A. Combining 2-deoxy-d-glucose with electron transport chain blockers. Cancer Biol. Ther. 2009, 8, 1237–1238. [Google Scholar] [CrossRef] [Green Version]
- Valera, V.; Ferretti, M.J.; Prabharasuth, D.D.; Chaimowitz, M.; Choudhury, M.; Phillips, J.L.; Konno, S. Is targeting glycolysis with 2-deoxyglucose a viable therapeutic approach to bladder cancer? Int. J. Cnacer Ther. Oncol. 2017, 5, 511. [Google Scholar]
- Kim, P.-J.; Lee, D.-Y.; Jeong, H. Centralized Modularity of N-Linked Glycosylation Pathways in Mammalian Cells. PLoS ONE 2009, 4, e7317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, P.; Schachter, H.; Taniguchi, N. Chapter 8. N-Glycans. In Essentials of Glycobiology, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Biberich, E. Synthesis, processing, and function of N-glycans in N-glycoproteins. Adv. Neurobiol. 2014, 9, 47–70. [Google Scholar]
- Kurtoglu, M.; Gao, N.; Shang, J.; Maher, J.C.; Lehrman, M.A.; Wangpaichitr, M.; Savaraj, N.; Lane, A.N.; Lampidis, T.J. Under normoxia, 2-deoxy-d-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol. Cancer Ther. 2007, 6, 3049–3058. [Google Scholar] [CrossRef] [Green Version]
- Berthe, A.; Zaffino, M.; Muller, C.; Foulquier, F.; Houdou, M.; Schulz, C.; Bost, F.; De Fay, E.; Mazerbourg, S.; Flament, S. Protein N-glycosylation alteration and glycolysis inhibition both contribute to the antiproliferative action of 2-deoxyglucose in breast cancer cells. Breast Cancer Res. Treat. 2018, 171, 581–591. [Google Scholar] [CrossRef]
- Emmett, M.R.; Wang, X.; Marshall, A.G.; Ji, Y.; Fokt, I.; Skora, S.; Conrad, C.A.; Priebe, W. 2-Deoxy-d-glucose inhibits N-glycosylation in glioblastoma multiforme-derived cancer stem cells. Neuro-Oncol. 2010, 12 (Suppl. 4), CB-03. [Google Scholar]
- Priebe, W.; Emmett, M.R.; Ji, J.; Fokt, I.; Skora, S.; Conrad, C.; Wang, X.; Marshall, A.G. 2-DG inhibits of N-glycosylation in glioblstaoma-derived cancer stem cells. Cancer Res. 2011. [Google Scholar] [CrossRef]
- Khaitan, D.; Chandna, S.; Arya, M.B.; Dwarakanath, B.S. Differential mechanisms of radiosensitization by 2-deoxy-d-glucose in the monolayers and multicellular spheroids of a human glioma cell line. Cancer Biol. Ther. 2006, 5, 1142–1151. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Diao, D.; Zhang, H.; Guo, Q.; Wu, X.; Song, Y.; Dang, C. High glucose-induced resistance to 5-fluorouracil in pancreatic cancer cells alleviated by 2-deoxy-d-glucose. Biomed. Rep. 2014, 2, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.C.; Wangpaichitr, M.; Savaraj, N.; Kurtoglu, M.; Lampidis, T.J. Hypoxia-inducible factor-1 confers resistance to the glycolytic inhibitor 2-deoxy-d-glucose. Mol. Cancer Ther. 2007, 6, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayakumar, A.; Rauvolfova, J.; Bao, H.; Fokt, I.; Skora, S.; Heimberger, A.; Priebe, W. Blockade of HIF-1 with a small molecule inhibitor WP1066 in melanoma. Cancer Res. 2013, 73, 3251. [Google Scholar]
- Stein, M.; Lin, H.; Jeyamohan, C.; Dvorzhinski, D.; Gounder, M.; Bray, K.; Eddy, S.; Goodin, S.; White, E.; Dipaola, R.S. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate 2010, 70, 1388–1394. [Google Scholar] [CrossRef] [Green Version]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, F.; Bradbury, C.M.; Kaushal, A.; Li, L.; Spitz, D.R.; Aft, R.L.; Gius, D. 2-Deoxy-d-glucose-induced cytotoxicity and radiosensitization in tumor cells is mediated via disruptions in thiol metabolism. Cancer Res. 2003, 63, 3413–3417. [Google Scholar]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer. 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Quintiliani, M. The Oxygen Effect in Radiation Inactivation of DNA and Enzymes. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1986, 50, 573–594. [Google Scholar] [CrossRef]
- Jain, V.K.; Gupta, I.; Lata, K. Energetics of Cellular Repair Processes in a Respiratory-Deficient Mutant of Yeast. Radiat. Res. 1982, 92, 463–473. [Google Scholar] [CrossRef]
- Dwarkanath, B.S.; Zolzer, F.; Chandana, S.; Bauch, T.; Adhikari, J.S.; Muller, W.U.; Streffer, C.; Jain, V. Heterogeneity in 2-deoxy-d-glucose-induced modifications in energetics and radiation responses of human tumor cell lines. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 1051–1061. [Google Scholar] [CrossRef]
- Dearling, J.L.J.; Qureshi, U.; Begent, R.H.J.; Pedley, R.B. Combining radioimmunotherapy with antihypoxia therapy 2-deoxy-d-glucose results in reduction of therapeutic efficacy. Clin. Cancer Res. 2007, 13, 1903–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Banerji, A.K.; Dwarakanath, B.S.; Tripathi, R.P.; Gupta, J.P.; Mathew, T.L.; Ravindranath, T.; Jain, V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther. Onkol. 2005, 181, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.; Sey, C.H.C.; Mairs, R.J. Radiosensitization of prostate cancer cells by 2-deoxy-d-glucose. Madridge J. Oncogen. 2018, 2, 30–34. [Google Scholar] [CrossRef]
- Jalota, A.; Kumar, M.; Das, B.C.; Yadav, A.K.; Chosdol, K.; Sinha, S. Synergistic increase in efficacy of a combination of 2-deoxy-d-glucose and cisplatin in normoxia and hypoxia: Switch from autophagy to apoptosis. Tumour Biol. 2016, 37, 12347–12358. [Google Scholar] [CrossRef]
- Simons, A.L.; Ahmad, I.M.; Mattson, D.M.; Dornfeld, K.J.; Spitz, D.R. 2-Deoxy-d-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007, 67, 3364–3370. [Google Scholar] [CrossRef] [Green Version]
- Bizjak, M.; Malavašič, P.; Dolinar, K.; Pohar, J.; Pirkmajer, S.; Pavlin, M. Combined treatment with Metformin and 2-deoxy glucose induces detachment of viable MDA-MB-231 breast cancer cells in vitro. Sci. Rep. 2017, 7, 1761. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.; Fu, K.; Jiang, C.; Huang, X.; Chan, W.C.; McKeithan, T. The Combination Of 2-DG and Metformin Inhibits The mTORC1 Pathway and Suppresses Aggressive B Cell Lymphoma Growth and Survival. Blood 2013, 122, 1665. [Google Scholar] [CrossRef]
- Zhu, J.; Zheng, Y.; Zhang, H.; Sun, H. Targeting cancer cell metabolism: The combination of metformin and 2-Deoxyglucose regulates apoptosis in ovarian cancer cells via p38 MAPK/JNK signaling pathway. Am. J. Transl. Res. 2016, 8, 4812–4821. [Google Scholar]
- Kim, E.H.; Lee, J.-H.; Oh, Y.; Koh, I.; Shim, J.-K.; Park, J.; Choi, J.; Yun, M.; Jeon, J.Y.; Huh, Y.M.; et al. Inhibition of glioblastoma tumorspheres by combined treatment with 2-deoxyglucose and metformin. Neuro-Oncol. 2017, 19, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Pattni, B.S.; Jhaveri, A.; Dutta, I.; Baleja, J.D.; Degterev, A.; Torchilin, V. Targeting energy metabolism of cancer cells: Combined administration of NCL-240 and 2-DG. Int. J. Pharm. 2017, 532, 149–156. [Google Scholar] [CrossRef]
- Wang, S.-Y.; Wei, Y.-H.; Shieh, D.-B.; Lin, L.-L.; Cheng, S.-P.; Wang, P.-W.; Chuang, J.-H. 2-Deoxy-d-Glucose Can Complement Doxorubicin and Sorafenib to Suppress the Growth of Papillary Thyroid Carcinoma Cells. PLoS ONE 2015, 10, e0130959. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.M.; Mustafa, E.H.; Mustafa, N.H.; Tahtamouni, L.H.; Abdalla, M.Y. 2DG enhances the susceptibility of breast cancer cells to doxorubicin. Cent. Eur. J. Biol. 2010, 5, 739–748. [Google Scholar] [CrossRef]
- Mustafa, E.H.; Mahmoud, H.T.; Al-Hudhud, M.Y.; Abdalla, M.Y.; Ahmad, I.M.; Yasin, S.R.; Elkarmi, A.Z.; Tahtamouni, L.H. 2-deoxy-d-Glucose Synergizes with Doxorubicin or l-Buthionine Sulfoximine to Reduce Adhesion and Migration of Breast Cancer Cells. Asian Pac. J. Cancer Prev. APJCP 2015, 16, 3213–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Fang, L.; Gibbs, S.; Huang, Y.; Dai, Z.; Wen, P.; Zheng, X.; Sadee, W.; Sun, D. Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemother. Pharmacol. 2007, 59, 495–505. [Google Scholar] [CrossRef]
- Reyes, R.; Wani, N.A.; Ghoshal, K.; Jacob, S.T.; Motiwala, T. Sorafenib and 2-Deoxyglucose Synergistically Inhibit Proliferation of both Sorafenib Sensitive and Resistant HCC Cells by Inhibiting ATP Production. Gene Expr. 2017, 17, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Ishige, N. 2-Deoxyglucose and sorafenib synergistically suppress the proliferation and motility of hepatocellular carcinoma cells. Oncol. Lett. 2017, 13, 800–804. [Google Scholar] [CrossRef] [Green Version]
- Maschek, G.; Savaraj, N.; Priebe, W.; Braunschweiger, P.G.; Hamilton, K.L.; Tidmarsh, G.F.; Young, L.R.D.; Lampidis, T.J. 2-deoxy-d-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004, 64, 31–34. [Google Scholar] [CrossRef] [Green Version]
- Zhelev, Z.; Ivanova, D.; Aoki, I.; Saga, T.; Bakalova, R. 2-Deoxy-d-glucose Sensitizes Cancer Cells to Barasertib and Everolimus by ROS-independent Mechanism(s). Anticancer Res. 2015, 35, 6623–6632. [Google Scholar]
- Goldberg, L.; Israeli, R.; Kloog, Y. FTS and 2-DG induce pancreatic cancer cell death and tumor shrinkage in mice. Cell Death Dis. 2012, 3, e284. [Google Scholar] [CrossRef]
- Fan, L.; Liu, C.; Gao, A.; Zhou, Y.; Li, J. Berberine combined with 2-deoxy-d-glucose synergistically enhances cancer cell proliferation inhibition via energy depletion and unfolded protein response disruption. Biochim. Biophys. Acta 2013, 1830, 5175–5183. [Google Scholar] [CrossRef]
- Liu, H.; Kurtoglu, M.; León-Annicchiarico, C.L.; Munoz-Pinedo, C.; Barredo, J.; Leclerc, G.; Merchan, J.; Liu, X.; Lampidis, T.J. Combining 2-deoxy-d-glucose with fenofibrate leads to tumor cell death mediated by simultaneous induction of energy and ER stress. Oncotarget 2016, 7, 36461–36473. [Google Scholar] [CrossRef] [PubMed]
- Hardie, G.D. Regulation of AMP-activated protein kinase by natural and synthetic activators. Acta Pharm. Sin. B 2016, 6, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Zielonka, J.; Dranka, B.P.; McAllister, D.; Mackinnon, A.C.; Joseph, J.; Kalyanaraman, B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012, 72, 2634–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Y.-Y.; Wang, T.; Chen, F.-Y.; Wu, Y.-L.; Shao, X.; Xiao, F.; Huang, H.-H.; Zhong, H.; Zhong, J.-H. Glycolytic inhibitor 2-deoxy-d-glucose suppresses cell proliferation and enhances methylprednisolone sensitivity in non-Hodgkin lymphoma cells through down-regulation of HIF-1α and c-MYC. Leuk. Lymphoma 2015, 56, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Li, K.; Liu, Y.; Wang, Z.Y.; Ruan, B.J.; Wang, L.; Xiang, A.; Wu, D.; Lu, Z. Co-delivery nanocarriers targeting folate receptor and encapsulating 2-deoxyglucose and α-tocopheryl succinate enhance anti-tumor effect in vivo. Int. J. Nanomed. 2017, 12, 5701–5715. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Yun, M.R.; Hong, Y.K.; Solca, F.; Kim, J.-H.; Kim, H.-J.; Cho, B.C. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol. Cancer Ther. 2013, 12, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Mathur, R.; Dwarakanath, B.S. The glycolytic inhibitor 2-deoxy-d-glucose enhances the efficacy of etoposide in ehrlich ascites tumor-bearing mice. Cancer Biol. Ther. 2005, 4, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Shen, Y.; Jin, F.; Miao, Y.; Qiu, X. Cancer Stem Cells in Small Cell Lung Cancer Cell Line H446: Higher Dependency on Oxidative Phosphorylation and Mitochondrial Substrate-Level Phosphorylation than Non-Stem Cancer Cells. PLoS ONE 2016, 11, e0154576. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, C.R.; Tilkens, S.B.; Guan, H.; Garner, J.A.; Or, P.M.Y.; Chan, A.M. Differential sensitivities of glioblastoma cell lines towards metabolic and signaling pathway inhibitions. Cancer Lett. 2013, 336, 299–306. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Cardoso-Carneiro, D.; Valbom, I.; Cury, F.P.; Silva, V.A.; Granja, S.; Reis, R.M.; Baltazar, F.; Martinho, O. Metabolic alterations underlying Bevacizumab therapy in glioblastoma cells. Oncotarget 2017, 8, 103657–103670. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; LeDoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming Trastuzumab Resistance in Breast Cancer by Targeting Dysregulated Glucose Metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef] [Green Version]
- Reddy, B.V.; Prasad, N.R. 2-deoxy-d-glucose combined with ferulic acid enhances radiation response in non-small cell lung carcinoma cells. Cent. Eur. J. Biol. 2011, 6, 743. [Google Scholar] [CrossRef] [Green Version]
- Al-Shammari, A.M.; Abdullah, A.H.; Allami, Z.M.; Yaseen, N.Y. 2-deoxyglucose and Newcastle Disease virus synergize to kill breast cancer cells by inhibition of glycolysis pathway through glyceraldehyde3-phosphate downregulation. Front. Mol. Biosci. 2019, 6, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Fath, M.A.; Scarbrough, P.M.; Watson, W.H.; Spitz, D.R. Combined inhibition of glycolysis, the pentose cycle, and thioredoxin metabolism selectively increases cytotoxicity and oxidative stress in human breast and prostate cancer. Redox Biol. 2015, 4, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, I.L.; Levy, M.M.; Kerr, D.S. The 2-deoxyglucose test as a supplement to fasting for detection of childhood hypoglycemia. Pediatri. Res. 1984, 18, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Strandberg, A.Y.; Pienimaki, T.; Pitkala, K.H.; Tilvis, R.S.; Salomaa, V.V.; Strandberg, T.E. Comparison of normal fsting and one-hour glucose levels as predictors of future diabetes during a 34-year follow-up. Ann. Med. 2013, 45, 336–340. [Google Scholar] [CrossRef]
- Lampidis, T.J.; Kurtoglu, M.; Maher, J.C.; Liu, H.; Krishan, A.; Sheft, V.; Szymanski, S.; Fokt, I.; Rudnicki, W.R.; Ginalski, K.; et al. Efficacy of 2-halogen substituted d-glucose analogs in blocking glycolysis and killing “hypoxic tumor cells”. Cancer Chemother. Pharmacol. 2006, 58, 725. [Google Scholar] [CrossRef]
- Wachsberger, P.R.; Gressen, E.L.; Bhala, A.; Bobyock, S.B.; Storck, C.; Coss, R.A.; Berd, D.; Leeper, D.B. Variability in glucose transporter-1 levels and hexokinase activity in human melanoma. Melanoma Res. 2002, 12, 35–43. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer. 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Datema, R.; Schwarz, R.T. Formation of 2-deoxyglucose-containing lipid-linked oligosaccharides. Interference with glycosylation of glycoproteins. Eur. J. Biochem. 1978, 90, 505–516. [Google Scholar] [CrossRef]
- Antanovic, L.; Fokt, I.; Szymanski, S.; Conrad, C.; Johansen, M.; Madden, T.; Priebe, W. 2-Deoxy-2-fluoro-d-mannose induces type II cell death in gliomas. Cancer Res. 2008, 68, 3359. [Google Scholar]
- Priebe, W.; Zielinski, R.; Fokt, I.; Felix, E.; Radjendirane, V.; Arumugam, J.; Tai Khuong, M.; Krasinski, M.; Skora, S. EXTH-07. Design and evaluation of WP1122, in inhibitor of glycolysis with increased CNS uptake. Neuro-Oncology 2018, 20, vi86. [Google Scholar] [CrossRef] [Green Version]
- Keith, M.; Zielinski, R.; Walker, C.M.; Le Roux, L.; Priebe, W.; Bankson, J.A.; Schellingerhout, D. Hyperpolarized pyruvate MR spectroscopy depicts glycolytic inhibition in a mouse model of glioma. Radiology 2019, 293, 168–173. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combined Therapy with 2-DG | Cancer Type | References |
|---|---|---|
| Preclinical Studies | ||
| Cisplatin | - head and neck carcinoma | [64,86,87] |
| - GBM | ||
| - bladder cancer | ||
| Metformin | - breast cancer | [88,89,90,91] |
| - B cell lymphoma cells | ||
| - ovarian cancer | ||
| - GBM | ||
| NCL-240 | - melanoma | [92] |
| - small lung carcinomas | ||
| - ovarian cancer | ||
| - breast cancer | ||
| Doxorubicin | - papillary thyroid carcinoma | [64,93,94,95] |
| - breast cancer | ||
| - bladder cancer | ||
| Daunorubicin | - colon cancer | [96] |
| - breast cancer | ||
| Gemcitabine | - bladder cancer | [64] |
| Sorafenib | - papillary thyroid carcinoma | [93,97,98] |
| - hepatocellular carcinoma | ||
| Adriamycin | - osteosarcoma | [99] |
| - non-small cell lung cancer | ||
| Barasertib and Everolimus | - leukemia | [100] |
| Salirasib | - pancreatic cancer | [101] |
| Paclitaxel | - osteosarcoma | [99] |
| - non-small cell lung cancer | ||
| Berberine | - lung cancer | [102] |
| Fenofibrate (FF) | - breast cancer | [103] |
| - melanoma | ||
| - osteosarcoma | ||
| Resveratrol | - neuroblastoma | [104] |
| Mito-Q, Mito-CP, Dec-TPP+ | - breast cancer | [105] |
| Methylprednisolone | - non-Hodgkin lymphoma | [106] |
| Alpha-tocopheryl succinate | - colon adenocarcinoma | [107] |
| - cervical carcinoma | ||
| - lung adenocarcinoma | ||
| Afatinib | - non-small cell lung cancer | [108] |
| Etoposide | - Ehrlich ascites tumor-bearing mice | [109] |
| Oligomycin | - small cell lung cancer | [110,111] |
| - GBM | ||
| Bevacizumab | - GBM | [112] |
| 5′-Fluorouracil | - pancreatic cancer | [73] |
| Trastuzumab | - breast cancer | [113] |
| Ferulic acid with irradiation | - non-small cell lung carcinoma | [114] |
| Radiotherapy | - breast cancer | [31,78,85] |
| - prostate cancer | ||
| - cervical cancer | ||
| Virotherapy (avian Newcastle disease virus (NDV)) | - breast cancer | [115] |
| Clinical Studies | ||
| Docetaxel | - breast cancer, | [77] |
| - lung cancer | ||
| - head and neck cancers | ||
| Dehydroepiandrosterone (DHEA) | - breast cancer | [116] |
| - prostate cancer | ||
| Radiotherapy | - GBM | [84] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 234. https://doi.org/10.3390/ijms21010234
Pajak B, Siwiak E, Sołtyka M, Priebe A, Zieliński R, Fokt I, Ziemniak M, Jaśkiewicz A, Borowski R, Domoradzki T, et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. International Journal of Molecular Sciences. 2020; 21(1):234. https://doi.org/10.3390/ijms21010234
Chicago/Turabian StylePajak, B., E. Siwiak, M. Sołtyka, A. Priebe, R. Zieliński, I. Fokt, M. Ziemniak, A. Jaśkiewicz, R. Borowski, T. Domoradzki, and et al. 2020. "2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents" International Journal of Molecular Sciences 21, no. 1: 234. https://doi.org/10.3390/ijms21010234