STAT3 for Cardiac Regenerative Medicine: Involvement in Stem Cell Biology, Pathophysiology, and Bioengineering


Abstract
:1. Pluripotent Stem Cells and Their Characteristics
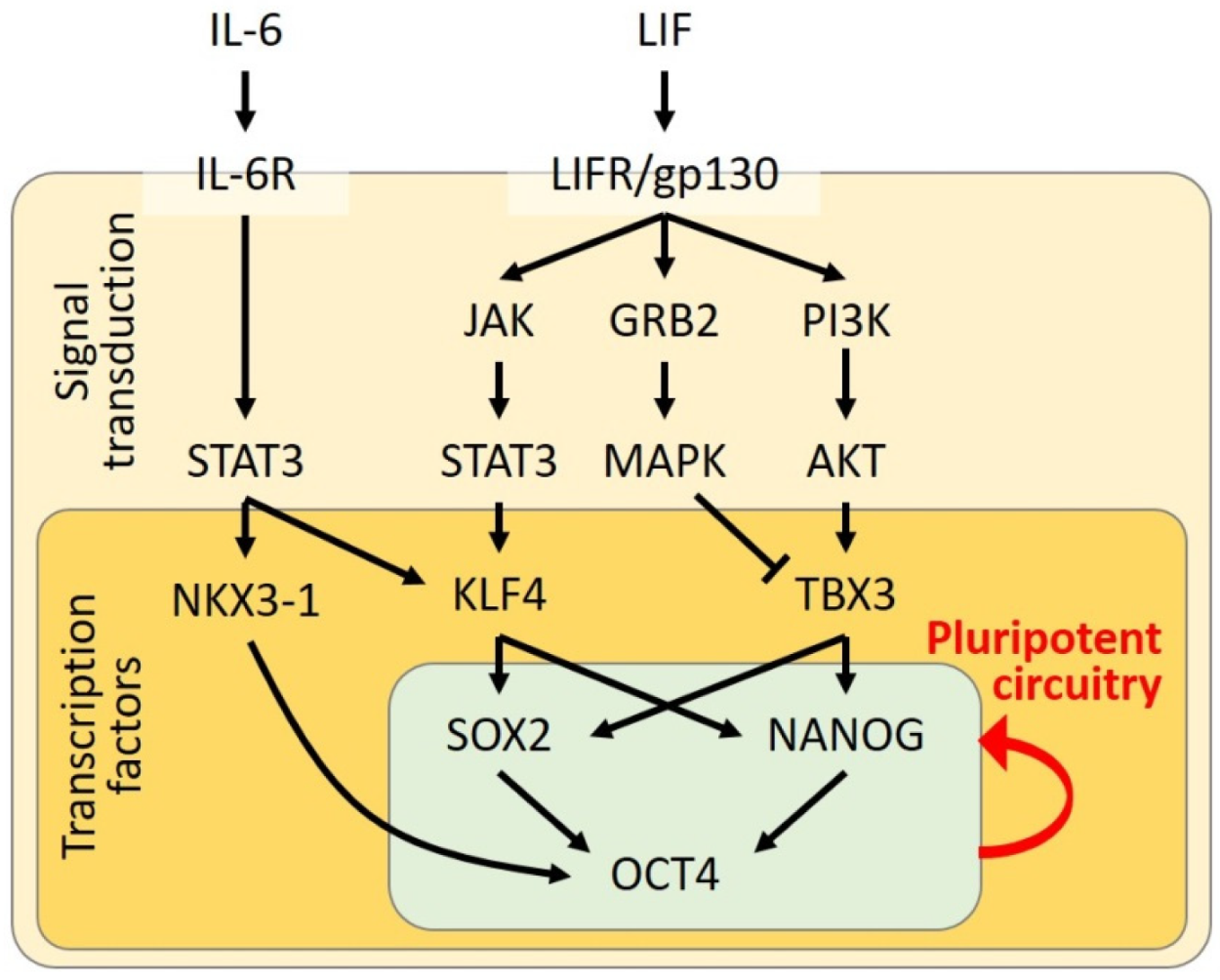
2. STAT3 in Maintenance of Pluripotency
3. STAT3 in Acquisition of Pluripotency
4. STAT3 in Cardiomyocyte Differentiation from Pluripotent Stem Cells
5. STAT3 in Heart Disease
5.1. STAT3 in Myocardial Infarction
5.2. STAT3 in Ischemic/Reperfusion Injury
5.3. STAT3 in Doxorubicin-Induced Cardiomyopathy
5.4. STAT3 in Cardiac Fibrosis and Hypertrophy
6. STAT3 Activation for Myocardial Regeneration
7. STAT3 Activation through Artificial Receptors for Myocardial Differentiation
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPKα | AMP-activated protein kinase α |
| BMSC | bone marrow stem cell or bone marrow stromal cell |
| BSA-FL | fluorescein-conjugated bovine serum albumin |
| DNMT | DNA methyltransferase |
| EB | embryoid body |
| EpiSC | epiblast stem cell |
| ESC | embryonic stem cell |
| FGF | fibroblast growth factor |
| FL | Fluorescein |
| G-CSF | granulocyte colony-stimulating factor |
| GCSFR | granulocyte colony-stimulating factor receptor |
| gp130 | glycoprotein 130 |
| GSK3β | glycogen synthase kinase 3β |
| HDAC | histone deacetylase |
| hESC | human embryonic stem cells |
| hiPSC | human induced pluripotent stem cell |
| hPSC | human pluripotent stem cell |
| IGF | insulin-like growth factor |
| IL-6 | interleukin 6 |
| Il11α | interleukin 11α |
| IL-13 | interleukin 13 |
| iPSC | induced pluripotent stem cell |
| JAK | Janus kinase |
| LIF | leukemia inhibitory factor |
| MAPK | mitogen-activated protein kinase |
| mEpiSC | mouse epiblast stem cell |
| mESC | mouse embryonic stem cell |
| miPSC | mouse induced pluripotent stem cell |
| MnSOD | manganese superoxide dismutase |
| mTOR | mammalian target of rapamycin |
| OSK | OCT4, SOX2, and KLF4 |
| OSKM | OCT4, SOX2, KLF4, and c-MYC |
| PI3K | phosphatidylinositol-3 kinase |
| PSC | pluripotent stem cell |
| scFv | single chain variable domain of antibody |
| STAT | signal transducer and activator of transcription |
References
- De Los Angeles, A.; Ferrari, F.; Xi, R.; Fujiwara, Y.; Benvenisty, N.; Deng, H.; Hochedlinger, K.; Jaenisch, R.; Lee, S.; Leitch, H.G. Hallmarks of pluripotency. Nature 2015, 525, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itskovitz-Eldor, J.; Schuldiner, M.; Karsenti, D.; Eden, A.; Yanuka, O.; Amit, M.; Soreq, H.; Benvenisty, N. Differentiation of human embryonic stem cells into embryoid bodies comprising the three embryonic germ layers. Mol. Med. 2000, 6, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuldiner, M.; Yanuka, O.; Itskovitz-Eldor, J.; Melton, D.A.; Benvenisty, N. Effects of eight growth factors on the differentiation of cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 11307–11312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adewumi, O.; Aflatoonian, B.; Ahrlund-Richter, L.; Amit, M.; Andrews, P.W.; Beighton, G.; Bello, P.A.; Benvenisty, N.; Berry, L.S.; Bevan, S. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007, 25, 803–816. [Google Scholar]
- Weinberger, L.; Ayyash, M.; Novershtern, N.; Hanna, J.H. Dynamic stem cell states: Naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 2016, 17, 155–169. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef] [Green Version]
- Bourillot, P.Y.; Aksoy, I.; Schreiber, V.; Wianny, F.; Schulz, H.; Hummel, O.; Hubner, N.; Savatier, P. Novel STAT3 target genes exert distinct roles in the inhibition of mesoderm and endoderm differentiation in cooperation with Nanog. Stem Cells 2009, 27, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Nakamura, T.; Nakao, K.; Arai, T.; Katsuki, M.; Heike, T.; Yokota, T. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 1999, 18, 4261–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, H.; Ogawa, K.; Shimosato, D.; Adachi, K. A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature 2009, 460, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Ying, Q.L.; Wray, J.; Nichols, J.; Batlle-Morera, L.; Doble, B.; Woodgett, J.; Cohen, P.; Smith, A. The ground state of embryonic stem cell self-renewal. Nature 2008, 453, 519–523. [Google Scholar] [CrossRef] [Green Version]
- Boeuf, H.; Hauss, C.; Graeve, F.D.; Baran, N.; Kedinger, C. Leukemia inhibitory factor-dependent transcriptional activation in embryonic stem cells. J. Cell Biol. 1997, 138, 1207–1217. [Google Scholar] [CrossRef]
- Burdon, T.; Smith, A.; Savatier, P. Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 2002, 12, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Niwa, H.; Burdon, T.; Chambers, I.; Smith, A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998, 12, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.; Smith, A. Capturing pluripotency. Cell 2008, 132, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Vallier, L.; Alexander, M.; Pedersen, R.A. Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J. Cell Sci. 2005, 118, 4495–4509. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.; Cheng, A.W.; Saha, K.; Kim, J.; Lengner, C.J.; Soldner, F.; Cassady, J.P.; Muffat, J.; Carey, B.W.; Jaenisch, R. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl. Acad. Sci. USA 2010, 107, 9222–9227. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.S.; Goke, J.; Ng, J.H.; Lu, X.; Gonzales, K.A.; Tan, C.P.; Tng, W.Q.; Hong, Z.Z.; Lim, Y.S.; Ng, H.H. Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell 2013, 13, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Aksoy, I.; Gonnot, F.; Osteil, P.; Aubry, M.; Hamela, C.; Rognard, C.; Hochard, A.; Voisin, S.; Fontaine, E. Reinforcement of STAT3 activity reprogrammes human embryonic stem cells to naive-like pluripotency. Nat. Commun. 2015, 6, 7095. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; van Oosten, A.L.; Theunissen, T.W.; Guo, G.; Silva, J.C.; Smith, A. Stat3 activation is limiting for reprogramming to ground state pluripotency. Cell Stem Cell 2010, 7, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- van Oosten, A.L.; Costa, Y.; Smith, A.; Silva, J.C. JAK/STAT3 signalling is sufficient and dominant over antagonistic cues for the establishment of naive pluripotency. Nat. Commun 2012, 3, 817. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Luo, Y.; Jiang, Z.; Ma, Y.; Lin, C.J.; Kim, C.; Carter, M.G.; Amano, T.; Park, J.; Kish, S. Jak/Stat3 signaling promotes somatic cell reprogramming by epigenetic regulation. Stem Cells 2012, 30, 2645–2656. [Google Scholar] [CrossRef]
- Mai, T.; Markov, G.J.; Brady, J.J.; Palla, A.; Zeng, H.; Sebastiano, V.; Blau, H.M. NKX3-1 is required for induced pluripotent stem cell reprogramming and can replace OCT4 in mouse and human iPSC induction. Nat. Cell Biol. 2018, 20, 900–908. [Google Scholar] [CrossRef]
- Zwi, L.; Caspi, O.; Arbel, G.; Huber, I.; Gepstein, A.; Park, I.H.; Gepstein, L. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation 2009, 120, 1513–1523. [Google Scholar] [CrossRef] [Green Version]
- Clifford, D.M.; Fisher, S.A.; Brunskill, S.J.; Doree, C.; Mathur, A.; Watt, S.; Martin-Rendon, E. Stem cell treatment for acute myocardial infarction. Cochrane Database Syst. Rev. 2012, CD006536. [Google Scholar]
- Dowell, J.D.; Rubart, M.; Pasumarthi, K.B.; Soonpaa, M.H.; Field, L.J. Myocyte and myogenic stem cell transplantation in the heart. Cardiovasc. Res. 2003, 58, 336–350. [Google Scholar] [CrossRef] [Green Version]
- Kattman, S.J.; Witty, A.D.; Gagliardi, M.; Dubois, N.C.; Niapour, M.; Hotta, A.; Ellis, J.; Keller, G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 2011, 8, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Mummery, C.; Ward-van Oostwaard, D.; Doevendans, P.; Spijker, R.; van den Brink, S.; Hassink, R.; van der Heyden, M.; Opthof, T.; Pera, M.; de la Riviere, A.B. Differentiation of human embryonic stem cells to cardiomyocytes: Role of coculture with visceral endoderm-like cells. Circulation 2003, 107, 2733–2740. [Google Scholar] [CrossRef] [Green Version]
- Murry, C.E.; Field, L.J.; Menasche, P. Cell-based cardiac repair: Reflections at the 10-year point. Circulation 2005, 112, 3174–3183. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hao, J.; Hong, C.C. Cardiac induction of embryonic stem cells by a small molecule inhibitor of Wnt/beta-catenin signaling. ACS Chem. Biol. 2011, 6, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.S.; Yoo, S.J.; Lee, J.E.; You, S.; Lee, H.T.; Yoon, H.S. Enhanced differentiation of human embryonic stem cells into cardiomyocytes by combining hanging drop culture and 5-azacytidine treatment. Differentiation 2006, 74, 149–159. [Google Scholar] [CrossRef]
- Takei, S.; Ichikawa, H.; Johkura, K.; Mogi, A.; No, H.; Yoshie, S.; Tomotsune, D.; Sasaki, K. Bone morphogenetic protein-4 promotes induction of cardiomyocytes from human embryonic stem cells in serum-based embryoid body development. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1793–H1803. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.H.; Wang, X.; Browne, C.; Zhang, Y.; Schinke, M.; Izumo, S.; Burcin, M. Wnt3a-induced mesoderm formation and cardiomyogenesis in human embryonic stem cells. Stem Cells 2009, 27, 1869–1878. [Google Scholar] [CrossRef]
- Ren, Y.; Lee, M.Y.; Schliffke, S.; Paavola, J.; Amos, P.J.; Ge, X.; Ye, M.; Zhu, S.; Senyei, G.; Lum, L. Small molecule Wnt inhibitors enhance the efficiency of BMP-4-directed cardiac differentiation of human pluripotent stem cells. J. Mol. Cell Cardiol. 2011, 51, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Cao, N.; Liu, Z.; Chen, Z.; Wang, J.; Chen, T.; Zhao, X.; Ma, Y.; Qin, L.; Kang, J.; Wei, B. Ascorbic acid enhances the cardiac differentiation of induced pluripotent stem cells through promoting the proliferation of cardiac progenitor cells. Cell Res. 2012, 22, 219–236. [Google Scholar] [CrossRef] [Green Version]
- Shimoji, K.; Yuasa, S.; Onizuka, T.; Hattori, F.; Tanaka, T.; Hara, M.; Ohno, Y.; Chen, H.; Egasgira, T.; Seki, T. G-CSF promotes the proliferation of developing cardiomyocytes in vivo and in derivation from ESCs and iPSCs. Cell Stem Cell 2010, 6, 227–237. [Google Scholar] [CrossRef] [Green Version]
- McDevitt, T.C.; Laflamme, M.A.; Murry, C.E. Proliferation of cardiomyocytes derived from human embryonic stem cells is mediated via the IGF/PI 3-kinase/Akt signaling pathway. J. Mol. Cell Cardiol. 2005, 39, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Demetri, G.D.; Griffin, J.D. Granulocyte colony-stimulating factor and its receptor. Blood 1991, 78, 2791–2808. [Google Scholar] [CrossRef] [Green Version]
- Welte, K.; Gabrilove, J.; Bronchud, M.H.; Platzer, E.; Morstyn, G. Filgrastim (r-metHuG-CSF): The first 10 years. Blood 1996, 88, 1907–1929. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Wright, D.E.; Weissman, I.L. Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc. Natl. Acad. Sci. USA 1997, 94, 1908–1913. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Huang, X.; Atakilit, A.; Zhu, Q.S.; Corey, S.J.; Sheppard, D. The Integrin alpha9beta1 contributes to granulopoiesis by enhancing granulocyte colony-stimulating factor receptor signaling. Immunity 2006, 25, 895–906. [Google Scholar] [CrossRef] [Green Version]
- Orlic, D.; Kajstura, J.; Chimenti, S.; Limana, F.; Jakoniuk, I.; Quaini, F.; Nadal-Ginard, B.; Bodine, D.M.; Leri, A.; Anversa, P. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc. Natl. Acad. Sci. USA 2001, 98, 10344–10349. [Google Scholar] [CrossRef] [Green Version]
- Minatoguchi, S.; Takemura, G.; Chen, X.H.; Wang, N.; Uno, Y.; Koda, M.; Arai, M.; Misao, Y.; Lu, C.; Suzuki, K. Acceleration of the healing process and myocardial regeneration may be important as a mechanism of improvement of cardiac function and remodeling by postinfarction granulocyte colony-stimulating factor treatment. Circulation 2004, 109, 2572–2580. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Imagawa, J.; Suzuki, Y.; Yogo, K.; Fukazawa, M.; Kuromaru, O.; Saito, Y. G-CSF treatment increases side population cell infiltration after myocardial infarction in mice. J. Mol. Cell Cardiol. 2004, 36, 707–710. [Google Scholar] [CrossRef]
- Kawada, H.; Fujita, J.; Kinjo, K.; Matsuzaki, Y.; Tsuma, M.; Miyatake, H.; Muguruma, Y.; Tsuboi, K.; Itabashi, Y.; Ikeda, Y. Nonhematopoietic mesenchymal stem cells can be mobilized and differentiate into cardiomyocytes after myocardial infarction. Blood 2004, 104, 3581–3587. [Google Scholar] [CrossRef]
- Orlic, D.; Kajstura, J.; Chimenti, S.; Jakoniuk, I.; Anderson, S.M.; Li, B.; Pickel, J.; McKay, R.; Nadal-Ginard, B.; Bodine, D.M. Bone marrow cells regenerate infarcted myocardium. Nature 2001, 410, 701–705. [Google Scholar] [CrossRef]
- Kocher, A.A.; Schuster, M.D.; Szabolcs, M.J.; Takuma, S.; Burkhoff, D.; Wang, J.; Homma, S.; Edwards, N.M.; Itescu, S. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat. Med. 2001, 7, 430–436. [Google Scholar] [CrossRef]
- Jackson, K.A.; Majka, S.M.; Wang, H.; Pocius, J.; Hartley, C.J.; Majesky, M.W.; Entman, M.L.; Michael, L.H.; Hirschi, K.K.; Goodell, M.A. Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J. Clin. Investg. 2001, 107, 1395–1402. [Google Scholar] [CrossRef] [Green Version]
- Balsam, L.B.; Wagers, A.J.; Christensen, J.L.; Kofidis, T.; Weissman, I.L.; Robbins, R.C. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature 2004, 428, 668–673. [Google Scholar] [CrossRef]
- Murry, C.E.; Soonpaa, M.H.; Reinecke, H.; Nakajima, H.; Nakajima, H.O.; Rubart, M.; Pasumarthi, K.B.; Virag, J.I.; Bartelmez, S.H.; Poppa, V. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 2004, 428, 664–668. [Google Scholar] [CrossRef]
- Norol, F.; Merlet, P.; Isnard, R.; Sebillon, P.; Bonnet, N.; Cailliot, C.; Carrion, C.; Ribeiro, M.; Charlotte, F.; Pradeau, P. Influence of mobilized stem cells on myocardial infarct repair in a nonhuman primate model. Blood 2003, 102, 4361–4368. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Li, J.; Wang, J.; Luo, X.; Ai, J.; Liu, Y.; Wang, N.; Liang, H.; Zhang, M.; Chen, N. microRNA-124 regulates cardiomyocyte differentiation of bone marrow-derived mesenchymal stem cells via targeting STAT3 signaling. Stem Cells 2012, 30, 1746–1755. [Google Scholar] [CrossRef]
- Ryan, J.M.; Barry, F.P.; Murphy, J.M.; Mahon, B.P. Mesenchymal stem cells avoid allogeneic rejection. J. Inflamm (Lond) 2005, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Harada, M.; Qin, Y.; Takano, H.; Minamino, T.; Zou, Y.; Toko, H.; Ohtsuka, M.; Matsuura, K.; Sano, M.; Nishi, J. G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat. Med. 2005, 11, 305–311. [Google Scholar] [CrossRef]
- Negoro, S.; Kunisada, K.; Tone, E.; Funamoto, M.; Oh, H.; Kishimoto, T.; Yamauchi-Takihara, K. Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc. Res. 2000, 47, 797–805. [Google Scholar] [CrossRef]
- Enomoto, D.; Obana, M.; Miyawaki, A.; Maeda, M.; Nakayama, H.; Fujio, Y. Cardiac-specific ablation of the STAT3 gene in the subacute phase of myocardial infarction exacerbated cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H471–H480. [Google Scholar] [CrossRef] [Green Version]
- Haghikia, A.; Missol-Kolka, E.; Tsikas, D.; Venturini, L.; Brundiers, S.; Castoldi, M.; Muckenthaler, M.U.; Eder, M.; Stapel, B.; Thum, T. Signal transducer and activator of transcription 3-mediated regulation of miR-199a-5p links cardiomyocyte and endothelial cell function in the heart: A key role for ubiquitin-conjugating enzymes. Eur. Heart J. 2011, 32, 1287–1297. [Google Scholar] [CrossRef]
- Benekli, M.; Baer, M.R.; Baumann, H.; Wetzler, M. Signal transducer and activator of transcription proteins in leukemias. Blood 2003, 101, 2940–2954. [Google Scholar] [CrossRef] [Green Version]
- Mullen, M.; Gonzalez-Perez, R.R. Leptin-Induced JAK/STAT Signaling and Cancer Growth. Vaccines (Basel) 2016, 4, 26. [Google Scholar] [CrossRef]
- Osugi, T.; Oshima, Y.; Fujio, Y.; Funamoto, M.; Yamashita, A.; Negoro, S.; Kunisada, K.; Izumi, M.; Nakaoka, Y.; Hirota, H. Cardiac-specific activation of signal transducer and activator of transcription 3 promotes vascular formation in the heart. J. Biol. Chem. 2002, 277, 6676–6681. [Google Scholar] [CrossRef] [Green Version]
- Smithgall, T.E.; Briggs, S.D.; Schreiner, S.; Lerner, E.C.; Cheng, H.; Wilson, M.B. Control of myeloid differentiation and survival by Stats. Oncogene 2000, 19, 2612–2618. [Google Scholar] [CrossRef] [Green Version]
- Negoro, S.; Kunisada, K.; Fujio, Y.; Funamoto, M.; Darville, M.I.; Eizirik, D.L.; Osugi, T.; Izumi, M.; Oshima, Y.; Nakaoka, Y. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation 2001, 104, 979–981. [Google Scholar] [CrossRef] [Green Version]
- Oshima, Y.; Fujio, Y.; Nakanishi, T.; Itoh, N.; Yamamoto, Y.; Negoro, S.; Tanaka, K.; Kishimoto, T.; Kawase, I.; Azuma, J. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc Res. 2005, 65, 428–435. [Google Scholar] [CrossRef]
- O’Sullivan, K.E.; Breen, E.P.; Gallagher, H.C.; Buggy, D.J.; Hurley, J.P. Understanding STAT3 signaling in cardiac ischemia. Basic Res. Cardiol. 2016, 111, 27. [Google Scholar] [CrossRef]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Fuchs, M.; Kaminski, K.; Schaefer, A.; Schieffer, B.; Hillmer, A.; Schmiedl, A.; Ding, Z.; Podewski, E. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004, 95, 187–195. [Google Scholar] [CrossRef]
- Boengler, K.; Ungefug, E.; Heusch, G.; Schulz, R. The STAT3 inhibitor stattic impairs cardiomyocyte mitochondrial function through increased reactive oxygen species formation. Curr. Pharm. Des. 2013, 19, 6890–6895. [Google Scholar] [CrossRef]
- Nebigil, C.G.; Desaubry, L. Updates in anthracycline-mediated cardiotoxicity. Front. Pharmacol. 2018, 9, 1262. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investg. 2014, 124, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Kunisada, K.; Negoro, S.; Tone, E.; Funamoto, M.; Osugi, T.; Yamada, S.; Okabe, M.; Kishimoto, T.; Yamauchi-Takihara, K. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc. Natl. Acad. Sci. USA 2000, 97, 315–319. [Google Scholar] [CrossRef] [Green Version]
- Rong, J.; Li, L.; Jing, L.; Fang, H.; Peng, S. JAK2/STAT3 pathway mediates protection of metallothionein against doxorubicin-induced cytotoxicity in mouse cardiomyocytes. Int. J. Toxicol. 2016, 35, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Guo, W.; Lin, S.Z.; Wang, Z.J.; Kan, J.T.; Chen, S.Y.; Zhu, Y.Z. Gp130-mediated STAT3 activation by S-propargyl-cysteine, an endogenous hydrogen sulfide initiator, prevents doxorubicin-induced cardiotoxicity. Cell Death. Dis. 2016, 7, e2339. [Google Scholar] [CrossRef] [Green Version]
- Melendez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Datta, R.; Bansal, T.; Rana, S.; Datta, K.; Datta Chaudhuri, R.; Chawla-Sarkar, M.; Sarkar, S. Myocyte-derived Hsp90 modulates collagen upregulation via biphasic activation of STAT-3 in fibroblasts during cardiac hypertrophy. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.T.; Lai, L.P.; Kuo, K.T.; Hwang, J.J.; Hsieh, C.S.; Hsu, K.L.; Tseng, C.D.; Tseng, Y.Z.; Chiang, F.T.; Lin, J.L. Angiotensin II activates signal transducer and activators of transcription 3 via Rac1 in atrial myocytes and fibroblasts: Implication for the therapeutic effect of statin in atrial structural remodeling. Circulation 2008, 117, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, H.; Hoshijima, M.; Gu, Y.; Nakamura, T.; Pradervand, S.; Hanada, T.; Hanakawa, Y.; Yoshimura, A.; Ross, J., Jr.; Chien, K.R. Suppressor of cytokine signaling-3 is a biomechanical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J. Clin. Investg. 2001, 108, 1459–1467. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, L.; Samanta, A.; Mahmoudi, S.M.; Buehler, T.; Cantilena, A.; Vincent, R.J.; Girgis, M.; Breeden, J.; Asante, S. STAT3 balances myocyte hypertrophy vis-a-vis autophagy in response to Angiotensin II by modulating the AMPKalpha/mTOR axis. PLoS ONE 2017, 12, e0179835. [Google Scholar]
- Altara, R.; Harmancey, R.; Didion, S.P.; Booz, G.W.; Zouein, F.A. Cardiac STAT3 deficiency impairs contractility and metabolic homeostasis in hypertension. Front. Pharmacol. 2016, 7, 436. [Google Scholar] [CrossRef] [Green Version]
- Hirota, H.; Chen, J.; Betz, U.A.K.; Rajewsky, K.; Gu, Y.; Ross, J.; Müller, W.; Chien, K.R. Loss of a gp130 cardiac muscle cell survival pathway Is a critical event in the onset of heart failure during biomechanical stress. Cell 1999, 97, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Uozumi, H.; Hiroi, Y.; Zou, Y.; Takimoto, E.; Toko, H.; Niu, P.; Shimoyama, M.; Yazaki, Y.; Nagai, R.; Komuro, I. gp130 plays a critical role in pressure overload-induced cardiac hypertrophy. J. Biol. Chem. 2001, 276, 23115–23119. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi-Takihara, K.; Kishimoto, T. A novel role for STAT3 in cardiac remodeling. Trends Cardiovasc. Med. 2000, 10, 298–303. [Google Scholar] [CrossRef]
- Herrera, S.C.; Bach, E.A. JAK/STAT signaling in stem cells and regeneration: From Drosophila to vertebrates. Development 2019, 146, dev167643. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Patel, P.H.; Kohlmaier, A.; Grenley, M.O.; McEwen, D.G.; Edgar, B.A. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 2009, 137, 1343–1355. [Google Scholar] [CrossRef] [Green Version]
- Oberpriller, J.O.; Oberpriller, J.C. Response of the adult newt ventricle to injury. J. Exp. Zool. 1974, 187, 249–253. [Google Scholar] [CrossRef]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef]
- Fang, Y.; Gupta, V.; Karra, R.; Holdway, J.E.; Kikuchi, K.; Poss, K.D. Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13416–13421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jopling, C.; Sleep, E.; Raya, M.; Marti, M.; Raya, A.; Izpisua Belmonte, J.C. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010, 464, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wang, D.; Renaud, G.; Wolfsberg, T.G.; Wilson, A.F.; Burgess, S.M. The stat3/socs3a pathway is a key regulator of hair cell regeneration in zebrafish. [corrected]. J. Neurosci. 2012, 32, 10662–10673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.F.; Wan, J.; Powell, C.; Ramachandran, R.; Myers, M.G., Jr.; Goldman, D. Leptin and IL-6 family cytokines synergize to stimulate Muller glia reprogramming and retina regeneration. Cell Rep. 2014, 9, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Du, X.J.; Bathgate, R.A.; Samuel, C.S.; Dart, A.M.; Summers, R.J. Cardiovascular effects of relaxin: From basic science to clinical therapy. Nat. Rev. Cardiol. 2010, 7, 48–58. [Google Scholar] [CrossRef]
- Obana, M.; Maeda, M.; Takeda, K.; Hayama, A.; Mohri, T.; Yamashita, T.; Nakaoka, Y.; Komuro, I.; Takeda, K.; Matsumiya, G. Therapeutic activation of signal transducer and activator of transcription 3 by interleukin-11 ameliorates cardiac fibrosis after myocardial infarction. Circulation 2010, 121, 684–691. [Google Scholar] [CrossRef] [Green Version]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [Green Version]
- Bicknell, K.A.; Coxon, C.H.; Brooks, G. Can the cardiomyocyte cell cycle be reprogrammed? J. Mol. Cell Cardiol. 2007, 42, 706–721. [Google Scholar] [CrossRef]
- Steinhauser, M.L.; Lee, R.T. Regeneration of the heart. EMBO Mol. Med. 2011, 3, 701–712. [Google Scholar] [CrossRef]
- Soonpaa, M.H.; Field, L.J. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am. J. Physiol. 1997, 272, H220–H226. [Google Scholar] [CrossRef]
- Walsh, S.; Ponten, A.; Fleischmann, B.K.; Jovinge, S. Cardiomyocyte cell cycle control and growth estimation in vivo--an analysis based on cardiomyocyte nuclei. Cardiovasc Res. 2010, 86, 365–373. [Google Scholar] [CrossRef]
- O’Meara, C.C.; Wamstad, J.A.; Gladstone, R.A.; Fomovsky, G.M.; Butty, V.L.; Shrikumar, A.; Gannon, J.B.; Boyer, L.A.; Lee, R.T. Transcriptional reversion of cardiac myocyte fate during mammalian cardiac regeneration. Circ. Res. 2015, 116, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, S.; Kikuno, R.; Tezuka, K.; Amann, E. Osteoblast-specific factor 2: Cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J. 1993, 294 Pt 1, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Stanton, L.W.; Garrard, L.J.; Damm, D.; Garrick, B.L.; Lam, A.; Kapoun, A.M.; Zheng, Q.; Protter, A.A.; Schreiner, G.F.; White, R.T. Altered patterns of gene expression in response to myocardial infarction. Circ. Res. 2000, 86, 939–945. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Oparil, S.; Feng, J.A.; Li, P.; Perry, G.; Chen, L.B.; Dai, M.; John, S.W.; Chen, Y.F. Effects of pressure overload on extracellular matrix expression in the heart of the atrial natriuretic peptide-null mouse. Hypertension 2003, 42, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Kühn, B.; del Monte, F.; Hajjar, R.J.; Chang, Y.S.; Lebeche, D.; Arab, S.; Keating, M.T. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat. Med. 2007, 13, 962–969. [Google Scholar] [CrossRef]
- Ince, H.; Petzsch, M.; Kleine, H.D.; Eckard, H.; Rehders, T.; Burska, D.; Kische, S.; Freund, M.; Nienaber, C.A. Prevention of left ventricular remodeling with granulocyte colony-stimulating factor after acute myocardial infarction: Final 1-year results of the Front-Integrated Revascularization and Stem Cell Liberation in Evolving Acute Myocardial Infarction by Granulocyte Colony-Stimulating Factor (FIRSTLINE-AMI) Trial. Circulation 2005, 112, I73–I80. [Google Scholar]
- Hedman, M.; Hartikainen, J.; Syvanne, M.; Stjernvall, J.; Hedman, A.; Kivela, A.; Vanninen, E.; Mussalo, H.; Kauppila, E.; Simula, S. Safety and feasibility of catheter-based local intracoronary vascular endothelial growth factor gene transfer in the prevention of postangioplasty and in-stent restenosis and in the treatment of chronic myocardial ischemia: Phase II results of the Kuopio Angiogenesis Trial (KAT). Circulation 2003, 107, 2677–2683. [Google Scholar]
- Kastrup, J.; Jorgensen, E.; Ruck, A.; Tagil, K.; Glogar, D.; Ruzyllo, W.; Botker, H.E.; Dudek, D.; Drvota, V.; Hesse, B. Direct intramyocardial plasmid vascular endothelial growth factor-A165 gene therapy in patients with stable severe angina pectoris A randomized double-blind placebo-controlled study: The Euroinject One trial. J. Am. Coll. Cardiol. 2005, 45, 982–988. [Google Scholar] [CrossRef] [Green Version]
- Losordo, D.W.; Vale, P.R.; Hendel, R.C.; Milliken, C.E.; Fortuin, F.D.; Cummings, N.; Schatz, R.A.; Asahara, T.; Isner, J.M.; Kuntz, R.E. Phase 1/2 placebo-controlled, double-blind, dose-escalating trial of myocardial vascular endothelial growth factor 2 gene transfer by catheter delivery in patients with chronic myocardial ischemia. Circulation 2002, 105, 2012–2018. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Hwang, H.Y.; Cho, K.R.; Park, E.A.; Lee, W.; Paeng, J.C.; Lee, D.S.; Kim, H.K.; Sohn, D.W.; Kim, K.B. Intramyocardial transfer of hepatocyte growth factor as an adjunct to CABG: Phase I clinical study. Gene Ther. 2013, 20, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiroldi, A.; Piccoli, M.; Cirillo, F.; Monasky, M.M.; Ciconte, G.; Pappone, C.; Anastasia, L. Cell-Based Therapies for Cardiac Regeneration: A Comprehensive Review of Past and Ongoing Strategies. Int. J. Mol. Sci. 2018, 19, 3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tendera, M.; Wojakowski, W.; Ruzyllo, W.; Chojnowska, L.; Kepka, C.; Tracz, W.; Musialek, P.; Piwowarska, W.; Nessler, J.; Buszman, P. Intracoronary infusion of bone marrow-derived selected CD34+CXCR4+ cells and non-selected mononuclear cells in patients with acute STEMI and reduced left ventricular ejection fraction: Results of randomized, multicentre Myocardial Regeneration by Intracoronary Infusion of Selected Population of Stem Cells in Acute Myocardial Infarction (REGENT) Trial. Eur. Heart J. 2009, 30, 1313–1321. [Google Scholar]
- Hare, J.M.; Traverse, J.H.; Henry, T.D.; Dib, N.; Strumpf, R.K.; Schulman, S.P.; Gerstenblith, G.; DeMaria, A.N.; Denktas, A.E.; Gammon, R.S. A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J. Am. Coll Cardiol. 2009, 54, 2277–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, S.; Domae, K.; Yoshikawa, Y.; Fukushima, S.; Nakamura, T.; Saito, A.; Sakata, Y.; Hamada, S.; Toda, K.; Pak, K. Phase I clinical trial of autologous stem cell-sheet transplantation therapy for treating cardiomyopathy. J. Am. Heart Assoc. 2017, 6, e003918. [Google Scholar] [CrossRef] [PubMed]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Inagawa, K.; Miyamoto, K.; Yamakawa, H.; Muraoka, N.; Sadahiro, T.; Umei, T.; Wada, R.; Katsumata, Y.; Kaneda, R.; Nakade, K. Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 1147–1156. [Google Scholar] [CrossRef]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.D.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, K.; Akiyama, M.; Tamura, F.; Isomi, M.; Yamakawa, H.; Sadahiro, T.; Muraoka, N.; Kojima, H.; Haginiwa, S.; Kurotsu, S. Direct in vivo reprogramming with sendai virus vectors improves cardiac function after myocardial infarction. Cell Stem Cell 2018, 22, 91–103.e5. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
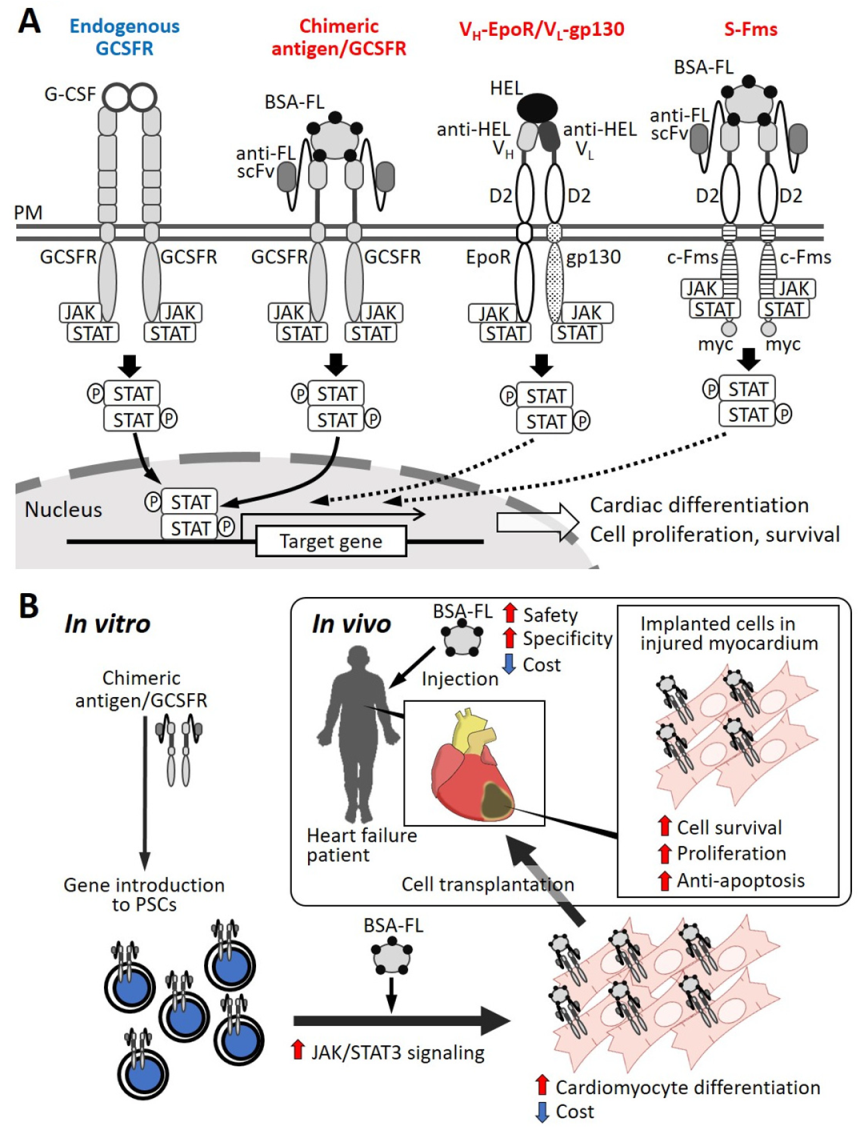
- Tsukamoto, T.; Sogo, T.; Ueyama, T.; Nakao, S.; Harada, Y.; Ihara, D.; Akagi, Y.; Kida, Y.S.; Hasegawa, K.; Nagamune, T. Chimeric G-CSF receptor-mediated STAT3 activation contributes to efficient induction of cardiomyocytes from mouse induced pluripotent stem cells. Biotechnol. J. 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Rendon, E.; Sweeney, D.; Lu, F.; Girdlestone, J.; Navarrete, C.; Watt, S.M. 5-Azacytidine-treated human mesenchymal stem/progenitor cells derived from umbilical cord, cord blood and bone marrow do not generate cardiomyocytes in vitro at high frequencies. Vox Sang. 2008, 95, 137–148. [Google Scholar] [CrossRef]
- Kawahara, M.; Hitomi, A.; Nagamune, T. S-Fms signalobody enhances myeloid cell growth and migration. Biotechnol. J. 2014, 9, 954–961. [Google Scholar] [CrossRef]
- Kawahara, M.; Shimo, Y.; Sogo, T.; Hitomi, A.; Ueda, H.; Nagamune, T. Antigen-mediated migration of murine pro-B Ba/F3 cells via an antibody/receptor chimera. J. Biotechnol. 2008, 133, 154–161. [Google Scholar] [CrossRef]
- Tanaka, K.; Kawahara, M.; Ueda, H.; Nagamune, T. Selection and growth regulation of genetically modified cells with hapten-specific antibody/receptor tyrosine kinase chimera. Biotechnol. Prog. 2009, 25, 1138–1145. [Google Scholar] [CrossRef]
- Kawahara, M.; Ueda, H.; Tsumoto, K.; Kumagai, I.; Mahoney, W.; Nagamune, T. A growth signal with an artificially induced erythropoietin receptor-gp130 cytoplasmic domain heterodimer. J. Biochem. 2001, 130, 305–312. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, H.; Shi, Y.; Chin, K.L.; Tang, D.C.; Rodgers, G.P. Identification of key genes responsible for cytokine-induced erythroid and myeloid differentiation and switching of hematopoietic stem cells by RAGE. Cell Res. 2006, 16, 923–939. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Watowich, S.S. Granulocyte colony-stimulating factor: Molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine 2008, 42, 277–288. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Ligand/Stimulus | Receptor/Effector | Cell/Tissue Type | Functions | Mechanisms | References |
|---|---|---|---|---|---|
| LIF | LIFR/gp130 | mouse early embryo | early development | possible role in visceral endoderm | [10] |
| LIF | LIFR/gp130 | mESC | pluripotency maintenance | induction of KLF4 expression subsequently activating SOX2 transcription | [11,12,13] |
| LIF | LIFR/gp130 | hESC | naïve pluripotency acquisition | NA | [22] |
| G-CSF | GCSFR/gp130 Y118F chimeric receptor | mEpiSC | naïve pluripotency acquisition | NA | [23,25] |
| LIF | LIFR/gp130 | MEF | reprogramming to miPSCs | demethylation and deacetylation of OCT4 and Nanog | [26] |
| IL-6 | IL6R | MEF, human fibroblast | reprogramming to miPSCs and hiPSCs | activation of activates endogenous OCT4 by NKX3-1 | [27] |
| G-CSF | GCSFR | mouse embryonic heart | cardiac development | cardiomyocyte proliferation | [40] |
| G-CSF | GCSFR | BMSC (mouse, rabbit, human) | cardioprotective against MI | mobilization to injured myocardium | [46,47,48,49,50,51,52] |
| G-CSF | GCSFR | BMSC (mouse, non-human primate) | cardioprotective against MI | cardiomyocyte survival as paracrine effect | [53,54,55] |
| NA | miR-124 | BMSC (rat) | pathological factor | inhibition of cardiomyocyte differentiation | [56] |
| G-CSF | GCSFR | mouse and rat cardiomyocyte | cardioprotective against MI | anti-apoptosis, prevention of ventricular remodeling | [58,59,60] |
| G-CSF | GCSFR | mouse and rat endothelial cell | cardioprotective against MI | cell survival, neovascularization | [58,59,60] |
| NA | miR-199-5p | mouse and rat cardiomyocyte | pathological factor | disruption of protein turnover | [61] |
| NA | miR-199-5p | mouse and rat endothelial cell | pathological factor | oxidative stress elevation | [61] |
| NA | NA | adult mouse heart | cardioprotective against I/R injury | decreasing of oxidative stress, apoptosis and mitochondrial dysfunction; increasing angiogenesis | [67,68,69] |
| NA | NA | adult mouse heart | cardioprotective against I/R injury | increase in antioxidants (metallothioneins, MnSOD): decrease in ROS production via complexes I and III activation | [66,67,70] |
| NA | NA | adult mouse heart | cardioprotective against doxorubicin-induced cardiomyopathy | cell survival, increase in antioxidants (metallothionein 1 and 2) | [76] |
| NA | gp130 | adult mouse heart | cardioprotective against doxorubicin-induced cardiomyopathy | in response to S-propargyl-cysteine (hydrogen sulfide initiator) | [77] |
| IL-6 | NA | rat cardiac fibroblast | physiological and pathological fibrosis | collagen synthesis | [78,79] |
| Angiotensin II /Rac1 | NA | rat cardiac fibroblast | fibrosis | collagen synthesis | [80] |
| LIF | LIFR/gp130 | mouse and rat cardiomyocyte | hypertrophy | cytokine-mediated hypertrophy and anti-apoptosis | [81] |
| Angiotensin II | NA | H9c2 cell line | anti-hypertrophy | inhibition of autophagy-related proteins; activation of AMPKα and mTOR | [82] |
| NA | NA | adult mouse heart | cardioprotection against hypertension | inhibition to shift energy metabolism from fatty acid oxidation to glycolysis | [83] |
| NA | gp130 | adult mouse heart | cardioprotection against early onset of dilated cardiomyopathy induced by pressure overload | anti-apoptosis | [84,85,86] |
| Il11α | NA | zebrafish heart | myocardial regeneration after injury | cardiomyocyte proliferation through cytokine production in endocardium and inflammatory cells | [91,92,93,94] |
| IL-11 | NA | adult mouse heart | cardioprotective against MI | Prevention of apoptosis, fibrosis and ventricular remodeling; neovascularization | [96] |
| IL-13 | NA | neonatal mouse heart | myocardial regeneration after injury | reversion of transcription profiles for cardiomyocyte development and maturation | [102] |
| Surrogate ligand (BSA-FL) | chimericantigen/GCSFR | miPSC | cardiomyocyte differentiation | NA | [122] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakao, S.; Tsukamoto, T.; Ueyama, T.; Kawamura, T. STAT3 for Cardiac Regenerative Medicine: Involvement in Stem Cell Biology, Pathophysiology, and Bioengineering. Int. J. Mol. Sci. 2020, 21, 1937. https://doi.org/10.3390/ijms21061937
Nakao S, Tsukamoto T, Ueyama T, Kawamura T. STAT3 for Cardiac Regenerative Medicine: Involvement in Stem Cell Biology, Pathophysiology, and Bioengineering. International Journal of Molecular Sciences. 2020; 21(6):1937. https://doi.org/10.3390/ijms21061937
Chicago/Turabian StyleNakao, Shu, Tasuku Tsukamoto, Tomoe Ueyama, and Teruhisa Kawamura. 2020. "STAT3 for Cardiac Regenerative Medicine: Involvement in Stem Cell Biology, Pathophysiology, and Bioengineering" International Journal of Molecular Sciences 21, no. 6: 1937. https://doi.org/10.3390/ijms21061937