In-Depth Analysis of Exoproteomes from Marine Bacteria by Shotgun Liquid Chromatography-Tandem Mass Spectrometry: the Ruegeria pomeroyi DSS-3 Case-Study


Abstract
:1. Introduction
2. Results and Discussion
2.1. A shotgun nano-LC-MS/MS strategy for a comprehensive exoproteome
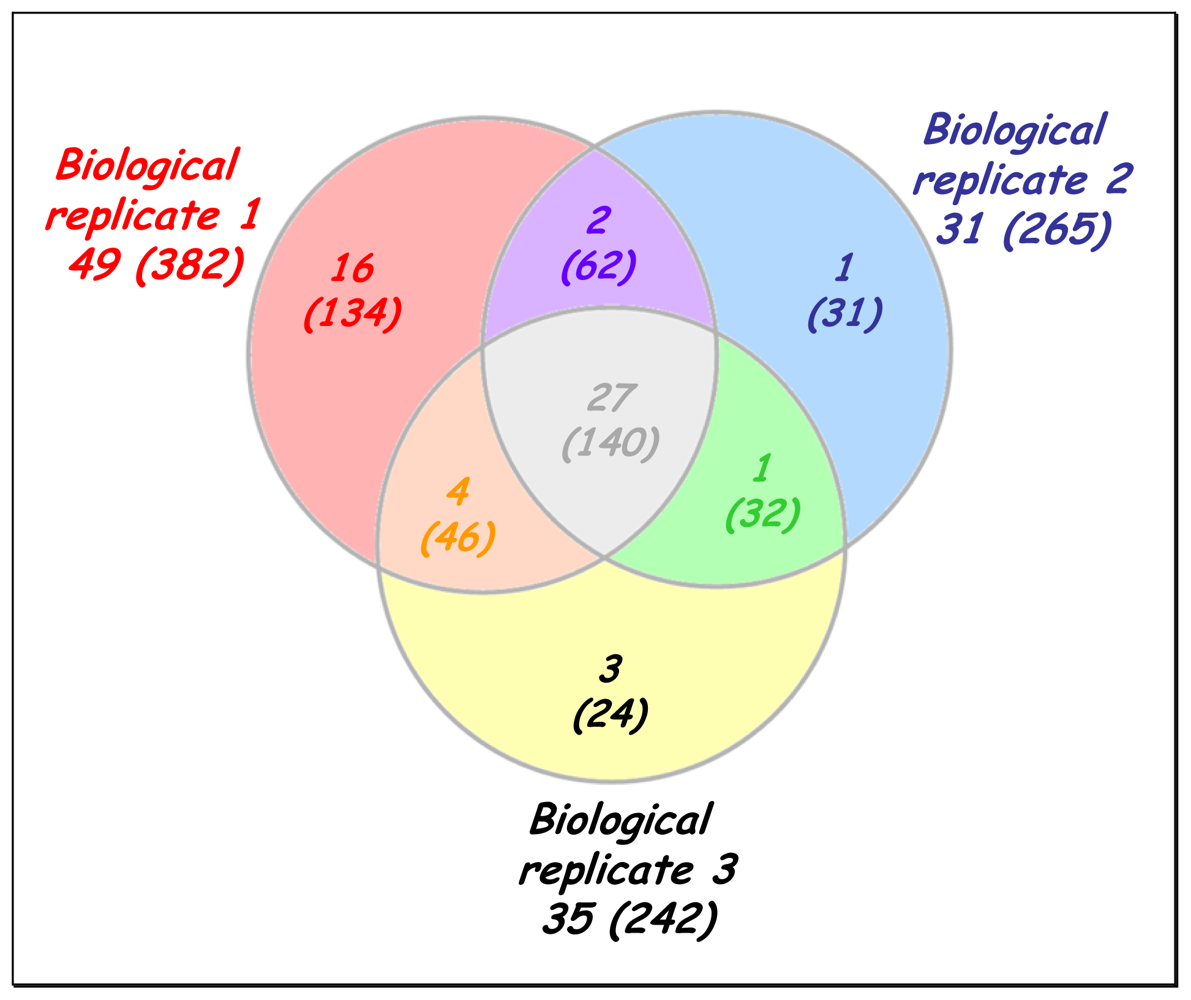
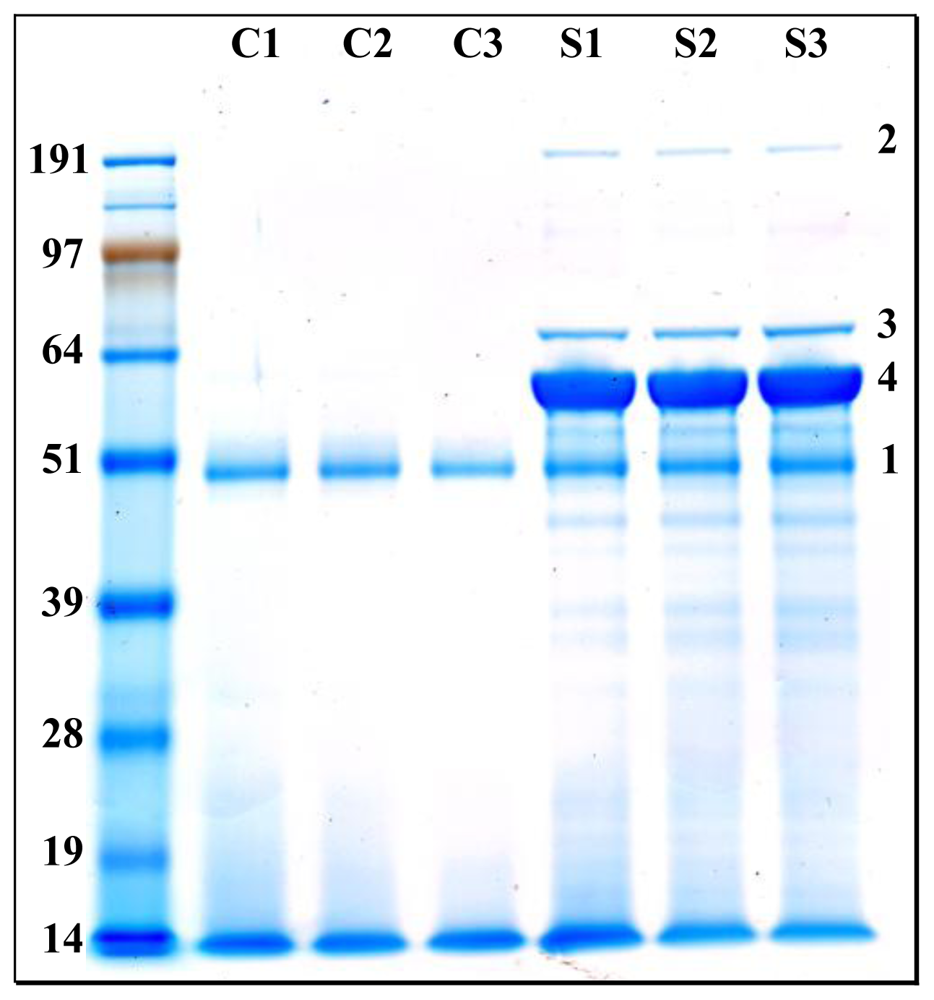
2.2. A broad exoproteome is revealed in a one-shot analysis
2.3. Confirmation of the nature and over-representation of the most abundant proteins
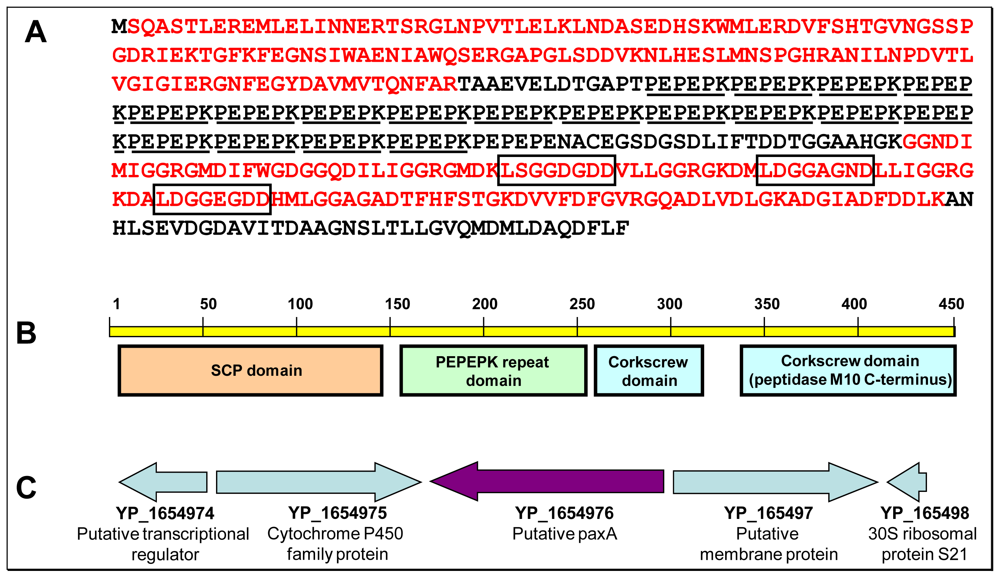
2.4. Structural and functional hints regarding YP_165496
2.5. Structure and role of the two other major secreted proteins
2.7. Other proteins identified with a signal peptide
3. Experimental
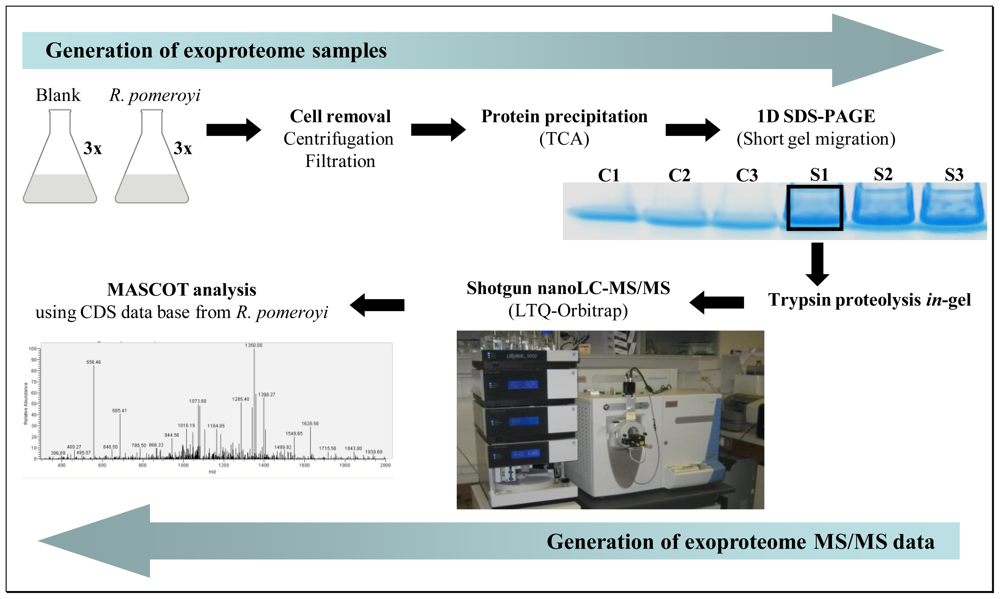
3.1. Exoproteome samples
3.2. Trypsin in-gel proteolysis and nano-LC-MS/MS analysis
3.3. MS/MS database search
3.4. Protein quantification
3.5. Protein and nucleic sequence analysis
4. Conclusions
Acknowledgements
- Samples Availability: Available from the authors.
References
- Thakur, NL; Jain, R; Natalio, F; Hamer, B; Thakur, AN; Muller, WE. Marine molecular biology: an emerging field of biological sciences. Biotechnol. Adv 2008, 26, 233–245. [Google Scholar]
- Yooseph, S; Sutton, G; Rusch, DB; Halpern, AL; Williamson, SJ; Remington, K; Eisen, JA; Heidelberg, KB; Manning, G; Li, W; et al. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol 2007, 5, e16. [Google Scholar]
- Brinkhoff, T; Giebel, HA; Simon, M. Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch. Microbiol 2008, 189, 531–539. [Google Scholar]
- Wagner-Dobler, I; Biebl, H. Environmental biology of the marine Roseobacter lineage. Annu. Rev. Microbiol 2006, 60, 255–280. [Google Scholar]
- Yi, H; Lim, YW; Chun, J. Taxonomic evaluation of the genera Ruegeria and Silicibacter: a proposal to transfer the genus Silicibacter Petursdottir and Kristjansson 1999 to the genus Ruegeria Uchino et al. 1999. Int. J. Syst. Evol. Microbiol 2007, 57, 815–819. [Google Scholar]
- Gonzalez, JM; Covert, JS; Whitman, WB; Henriksen, JR; Mayer, F; Scharf, B; Schmitt, R; Buchan, A; Fuhrman, JA; Kiene, RP; Moran, MA. Silicibacter pomeroyi sp. nov. and Roseovarius nubinhibens sp. nov., dimethylsulfoniopropionate-demethylating bacteria from marine environments. Int. J. Syst. Evol. Microbiol 2003, 53, 1261–1269. [Google Scholar]
- Moran, MA; Buchan, A; Gonzalez, JM; Heidelberg, JF; Whitman, WB; Kiene, RP; Henriksen, JR; King, GM; Belas, R; Fuqua, C; et al. Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature 2004, 432, 910–913. [Google Scholar]
- Burgmann, H; Howard, EC; Ye, W; Sun, F; Sun, S; Napierala, S; Moran, MA. Transcriptional response of Silicibacter pomeroyi DSS-3 to dimethylsulfoniopropionate (DMSP). Environ. Microbiol 2007, 9, 2742–2755. [Google Scholar]
- Lauro, FM; McDougald, D; Thomas, T; Williams, TJ; Egan, S; Rice, S; DeMaere, MZ; Ting, L; Ertan, H; Johnson, J; Ferriera, S; Lapidus, A; Anderson, I; Kyrpides, N; Munk, AC; Detter, C; Han, CS; Brown, MV; Robb, FT; Kjelleberg, S; Cavicchioli, R. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 15527–15533. [Google Scholar]
- Antelmann, H; Tjalsma, H; Voigt, B; Ohlmeier, S; Bron, S; van Dijl, JM; Hecker, M. A proteomic view on genome-based signal peptide predictions. Genome Res 2001, 11, 1484–1502. [Google Scholar]
- Preston, GM; Studholme, DJ; Caldelari, I. Profiling the secretomes of plant pathogenic Proteobacteria. FEMS Microbiol. Rev 2005, 29, 331–360. [Google Scholar]
- Desvaux, M; Hébraud, M; Talon, R; Henderson, IR. Secretion and subcellular localizations of bacterial proteins: a semantic awareness issue. Trends Microbiol 2009, 17, 139–145. [Google Scholar]
- Kazemi-Pour, N; Condemine, G; Hugouvieux-Cotte-Pattat, N. The secretome of the plant pathogenic bacterium Erwinia chrysanthemi. Proteomics 2004, 4, 3177–3186. [Google Scholar]
- Clair, G; Roussi, S; Armengaud, J; Duport, C. Expanding the known repertoire of virulence factors produced by Bacillus cereus through early secretome profiling in three redox conditions. Mol. Cell. Proteomics 2010. [Google Scholar] [CrossRef]
- Gohar, M; Gilois, N; Graveline, R; Garreau, C; Sanchis, V; Lereclus, D. A comparative study of Bacillus cereus, Bacillus thuringiensis and Bacillus anthracis extracellular proteomes. Proteomics 2005, 5, 3696–3711. [Google Scholar]
- Kalkum, M; Lyon, GJ; Chait, BT. Detection of secreted peptides by using hypothesis-driven multistage mass spectrometry. Proc. Natl. Acad. Sci. USA 2003, 100, 2795–2800. [Google Scholar]
- Saier, MH, Jr. Protein secretion and membrane insertion systems in gram-negative bacteria. J. Membr. Biol 2006, 214, 75–90. [Google Scholar]
- Choo, KH; Tan, TW; Ranganathan, S. A comprehensive assessment of N-terminal signal peptides prediction methods. BMC Bioinformatics 2009, 10, S2. [Google Scholar]
- Bendtsen, JD; Nielsen, H; von Heijne, G; Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol 2004, 340, 783–795. [Google Scholar]
- Emanuelsson, O; Brunak, S; von Heijne, G; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc 2007, 2, 953–971. [Google Scholar]
- Erickson, BK; Mueller, R; Verberkmoes, NC; Shah, M; Singer, SW; Thelen, M; Banfield, JF; Hettich, RL. Computational Prediction and Experimental Validation of Signal Peptide Cleavages in the Extracellular Proteome of a Natural Microbial Community. J. Proteome Res 2010, 9, 2148–2159. [Google Scholar]
- Leversen, NA; de Souza, GA; Malen, H; Prasad, S; Jonassen, I; Wiker, HG. Evaluation of signal peptide prediction algorithms for identification of mycobacterial signal peptides using sequence data from proteomic methods. Microbiology 2009, 155, 2375–2383. [Google Scholar]
- Zhou, M; Boekhorst, J; Francke, C; Siezen, R. LocateP: Genome-scale subcellular-location predictor for bacterial proteins. BMC Bioinformatics 2008, 9, 173. [Google Scholar]
- Josic, D; Kovac, S. Application of proteomics in biotechnology—microbial proteomics. Biotechnol. J 2008, 3, 496–509. [Google Scholar]
- Keller, M; Hettich, R. Environmental proteomics: a paradigm shift in characterizing microbial activities at the molecular level. Microbiol. Mol. Biol. Rev 2009, 73, 62–70. [Google Scholar]
- Schneider, T; Riedel, K. Environmental proteomics: analysis of structure and function of microbial communities. Proteomics 2010, 10, 785–798. [Google Scholar]
- Armengaud, J. Proteogenomics and systems biology: quest for the ultimate missing parts. Expert Rev. Proteomics 2010, 7, 65–77. [Google Scholar]
- Dumas, E; Desvaux, M; Chambon, C; Hébraud, M. Insight into the core and variant exoproteomes of Listeria monocytogenes species by comparative subproteomic analysis. Proteomics 2009, 9, 3136–3155. [Google Scholar]
- Westers, L; Westers, H; Zanen, G; Antelmann, H; Hecker, M; Noone, D; Devine, KM; van Dijl, JM; Quax, WJ. Genetic or chemical protease inhibition causes significant changes in the Bacillus subtilis exoproteome. Proteomics 2008, 8, 2704–2713. [Google Scholar]
- Shah, P; Atwood, JA; Orlando, R; El Mubarek, H; Podila, GK; Davis, MR. Comparative proteomic analysis of Botrytis cinerea secretome. J. Proteome Res 2009, 8, 1123–1130. [Google Scholar]
- Tunica, D; Yin, X; Sidibe, A; Stegemann, C; Nissum, M; Zeng, L; Brunet, M; Mayr, M. Proteomic analysis of the secretome of human umbilical vein endothelial cells using a combination of free-flow electrophoresis and nanoflow LC-MS/MS. Proteomics 2009, 9, 4991–4996. [Google Scholar]
- Evans, FF; Raftery, MJ; Egan, S; Kjelleberg, S. Profiling the secretome of the marine bacterium Pseudoalteromonas tunicata using amine-specific isobaric tagging (iTRAQ). J. Proteome Res 2007, 6, 967–975. [Google Scholar]
- Liu, H; Sadygov, RG; Yates, JR, III. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem 2004, 76, 4193–4201. [Google Scholar]
- Mueller, LN; Brusniak, M-Y; Mani, DR; Aebersold, R. An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res 2008, 7, 51–61. [Google Scholar]
- Kuhnert, P; Heyberger-Meyer, B; Nicolet, J; Frey, J. Characterization of PaxA and its operon: a cohemolytic RTX toxin determinant from pathogenic Pasteurella aerogenes. Infect. Immun 2000, 68, 6–12. [Google Scholar]
- Rodriguez, J; Gupta, N; Smith, RD; Pevzner, PA. Does trypsin cut before proline? J. Proteome Res 2008, 7, 300–305. [Google Scholar]
- Armengaud, J. A perfect genome annotation is within reach with the proteomics and genomics alliance. Curr. Opin. Microbiol 2009, 12, 292–300. [Google Scholar]
- Bradshaw, RA; Brickey, WW; Walker, KW. N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem. Sci 1998, 23, 263–267. [Google Scholar]
- Frottin, F; Martinez, A; Peynot, P; Mitra, S; Holz, RC; Giglione, C; Meinnel, T. The proteomics of N-terminal methionine cleavage. Mol. Cell. Proteomics 2006, 5, 2336–2349. [Google Scholar]
- Giglione, C; Vallon, O; Meinnel, T. Control of protein life-span by N-terminal methionine excision. EMBO J 2003, 22, 13–23. [Google Scholar]
- Yeats, C; Bentley, S; Bateman, A. New knowledge from old: in silico discovery of novel protein domains in Streptomyces coelicolor. BMC Microbiol 2003, 3, 3. [Google Scholar]
- Lally, ET; Hill, RB; Kieba, IR; Korostoff, J. The interaction between RTX toxins and target cells. Trends Microbiol 1999, 7, 356–361. [Google Scholar]
- Li, L; Rock, JL; Nelson, DR. Identification and characterization of a repeat-in-toxin gene cluster in Vibrio anguillarum. Infect. Immun 2008, 76, 2620–2632. [Google Scholar]
- Lin, W; Fullner, KJ; Clayton, R; Sexton, JA; Rogers, MB; Calia, KE; Calderwood, SB; Fraser, C; Mekalanos, JJ. Identification of a Vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc. Natl. Acad. Sci. USA 1999, 96, 1071–1076. [Google Scholar]
- Sasaki, H; Kawamoto, E; Tanaka, Y; Sawada, T; Kunita, S; Yagami, K-i. Identification and characterization of hemolysin-like proteins similar to RTX toxin in Pasteurella pneumotropica. J. Bacteriol 2009, 191, 3698–3705. [Google Scholar]
- Gogarten, JP; Senejani, AG; Zhaxybayeva, O; Olendzenski, L; Hilario, E. INTEINS: structure, function, and evolution. Annu. Rev. Microbiol 2002, 56, 263–287. [Google Scholar]
- Perler, FB. InBase: the intein database. Nucleic Acids Res 2002, 30, 383–384. [Google Scholar]
- Persson, OP; Pinhassi, J; Riemann, L; Marklund, B-I; Rhen, M; Normark, S; González, JM; Hagström, Å. High abundance of virulence gene homologues in marine bacteria. Environ. Microbiol 2009, 11, 1348–1357. [Google Scholar]
- Moran, MA; Belas, R; Schell, MA; Gonzalez, JM; Sun, F; Sun, S; Binder, BJ; Edmonds, J; Ye, W; Orcutt, B; Howard, EC; Meile, C; Palefsky, W; Goesmann, A; Ren, Q; Paulsen, I; Ulrich, LE; Thompson, LS; Saunders, E; Buchan, A. Ecological genomics of marine Roseobacters. Appl. Environ. Microbiol 2007, 73, 4559–4569. [Google Scholar]
- Baudet, M; Ortet, P; Gaillard, J-C; Fernandez, B; Guérin, P; Enjalbal, C; Subra, G; de Groot, A; Barakat, M; Dedieu, A; Armengaud, J. Proteomics-based refinement of Deinococcus deserti genome annotation reveals an unwonted use of non-canonical translation initiation codons. Mol. Cell. Proteomics 2010, 9, 415–426. [Google Scholar]
- Stancik, LM; Stancik, DM; Schmidt, B; Barnhart, DM; Yoncheva, YN; Slonczewski, JL. pH-dependent expression of periplasmic proteins and amino acid catabolism in Escherichia coli. J. Bacteriol 2002, 184, 4246–4258. [Google Scholar]
- Handa, N; Terada, T; Doi-Katayama, Y; Hirota, H; Tame, JRH; Park, S-Y; Kuramitsu, S; Shirouzu, M; Yokoyama, S. Crystal structure of a novel polyisoprenoid-binding protein from Thermus thermophilus HB8. Protein Sci 2005, 14, 1004–1010. [Google Scholar]
- de Groot, A; Dulermo, R; Ortet, P; Blanchard, L; Guerin, P; Fernandez, B; Vacherie, B; Dossat, C; Jolivet, E; Siguier, P; Chandler, M; Barakat, M; Dedieu, A; Barbe, V; Heulin, T; Sommer, S; Achouak, W; Armengaud, J. Alliance of proteomics and genomics to unravel the specificities of Sahara bacterium Deinococcus deserti. PLoS Genet 2009, 5, e1000434. [Google Scholar]
- Dupierris, V; Masselon, C; Court, M; Kieffer-Jaquinod, S; Bruley, C. A toolbox for validation of mass spectrometry peptides identification and generation of database: IRMa. Bioinformatics 2009, 25, 1980–1981. [Google Scholar]
- Zivanovic, Y; Armengaud, J; Lagorce, A; Leplat, C; Guerin, P; Dutertre, M; Anthouard, V; Forterre, P; Wincker, P; Confalonieri, F. Genome analysis and genome-wide proteomics of Thermococcus gammatolerans, the most radioresistant organism known amongst the Archaea. Genome Biol 2009, 10, R70. [Google Scholar]
- Nogueira, T; Rankin, DJ; Touchon, M; Taddei, F; Brown, SP; Rocha, EPC. Horizontal gene transfer of the secretome drives the evolution of bacterial cooperation and virulence. Curr. Biol 2009, 19, 1683–1691. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Function | # peptides | # MS/MS | Signal Peptide prediction |
|---|---|---|---|---|
| Most abundant proteins detected | ||||
| YP_165496 | PaxA, putative | 45 | 2610 | absence |
| YP_165625 | type I secretion target repeat-containing protein | 48 | 359 | absence |
| YP_168868 | type I secretion target repeat-containing protein | 37 | 164 | absence |
| Proteins ABC and TRAP transporters | ||||
| YP_168968 | oligopeptide/dipeptide ABC transporter, periplasmic | 13 | 58 | presence |
| YP_168022 | peptide/opine/nickel uptake ABC transporter periplasmic | 16 | 54 | presence |
| YP_168201 | peptide/nickel/opine uptake ABC transporter periplasmic | 10 | 42 | presence * |
| YP_165781 | glutamate/glutamine/aspartate/asparagine ABC transporter, periplasmic | 10 | 39 | presence * |
| YP_168669 | polyamine ABC trasnporter, periplasmic polyamine-binding protein | 6 | 28 | presence |
| YP_165868 | sugar ABC transporter, periplasmic sugar-binding protein | 7 | 24 | presence |
| YP_167838 | TRAP transporter solute receptor DctP family protein | 6 | 19 | presence |
| YP_167174 | branched-chain amino acid ABC transporter, periplasmic | 4 | 14 | presence |
| YP_167412 | TRAP transporter solute receptor TAXI family protein | 5 | 13 | presence |
| YP_165067 | TRAP dicarboxylate transporter, DctP subunit | 4 | 13 | presence |
| YP_165642 | sugar ABC transporter, periplasmic sugar-binding protein | 3 | 12 | presence |
| YP_166898 | oligopeptide/dipeptide ABC transporter, periplasmic | 2 | 10 | presence * |
| YP_166382 | glycine betaine/proline ABC transporter, periplasmic | 2 | 5 | presence |
| YP_165960 | oligopeptide ABC transporter, periplasmic | 3 | 4 | presence |
| YP_166114 | xylose ABC transporter, periplasmic xylose-binding protein | 2 | 2 | presence |
| YP_167025 | ABC transporter, periplasmic substrate-binding protein | 2 | 2 | presence |
| Other proteins with predicted signal peptide | ||||
| YP_165456 | hypothetical protein SPO0186 | 10 | 37 | presence |
| YP_167495 | 3-oxoacyl-(acyl carrier protein) synthase II | 4 | 21 | presence |
| YP_167459 | peptidyl-prolyl cis-trans isomerase, cyclophilin-type | 6 | 20 | presence |
| YP_165903 | bmp family protein | 4 | 14 | presence |
| YP_165402 | cytochrome c family protein | 4 | 12 | presence |
| YP_165589 | acetyl-CoA acetyltransferase | 4 | 9 | presence |
| YP_168626 | outer membrane porin | 3 | 7 | presence |
| YP_167817 | solute-binding family 7 protein | 2 | 5 | presence |
| YP_167786 | solute-binding family 7 protein | 3 | 4 | presence |
| YP_167503 | hypothetical protein SPO2279 | 2 | 4 | presence |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Christie-Oleza, J.A.; Armengaud, J. In-Depth Analysis of Exoproteomes from Marine Bacteria by Shotgun Liquid Chromatography-Tandem Mass Spectrometry: the Ruegeria pomeroyi DSS-3 Case-Study. Mar. Drugs 2010, 8, 2223-2239. https://doi.org/10.3390/md8082223
Christie-Oleza JA, Armengaud J. In-Depth Analysis of Exoproteomes from Marine Bacteria by Shotgun Liquid Chromatography-Tandem Mass Spectrometry: the Ruegeria pomeroyi DSS-3 Case-Study. Marine Drugs. 2010; 8(8):2223-2239. https://doi.org/10.3390/md8082223
Chicago/Turabian StyleChristie-Oleza, Joseph Alexander, and Jean Armengaud. 2010. "In-Depth Analysis of Exoproteomes from Marine Bacteria by Shotgun Liquid Chromatography-Tandem Mass Spectrometry: the Ruegeria pomeroyi DSS-3 Case-Study" Marine Drugs 8, no. 8: 2223-2239. https://doi.org/10.3390/md8082223