Modified Vaccinia Virus Ankara (MVA) as Production Platform for Vaccines against Influenza and Other Viral Respiratory Diseases


 ,
,
Abstract
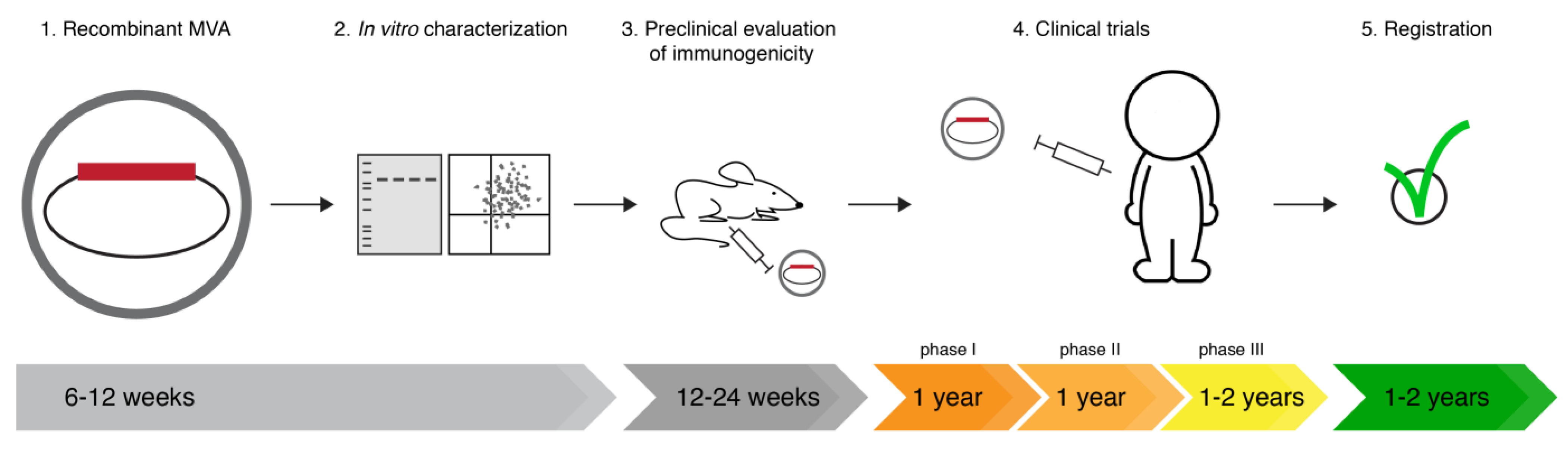
:1. Introduction
2. Targets for Influenza Vaccine Development
2.1. Conventional Influenza Vaccine
2.2. Induction of HA Stalk-Specific Antibodies
2.3. Antibody Response against NA and M2
2.4. Broadly Reactive T Cell Responses against Influenza Viruses
2.5. Universal Influenza Vaccine
3. MVA
3.1. The Development of the Attenuated Vaccinia Virus Strain MVA
3.2. Advantages of MVA as Viral Vector
4. MVA as an Influenza Vaccine
4.1. MVA-HA
4.1.1. Vaccines against A/H5N1 Viruses
4.1.2. MVA-Based Vaccines against H1N1 Viruses
4.2. MVA-HA+NP
4.3. MVA-NP+M1
4.4. MVA Expressing Other Combinations of Influenza Virus Proteins

{kind=link}
{kind=link}
| MVA vaccine | Response | Model | Protective efficacy after challenge | Literature | |
|---|---|---|---|---|---|
| MVA-NA-Ca/09 | B cells | mice | Partial homologous protection | [115] | |
| MVA-HA-HK/97 | B cells | mice | Homologous protection | [110] | |
| MVA-HA-VN/04 | B cells | mice | Cross-clade protection | [106,110,111] | |
| chickens | Homologous protection | [83] | |||
| macaques | Cross-clade protection | [38,113] | |||
| MVA-HA-IN/05 | B cells | mice | Cross-clade protection | [111] | |
| MVA-HA-TT/05 | B cells | mice | Partial cross-clade protection | [111] | |
| MVA-HA-AN/05 | B cells | mice | Partial cross-clade protection | [111] | |
| MVA-HA-CE/06 | B cells | mice | Partial cross-clade protection | [111] | |
| MVA-HA-Ca/09 | B cells | mice | Homologous protection and to some extent heterosubtypic protection against swine viruses | [115,116] | |
| ferret | Intrasubtypic protection | [117] | |||
| MVA-HAstalk | B cells | mice | No protection | [127] | |
| MVA-HAstalk/M2e | B cells | mice | No protection | [127] | |
| MVA-HAstalk/M2e+NP | B cells and T cells | mice | Heterologous protection | [127] | |
| MVA-HAstalk+NP | B cells and T cells | mice | Heterologous protection | [127] | |
| MVA-HA+NP | B cells and T cells | mice | Homologous protection | [85,106,118] | |
| MVA-NP | B cells and T cells | mice | Heterologous protection | [127] | |
| MVA-NP+M1 | T cells | mice | Partial heterologous protection* | [123,125] | |
| chickens | Heterologous protection* | [124,125] | |||
| pigs | Not tested with challenge | [125] | |||
| humans | Intrasubtypic protection, safe in elderly | [119,120,121,122,126] | |||
| MVA-M1 | ** | mice | No protection | [127] | |
| MVA-M2 | ** | mice | No protection | [127] | |
| MVA-PB1 | mice | No protection | [127] | ||
| MVA-HA-Eq/Ky81 (A/Equine/Kentucky/1/81) MVA-NP-Eq/Ky81 | HA: B-cells not tested | horses | HA: Homologous protectionNP: Partial homologous protectionLiterature: | [128] | |
5. MVA-Based Vaccine against Other Respiratory Diseases
5.1. Respiratory Diseases Caused by Viruses of the Paramyxoviridae Family
6. Future Perspectives


7. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References and Notes
- World Health Organization. Recommended composition of influenza virus vaccines for use in the 2014–2015 northern hemisphere influenza season; Geneva, Switzerland, 2014. [Google Scholar]
- Richard, M.; Schrauwen, E.J.; de Graaf, M.; Bestebroer, T.M.; Spronken, M.I.; van Boheemen, S.; de Meulder, D.; Lexmond, P.; Linster, M.; Herfst, S.; et al. Limited airborne transmission of H7N9 influenza A virus between ferrets. Nature 2013, 501, 560–563. [Google Scholar] [CrossRef]
- Herfst, S.; Schrauwen, E.J.; Linster, M.; Chutinimitkul, S.; de Wit, E.; Munster, V.J.; Sorrell, E.M.; Bestebroer, T.M.; Burke, D.F.; Smith, D.J.; et al. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 2012, 336, 1534–1541. [Google Scholar] [CrossRef]
- World Health Organization. Information sheet—Observed rate of vaccine reactions—Influenza vaccine; Geneva, Switzerland, 2012. [Google Scholar]
- Osterholm, M.T.; Kelley, N.S.; Sommer, A.; Belongia, E.A. Efficacy and effectiveness of influenza vaccines: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 36–44. [Google Scholar] [CrossRef]
- Partridge, J.; Kieny, M.P. Global production capacity of seasonal influenza vaccine in 2011. Vaccine 2013, 31, 728–731. [Google Scholar] [CrossRef]
- Broadbent, A.J.; Subbarao, K. Influenza virus vaccines: Lessons from the 2009 H1N1 pandemic. Curr. Opin. Virol. 2011, 1, 254–262. [Google Scholar] [CrossRef]
- He, X.S.; Holmes, T.H.; Zhang, C.; Mahmood, K.; Kemble, G.W.; Lewis, D.B.; Dekker, C.L.; Greenberg, H.B.; Arvin, A.M. Cellular immune responses in children and adults receiving inactivated or live attenuated influenza vaccines. J. Virol. 2006, 80, 11756–11766. [Google Scholar] [CrossRef]
- Forrest, B.D.; Pride, M.W.; Dunning, A.J.; Capeding, M.R.; Chotpitayasunondh, T.; Tam, J.S.; Rappaport, R.; Eldridge, J.H.; Gruber, W.C. Correlation of cellular immune responses with protection against culture-confirmed influenza virus in young children. Clin. Vaccine Immunol. 2008, 15, 1042–1053. [Google Scholar] [CrossRef]
- Krammer, F.; Palese, P. Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr. Opin. Virol. 2013, 3, 521–530. [Google Scholar] [CrossRef]
- Corti, D.; Suguitan, A.L.; Pinna, D.; Silacci, C.; Fernandez-Rodriguez, B.M.; Vanzetta, F.; Santos, C.; Luke, C.J.; Torres-Velez, F.J.; Temperton, N.J.; et al. Heterosubtypic neutralizing antibodies are produced by individuals immunized with a seasonal influenza vaccine. J. Clin. Invest. 2010, 120, 1663–1673. [Google Scholar] [CrossRef] [Green Version]
- Krause, J.C.; Tsibane, T.; Tumpey, T.M.; Huffman, C.J.; Basler, C.F.; Crowe, J.E., Jr. A broadly neutralizing human monoclonal antibody that recognizes a conserved, novel epitope on the globular head of the influenza H1N1 virus hemagglutinin. J. Virol. 2011, 85, 10905–10908. [Google Scholar] [CrossRef]
- Krause, J.C.; Tsibane, T.; Tumpey, T.M.; Huffman, C.J.; Albrecht, R.; Blum, D.L.; Ramos, I.; Fernandez-Sesma, A.; Edwards, K.M.; Garcia-Sastre, A.; et al. Human monoclonal antibodies to pandemic 1957 H2N2 and pandemic 1968 H3N2 influenza viruses. J. Virol. 2012, 86, 6334–6340. [Google Scholar] [CrossRef]
- Ekiert, D.C.; Kashyap, A.K.; Steel, J.; Rubrum, A.; Bhabha, G.; Khayat, R.; Lee, J.H.; Dillon, M.A.; O'Neil, R.E.; Faynboym, A.M.; et al. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature 2012, 489, 526–532. [Google Scholar] [CrossRef]
- Dreyfus, C.; Laursen, N.S.; Kwaks, T.; Zuijdgeest, D.; Khayat, R.; Ekiert, D.C.; Lee, J.H.; Metlagel, Z.; Bujny, M.V.; Jongeneelen, M.; et al. Highly conserved protective epitopes on influenza B viruses. Science 2012, 337, 1343–1348. [Google Scholar] [CrossRef]
- Lee, P.S.; Yoshida, R.; Ekiert, D.C.; Sakai, N.; Suzuki, Y.; Takada, A.; Wilson, I.A. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc. Natl. Acad. Sci. USA 2012, 109, 17040–17045. [Google Scholar]
- Okuno, Y.; Isegawa, Y.; Sasao, F.; Ueda, S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J. Virol. 1993, 67, 2552–2558. [Google Scholar]
- Throsby, M.; van den Brink, E.; Jongeneelen, M.; Poon, L.L.; Alard, P.; Cornelissen, L.; Bakker, A.; Cox, F.; van Deventer, E.; Guan, Y.; et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One 2008, 3, e3942. [Google Scholar] [CrossRef] [Green Version]
- Ekiert, D.C.; Bhabha, G.; Elsliger, M.A.; Friesen, R.H.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I.A. Antibody recognition of a highly conserved influenza virus epitope. Science 2009, 324, 246–251. [Google Scholar] [CrossRef]
- Margine, I.; Hai, R.; Albrecht, R.A.; Obermoser, G.; Harrod, A.C.; Banchereau, J.; Palucka, K.; Garcia-Sastre, A.; Palese, P.; Treanor, J.J.; et al. H3N2 influenza virus infection induces broadly reactive hemagglutinin stalk antibodies in humans and mice. J. Virol. 2013, 87, 4728–4737. [Google Scholar]
- Kashyap, A.K.; Steel, J.; Oner, A.F.; Dillon, M.A.; Swale, R.E.; Wall, K.M.; Perry, K.J.; Faynboym, A.; Ilhan, M.; Horowitz, M.; et al. Combinatorial antibody libraries from survivors of the Turkish H5N1 avian influenza outbreak reveal virus neutralization strategies. Proc. Natl. Acad. Sci. USA 2008, 105, 5986–5991. [Google Scholar] [CrossRef]
- Sui, J.; Hwang, W.C.; Perez, S.; Wei, G.; Aird, D.; Chen, L.M.; Santelli, E.; Stec, B.; Cadwell, G.; Ali, M.; et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009, 16, 265–273. [Google Scholar] [CrossRef]
- Friesen, R.H.; Koudstaal, W.; Koldijk, M.H.; Weverling, G.J.; Brakenhoff, J.P.; Lenting, P.J.; Stittelaar, K.J.; Osterhaus, A.D.; Kompier, R.; Goudsmit, J. New class of monoclonal antibodies against severe influenza: Prophylactic and therapeutic efficacy in ferrets. PLoS One 2010, 5, e9106. [Google Scholar]
- Kashyap, A.K.; Steel, J.; Rubrum, A.; Estelles, A.; Briante, R.; Ilyushina, N.A.; Xu, L.; Swale, R.E.; Faynboym, A.M.; Foreman, P.K.; et al. Protection from the 2009 H1N1 pandemic influenza by an antibody from combinatorial survivor-based libraries. PLoS Pathog. 2010, 6, e1000990. [Google Scholar] [CrossRef]
- Friesen, R.H.; Lee, P.S.; Stoop, E.J.; Hoffman, R.M.; Ekiert, D.C.; Bhabha, G.; Yu, W.; Juraszek, J.; Koudstaal, W.; Jongeneelen, M.; et al. A common solution to group 2 influenza virus neutralization. Proc. Natl. Acad. Sci. USA 2014, 111, 445–450. [Google Scholar] [CrossRef]
- Krammer, F.; Hai, R.; Yondola, M.; Tan, G.S.; Leyva-Grado, V.H.; Ryder, A.B.; Miller, M.S.; Rose, J.K.; Palese, P.; Garcia-Sastre, A.; et al. Assessment of influenza virus hemagglutinin stalk-based immunity in ferrets. J. Virol. 2014, 88, 3432–3442. [Google Scholar] [CrossRef]
- Hai, R.; Krammer, F.; Tan, G.S.; Pica, N.; Eggink, D.; Maamary, J.; Margine, I.; Albrecht, R.A.; Palese, P. Influenza viruses expressing chimeric hemagglutinins: Globular head and stalk domains derived from different subtypes. J. Virol. 2012, 86, 5774–5781. [Google Scholar] [CrossRef]
- Margine, I.; Krammer, F.; Hai, R.; Heaton, N.S.; Tan, G.S.; Andrews, S.A.; Runstadler, J.A.; Wilson, P.C.; Albrecht, R.A.; Garcia-Sastre, A.; et al. Hemagglutinin stalk-based universal vaccine constructs protect against group 2 influenza A viruses. J. Virol. 2013, 87, 10435–10446. [Google Scholar] [CrossRef]
- Goff, P.H.; Eggink, D.; Seibert, C.W.; Hai, R.; Martinez-Gil, L.; Krammer, F.; Palese, P. Adjuvants and immunization strategies to induce influenza virus hemagglutinin stalk antibodies. PLoS One 2013, 8, e79194. [Google Scholar]
- Krammer, F.; Margine, I.; Hai, R.; Flood, A.; Hirsh, A.; Tsvetnitsky, V.; Chen, D.; Palese, P. H3 stalk-based chimeric hemagglutinin influenza virus constructs protect mice from H7N9 challenge. J. Virol. 2014, 88, 2340–2343. [Google Scholar] [CrossRef]
- Eggink, D.; Goff, P.H.; Palese, P. Guiding the immune response against influenza virus hemagglutinin toward the conserved stalk domain by hyperglycosylation of the globular head domain. J. Virol. 2014, 88, 699–704. [Google Scholar] [CrossRef]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef]
- Marcelin, G.; Bland, H.M.; Negovetich, N.J.; Sandbulte, M.R.; Ellebedy, A.H.; Webb, A.D.; Griffin, Y.S.; DeBeauchamp, J.L.; McElhaney, J.E.; Webby, R.J. Inactivated seasonal influenza vaccines increase serum antibodies to the neuraminidase of pandemic influenza A(H1N1) 2009 virus in an age-dependent manner. J. Infect. Dis. 2010, 202, 1634–1638. [Google Scholar] [CrossRef]
- Marcelin, G.; DuBois, R.; Rubrum, A.; Russell, C.J.; McElhaney, J.E.; Webby, R.J. A contributing role for anti-neuraminidase antibodies on immunity to pandemic H1N1 2009 influenza A virus. PLoS One 2011, 6, e26335. [Google Scholar]
- Marcelin, G.; Sandbulte, M.R.; Webby, R.J. Contribution of antibody production against neuraminidase to the protection afforded by influenza vaccines. Rev. Med. Virol. 2012, 22, 267–279. [Google Scholar] [CrossRef]
- Chen, Z.; Kadowaki, S.; Hagiwara, Y.; Yoshikawa, T.; Matsuo, K.; Kurata, T.; Tamura, S. Cross-protection against a lethal influenza virus infection by DNA vaccine to neuraminidase. Vaccine 2000, 18, 3214–3222. [Google Scholar] [CrossRef]
- Sandbulte, M.R.; Jimenez, G.S.; Boon, A.C.; Smith, L.R.; Treanor, J.J.; Webby, R.J. Cross-reactive neuraminidase antibodies afford partial protection against H5N1 in mice and are present in unexposed humans. PLoS Med. 2007, 4, e59. [Google Scholar] [CrossRef]
- Bosch, B.J.; Bodewes, R.; de Vries, R.P.; Kreijtz, J.H.; Bartelink, W.; van Amerongen, G.; Rimmelzwaan, G.F.; de Haan, C.A.; Osterhaus, A.D.; Rottier, P.J. Recombinant soluble, multimeric HA and NA exhibit distinctive types of protection against pandemic swine-origin 2009 A(H1N1) influenza virus infection in ferrets. J. Virol. 2010, 84, 10366–10374. [Google Scholar] [CrossRef]
- Zebedee, S.L.; Lamb, R.A. Influenza A virus M2 protein: Monoclonal antibody restriction of virus growth and detection of M2 in virions. J. Virol. 1988, 62, 2762–2772. [Google Scholar]
- Jegerlehner, A.; Schmitz, N.; Storni, T.; Bachmann, M.F. Influenza A vaccine based on the extracellular domain of M2: Weak protection mediated via antibody-dependent NK cell activity. J. Immunol. 2004, 172, 5598–5605. [Google Scholar] [CrossRef]
- Song, J.M.; Wang, B.Z.; Park, K.M.; van Rooijen, N.; Quan, F.S.; Kim, M.C.; Jin, H.T.; Pekosz, A.; Compans, R.W.; Kang, S.M. Influenza virus-like particles containing M2 induce broadly cross protective immunity. PLoS One 2011, 6, e14538. [Google Scholar] [CrossRef]
- El Bakkouri, K.; Descamps, F.; de Filette, M.; Smet, A.; Festjens, E.; Birkett, A.; van Rooijen, N.; Verbeek, S.; Fiers, W.; Saelens, X. Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J. Immunol. 2011, 186, 1022–1031. [Google Scholar] [CrossRef]
- Wu, F.; Huang, J.H.; Yuan, X.Y.; Huang, W.S.; Chen, Y.H. Characterization of immunity induced by M2e of influenza virus. Vaccine 2007, 25, 8868–8873. [Google Scholar] [CrossRef]
- Heinen, P.P.; Rijsewijk, F.A.; de Boer-Luijtze, E.A.; Bianchi, A.T. Vaccination of pigs with a DNA construct expressing an influenza virus M2-nucleoprotein fusion protein exacerbates disease after challenge with influenza A virus. J. Gen. Virol. 2002, 83, 1851–1859. [Google Scholar]
- Kim, M.C.; Lee, J.S.; Kwon, Y.M.; O, E.; Lee, Y.J.; Choi, J.G.; Wang, B.Z.; Compans, R.W.; Kang, S.M. Multiple heterologous M2 extracellular domains presented on virus-like particles confer broader and stronger M2 immunity than live influenza A virus infection. Antivir. Res. 2013, 99, 328–335. [Google Scholar] [CrossRef]
- Tompkins, S.M.; Zhao, Z.S.; Lo, C.Y.; Misplon, J.A.; Liu, T.; Ye, Z.; Hogan, R.J.; Wu, Z.; Benton, K.A.; Tumpey, T.M.; Epstein, S.L. Matrix protein 2 vaccination and protection against influenza viruses, including subtype H5N1. Emerg. Infect. Dis. 2007, 13, 426–435. [Google Scholar] [CrossRef]
- Neirynck, S.; Deroo, T.; Saelens, X.; Vanlandschoot, P.; Jou, W.M.; Fiers, W. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat. Med. 1999, 5, 1157–1163. [Google Scholar] [CrossRef]
- Turley, C.B.; Rupp, R.E.; Johnson, C.; Taylor, D.N.; Wolfson, J.; Tussey, L.; Kavita, U.; Stanberry, L.; Shaw, A. Safety and immunogenicity of a recombinant M2e-flagellin influenza vaccine (STF2.4xM2e) in healthy adults. Vaccine 2011, 29, 5145–5152. [Google Scholar] [CrossRef]
- Schotsaert, M.; de Filette, M.; Fiers, W.; Saelens, X. Universal M2 ectodomain-based influenza A vaccines: Preclinical and clinical developments. Expert. Rev. Vaccines 2009, 8, 499–508. [Google Scholar] [CrossRef]
- Hillaire, M.L.; Rimmelzwaan, G.F.; Kreijtz, J.H. Clearance of influenza virus infections by T cells: Risk of collateral damage? Curr. Opin. Virol. 2013, 3, 430–437. [Google Scholar] [CrossRef]
- McMichael, A.J.; Gotch, F.M.; Noble, G.R.; Beare, P.A. Cytotoxic T-cell immunity to influenza. N. Engl. J. Med. 1983, 309, 13–17. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Bennink, J.R.; Smith, G.L.; Moss, B. Influenza A virus nucleoprotein is a major target antigen for cross-reactive anti-influenza A virus cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 1985, 82, 1785–1789. [Google Scholar] [CrossRef]
- McMichael, A.J.; Michie, C.A.; Gotch, F.M.; Smith, G.L.; Moss, B. Recognition of influenza A virus nucleoprotein by human cytotoxic T lymphocytes. J. Gen. Virol. 1986, 67, 719–726. [Google Scholar] [CrossRef]
- Gotch, F.; McMichael, A.; Smith, G.; Moss, B. Identification of viral molecules recognized by influenza-specific human cytotoxic T lymphocytes. J. Exp. Med. 1987, 165, 408–416. [Google Scholar] [CrossRef]
- Jameson, J.; Cruz, J.; Terajima, M.; Ennis, F.A. Human CD8+ and CD4+ T lymphocyte memory to influenza A viruses of swine and avian species. J. Immunol. 1999, 162, 7578–7583. [Google Scholar]
- Kreijtz, J.H.; Bodewes, R.; van Amerongen, G.; Kuiken, T.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Primary influenza A virus infection induces cross-protective immunity against a lethal infection with a heterosubtypic virus strain in mice. Vaccine 2007, 25, 612–620. [Google Scholar] [CrossRef]
- Kreijtz, J.H.; de Mutsert, G.; van Baalen, C.A.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Cross-recognition of avian H5N1 influenza virus by human cytotoxic T-lymphocyte populations directed to human influenza A virus. J. Virol. 2008, 82, 5161–5166. [Google Scholar] [CrossRef]
- Hillaire, M.L.; Vogelzang-van Trierum, S.E.; Kreijtz, J.H.; de Mutsert, G.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Human T-cells directed to seasonal influenza A virus cross-react with 2009 pandemic influenza A (H1N1) and swine-origin triple-reassortant H3N2 influenza viruses. J. Gen. Virol. 2013, 94, 583–592. [Google Scholar] [CrossRef]
- van de Sandt, C.E.; Kreijtz, J.H.; de Mutsert, G.; Geelhoed-Mieras, M.M.; Hillaire, M.L.; Vogelzang-van Trierum, S.E.; Osterhaus, A.D.; Fouchier, R.A.; Rimmelzwaan, G.F. Human cytotoxic T lymphocytes directed to seasonal influenza A viruses cross-react with the newly emerging H7N9 virus. J. Virol. 2014, 88, 1684–1693. [Google Scholar] [CrossRef]
- Hillaire, M.L.; Osterhaus, A.D.; Rimmelzwaan, G.F. Induction of virus-specific cytotoxic T lymphocytes as a basis for the development of broadly protective influenza vaccines. J. Biomed. Biotechnol. 2011, 2011, 939860. [Google Scholar]
- Brown, D.M.; Dilzer, A.M.; Meents, D.L.; Swain, S.L. CD4 T cell-mediated protection from lethal influenza: Perforin and antibody-mediated mechanisms give a one-two punch. J. Immunol. 2006, 177, 2888–2898. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Verhoeven, D.; Page, C.A.; Turner, D.; Farber, D.L. Memory CD4 T cells direct protective responses to influenza virus in the lungs through helper-independent mechanisms. J. Virol. 2010, 84, 9217–9226. [Google Scholar] [CrossRef]
- Alam, S.; Sant, A.J. Infection with seasonal influenza virus elicits CD4 T cells specific for genetically conserved epitopes that can be rapidly mobilized for protective immunity to pandemic H1N1 influenza virus. J. Virol. 2011, 85, 13310–13321. [Google Scholar] [CrossRef]
- McKinstry, K.K.; Strutt, T.M.; Kuang, Y.; Brown, D.M.; Sell, S.; Dutton, R.W.; Swain, S.L. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J. Clin. Invest. 2012, 122, 2847–2856. [Google Scholar] [CrossRef]
- Wilkinson, T.M.; Li, C.K.; Chui, C.S.; Huang, A.K.; Perkins, M.; Liebner, J.C.; Lambkin-Williams, R.; Gilbert, A.; Oxford, J.; Nicholas, B.; et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. 2012, 18, 274–280. [Google Scholar] [CrossRef]
- Hillaire, M.L.; van Trierum, S.E.; Bodewes, R.; van Baalen, C.A.; van Binnendijk, R.S.; Koopmans, M.P.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Characterization of the human CD8(+) T cell response following infection with 2009 pandemic influenza H1N1 virus. J. Virol. 2011, 85, 12057–12061. [Google Scholar] [CrossRef]
- Sridhar, S.; Begom, S.; Bermingham, A.; Hoschler, K.; Adamson, W.; Carman, W.; Bean, T.; Barclay, W.; Deeks, J.J.; Lalvani, A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 2013, 19, 1305–1312. [Google Scholar] [CrossRef]
- Zheng, M.; Luo, J.; Chen, Z. Development of universal influenza vaccines based on influenza virus M and NP genes. Infection 2014, 42, 251–262. [Google Scholar] [CrossRef]
- Mayr, A.; Munz, E. Veränderung von Vaccinevirus durch Dauerpassagen in Hühnerembryofibroblastenkulturen. (in German). Zentralbl. Bakteriol. B. 1964, I, 24–35. [Google Scholar]
- Mayr, A.; Stickl, H.; Müller, H. Der Pockenimpfstamm MVA: Marker, genetische Struktur, Erfahrung mit der parenteralen Schutzimpfung und Verhalten im abwehrgeschwächten Organismus. (in German). Zentralbl. Bakteriol. B. 1978, 167, 375–390. [Google Scholar]
- Stickl, H.; Hochstein-Mintzel, V.; Mayr, A. MVA-Stufenimpfung gegen Pocken. Dtsch. Med. Wochenschr. 1974, 99, 2386–2392. [Google Scholar] [CrossRef]
- Antoine, G.; Scheiflinger, F.; Dorner, F.; Falkner, F.G. The Complete Genomic Sequence of the Modified Vaccinia Ankara Strain: Comparison with Other Orthopoxviruses. Virology 1998, 244, 365–396. [Google Scholar] [CrossRef]
- Meisinger-Henschel, C.; Späth, M.; Lukassen, S.; Wolferstätter, M.; Kachelriess, H.; Baur, K.; Dirmeier, U.; Wagner, M.; Chaplin, P.; Suter, M.; et al. Introduction of the Six Major Genomic Deletions of Modified Vaccinia Virus Ankara (MVA) into the Parental Vaccinia Virus Is Not Sufficient To Reproduce an MVA-Like Phenotype in Cell Culture and in Mice. J. Virol. 2010, 84, 9907–9919. [Google Scholar] [CrossRef]
- Meyer, H.; Sutter, G.; Mayr, A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J. Gen. Virol. 1991, 72, 1031–1038. [Google Scholar] [CrossRef]
- Carroll, M.W.; Moss, B. Host Range and Cytopathogenicity of the Highly Attenuated MVA Strain of Vaccinia Virus: Propagation and Generation of Recombinant Viruses in a Nonhuman Mammalian Cell Line. Virology 1997, 238, 198–211. [Google Scholar] [CrossRef]
- Drexler, I.; Heller, K.; Wahren, B.; Erfle, V.; Sutter, G. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J. Gen. Virol. 1998, 79, 347–352. [Google Scholar]
- Sutter, G.; Moss, B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. USA 1992, 89, 10847–10851. [Google Scholar] [CrossRef]
- Kibler, K.V.; Shors, T.; Perkins, K.B.; Zeman, C.C.; Banaszak, M.P.; Biesterfeldt, J.; Langland, J.O.; Jacobs, B.L. Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J. Virol. 1997, 71, 1992–2003. [Google Scholar]
- Somogyi, P.; Frazier, J.; Skinner, M.A. Fowlpox Virus Host Range Restriction: Gene Expression, DNA Replication, and Morphogenesis in Nonpermissive Mammalian Cells. Virology 1993, 197, 439–444. [Google Scholar] [CrossRef]
- Tartaglia, J. Highly Attenuated Poxvirus Vectors. AIDS Res. Hum. Retroviruses 1992, 8, 16. [Google Scholar]
- Ramírez, J.C.; Gherardi, M.M.; Esteban, M. Biology of Attenuated Modified Vaccinia Virus Ankara Recombinant Vector in Mice: Virus Fate and Activation of B- and T-Cell Immune Responses in Comparison with the Western Reserve Strain and Advantages as a Vaccine. J. Virol. 2000, 74, 923–933. [Google Scholar] [CrossRef]
- Stittelaar, K.J.; Wyatt, L.S.; de Swart, R.L.; Vos, H.W.; Groen, J.; van Amerongen, G.; van Binnendijk, R.S.; Rozenblatt, S.; Moss, B.; Osterhaus, A.D.M.E. Protective Immunity in Macaques Vaccinated with a Modified Vaccinia Virus Ankara-Based Measles Virus Vaccine in the Presence of Passively Acquired Antibodies. J. Virol. 2000, 74, 4236–4243. [Google Scholar] [CrossRef]
- Veits, J.; Romer-Oberdorfer, A.; Helferich, D.; Durban, M.; Suezer, Y.; Sutter, G.; Mettenleiter, T.C. Protective efficacy of several vaccines against highly pathogenic H5N1 avian influenza virus under experimental conditions. Vaccine 2008, 26, 1688–1696. [Google Scholar] [CrossRef]
- Werner, G.T.; Jentzsch, U.; Metzger, E.; Simon, J. Studies on poxvirus infections in irradiated animals. Arch. Virol. 1980, 64, 247–256. [Google Scholar] [CrossRef]
- Sutter, G.; Wyatt, L.S.; Foley, P.L.; Bennink, J.R.; Moss, B. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine 1994, 12, 1032–1040. [Google Scholar] [CrossRef]
- Drexler, I.; Staib, C.; Sutter, G. Modified vaccinia virus Ankara as antigen delivery system: How can we best use its potential? Curr. Opin. Biotechnol. 2004, 15, 506–512. [Google Scholar] [CrossRef]
- Gasteiger, G.; Kastenmuller, W.; Ljapoci, R.; Sutter, G.; Drexler, I. Cross-Priming of Cytotoxic T Cells Dictates Antigen Requisites for Modified Vaccinia Virus Ankara Vector Vaccines. J. Virol. 2007, 81, 11925–11936. [Google Scholar] [CrossRef]
- Gorse, G.J.; Newman, M.J.; deCamp, A.; Hay, C.M.; de Rosa, S.C.; Noonan, E.; Livingston, B.D.; Fuchs, J.D.; Kalams, S.A.; Cassis-Ghavami, F.L.; et al. DNA and Modified Vaccinia Virus Ankara Vaccines Encoding Multiple Cytotoxic and Helper T-Lymphocyte Epitopes of Human Immunodeficiency Virus Type 1 (HIV-1) Are Safe but Weakly Immunogenic in HIV-1-Uninfected, Vaccinia Virus-Naive Adults. Clin. Vaccine Immunol. 2012, 19, 649–658. [Google Scholar] [CrossRef]
- Schliehe, C.; Bitzer, A.; van den Broek, M.; Groettrup, M. Stable Antigen Is Most Effective for Eliciting CD8+ T-Cell Responses after DNA Vaccination and Infection with Recombinant Vaccinia Virus in Vivo. J. Virol. 2012, 86, 9782–9793. [Google Scholar] [CrossRef]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of Antigen Processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef]
- Carroll, M.W.; Overwijk, W.W.; Chamberlain, R.S.; Rosenberg, S.A.; Moss, B.; Restifo, N.P. Highly attenuated modified vaccinia virus Ankara (MVA) as an effective recombinant vector: A Murine tumor model. Vaccine 1997, 15, 387–394. [Google Scholar] [CrossRef]
- Hirsch, V.M.; Fuerst, T.R.; Sutter, G.; Carroll, M.W.; Yang, L.C.; Goldstein, S.; Piatak, M.; Elkins, W.R.; Alvord, W.G.; Montefiori, D.C.; et al. Patterns of viral replication correlate with outcome in simian immunodeficiency virus (SIV)-infected macaques: Effect of prior immunization with a trivalent SIV vaccine in modified vaccinia virus Ankara. J. Virol. 1996, 70, 3741–3752. [Google Scholar]
- Blanchard, T.J.; Alcami, A.; Andrea, P.; Smith, G.L. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: Implications for use as a human vaccine. J. Gen. Virol. 1998, 79, 1159–1167. [Google Scholar]
- Büttner, M.; Czerny, C.; Lehner, K.H.; Wertz, K. Interferon induction in peripheral blood mononuclear leukocytes of man and farm animals by poxvirus vector candidates and some poxvirus constructs. Vet. Immunol. Immunopathol. 1995, 46, 237–250. [Google Scholar] [CrossRef]
- Delaloye, J.; Roger, T.; Steiner-Tardivel, Q.-G.; Le Roy, D.; Knaup Reymond, M.; Akira, S.; Petrilli, V.; Gomez, C.E.; Perdiguero, B.; Tschopp, J.; et al. Innate Immune Sensing of Modified Vaccinia Virus Ankara (MVA) Is Mediated by TLR2-TLR6, MDA-5 and the NALP3 Inflammasome. PLoS Pathog. 2009, 5, e1000480. [Google Scholar] [CrossRef]
- Fleige, H.; Ravens, S.; Moschovakis, G.L.; Bölter, J.; Willenzon, S.; Sutter, G.; Häussler, S.; Kalinke, U.; Prinz, I.; Förster, R. IL-17–induced CXCL12 recruits B cells and induces follicle formation in BALT in the absence of differentiated FDCs. J. Exp. Med. 2014, 211, 643–651. [Google Scholar] [CrossRef]
- Förster, R.; Wolf, G.; Mayr, A. Highly attenuated poxviruses induce functional priming of neutrophils in vitro. Arch. Virol. 1994, 136, 219–226. [Google Scholar] [CrossRef]
- Halle, S.; Dujardin, H.C.; Bakocevic, N.; Fleige, H.; Danzer, H.; Willenzon, S.; Suezer, Y.; Hämmerling, G.; Garbi, N.; Sutter, G.; et al. Induced bronchus-associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J. Exp. Med. 2009, 206, 2593–2601. [Google Scholar] [CrossRef]
- Lehmann, M.H.; Kastenmuller, W.; Kandemir, J.D.; Brandt, F.; Suezer, Y.; Sutter, G. Modified Vaccinia Virus Ankara Triggers Chemotaxis of Monocytes and Early Respiratory Immigration of Leukocytes by Induction of CCL2 Expression. J. Virol. 2009, 83, 2540–2552. [Google Scholar] [CrossRef]
- Waibler, Z.; Anzaghe, M.; Ludwig, H.; Akira, S.; Weiss, S.; Sutter, G.; Kalinke, U. Modified Vaccinia Virus Ankara Induces Toll-Like Receptor-Independent Type I Interferon Responses. J. Virol. 2007, 81, 12102–12110. [Google Scholar] [CrossRef]
- Gilbert, S.C. Clinical development of Modified Vaccinia virus Ankara vaccines. Vaccine 2013, 31, 4241–4246. [Google Scholar] [CrossRef]
- Gómez, C.E.; Perdiguero, B.; García-Arriaza, J.; Esteban, M. Clinical applications of attenuated MVA poxvirus strain. Expert Rev. Vaccine. 2013, 12, 1395–1416. [Google Scholar] [CrossRef]
- Kreijtz, J.H.C.M.; Gilbert, S.C.; Sutter, G. Poxvirus vectors. Vaccine 2013, 31, 4217–4219. [Google Scholar] [CrossRef]
- European Medicines Agency Authorization Procedures. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/q_and_a/q_and_a_detail_000080.jsp (accessed on 17 March 2014).
- Saxena, M.; Van, T.T.; Baird, F.J.; Coloe, P.J.; Smooker, P.M. Pre-existing immunity against vaccine vectors--friend or foe? Microbiology 2013, 159, 1–11. [Google Scholar] [CrossRef]
- Brewoo, J.N.; Powell, T.D.; Jones, J.C.; Gundlach, N.A.; Young, G.R.; Chu, H.; Das, S.C.; Partidos, C.D.; Stinchcomb, D.T.; Osorio, J.E. Cross-protective immunity against multiple influenza virus subtypes by a novel modified vaccinia Ankara (MVA) vectored vaccine in mice. Vaccine 2013, 31, 1848–1855. [Google Scholar]
- World Health Organization. Cumulative number of confirmed human cases for avian influenza A(H5N1) reported to WHO, 2003–2014; Geneva, Switzerland, 2014. [Google Scholar]
- Imai, M.; Herfst, S.; Sorrell, E.M.; Schrauwen, E.J.; Linster, M.; de Graaf, M.; Fouchier, R.A.; Kawaoka, Y. Transmission of influenza A/H5N1 viruses in mammals. Virus Res. 2013, 178, 15–20. [Google Scholar] [CrossRef]
- Rimmelzwaan, G.F.; Kreijtz, J.H.C.M.; Suezer, Y.; Schwantes, A.; Osterhaus, A.D.; Sutter, G. Preclinical evaluation of influenza vaccines based on replication-deficient poxvirus vector MVA. Procedia Vaccinol. 2011, 4, 5. [Google Scholar]
- Kreijtz, J.H.; Suezer, Y.; van Amerongen, G.; de Mutsert, G.; Schnierle, B.S.; Wood, J.M.; Kuiken, T.; Fouchier, R.A.; Lower, J.; Osterhaus, A.D.; et al. Recombinant modified vaccinia virus Ankara-based vaccine induces protective immunity in mice against infection with influenza virus H5N1. J. Infect. Dis. 2007, 195, 1598–1606. [Google Scholar] [CrossRef]
- Hessel, A.; Schwendinger, M.; Holzer, G.W.; Orlinger, K.K.; Coulibaly, S.; Savidis-Dacho, H.; Zips, M.L.; Crowe, B.A.; Kreil, T.R.; Ehrlich, H.J.; et al. Vectors based on modified vaccinia Ankara expressing influenza H5N1 hemagglutinin induce substantial cross-clade protective immunity. PLoS One 2011, 6, e16247. [Google Scholar]
- Kreijtz, J.H.; Suezer, Y.; de Mutsert, G.; van den Brand, J.M.; van Amerongen, G.; Schnierle, B.S.; Kuiken, T.; Fouchier, R.A.; Lower, J.; Osterhaus, A.D.; et al. Preclinical evaluation of a modified vaccinia virus Ankara (MVA)-based vaccine against influenza A/H5N1 viruses. Vaccine 2009, 27, 6296–6299. [Google Scholar] [CrossRef]
- Kreijtz, J.H.; Suezer, Y.; de Mutsert, G.; van den Brand, J.M.; van Amerongen, G.; Schnierle, B.S.; Kuiken, T.; Fouchier, R.A.; Lower, J.; Osterhaus, A.D.; et al. Recombinant modified vaccinia virus Ankara expressing the hemagglutinin gene confers protection against homologous and heterologous H5N1 influenza virus infections in macaques. J. Infect. Dis. 2009, 199, 405–413. [Google Scholar] [CrossRef]
- Kreijtz, J.H.; Suezer, Y.; de Mutsert, G.; van Amerongen, G.; Schwantes, A.; van den Brand, J.M.; Fouchier, R.A.; Lower, J.; Osterhaus, A.D.; Sutter, G.; et al. MVA-based H5N1 vaccine affords cross-clade protection in mice against influenza A/H5N1 viruses at low doses and after single immunization. PLoS One 2009, 4, e7790. [Google Scholar]
- Hessel, A.; Schwendinger, M.; Fritz, D.; Coulibaly, S.; Holzer, G.W.; Sabarth, N.; Kistner, O.; Wodal, W.; Kerschbaum, A.; Savidis-Dacho, H.; et al. A pandemic influenza H1N1 live vaccine based on modified vaccinia Ankara is highly immunogenic and protects mice in active and passive immunizations. PLoS One 2010, 5, e12217. [Google Scholar]
- Castrucci, M.R.; Facchini, M.; di Mario, G.; Garulli, B.; Sciaraffia, E.; Meola, M.; Fabiani, C.; de Marco, M.A.; Cordioli, P.; Siccardi, A.; et al. Modified vaccinia virus Ankara expressing the hemagglutinin of pandemic (H1N1) 2009 virus induces cross-protective immunity against Eurasian 'avian-like' H1N1 swine viruses in mice. Influenza Other Respir. Viruses 2014, 8, 367–375. [Google Scholar] [CrossRef]
- Kreijtz, J.H.; Suzer, Y.; Bodewes, R.; Schwantes, A.; van Amerongen, G.; Verburgh, R.J.; de Mutsert, G.; van den Brand, J.; van Trierum, S.E.; Kuiken, T.; et al. Evaluation of a modified vaccinia virus Ankara (MVA)-based candidate pandemic influenza A/H1N1 vaccine in the ferret model. J. Gen. Virol. 2010, 91, 2745–2752. [Google Scholar] [CrossRef]
- Bender, B.S.; Rowe, C.A.; Taylor, S.F.; Wyatt, L.S.; Moss, B.; Small, P.A., Jr. Oral immunization with a replication-deficient recombinant vaccinia virus protects mice against influenza. J. Virol. 1996, 70, 6418–6424. [Google Scholar]
- Berthoud, T.K.; Hamill, M.; Lillie, P.J.; Hwenda, L.; Collins, K.A.; Ewer, K.J.; Milicic, A.; Poyntz, H.C.; Lambe, T.; Fletcher, H.A.; et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP+M1. Clin. Infect. Dis. 2011, 52, 1–7. [Google Scholar] [CrossRef]
- Powell, T.J.; Peng, Y.; Berthoud, T.K.; Blais, M.E.; Lillie, P.J.; Hill, A.V.; Rowland-Jones, S.L.; McMichael, A.J.; Gilbert, S.C.; Dong, T. Examination of influenza specific T cell responses after influenza virus challenge in individuals vaccinated with MVA-NP+M1 vaccine. PLoS One 2013, 8, e62778. [Google Scholar]
- Antrobus, R.D.; Lillie, P.J.; Berthoud, T.K.; Spencer, A.J.; McLaren, J.E.; Ladell, K.; Lambe, T.; Milicic, A.; Price, D.A.; Hill, A.V.; et al. A T cell-inducing influenza vaccine for the elderly: Safety and immunogenicity of MVA-NP+M1 in adults aged over 50 years. PLoS One 2012, 7, e48322. [Google Scholar]
- Lillie, P.J.; Berthoud, T.K.; Powell, T.J.; Lambe, T.; Mullarkey, C.; Spencer, A.J.; Hamill, M.; Peng, Y.; Blais, M.E.; Duncan, C.J.; et al. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin. Infect. Dis. 2012, 55, 19–25. [Google Scholar] [CrossRef]
- Lambe, T.; Carey, J.B.; Li, Y.; Spencer, A.J.; van Laarhoven, A.; Mullarkey, C.E.; Vrdoljak, A.; Moore, A.C.; Gilbert, S.C. Immunity against heterosubtypic influenza virus induced by adenovirus and MVA expressing nucleoprotein and matrix protein-1. Sci. Rep. 2013, 3, 1443. [Google Scholar]
- Boyd, A.C.; Ruiz-Hernandez, R.; Peroval, M.Y.; Carson, C.; Balkissoon, D.; Staines, K.; Turner, A.V.; Hill, A.V.; Gilbert, S.C.; Butter, C. Towards a universal vaccine for avian influenza: Protective efficacy of modified Vaccinia virus Ankara and Adenovirus vaccines expressing conserved influenza antigens in chickens challenged with low pathogenic avian influenza virus. Vaccine 2013, 31, 670–675. [Google Scholar] [CrossRef]
- Mullarkey, C.E.; Boyd, A.; van Laarhoven, A.; Lefevre, E.A.; Veronica Carr, B.; Baratelli, M.; Molesti, E.; Temperton, N.J.; Butter, C.; Charleston, B.; et al. Improved adjuvanting of seasonal influenza vaccines: Preclinical studies of MVA-NP+M1 coadministration with inactivated influenza vaccine. Eur. J. Immunol. 2013, 43, 1940–1952. [Google Scholar] [Green Version]
- Antrobus, R.D.; Berthoud, T.K.; Mullarkey, C.E.; Hoschler, K.; Coughlan, L.; Zambon, M.; Hill, A.V.; Gilbert, S.C. Coadministration of seasonal influenza vaccine and MVA-NP+M1 simultaneously achieves potent humoral and cell-mediated responses. Mol. Ther. 2014, 22, 233–238. [Google Scholar] [CrossRef]
- Hessel, A.; Savidis-Dacho, H.; Coulibaly, S.; Portsmouth, D.; Kreil, T.R.; Crowe, B.A.; Schwendinger, M.G.; Pilz, A.; Barrett, P.N.; Falkner, F.G.; et al. MVA vectors expressing conserved influenza proteins protect mice against lethal challenge with H5N1, H9N2 and H7N1 viruses. PLoS One 2014, 9, e88340. [Google Scholar]
- Breathnach, C.C.; Clark, H.J.; Clark, R.C.; Olsen, C.W.; Townsend, H.G.G.; Lunn, D.P. Immunization with recombinant modified vaccinia Ankara (rMVA) constructs encoding the HA or NP gene protects ponies from equine influenza virus challenge. Vaccine 2006, 24, 1180–1190. [Google Scholar] [CrossRef]
- Collins, P.L.; Melero, J.A. Progress in understanding and controlling respiratory syncytial virus: Still crazy after all these years. Virus Res. 2011, 162, 80–99. [Google Scholar] [CrossRef]
- Schomacker, H.; Schaap-Nutt, A.; Collins, P.L.; Schmidt, A.C. Pathogenesis of acute respiratory illness caused by human parainfluenza viruses. Curr. Opin. Virol. 2012, 2, 294–299. [Google Scholar] [CrossRef]
- van Bleek, G.M.; Osterhaus, A.D.M.E.; de Swart, R.L. RSV 2010: Recent advances in research on respiratory syncytial virus and other pneumoviruses. Vaccine 2011, 29, 7285–7291. [Google Scholar] [CrossRef]
- Wyatt, L.S.; Shors, S.T.; Murphy, B.R.; Moss, B. Development of a replication-deficient recombinant vaccinia virus vaccine effective against parainfluenza virus 3 infection in an animal model. Vaccine 1996, 14, 1451–1458. [Google Scholar] [CrossRef]
- Durbin, A.P.; Cho, C.J.; Elkins, W.R.; Wyatt, L.S.; Moss, B.; Murphy, B.R. Comparison of the Immunogenicity and Efficacy of a Replication-Defective Vaccinia Virus Expressing Antigens of Human Parainfluenza Virus Type 3 (HPIV3) with Those of a Live Attenuated HPIV3 Vaccine Candidate in Rhesus Monkeys Passively Immunized with PIV3 Antibodies. J. Infect. Dis. 1999, 179, 1345–1351. [Google Scholar] [CrossRef]
- Durbin, A.P.; Wyatt, L.S.; Siew, J.; Moss, B.; Murphy, B.R. The immunogenicity and efficacy of intranasally or parenterally administered replication-deficient vaccinia-parainfluenza virus type 3 recombinants in rhesus monkeys. Vaccine 1998, 16, 1324–1330. [Google Scholar] [CrossRef]
- Wyatt, L.S.; Whitehead, S.S.; Venanzi, K.A.; Murphy, B.R.; Moss, B. Priming and boosting immunity to respiratory syncytial virus by recombinant replication-defective vaccinia virus MVA. Vaccine 1999, 18, 392–397. [Google Scholar] [CrossRef]
- Olszewska, W.; Suezer, Y.; Sutter, G.; Openshaw, P.J.M. Protective and disease-enhancing immune responses induced by recombinant modified vaccinia Ankara (MVA) expressing respiratory syncytial virus proteins. Vaccine 2004, 23, 215–221. [Google Scholar] [CrossRef]
- De Waal, L.; Wyatt, L.S.; Yüksel, S.; van Amerongen, G.; Moss, B.; Niesters, H.G.M.; Osterhaus, A.D.M.E.; de Swart, R.L. Vaccination of infant macaques with a recombinant modified vaccinia virus Ankara expressing the respiratory syncytial virus F and G genes does not predispose for immunopathology. Vaccine 2004, 22, 923–926. [Google Scholar] [CrossRef]
- Taylor, G. Bovine Model of Respiratory Syncytial Virus Infection. In Challenges and Opportunities for Respiratory Syncytial Virus Vaccines; Anderson, L.J., Graham, B.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 372, pp. 327–345. [Google Scholar]
- Antonis, A.F.G.; van der Most, R.G.; Suezer, Y.; Stockhofe-Zurwieden, N.; Daus, F.; Sutter, G.; Schrijver, R.S. Vaccination with recombinant modified vaccinia virus Ankara expressing bovine respiratory syncytial virus (bRSV) proteins protects calves against RSV challenge. Vaccine 2007, 25, 4818–4827. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Micro. 2013, 11, 836–848. [Google Scholar] [CrossRef]
- Bisht, H.; Roberts, A.; Vogel, L.; Bukreyev, A.; Collins, P.L.; Murphy, B.R.; Subbarao, K.; Moss, B. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc. Natl. Acad. Sci. USA 2004, 101, 6641–6646. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, L.; Qin, C.; Ba, L.; Yi, C.E.; Zhang, F.; Wei, Q.; He, T.; Yu, W.; Yu, J.; et al. Recombinant Modified Vaccinia Virus Ankara Expressing the Spike Glycoprotein of Severe Acute Respiratory Syndrome Coronavirus Induces Protective Neutralizing Antibodies Primarily Targeting the Receptor Binding Region. J. Virol. 2005, 79, 2678–2688. [Google Scholar] [CrossRef]
- Song, F.; Fux, R.; Provacia, L.B.; Volz, A.; Eickmann, M.; Becker, S.; Osterhaus, A.D.M.E.; Haagmans, B.L.; Sutter, G. Middle East Respiratory Syndrome Coronavirus Spike Protein Delivered by Modified Vaccinia Virus Ankara Efficiently Induces Virus-Neutralizing Antibodies. J. Virol. 2013, 87, 11950–11954. [Google Scholar] [CrossRef]
- Kremer, M.; Volz, A.; Kreijtz, J.H.C.M.; Fux, R.; Lehmann, M.H.; Sutter, G. Easy and efficient protocols for working with recombinant vaccinia virus MVA. Methods Mol. Biol. 2012, 890, 59–93. [Google Scholar] [CrossRef]
- Lohr, V.; Rath, A.; Genzel, Y.; Jordan, I.; Sandig, V.; Reichl, U. New avian suspension cell lines provide production of influenza virus and MVA in serum-free media: Studies on growth, metabolism and virus propagation. Vaccine 2009, 27, 4975–4982. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on quality, non-clinical and clinical aspects of live recombinant viral vectored vaccines; London, UK, 2010. [Google Scholar]
- Harrop, R.; John, J.; Carroll, M.W. Recombinant viral vectors: Cancer vaccines. Adv. Drug Deliv. Rev. 2006, 58, 931–947. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Altenburg, A.F.; Kreijtz, J.H.C.M.; De Vries, R.D.; Song, F.; Fux, R.; Rimmelzwaan, G.F.; Sutter, G.; Volz, A. Modified Vaccinia Virus Ankara (MVA) as Production Platform for Vaccines against Influenza and Other Viral Respiratory Diseases. Viruses 2014, 6, 2735-2761. https://doi.org/10.3390/v6072735
Altenburg AF, Kreijtz JHCM, De Vries RD, Song F, Fux R, Rimmelzwaan GF, Sutter G, Volz A. Modified Vaccinia Virus Ankara (MVA) as Production Platform for Vaccines against Influenza and Other Viral Respiratory Diseases. Viruses. 2014; 6(7):2735-2761. https://doi.org/10.3390/v6072735
Chicago/Turabian StyleAltenburg, Arwen F., Joost H. C. M. Kreijtz, Rory D. De Vries, Fei Song, Robert Fux, Guus F. Rimmelzwaan, Gerd Sutter, and Asisa Volz. 2014. "Modified Vaccinia Virus Ankara (MVA) as Production Platform for Vaccines against Influenza and Other Viral Respiratory Diseases" Viruses 6, no. 7: 2735-2761. https://doi.org/10.3390/v6072735