
HostPhinder: A Phage Host Prediction Tool

Abstract
:1. Introduction
2. Materials and Methods
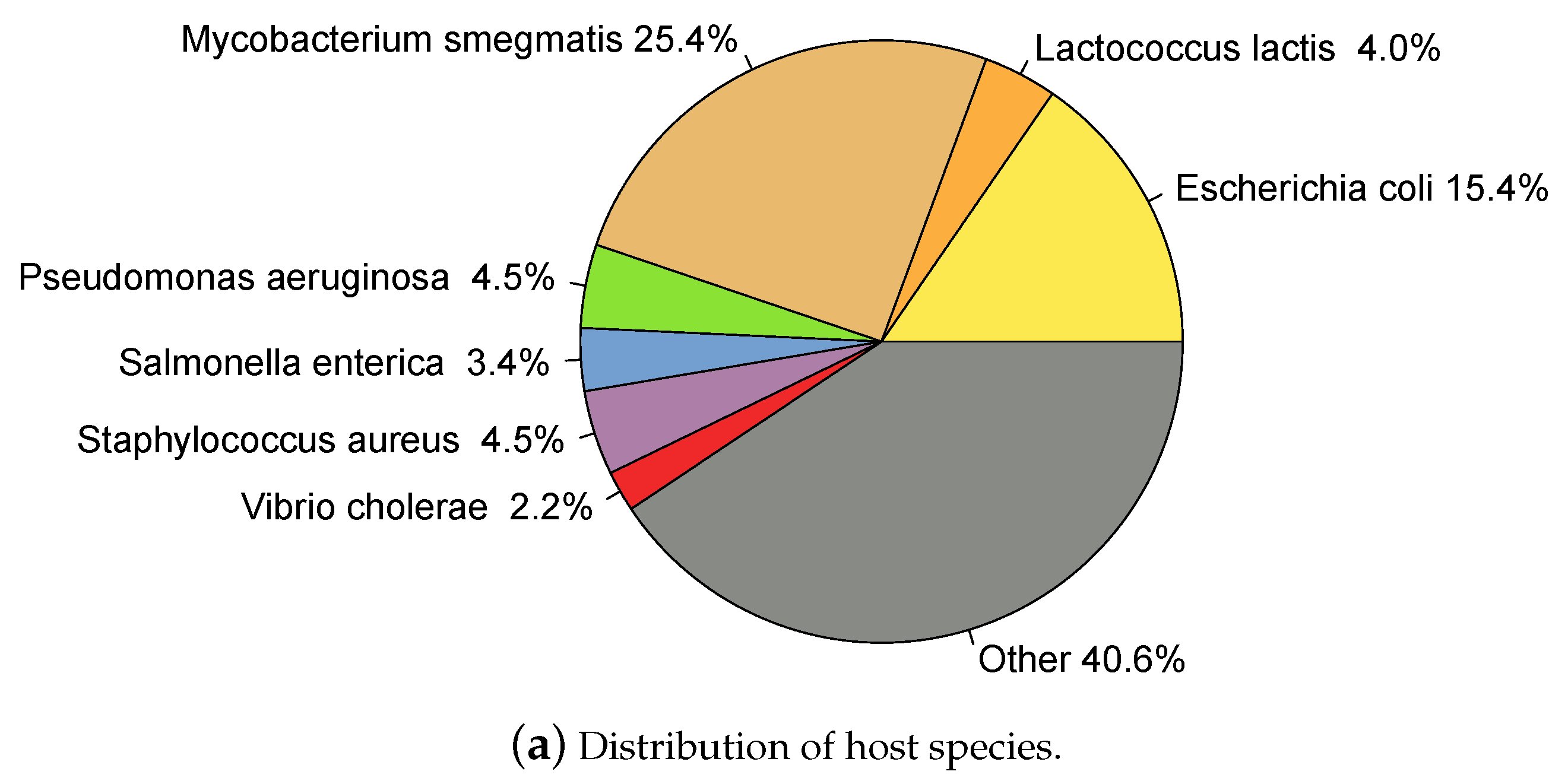
2.1. Whole Genome Phage Sequences from Public Databases
2.2. Data Partitioning and Clustering
2.3. K-mer-Based Resemblance Measures
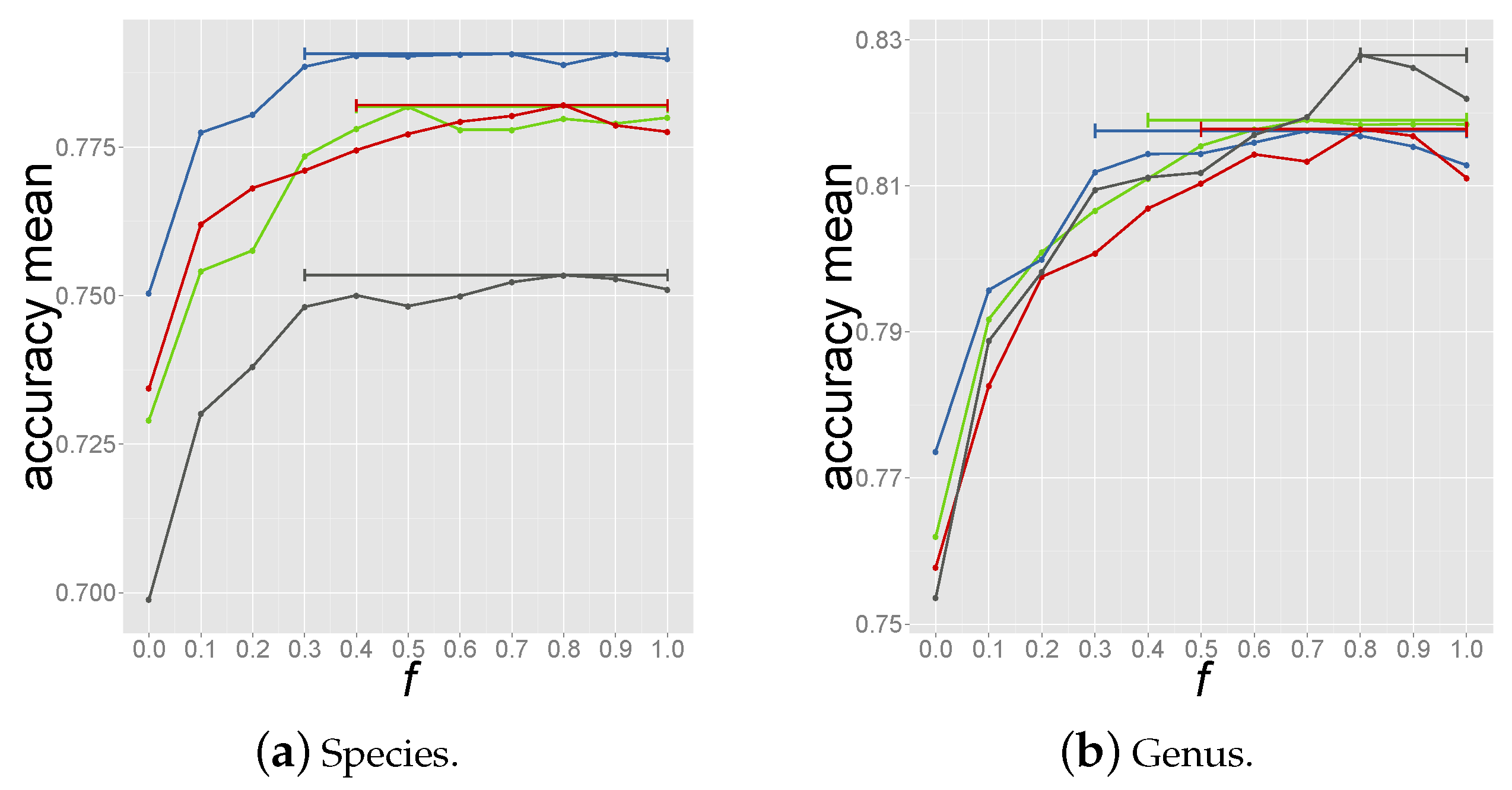
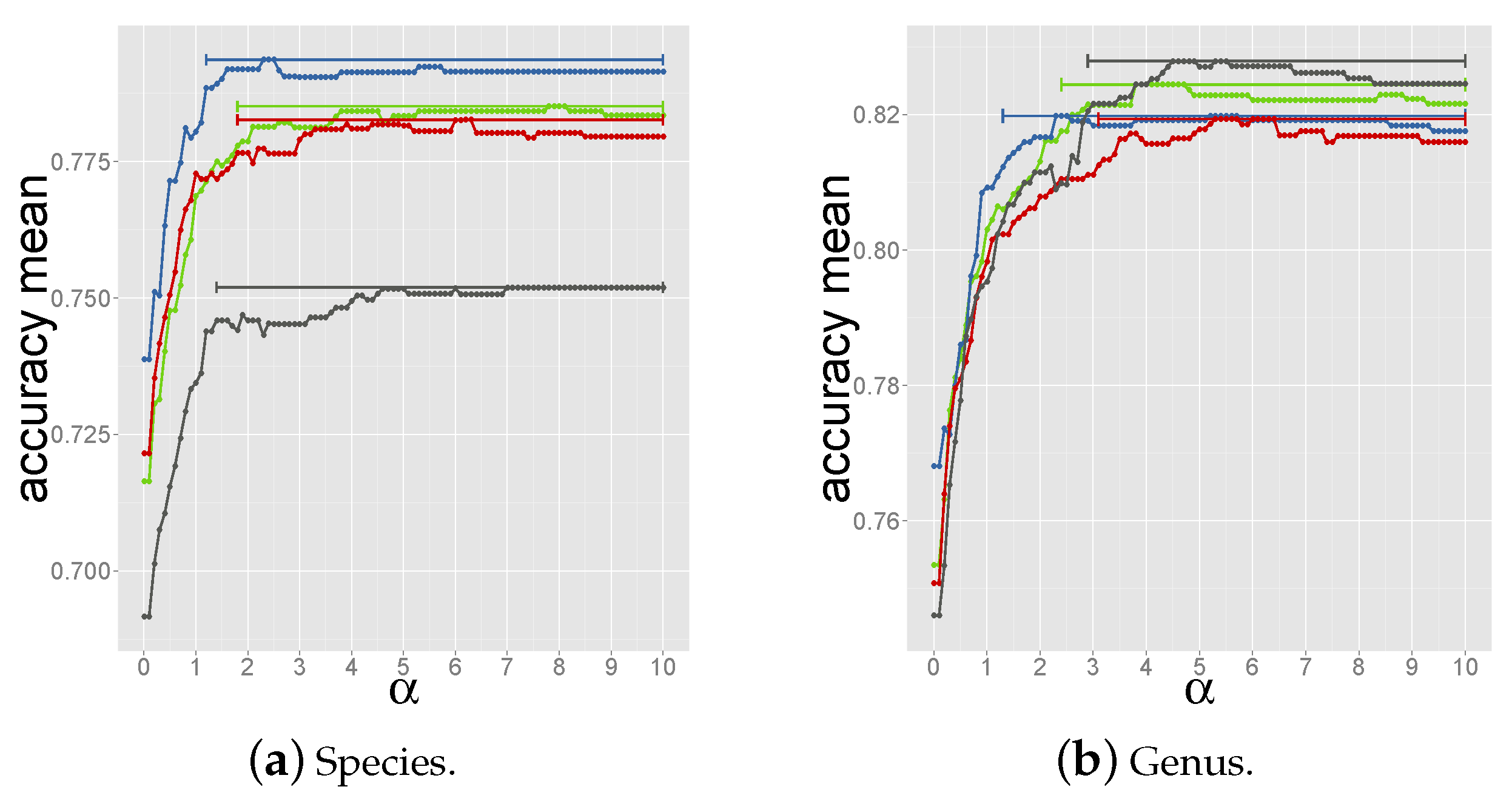
2.4. Determining the Measure and Selection Criteria for Final Prediction
2.4.1. Criterion 1: Host of Best-Matching Reference Phage
2.4.2. Criterion 2: Majority Host among Top-10 Reference Phages
2.4.3. Criterion 3: Majority Host among Reference Phages above Coverage Threshold
2.4.4. Criterion 4: Summing up Normalized Coverage Values of Phages with Same Host
2.5. Programming Language and Speed of Execution
2.6. BLAST Evaluation
2.7. Establishing an Evaluation Set of Predicted Prophages
2.8. Host Prediction of INTESTI Bacteriophage Cocktail
3. Results
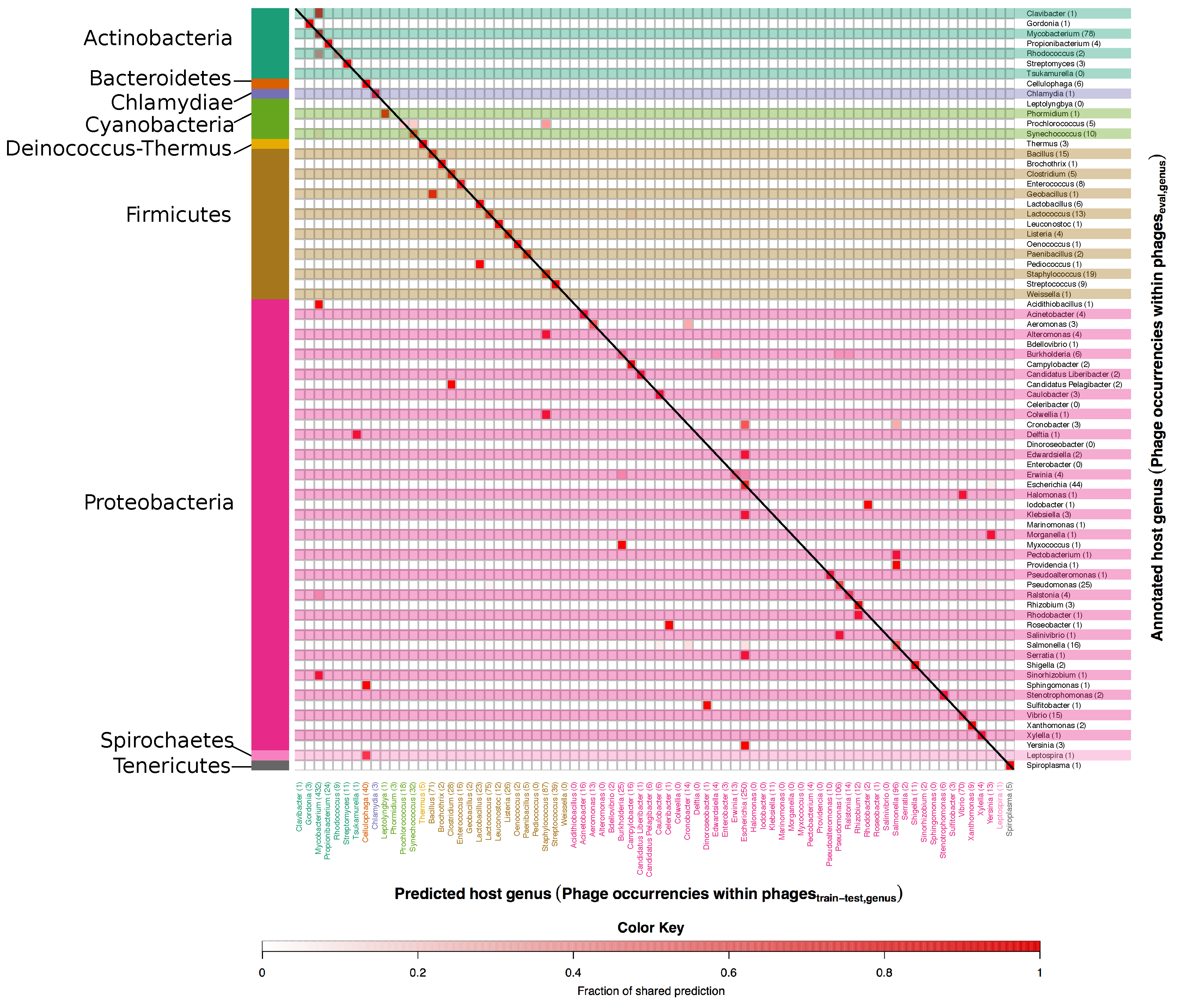
3.1. Developing and Benchmarking the HostPhinder Method
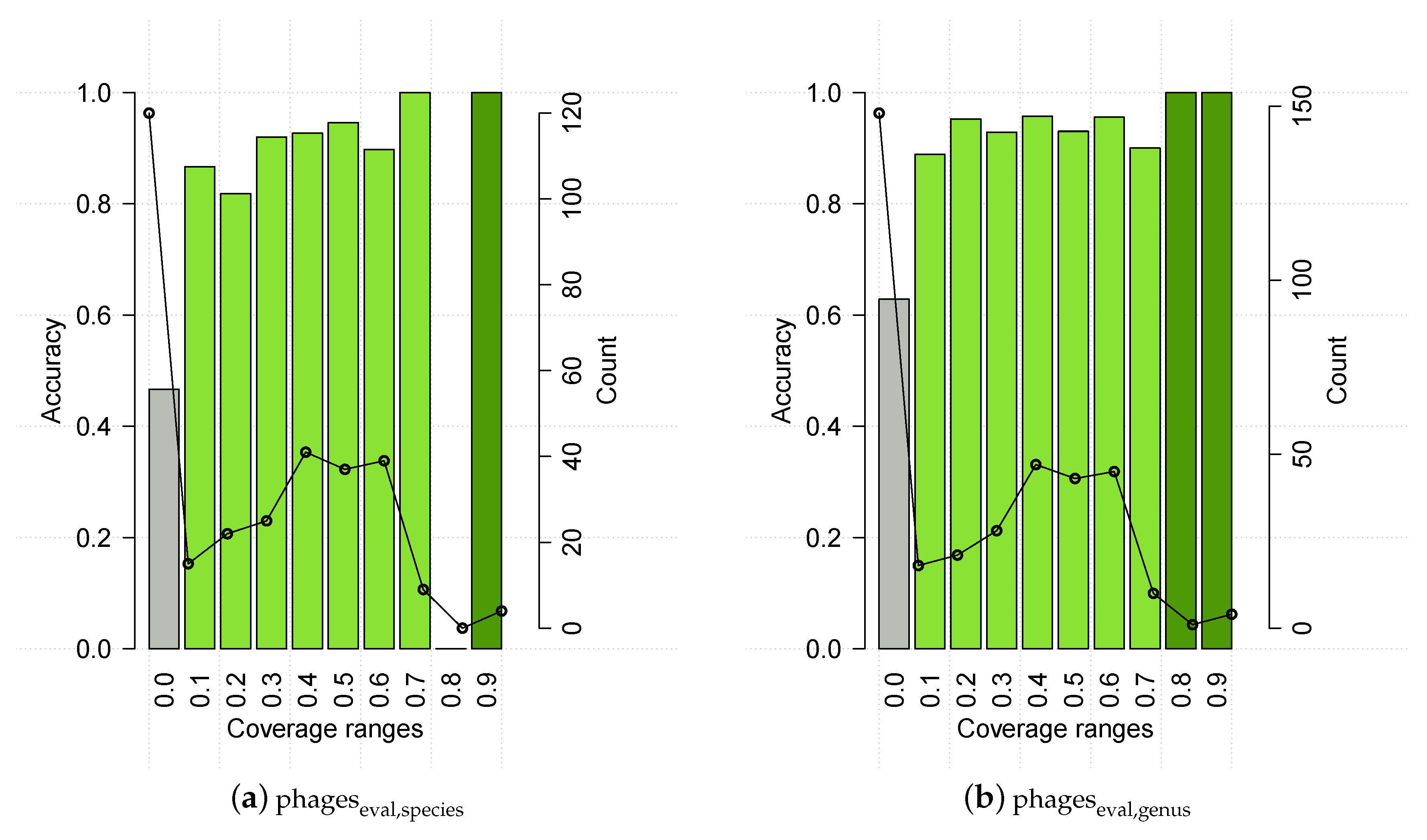
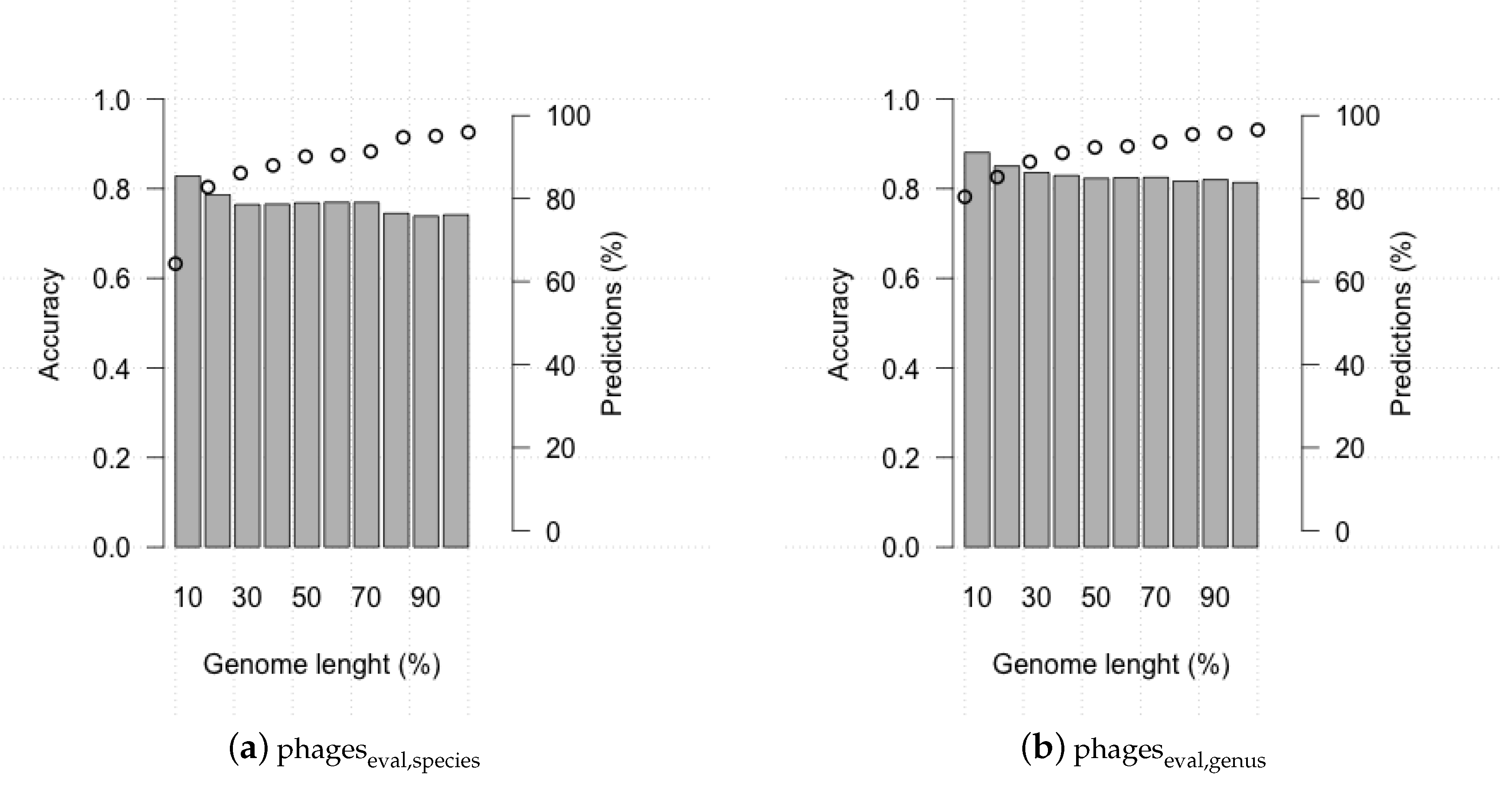
3.2. Evaluating HostPhinder’s Performance on Complete and Partial Genomes
3.3. Comparing HostPhinder to BLAST
3.4. HostPhinder’s Performance on Predicted Prophages and Establishment of Confidence Threshold
3.5. Host Analysis of Phages from Therapeutic Phage Cocktail from the Georgian George Eliava Institute
4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- HostPhinder web service. Available online: http://cge.cbs.dtu.dk/services/HostPhinder (accessed on 1 April 2016).
- HostPhinder Docker image. Available online: https://registry.hub.docker.com/u/julvi/hostphinder (accessed on 1 April 2016).
- Kapi, A. The evolving threat of antimicrobial resistance: Options for action. Indian J. Med. Res. 2014, 139, 182–183. [Google Scholar]
- WHO. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Harper, D.; Anderson, J.; Enright, M. Phage therapy: Delivering on the promise. Ther. Deliv. 2011, 2, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, M.; Adamia, R. Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotechnol. 2010, 28, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, M.; Adamia, R. Phage therapy experience at the Eliava Institute. Méd. Mal. Infect. 2008, 38, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Miedzybrodzki, R.; Borysowski, J.; Weber-Dabrowska, B.; Fortuna, W.; Letkiewicz, S.; Szufnarowski, K.; Pawelczyk, Z.; Rogóz, P.; Klak, M.; Wojtasik, E.; et al. Charpter 3—Clinical aspects of phage therapy. Adv. Virus Res. 2012, 83, 73–121. [Google Scholar] [PubMed]
- Weber-Dąbrowska, B.; Mulczyk, M.; Górski, A. Bacteriophage therapy of bacterial infections: An update of our institute’s experience. In Inflammation; Springer: Netherlands, 2001; pp. 201–209. [Google Scholar]
- Biswas, B.; Adhya, S.; Washart, P.; Paul, B.; Trostel, A.N.; Powell, B.; Carlton, R.; Merril, C.R. Bacteriophage therapy rescues mice bacteremic from a clinical isolate of vancomycin-resistant Enterococcus faecium. Infect. Immun. 2002, 70, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, R.; Parlato, M.; Borriello, G.; Salvatore, P.; Iannelli, D. Experimental phage therapy against Staphylococcus aureus in mice. Antimicrob. Agents Chemother. 2007, 51, 2765–2773. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.W.; Huggins, M. Successful treatment of experimental Escherichia coli infections in mice using phage: Its general superiority over antibiotics. J. Gen. Microbiol. 1982, 128, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Hawkins, C.; Änggård, E.; Harper, D. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; A preliminary report of efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, S.; Uchiyama, J.; Takemura-Uchiyama, I.; Daibata, M. Perspective: The age of the phage. Nature 2014, 509. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S. Phage therapy gets revitalized. Nature 2014, 510. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A. Using lytic bacteriophages to eliminate or significantly reduce contamination of food by foodborne bacterial pathogens. J. Sci. Food Agric. 2013, 93, 3137–3146. [Google Scholar] [CrossRef] [PubMed]
- Guenther, S.; Huwyler, D.; Richard, S.; Loessner, M.J. Virulent bacteriophage for efficient biocontrol of Listeria monocytogenes in ready-to-eat foods. Appl. Environ. Microbiol. 2009, 75, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, C.L.; Atterbury, R.; El-Shibiny, A.; Connerton, P.; Dillon, E.; Scott, A.; Connerton, I. Bacteriophage therapy to reduce Campylobacter jejuni colonization of broiler chickens. Appl. Environ. Microbiol. 2005, 71, 6554–6563. [Google Scholar] [CrossRef] [PubMed]
- McLean, S.K.; Dunn, L.A.; Palombo, E.A. Phage inhibition of Escherichia coli in ultrahigh-temperature-treated and raw milk. Foodborne Pathog. Dis. 2013, 10, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Sorek, R. The phage-host arms race: Shaping the evolution of microbes. Bioessays 2011, 33, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Garneau, J.E.; Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 2010, 64, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Fineran, P.C.; Blower, T.R.; Foulds, I.J.; Humphreys, D.P.; Lilley, K.S.; Salmond, G.P. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc. Natl. Acad. Sci. USA 2009, 106, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A. Codon bias is a major factor explaining phage evolution in translationally biased hosts. J. Mol. Evol. 2008, 66, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Evans, T.J.; Przybilski, R.; Fineran, P.C.; Salmond, G.P. Viral evasion of a bacterial suicide system by RNA-based molecular mimicry enables infectious altruism. PLoS Genet. 2012, 8, e1003023. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.S.; Hartman, H.; Levin, S.A. Coevolutionary arms races between bacteria and bacteriophage. Proc. Natl. Acad. Sci. USA 2005, 102, 9535–9540. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Turner, P.E.; Burch, C.L. Pleiotropic costs of niche expansion in the RNA bacteriophage Φ6. Genetics 2006, 172, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Amarillas, L.; Cháidez-Quiroz, C.; Sañudo-Barajas, A.; León-Félix, J. Complete genome sequence of a polyvalent bacteriophage, phiKP26, active on Salmonella and Escherichia coli. Arch. Virol. 2013, 158, 2395–2398. [Google Scholar] [CrossRef] [PubMed]
- Loessner, M.J.; Neugirg, E.; Zink, R.; Scherer, S. Isolation, classification and molecular characterization of bacteriophages for Enterobacter species. J. Gen. Microbiol. 1993, 139, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Meaden, S. Understanding bacteriophage specificity in natural microbial communities. Viruses 2013, 5, 806–823. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.R. Diversity among the tailed-bacteriophages that infect the Enterobacteriaceae. Res. Microbiol. 2008, 159, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Edwards, R. The Phage Proteomic Tree: A genome-based taxonomy for phage. J. Bacteriol. 2002, 184, 4529–4535. [Google Scholar] [CrossRef] [PubMed]
- Jacobs-Sera, D.; Marinelli, L.J.; Bowman, C.; Broussard, G.W.; Bustamante, C.G.; Boyle, M.M.; Petrova, Z.O.; Dedrick, R.M.; Pope, W.H.; Advancing, S.E.A.P.H.; et al. On the nature of mycobacteriophage diversity and host preference. Virology 2012, 434, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Cosentino, S.; Lukjancenko, O.; Saputra, D.; Rasmussen, S.; Hasman, H.; Sicheritz-Pontén, T.; Aarestrup, F.M.; Ussery, D.W.; Lund, O. Benchmarking of methods for genomic taxonomy. J. Clin. Microbiol. 2014, 52, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W. Bacteriophage genomics. Curr. Opin. Microbiol. 2003, 6, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.G.; Hatfull, G.F.; Hendrix, R.W. Imbroglios of viral taxonomy: Genetic exchange and failings of phenetic approaches. J. Bacteriol. 2002, 184, 4891–4905. [Google Scholar] [CrossRef] [PubMed]
- Zschach, H.; Joensen, K.G.; Lindhard, B.; Lund, O.; Goderdzishvili, M.; Chkonia, I.; Jgenti, G.; Kvatadze, N.; Alavidze, Z.; Kutter, E.M.; et al. What can we learn from a metagenomic analysis of a Georgian bacteriophage cocktail? Viruses 2015, 7, 6570–6589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Phages.ids - VBI mirrors page. Available online: http://mirrors.vbi.vt.edu/mirrors/ftp.ncbi.nih.gov/genomes/IDS/Phages.ids (accessed on 1 April 2016).
- NCBI viral Genome Resource. Available online: http://www.ncbi.nlm.nih.gov/genomes/GenomesHome.cgi (accessed on 1 April 2016).
- EMBL EBI phage genomes list. Available online: http://www.ebi.ac.uk/genomes/phage.html (accessed on 1 April 2016).
- phagesdb for Mycobacteriophages. Available online: http://phagesdb.org/ (accessed on 1 April 2016).
- phagesdb for Arthrobacter. Available online: http://arthrobacter.phagesdb.org/ (accessed on 1 April 2016).
- phagesdb for Bacillus. Available online: http://bacillus.phagesdb.org/ (accessed on 1 April 2016).
- phagesdb for Streptomyces. Available online: http://streptomyces.phagesdb.org/ (accessed on 1 April 2016).
- Euzéby, J.P. List of Bacterial Names with Standing in Nomenclature: A folder available on the Internet. Int. J. Syst. Bacteriol. 1997, 47, 590–592. [Google Scholar] [CrossRef] [PubMed]
- HostPhinder Github repository. Available online: https://github.com/julvi/HostPhinder (accessed on 1 April 2016).
- Hobohm, U.; Scharf, M.; Schneider, R.; Sander, C. Selection of representative protein data sets. Protein Sci. 1992, 1, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Bonferroni, C.E. Teoria Statistica Delle Classi e Calcolo Delle Probabilita; Libreria Internazionale Seeber: Firenze, Italy, 1936. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Akhter, S.; Aziz, R.K.; Edwards, R.A. PhiSpy: A novel algorithm for finding prophages in bacterial genomes that combines similarity-and composition-based strategies. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- NCBI complete bacterial genomes. Available online: ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/ (accessed on 1 April 2016).
- Phantome manually verified prophages, dating 14 March 2012. Available online: http://www.phantome.org/Downloads/Prophages/PhiSpy/Manually_Verified/ (accessed on 1 April 2016).
- Dang, V.T.; Sullivan, M.B. Emerging methods to study bacteriophage infection at the single-cell level. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Martínez-García, M.; Santos, F.; Moreno-Paz, M.; Parro, V.; Antón, J. Unveiling viral-host interactions within the ‘microbial dark matter’. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.J.; Allen, L.Z.; Lorenzi, H.A.; Fadrosh, D.W.; Brami, D.; Thiagarajan, M.; McCrow, J.P.; Tovchigrechko, A.; Yooseph, S.; Venter, J.C. Metagenomic exploration of viruses throughout the Indian Ocean. PLoS ONE 2012, 7, e42047. [Google Scholar]
- Edwards, R.A.; McNair, K.; Faust, K.; Raes, J.; Dutilh, B.E. Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev. 2016, 40, 258–272. [Google Scholar]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar]
- Kawulok, J.; Deorowicz, S. CoMeta: Classification of metagenomes using k-mers. PLoS ONE 2015, 10, e0121453. [Google Scholar]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15. [Google Scholar] [CrossRef]
- Edwards, R.A.; Olson, R.; Disz, T.; Pusch, G.D.; Vonstein, V.; Stevens, R.; Overbeek, R. Real Time Metagenomics: Using k-mers to annotate metagenomes. Bioinformatics 2012, 28, 3316–3317. [Google Scholar] [PubMed]
- Marinelli, L.J.; Fitz-Gibbon, S.; Hayes, C.; Bowman, C.; Inkeles, M.; Loncaric, A.; Russell, D.A.; Jacobs-Sera, D.; Cokus, S.; Pellegrini, M.; et al. Propionibacterium acnes bacteriophages display limited genetic diversity and broad killing activity against bacterial skin isolates. MBio 2012, 3. [Google Scholar] [CrossRef]
- Liu, J.; Yan, R.; Zhong, Q.; Ngo, S.; Bangayan, N.J.; Nguyen, L.; Lui, T.; Liu, M.; Erfe, M.C.; Craft, N.; et al. The diversity and host interactions of Propionibacterium acnes bacteriophages on human skin. ISME J. 2015, 9, 2078–2093. [Google Scholar] [CrossRef] [PubMed]
- Farrar, M.D.; Howson, K.M.; Bojar, R.A.; West, D.; Towler, J.C.; Parry, J.; Pelton, K.; Holland, K.T. Genome sequence and analysis of a Propionibacterium acnes bacteriophage. J. Bacteriol. 2007, 189, 4161–4167. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.; Suissa, M.; Chiswell, D.; Azriel, A.; Berman, B.; Shahar, D.; Reznick, S.; Sharf, R.; Wyse, J.; Bar-On, T.; et al. A bacteriophage reagent for Salmonella: Molecular studies on Felix 01. Int. J. Food Microbiol. 2002, 74, 217–227. [Google Scholar] [CrossRef]
- Ford, M.E.; Sarkis, G.J.; Belanger, A.E.; Hendrix, R.W.; Hatfull, G.F. Genome structure of mycobacteriophage D29: Implications for phage evolution. J. Mol. Biol. 1998, 279, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Schwudke, D.; Ergin, A.; Michael, K.; Volkmar, S.; Appel, B.; Knabner, D.; Konietzny, A.; Strauch, E. Broad-host-range Yersinia phage PY100: Genome sequence, proteome analysis of virions, and DNA packaging strategy. J. Bacteriol. 2008, 190, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.; Elliott, J.M.; Ramanculov, E.; Chain, P.S.; Chu, M.C.; Molineux, I.J. The genome sequence of Yersinia pestis bacteriophage φA1122 reveals an intimate history with the coliphage T3 and T7 genomes. J. Bacteriol. 2003, 185, 5248–5262. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Cui, Y.; Yan, Y.; Du, Z.; Tan, Y.; Yang, H.; Bi, Y.; Zhang, P.; Zhou, L.; Zhou, D.; et al. Outer membrane proteins Ail and OmpF of Yersinia pestis are involved in the adsorption of T7-related bacteriophage Yep-phi. J. Virol. 2013, 87, 12260–12269. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.W.; Kim, K.H. Comparative genomic analysis of bacteriophage EP23 infecting Shigella sonnei and Escherichia coli. J. Microbiol. 2011, 49, 927–934. [Google Scholar] [CrossRef] [PubMed]
- De Lappe, N.; Doran, G.; O’Connor, J.; O’Hare, C.; Cormican, M. Characterization of bacteriophages used in the Salmonella enterica serovar Enteritidis phage-typing scheme. J. Med. Microbiol. 2009, 58, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Hood, A. Phage typing of Staphylococcus aureus. J. Hyg. 1953, 51, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bielke, L.; Higgins, S.; Donoghue, A.; Donoghue, D.; Hargis, B. Salmonella host range of bacteriophages that infect multiple genera. Poult. Sci. 2007, 86, 2536–2540. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.C.; Schrader, H.S.; Rieland, B.; Thompson, T.L.; Lee, K.W.; Nickerson, K.W.; Kokjohn, T.A. Prevalence of broad-host-range lytic bacteriophages of Sphaerotilus natans, Escherichia coli, and Pseudomonas aeruginosa. Appl. Environ. Microbiol. 1998, 64, 575–580. [Google Scholar] [PubMed]
- Olsen, R.H.; Siak, J.S.; Gray, R.H. Characteristics of PRD1, a plasmid-dependent broad host range DNA bacteriophage. J. Virol. 1974, 14, 689–699. [Google Scholar] [PubMed]
- Carson, L.; Gorman, S.P.; Gilmore, B.F. The use of lytic bacteriophages in the prevention and eradication of biofilms of Proteus mirabilis and Escherichia coli. FEMS Immunol. Med. Microbiol. 2010, 59, 447–455. [Google Scholar] [CrossRef] [PubMed]










| Score | z | frac | frac | Coverage | |
|---|---|---|---|---|---|
| Species (%) | |||||
| Genus (%) |
| Method | Criterion 1 (First Host) | Criterion 2 (Majority Host among Top-10) | Criterion 3 (Coverage Threshold, ) | Criterion 4 (Summing up Normalized Coverage Values, ) |
|---|---|---|---|---|
| Accuracy, Species (%) | ||||
| Accuracy, Genus (%) |
| Species | Representation in | Genus | Representation in |
|---|---|---|---|
| Enterococcus faecalis | 15 | Acinetobacter | 16 |
| Listeria monocytogenes | 21 | Listeria | 26 |
| Propionibacterium acnes | 21 | Propionibacterium | 24 |
| Vibrio cholerae | 35 | Streptococcus | 39 |
| Streptomyces | 11 | ||
| Thermus | 5 |
| BLAST | HostPhinder | |
|---|---|---|
| No. of predictions, training on | 90% | 97% |
| No. of predictions, training on | 91% | 96% |
| Accuracy on common predictions (GENERA) (%) | 84.66 ± 0.188 | 85.13 ± 0.176 |
| Accuracy on common predictions (SPECIES) (%) | 76.92 ± 0.252 | 78.69 ± 0.237 |

{kind=link}
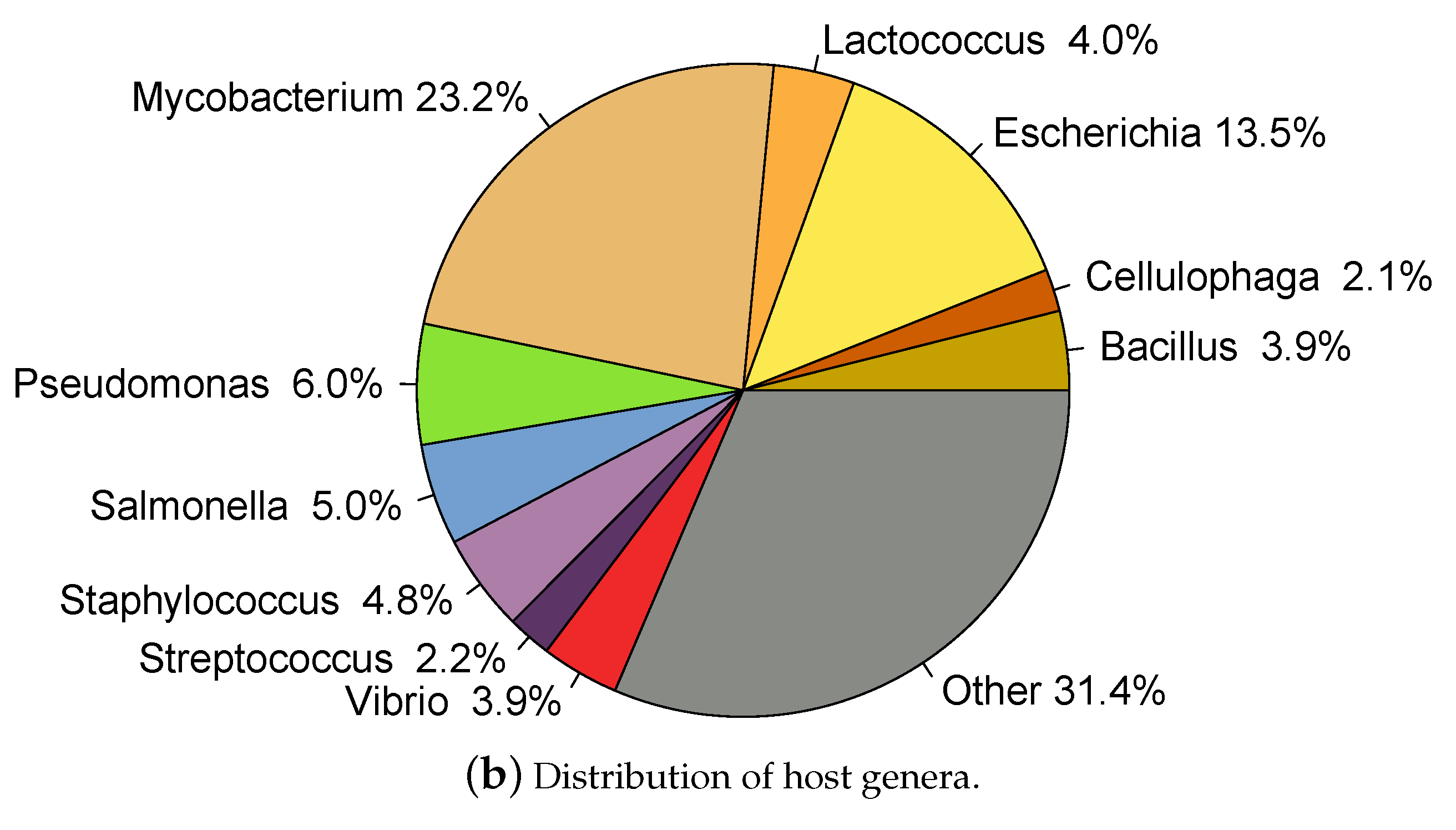{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villarroel, J.; Kleinheinz, K.A.; Jurtz, V.I.; Zschach, H.; Lund, O.; Nielsen, M.; Larsen, M.V. HostPhinder: A Phage Host Prediction Tool. Viruses 2016, 8, 116. https://doi.org/10.3390/v8050116
Villarroel J, Kleinheinz KA, Jurtz VI, Zschach H, Lund O, Nielsen M, Larsen MV. HostPhinder: A Phage Host Prediction Tool. Viruses. 2016; 8(5):116. https://doi.org/10.3390/v8050116
Chicago/Turabian StyleVillarroel, Julia, Kortine Annina Kleinheinz, Vanessa Isabell Jurtz, Henrike Zschach, Ole Lund, Morten Nielsen, and Mette Voldby Larsen. 2016. "HostPhinder: A Phage Host Prediction Tool" Viruses 8, no. 5: 116. https://doi.org/10.3390/v8050116