
A New Epitope Selection Method: Application to Design a Multi-Valent Epitope Vaccine Targeting HRAS Oncogene in Squamous Cell Carcinoma


Abstract
:1. Introduction
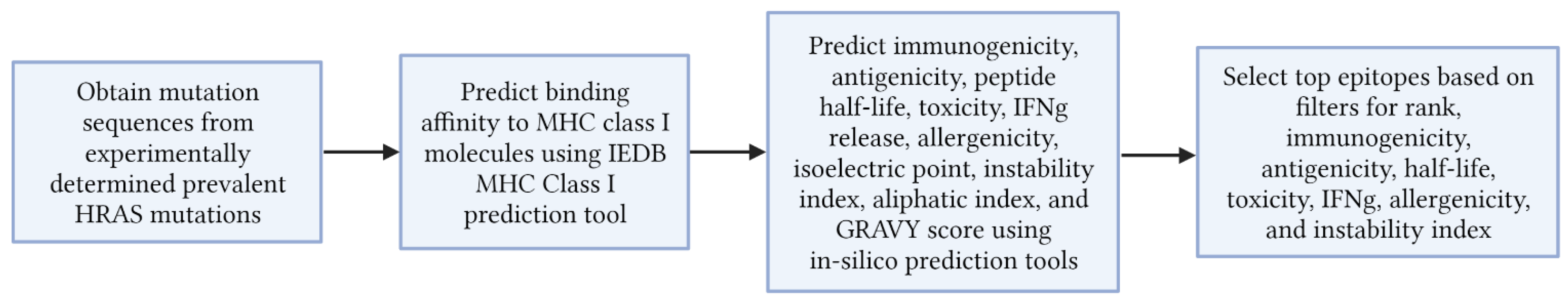
2. Materials and Methods
2.1. Obtaining Mutated HRAS Peptide Sequences
2.2. MHC Class I Binding Affinity Determination
2.3. Selectin of Top Epitopes
2.4. Murine Model Binding Affinity and Three-Dimensional Analysis
3. Results
3.1. Immune Epitope Database (IEDB) Binding Affinity Analysis
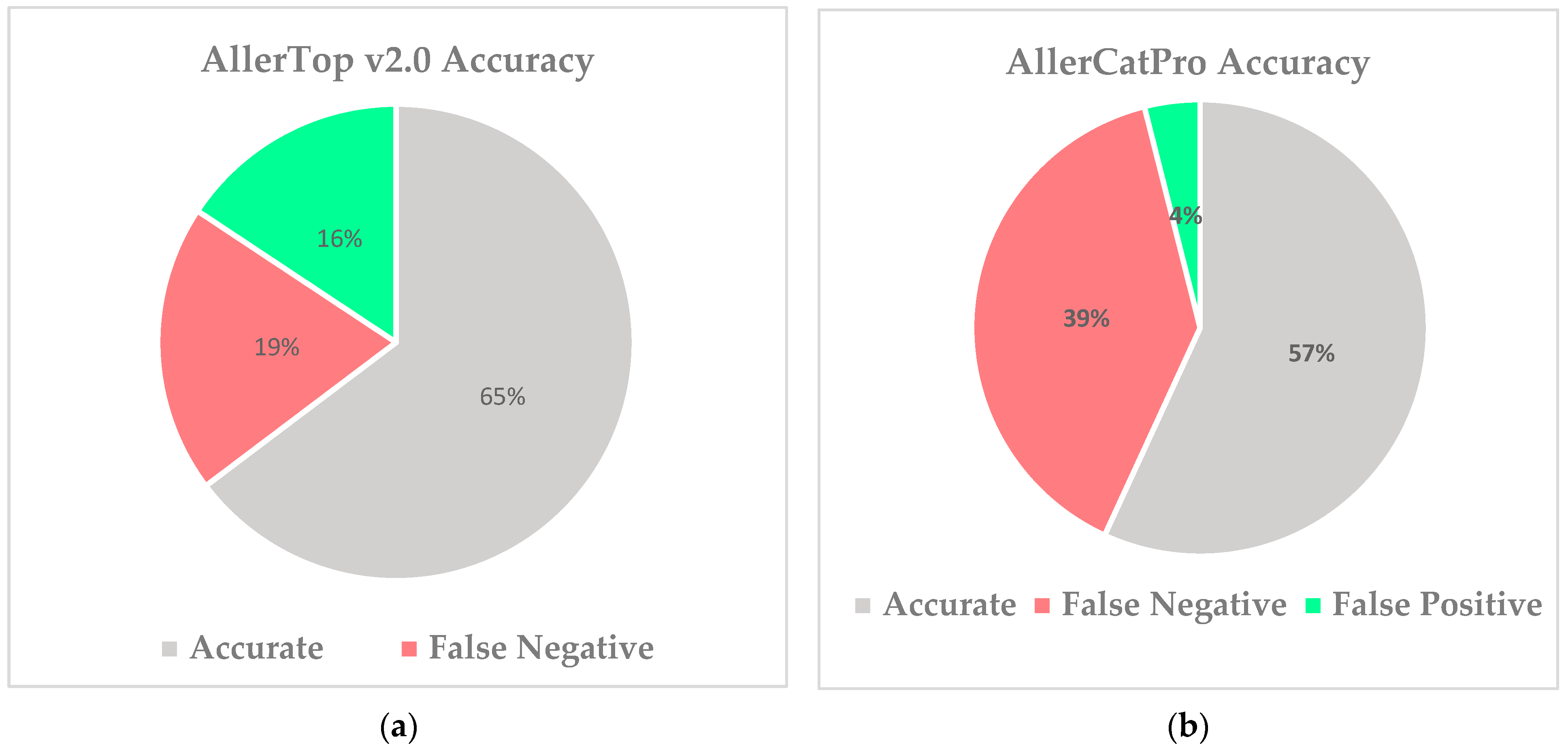
3.2. Determining Superior Allergenicity Predictor
3.3. Replacement of “Cell Permeability” Parameter
3.4. Formula Derivation Using a Two-Variable Equation
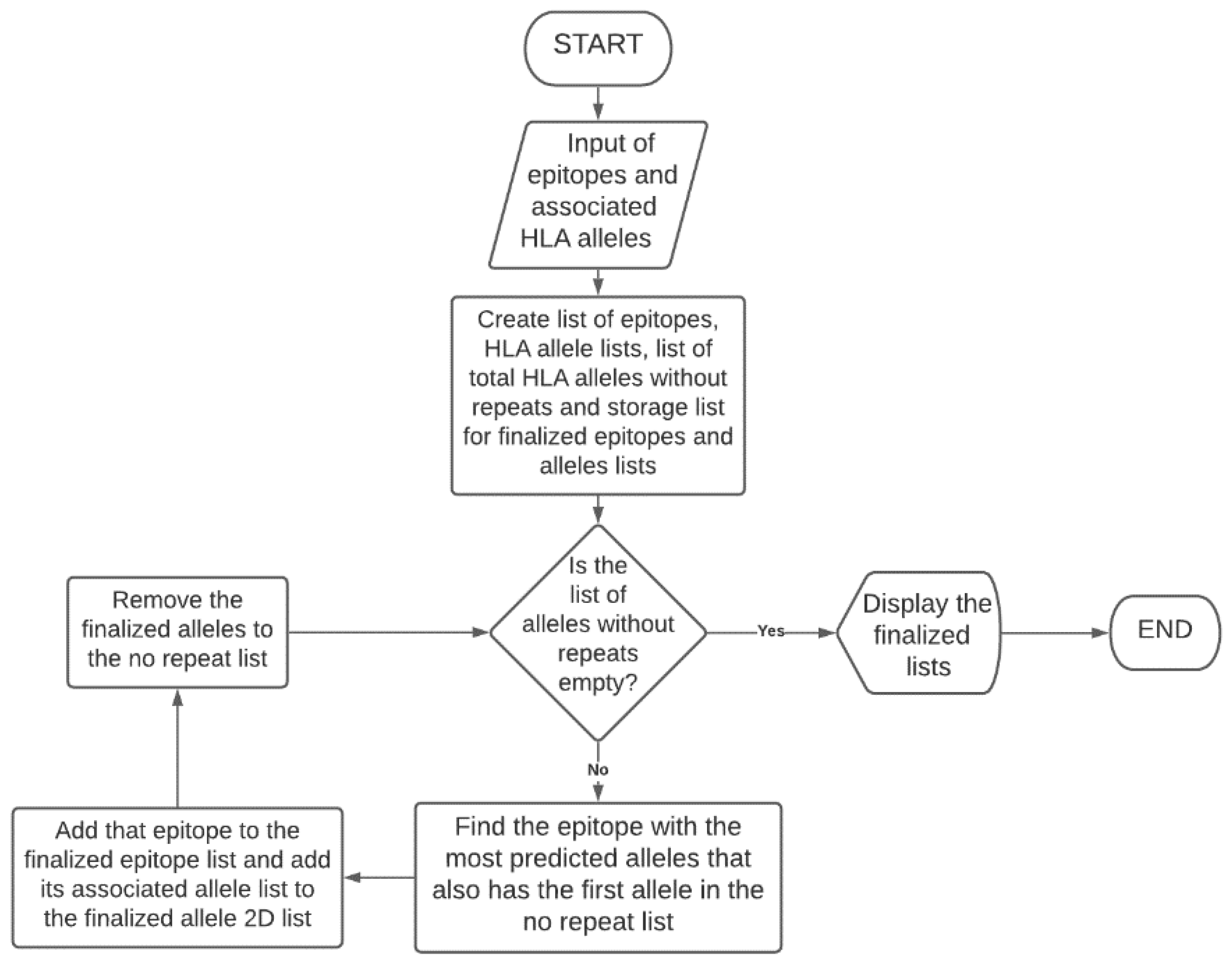
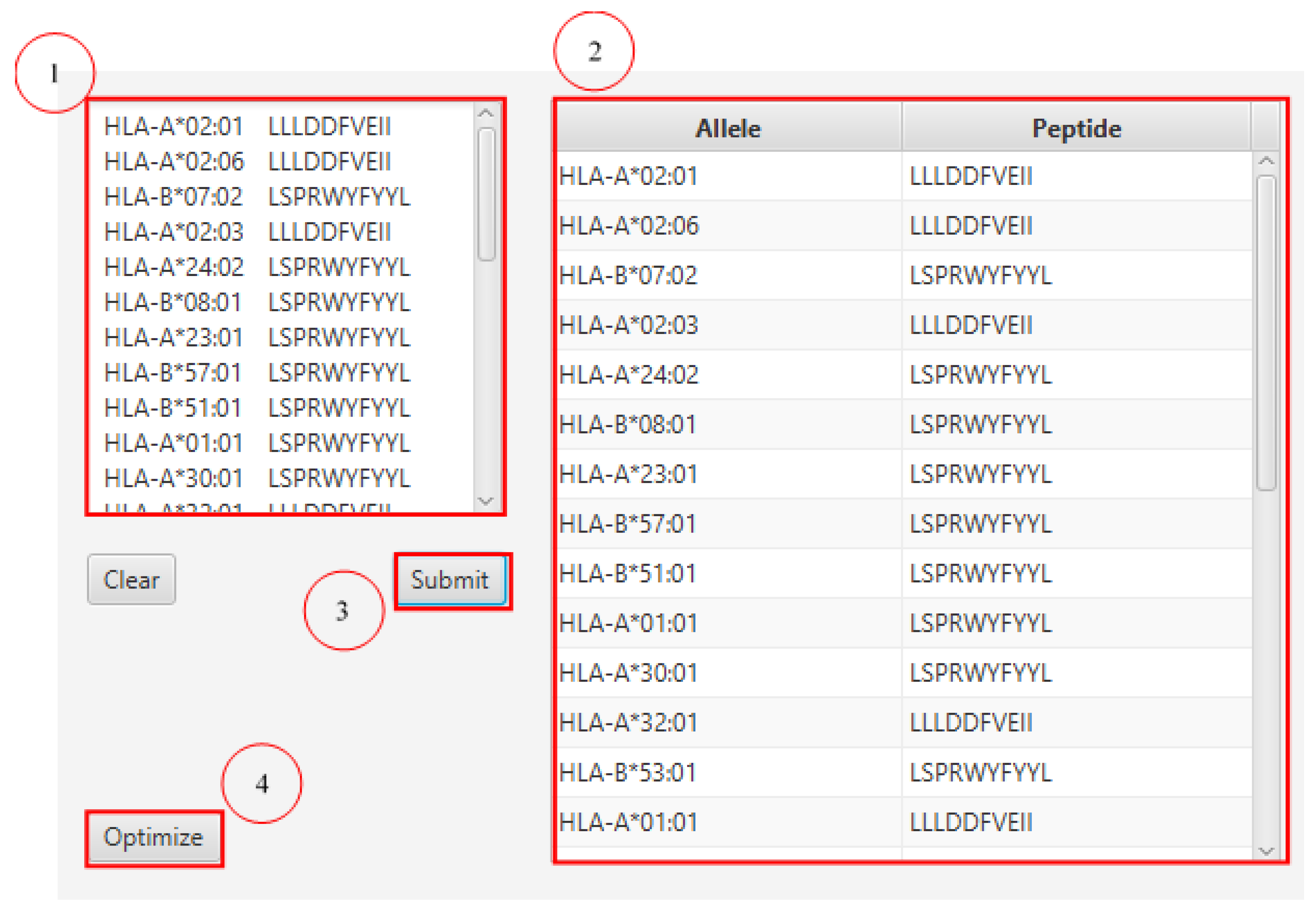
3.5. Epitope Optimization for Maximum Population Coverage
3.6. Consideration of a Three-Variable Filtration Method
3.7. Validation of Epitope Selection Method Using Experimental Immunogenic Epitopes
3.8. Finalized Filtration
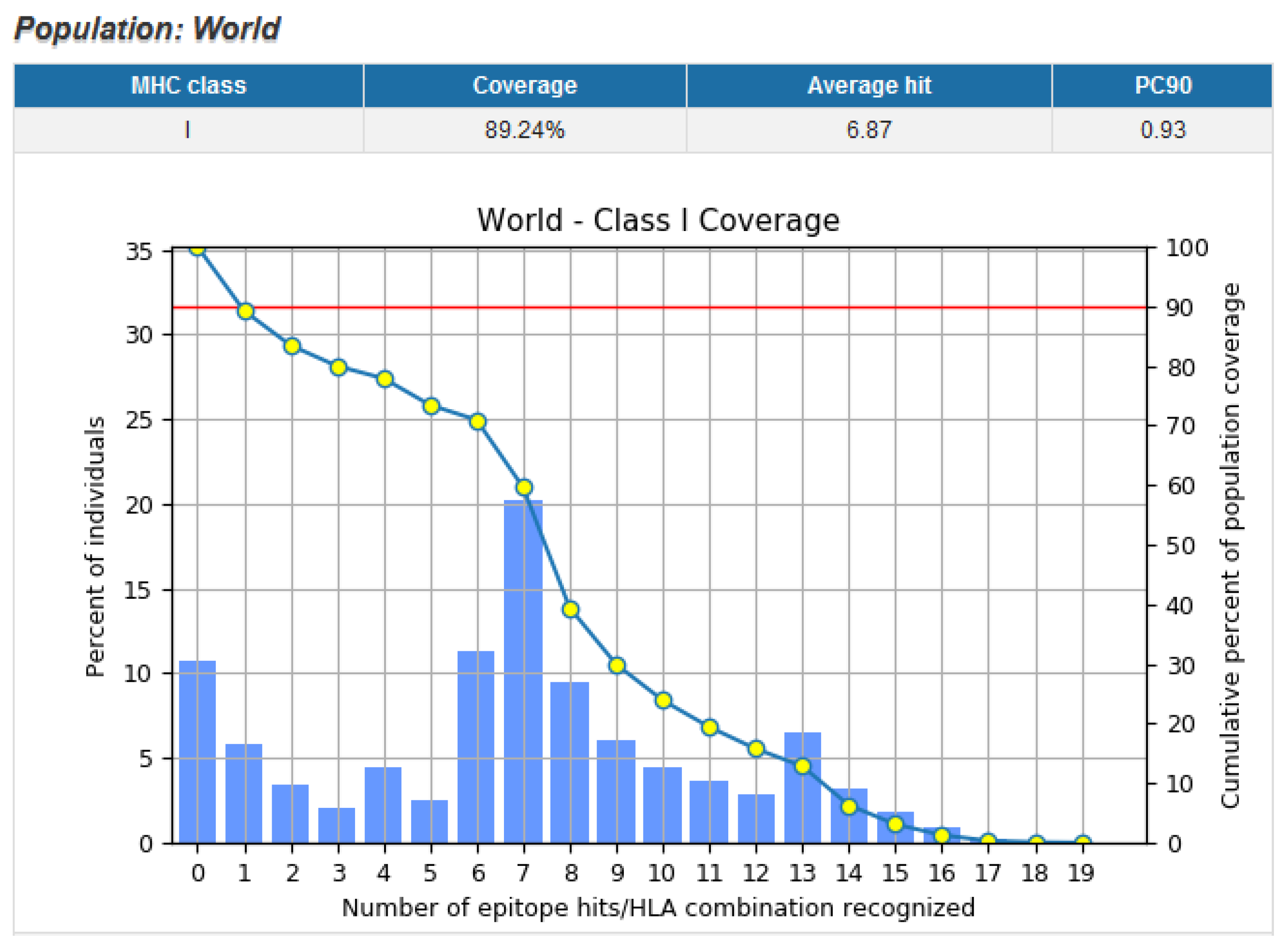
3.9. Population Coverage of Top Epitopes
3.10. Top Epitope Selection without IFNγ Release Parameter
3.11. Murine MHC Binding Affinity
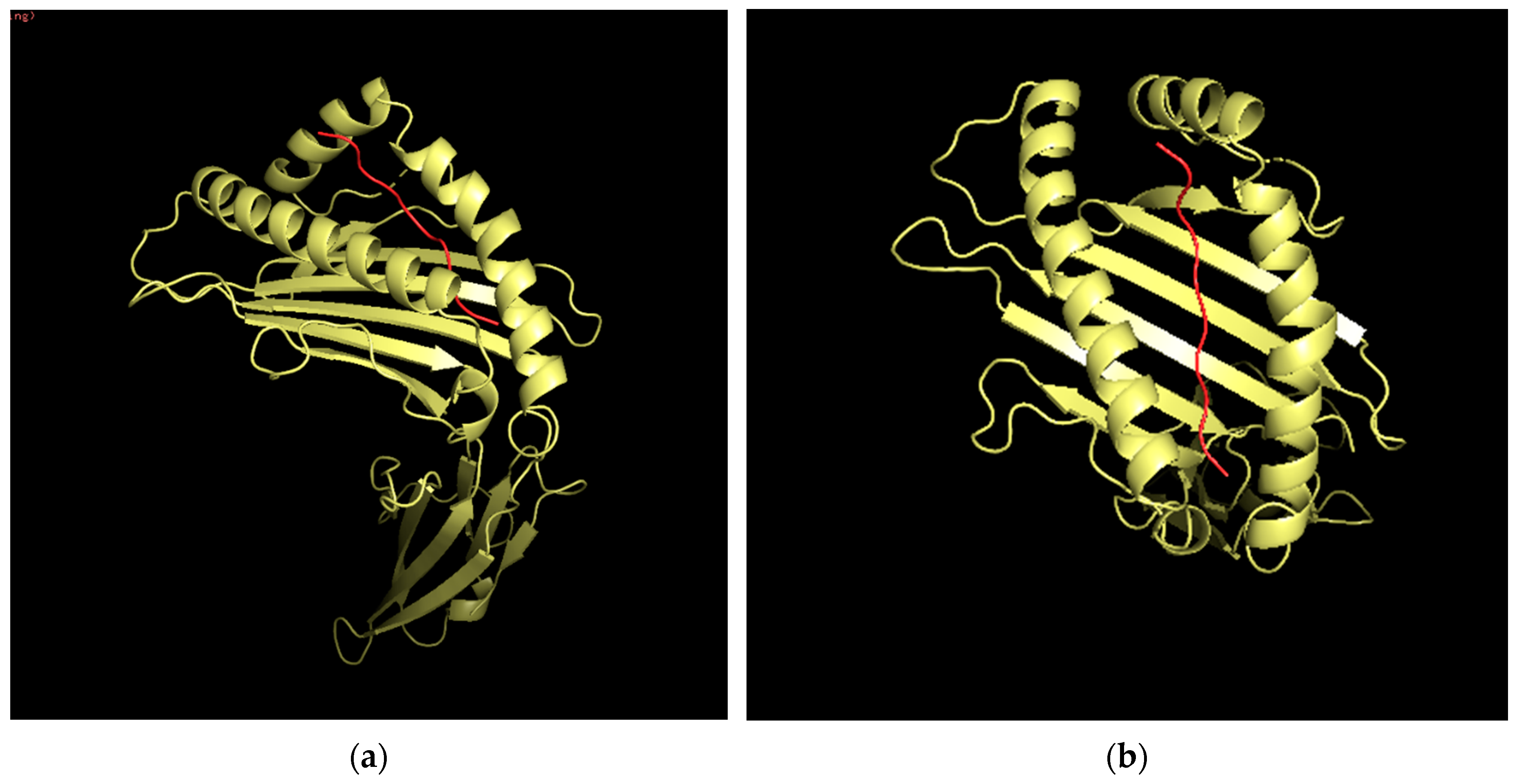
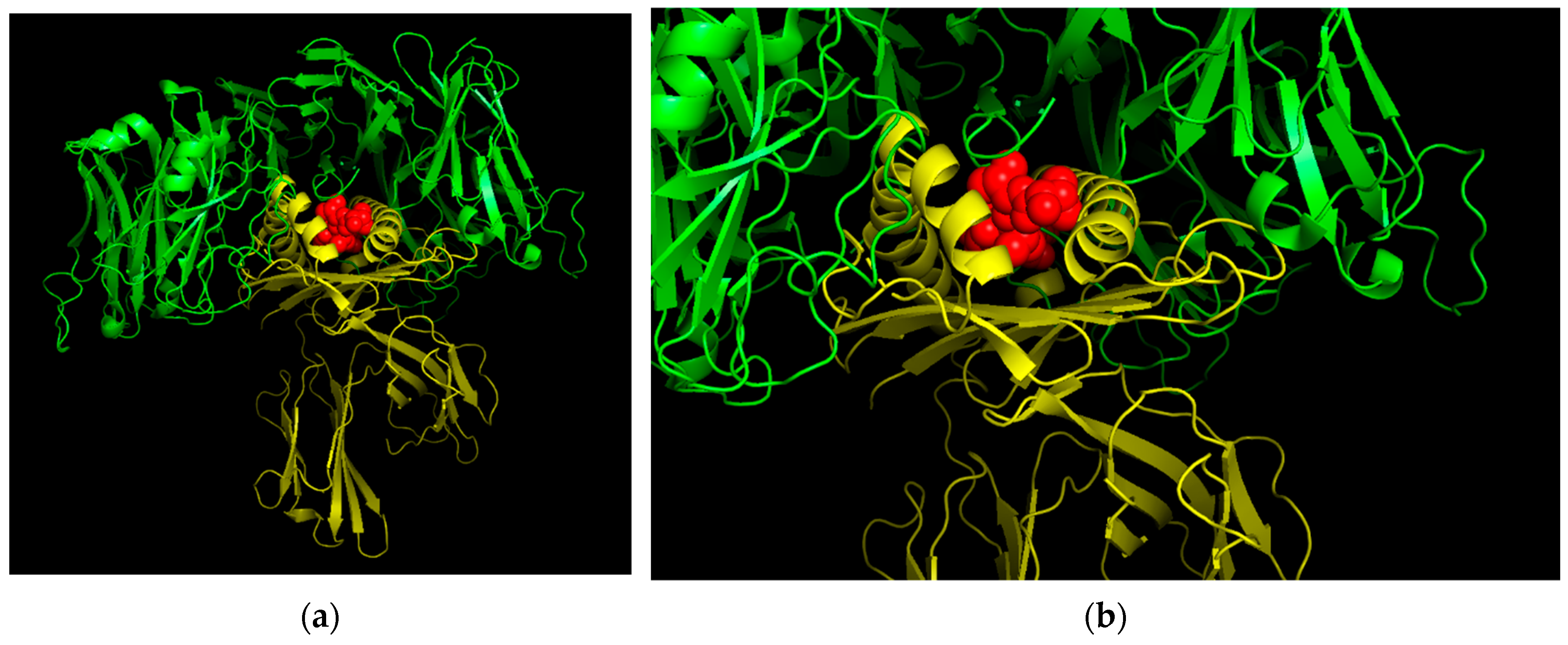
3.12. Three-Dimensional (3D) Structural Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fania, L.; Didona, D.; Di Pietro, F.; Verkhovskaia, S.; Morese, R.; Paolino, G.; Donati, M.; Ricci, F.; Coco, V.; Ricci, F.; et al. Cutaneous Squamous Cell Carcinoma: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2021, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine. HRAS Gene. Retrieved from Medline Plus. Available online: https://medlineplus.gov/genetics/gene/hras/ (accessed on 18 August 2020).
- Scott, A.J.; Lieu, C.H.; Messersmith, W.A. Therapeutic Approaches to RAS Mutation. Cancer J. 2016, 22, 165–174. [Google Scholar] [CrossRef] [Green Version]
- The AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, E.; Masago, K.; Fujita, S.; Hanai, N.; Yatabe, Y. FrequentKRASandHRASmutations in squamous cell papillomas of the head and neck. J. Pathol. Clin. Res. 2020, 6, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Gilardi, M.; Wang, Z.; Proietto, M.; Chillà, A.; Calleja-Valera, J.L.; Goto, Y.; Vanoni, M.; Janes, M.R.; Mikulski, Z.; Gualberto, A.; et al. Tipifarnib as a Precision Therapy for HRAS-Mutant Head and Neck Squamous Cell Carcinomas. Mol. Cancer Ther. 2020, 19, 1784–1796. [Google Scholar] [CrossRef]
- Rahma, O.E.; Hamilton, J.M.; Wojtowicz, M.; Dakheel, O.; Bernstein, S.; Liewehr, D.J.; Steinberg, S.M.; Khleif, S.N. The immunological and clinical effects of mutated ras peptide vaccine in combination with IL-2, GM-CSF, or both in patients with solid tumors. J. Transl. Med. 2014, 12, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qamar, M.T.U.; Saleem, S.; Ashfaq, U.A.; Bari, A.; Anwar, F.; Alqahtani, S. Epitope-based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: An immune-informatics study. J. Transl. Med. 2019, 17, 1–14. [Google Scholar] [CrossRef]
- Bhatt, T.K.; Nimesh, S. Chapter 9—Design of vaccine constructs. In The Design and Development of Novel Drugs and Vaccines: Principles and Protocols; Academic Press: Cambridge, MA, USA, 2021; pp. 109–119. [Google Scholar]
- Stephens, A.J.; Burgess-Brown, N.A.; Jiang, S. Beyond Just Peptide Antigens: The Complex World of Peptide-Based Cancer Vaccines. Front. Immunol. 2021, 12, 696791. [Google Scholar] [CrossRef] [PubMed]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information. HRAS HRas Proto-Oncogene, GTPase [Homo sapiens (Human)]. Retrieved from National Library of Medicine Gene Viewer. Available online: https://www.ncbi.nlm.nih.gov/gene/3265 (accessed on 19 July 2021).
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, G.; Rego, S.; Nguyenkhoa, V.; Dakshanamurthy, S. Design of t cell epitope-based vaccine candidate for SARS-CoV-2 targeting nucleocapsid and spike protein escape variants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Parn, S.; Jabbour, G.; Nguyenkhoa, V.; Dakshanamurthy, S. Design of Peptide vaccine for COVID19: CD8+ and CD4+ T cell epitopes from SARS-CoV-2 open-reading-frame protein variants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Karosiene, E.; Edwards, L.; Grifoni, A.; Paul, S.; Andreatta, M.; Weiskopf, D.; Sidney, J.; Nielsen, M.; Peters, B.; et al. Predicting HLA CD4 Immunogenicity in Human Populations. Front. Immunol. 2018, 9, 1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S.; Open Source Drug Discovery Consortium. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Dhanda, S.K.; Vir, P.; Raghava, G.P.S. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- DeVette, C.; Andreatta, M.; Bardet, W.; Cate, S.J.; Jurtz, V.I.; Jackson, K.W.; Welm, A.L.; Nielsen, M.; Hildebrand, W.H. NetH2pan: A Computational Tool to Guide MHC Peptide Prediction on Murine Tumors. Cancer Immunol. Res. 2018, 6, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Bui, H.H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 17, 153. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Singh, H.P.; Tuknait, A.; Chaudhary, K.; Singh, B.; Kumaran, S.S.; Raghava, G.P.S. PEPstrMOD: Structure prediction of peptides containing natural, non-natural and modified residues. Biol. Direct 2015, 10, 73. [Google Scholar] [CrossRef] [Green Version]
- London, N.; Raveh, B.; Cohen, E.; Fathi, G.; Schueler-Furman, O. Rosetta FlexPepDock web server—high resolution modeling of peptide–protein interactions. Nucleic Acids Res. 2011, 39, W249–W253. [Google Scholar] [CrossRef] [Green Version]
- Maurer-Stroh, S.; Krutz, N.L.; Kern, P.S.; Gunalan, V.; Nguyen, M.N.; Limviphuvadh, V.; Eisenhaber, F.; Gerberick, G.F. AllerCatPro—prediction of protein allergenicity potential from the protein sequence. Bioinformatics 2019, 35, 3020–3027. [Google Scholar] [CrossRef] [Green Version]
- Gautam, A.; Chaudhary, K.; Kumar, R.; Sharma, A.; Kapoor, P.; Tyagi, A.; Open source Drug Discovery Consortium; Raghava, G.P.S. In silico approaches for designing highly effective cell penetrating peptides. J. Transl. Med. 2013, 11, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Jiang, H.; Qiu, M.; Liu, L.; Zou, S.; Li, Y.; Guo, Q.; Han, N.; Sun, Y.; Wang, K.; et al. Multi-Epitope Vaccine Design Using an Immunoinformatic Approach for SARS-CoV-2. Pathogens 2021, 10, 737. [Google Scholar] [CrossRef]
- Fleri, W.; Paul, S.; Dhanda, S.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource in Epitope Discovery and Synthetic Vaccine Design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadeer, A.A.; Ahmed, S.F.; McKay, M.R. Landscape of epitopes targeted by T cells in 852 individuals recovered from COVID-19: Meta-analysis, immunoprevalence, and web platform. Cell Rep. Med. 2021, 2, 100312. [Google Scholar] [CrossRef]
- Snyder, T.M.; Gittelman, R.M.; Klinger, M.; May, D.H.; Osborne, E.J.; Taniguchi, R.; Zahid, H.J.; Kaplan, I.M.; Dines, J.N.; Noakes, M.T.; et al. Magnitude and Dynamics of the T-Cell Response to SARS-CoV-2 Infection at Both Individual and Population Levels. medRxiv 2020. [Google Scholar] [CrossRef]
- Mathur, D.; Singh, S.; Mehta, A.; Agrawal, P.; Raghava, G.P.S. In silico approaches for predicting the half-life of natural and modified peptides in blood. PLoS ONE 2018, 13, e0196829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
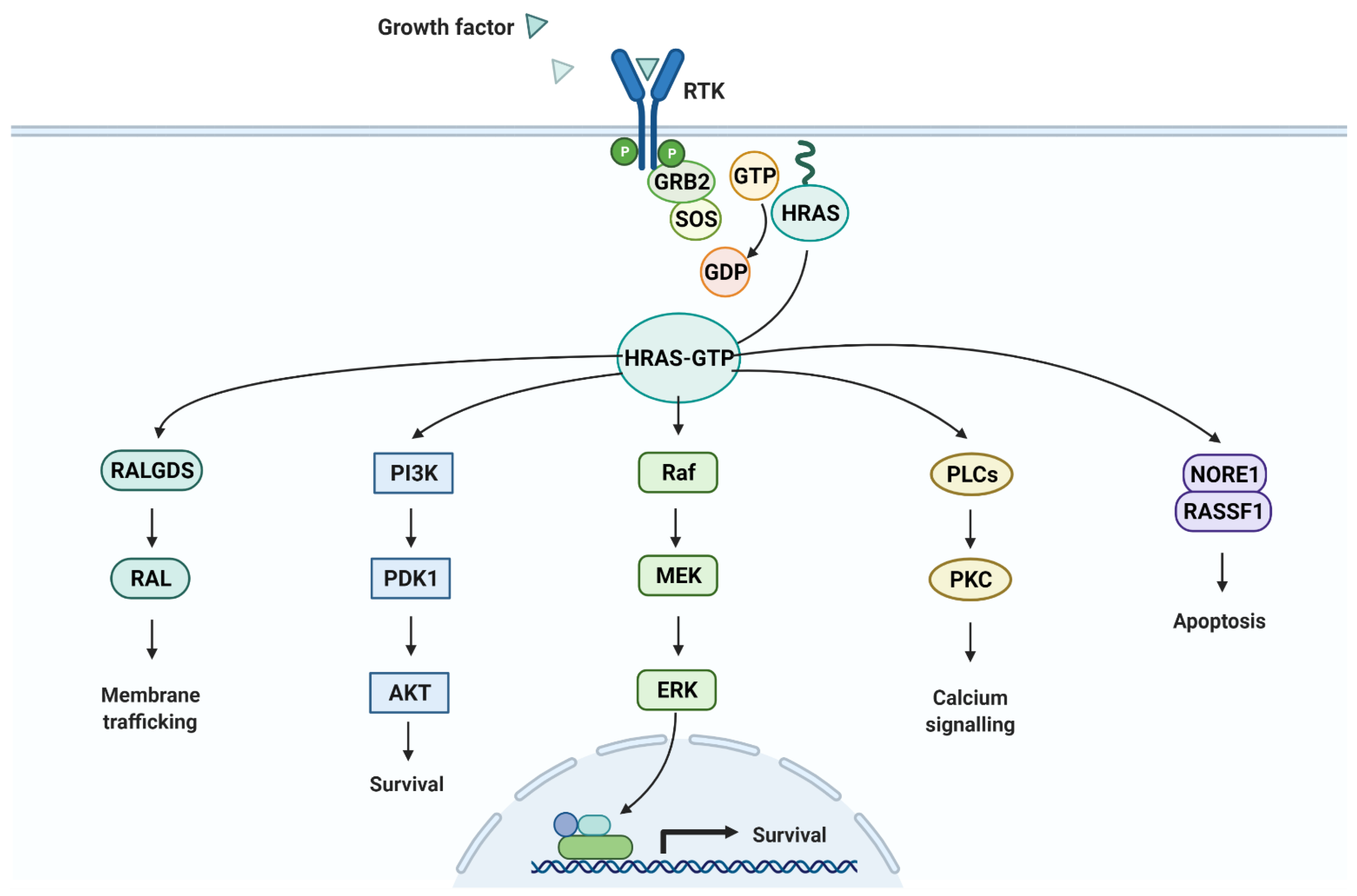
- “Ras Pathway”, by BioRender.com. 2021. Available online: https://app.biorender.com/biorender-templates (accessed on 24 September 2021).
- Guruprasad, K.; Reddy, B.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef] [PubMed]
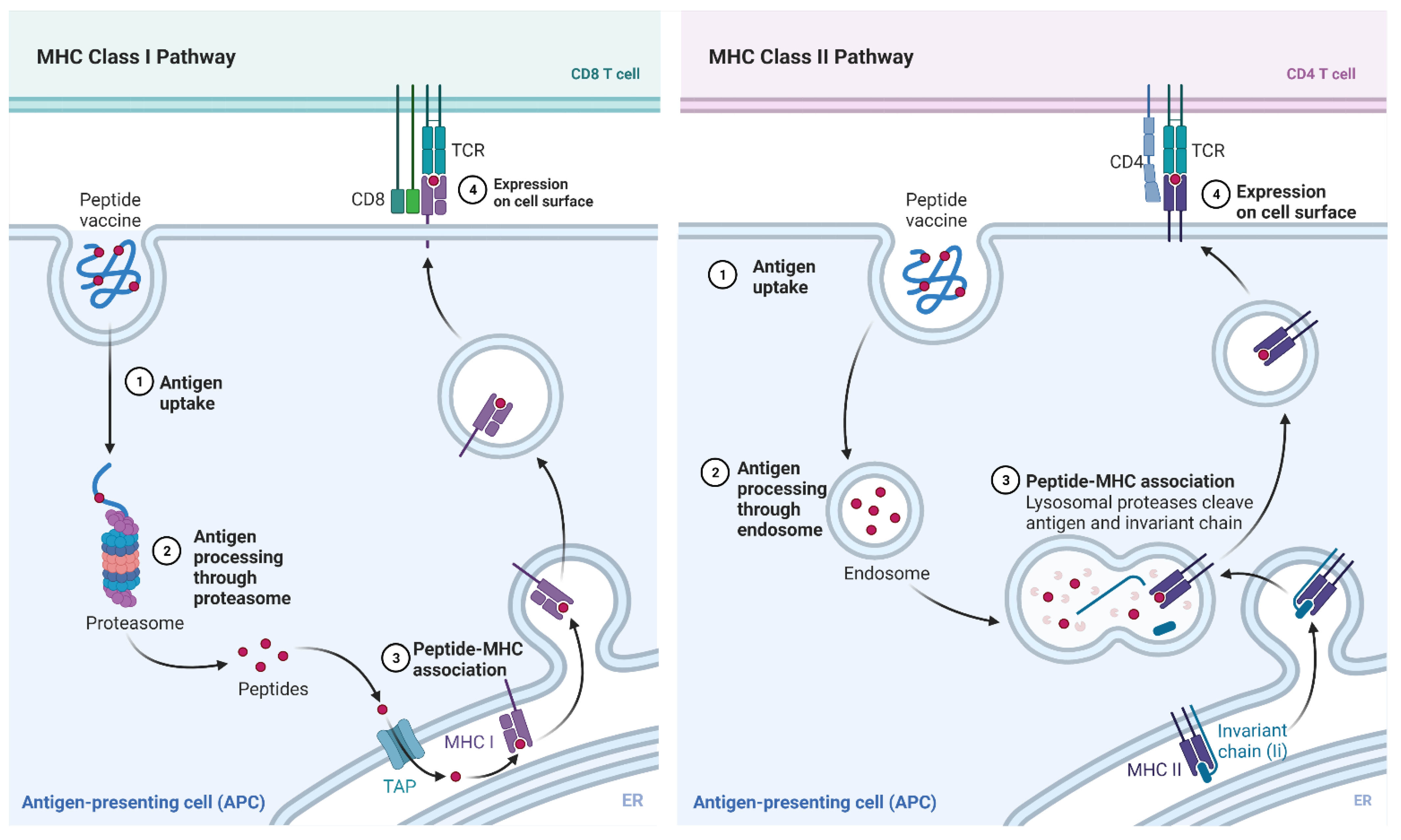
- “MHC Class I and II Pathways”, by BioRender.com. 2021. Available online: https://app.biorender.com/biorender-templates (accessed on 24 September 2021).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Tool Name | Tool Link | Threshold |
|---|---|---|---|
| Rank | Immune Epitope Database (IEDB) NetMHCpan EL 4.1 | http://tools.iedb.org/mhci/ (accessed on 24 September 2021) | <10 |
| Immunogenicity | IEDB Immunogenicity | http://tools.iedb.org/immunogenicity/ (accessed on 24 September 2021) | >0 |
| Antigenicity | VaxiJen | http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html (accessed on 24 September 2021) | >0.4 |
| Half-Life | ProtParam | https://web.expasy.org/protparam/ (accessed on 24 September 2021) | >1 h |
| Toxicity | ToxinPred | https://webs.iiitd.edu.in/raghava/toxinpred/ (accessed on 24 September 2021) | Non-Toxic |
| IFNγ | IFNepitope | https://webs.iiitd.edu.in/raghava/ifnepitope/predict.php (accessed on 24 September 2021) | Positive |
| Allergenicity | Allertop v2.0 | https://www.ddg-pharmfac.net/AllerTOP/ (accessed on 24 September 2021) | Non-Allergen |
| Isoelectric Point | ProtParam | https://web.expasy.org/protparam/ (accessed on 24 September 2021) | N/A |
| Instability Index | ProtParam | https://web.expasy.org/protparam/ (accessed on 24 September 2021) | <40 |
| Aliphatic Index | ProtParam | https://web.expasy.org/protparam/ (accessed on 24 September 2021) | N/A |
| GRAVY Score | ProtParam | https://web.expasy.org/protparam/ (accessed on 24 September 2021) | N/A |
| Mutation | Peptide | HLA Alleles |
|---|---|---|
| G12C + G13D | VVVGACDVGK | HLA-A*68:01,HLA-A*11:01,HLA-A*03:01,HLA-A*30:01,HLA-A*31:01 |
| G12D + G13C | KLVVVGADC | HLA-A*02:01 |
| LVVVGADCV | HLA-A*68:02,HLA-A*02:06,HLA-A*02:03,HLA-A*02:01 | |
| VVVGADCVGK | HLA-A*68:01,HLA-A*11:01,HLA-A*03:01,HLA-A*30:01,HLA-A*31:01 | |
| G12D + G13D | KLVVVGADDV | HLA-A*02:03,HLA-A*02:01 |
| VVGADDVGK | HLA-A*11:01,HLA-A*68:01,HLA-A*03:01,HLA-A*30:01 | |
| VVVGADDVGK | HLA-A*68:01,HLA-A*11:01,HLA-A*03:01 | |
| G12D + G13R | KLVVVGADR | HLA-A*31:01,HLA-A*03:01,HLA-A*68:01,HLA-A*33:01,HLA-A*11:01 |
| LVVVGADRV | HLA-A*68:02,HLA-A*02:06,HLA-A*02:03,HLA-A*02:01,HLA-B*51:01 | |
| G12D + G13S | KLVVVGADSV | HLA-A*02:03,HLA-A*02:01,HLA-A*02:06 |
| LVVVGADSV | HLA-A*02:06,HLA-A*68:02,HLA-A*02:03,HLA-B*51:01,HLA-A*02:01,HLA-A*26:01,HLA-B*35:01 | |
| G12S + G13C | KLVVVGASC | HLA-A*02:06,HLA-A*02:03,HLA-A*02:01,HLA-B*15:01,HLA-A*32:01 |
| G13D | VVVGAGDVGK | HLA-A*11:01,HLA-A*68:01,HLA-A*03:01,HLA-A*30:01,HLA-A*31:01 |
| Q61L | DTAGLEEYSA | HLA-A*68:02,HLA-A*26:01 |
| Q61L + E62G | AGLGEYSAM | HLA-B*15:01,HLA-A*30:02,HLA-B*35:01,HLA-B*08:01,HLA-A*02:06,HLA-B*07:02,HLA-B*51:01,HLA-A*26:01 |
| DTAGLGEYSA | HLA-A*68:02,HLA-A*68:01,HLA-A*26:01 |
| Area | Percent Coverage with IFNγ Filter | Percent Coverage without IFNγ Filter |
|---|---|---|
| Central Africa | 68.24 | 86.04 |
| Central America | 2.78 | 7.76 |
| East Africa | 74.1 | 90.78 |
| East Asia | 85.33 | 98.18 |
| Europe | 94.32 | 99.68 |
| North Africa | 82.04 | 96.03 |
| North America | 90.7 | 99.06 |
| Northeast Asia | 83.73 | 94.7 |
| Oceania | 63.69 | 94.71 |
| South Africa | 75.77 | 93.03 |
| South America | 71.3 | 88.3 |
| South Asia | 83.44 | 94.73 |
| Southeast Asia | 72.0 | 94.56 |
| Southwest Asia | 80.94 | 92.5 |
| West Africa | 81.05 | 95.49 |
| West Indies | 88.11 | 98.98 |
| World | 89.24 | 98.55 |
| Region Average | 74.85 | 89.41 |
| Standard Deviation | 20.28 | 25.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savsani, K.; Jabbour, G.; Dakshanamurthy, S. A New Epitope Selection Method: Application to Design a Multi-Valent Epitope Vaccine Targeting HRAS Oncogene in Squamous Cell Carcinoma. Vaccines 2022, 10, 63. https://doi.org/10.3390/vaccines10010063
Savsani K, Jabbour G, Dakshanamurthy S. A New Epitope Selection Method: Application to Design a Multi-Valent Epitope Vaccine Targeting HRAS Oncogene in Squamous Cell Carcinoma. Vaccines. 2022; 10(1):63. https://doi.org/10.3390/vaccines10010063
Chicago/Turabian StyleSavsani, Kush, Gabriel Jabbour, and Sivanesan Dakshanamurthy. 2022. "A New Epitope Selection Method: Application to Design a Multi-Valent Epitope Vaccine Targeting HRAS Oncogene in Squamous Cell Carcinoma" Vaccines 10, no. 1: 63. https://doi.org/10.3390/vaccines10010063