 Lilian A. Martinez-Carrera
Lilian A. Martinez-Carrera Brunhilde Wirth
Brunhilde Wirth- Institute of Human Genetics, Institute for Genetics and Center for Molecular Medicine of The University of Cologne, Cologne, Germany
Spinal muscular atrophies (SMAs) are characterized by degeneration of spinal motor neurons and muscle weakness. Autosomal recessive SMA is the most common form and is caused by homozygous deletions/mutations of the SMN1 gene. However, families with dominant inherited SMA have been reported, for most of them the causal gene remains unknown. Recently, we and others have identified heterozygous mutations in BICD2 as causative for autosomal dominant SMA, lower extremity-predominant, 2 (SMALED2) and hereditary spastic paraplegia (HSP). BICD2 encodes the Bicaudal D2 protein, which is considered to be a golgin, due to its coiled-coil (CC) structure and interaction with the small GTPase RAB6A located at the Golgi apparatus. Golgins are resident proteins in the Golgi apparatus and form a matrix that helps to maintain the structure of this organelle. Golgins are also involved in the regulation of vesicle transport. In vitro overexpression experiments and studies of fibroblast cell lines derived from patients, showed fragmentation of the Golgi apparatus. In the current review, we will discuss possible causes for this disruption, and the consequences at cellular level, with a view to better understand the pathomechanism of this disease.
Spinal muscular atrophy (SMA) is a diverse group of genetic disorders characterized by aberrant development and/or loss of spinal motor neurons, and muscle weakness without sensory neuron involvement (Wee et al., 2010). SMA is usually classified by the pattern of weakness (i.e., proximal or distal) and mode of inheritance (autosomal recessive, autosomal dominant, X-linked).
Autosomal recessive SMA linked to chromosome 5 (5q-SMA) is the most common form accounting for up to 95% of the SMA cases, and is caused by homozygous deletion/mutation of the survival motor neuron 1 (SMN1) gene, localized on chromosome 5q12-q13 (Lefebvre et al., 1995; Wirth, 2000). The 5q-SMA is classified into different types based on onset and severity of symptoms. The most severe form of 5q-SMA (type I) has an onset within the first 6 months of life, permanent inability to seat without support and usually death within 2 years (Iannaccone et al., 1993; Rudnik-Schöneborn et al., 1996). The intermediate form of 5q-SMA (type II) has an onset between 6 and 18 months of life, ability to seat but not to walk and death usually occurs after the age of 2 years. A milder form is the 5q-SMA type III with an onset at around 3 years of age, ability to stand and walk but because of progressive muscle weakness, the patients mostly are wheelchair-bound as the disease progresses (Zerres and Rudnik-Schöneborn, 1995; Zerres et al., 1995). The 5q-SMA type IV, also called “adult SMA,” is considered the mildest and less severe form with a late onset of around 30 years of age (Zerres et al., 1995). All the mentioned forms of 5q-SMA commonly share proximal muscle weakness with slow progression in case of type II-IV SMA.
In contrast to autosomal recessive 5q-SMA, the autosomal dominant SMA (ADSMA) forms are less frequent, milder, with vague progression and probably, even under-diagnosed. In approx. 70% of individuals with ADSMA the genetic cause is still unknown. However, due to remarkable advances in next generation sequencing and gene discovery, the number of newly described causative genes increases continuously.
Beside autosomal dominant SMA lower extremity-predominant 1 (SMALED1, OMIM #58600) caused by mutations in dynein cytoplasmic 1 heavy chain 1 protein (DYNC1H1; OMIM *600112), we and others have recently identified heterozygous variants in BICD2 (OMIM *609797) as causative for autosomal dominant SMA, lower extremity-predominant, 2, (SMALED2; OMIM #615520) and hereditary spastic paraplegia (HSP) (Neveling et al., 2013; Oates et al., 2013; Peeters et al., 2013).
Clinical Features of Individuals Carrying Mutations in BICD2
Individuals carrying heterozygous missense variants in BICD2 exhibit muscle weakness and atrophy predominantly of the proximal lower limbs. However, in some cases the distal lower limbs are also affected, and in few others upper limbs are additionally compromised. The initial reasons for clinical counseling are commonly the difficulties in walking (waddling gait and toe walking), and delayed motor milestones. Contractures are reported frequently, and few patients present congenital hip dysplasia. Notoriously, many patients show evident wasting of the lower limbs and a very broad upper body, which resembles a bodybuilder-like shape. Symptoms are usually present at birth or appear in early childhood, but some cases with adult onset have been also described (Table 1; Neveling et al., 2013; Oates et al., 2013; Peeters et al., 2013; Synofzik et al., 2014; Rossor et al., 2015). The course of disease is slowly progressive or non-progressive. Due to the mild phenotype, patients are sometimes diagnosed as SMA type IV, the mildest form of the classical 5q-SMA. However, most cases of SMALED2 present a congenital or early onset with foot deformities and joint contractures, and difficulties when began walking (Frijns et al., 1994; Adams et al., 1998; Oates et al., 2012; Neveling et al., 2013). One infant who died of other causes at 14 months of age showed in the post-mortem examination decreased number of anterior horn cells in the lumber and cervical spine with no peripheral nerve pathology confirming a SMA (Oates et al., 2012).
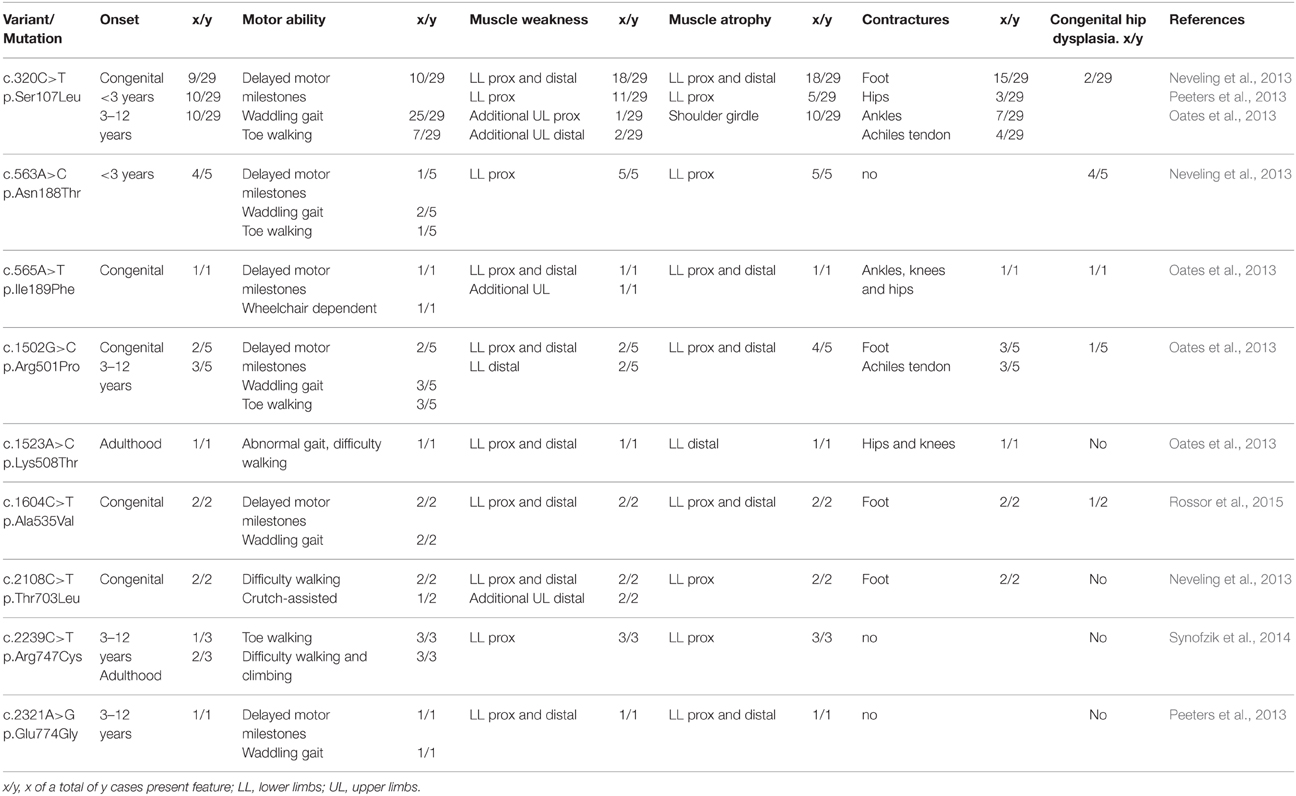
Table 1. Clinical summary of SMALED2 patients carrying BICD2 substitutions.
Two families affected by an HSP phenotype associated to BICD2 variants, have been described (Oates et al., 2013; Novarino et al., 2014). In the first family, the affected individuals presented features in adulthood, and showed lower-limb spasticity and hyperreflexia as is typical for HSP. It is also reported, a slow progression of contractures, weakness, and wasting (Oates et al., 2013). Regarding the association of this specific variant with the development of HSP but not SMALED2, it would be of particular interest to exclude the involvement of secondary disease-causing genes, resulting then in a multigenic disease, instead of a monogenic form. The second family with HPS linked to BICD2 is consanguineous, and the affected individuals carry the homozygous variant in BICD2, c.G1823A, p.S608L (Novarino et al., 2014). This variant was identified by whole-exome sequencing in combination with network analysis, and is the only homozygous variant reported in BICD2 so far.
Numerous experimental attempts have been made leading to a better understanding of the physiological role of BICD2 and yielded first insights into possible effects of variants in BICD2 on a cellular and molecular level (Table 2). However, further investigation is needed to fully understand the pathomechanism of how mutations in BICD2 can cause SMALED2 and rarely HSP-like phenotype. Next, we summarize the mutation spectrum and functional knowledge about BICD2, and focus on Golgi fragmentation as a cellular consequence of certain BICD2 mutations, as a possible mechanism to explain failure in neuronal development and maintenance.

Table 2. Summary of possible molecular mechanism involved in SMALED2 due to BICD2 substitutions.
Genetic Spectrum of Pathogenic Variants Found in BICD2 in Patients with SMALED2
BICD2 localizes in chromosomal region 9q22.31 and encodes a canonical isoform of 824 amino acids. All the variants found in patients with SMALED2 are heterozygous and lead to single amino acid substitutions (Figure 1). The most common variant reported is the c.320C>T, p.Ser107Leu, found in 29 of 49 cases corresponding to 5 of 13 families described with SMALED2. This variant is located within a CpG dinucleotide (Neveling et al., 2013; Oates et al., 2013; Peeters et al., 2013; Bansagi et al., 2015; Rossor et al., 2015). The cytosine-guanine (CpG) dinucleotide is considered a hotspot for pathological variants. Cytosine is subject for methylation however, spontaneous deamination of 5-methylcytosine may occur, yielding thymine instead (Shen et al., 1994).
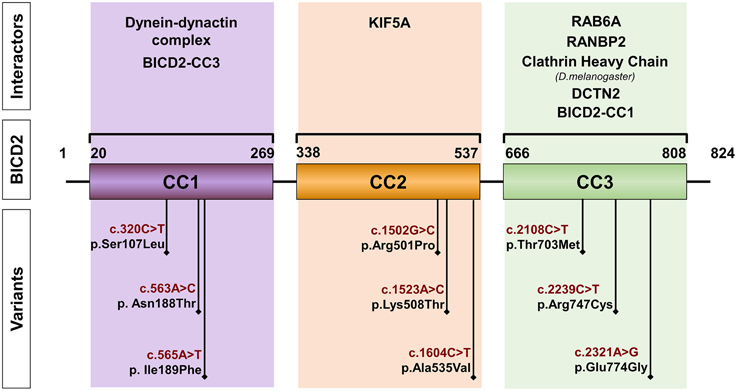
Figure 1. Schematic representation of BICD2. The diagram depicts the coiled-coil domains (CC) of BICD2 indicating the corresponding interacting partners. The position of variants, found in patients with SMALED2 is shown (Neveling et al., 2013; Oates et al., 2013; Peeters et al., 2013; Synofzik et al., 2014; Rossor et al., 2015).
BICD2 is Highly Conserved Among Species
Bicaudal-D (BicD) was identified first in Drosophila melanogaster in a mutant screen for dominant maternal-effect proteins. Loss-of-function mutations in BicD interfere with the determination of the oocyte, while gain-of-function substitutions (e.g., p.Glu224Lys, p.Phe684Ile) disrupt the establishment of anterior and posterior polarity giving rise to bicaudal (two tails) embryos (Mohler and Wieschaus, 1986; Steward, 1987; Schüpbach and Wieschaus, 1991). BicD null mutations are recessively lethal (Ran et al., 1994). Further studies show that BicD is a component of dynein-based transport and is implicated in the transport of mRNA (e.g., clathrin heavy chain and osk) to specific cellular regions of the fly during oogenesis and embryogenesis (Figure 2; Bullock and Ish-Horowicz, 2001). Experimental evidence suggested that BicD, together with clathrin heavy chain, mediates endocytosis during oocyte development (Vazquez-Pianzola et al., 2014). BicD is also involved in the regulation of bi-directional transport of lipid droplets (Larsen et al., 2008).
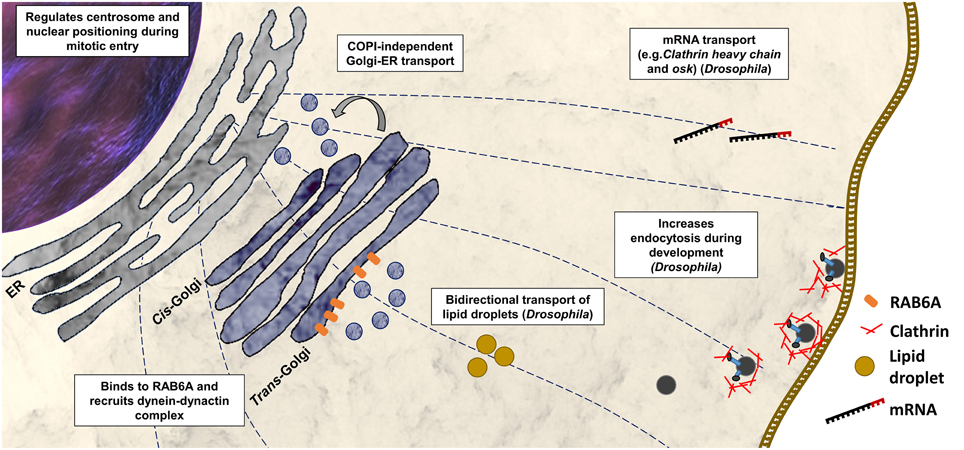
Figure 2. Functions of BICD2 in the cell. Cellular process with BICD2 involvement as described from studies in mammals or Drosophila are shown. BICD2 binds to RAB6A and recruits the dynein-dynactin complex to the trans-Golgi to facilitate COPI-independent Golgi-ER transport (Hoogenraad et al., 2001, 2003; Matanis et al., 2002). Prior mitosis (during G2 phase), BICD2 binds to a component of the nuclear pore complex, the RANBP2, and recruits dynein-dynactin to keep centrosomes tethered to the nucleus (Splinter et al., 2012). Studies in D. melanogaster have shown that BicD participates in mRNA transport (Bullock and Ish-Horowicz, 2001), endocytosis together with clathrin heavy chain (Vazquez-Pianzola et al., 2014), and in the regulation of bi-directional transport of lipid droplets (Larsen et al., 2008).
There is only one BicD gene in invertebrates, while mammals have two homologs BICD1 and BICD2. BICD1 and BICD2 interact with the dynein-dynactin motor complex (Hoogenraad et al., 2001), and both have been associated with Golgi-ER transport (Matanis et al., 2002). Studies in Bicd2 knockout mice revealed, that BICD2 has an important role in neuronal cell migration (Figure 2) (Jaarsma et al., 2014). The Bicd2 knockout mice present severe developmental defects of the cerebellar cortex, however other brain regions and spinal cord appear normal (Jaarsma et al., 2014).
BICD2 Structure and Organization
Studies in Drosophila revealed that more than half of the BicD protein consists of heptad repeats. A heptad repeat is a repeating pattern of seven amino acids where hydrophobic residues are preferentially located at positions 1 and 4 (McLachlan and Karn, 1983). These heptad repeats mediate the packaging of one helix against another, forming coiled-coil (CC) structures made up of two or three protein molecules and resulting in homo- or heterodimers or in multimers (Bruccoleri et al., 1986). The BICD protein is predicted to consist of CC domains: the N-terminal domain contains the coiled-coil segment 1 (CC1) and the coiled-coil segment 2 (CC2), and the C-terminal contains the coiled-coil segment 3 (CC3). Studies in Drosophila and mammals suggested, that BICD interacts with itself forming homodimers (Oh et al., 2000; Hoogenraad et al., 2001). In the case of BICD2, the segment 1 of the N-terminal domain (CC1, amino acids 10–321) binds to the C-terminal segment (CC3, amino acids 706–810) (Hoogenraad et al., 2001). A hypothetical model proposes that as a result of this interaction between segments, the soluble free BICD2 folds up, and only when C-terminal segment engages in an interaction with other proteins, the N-terminal segment of the BICD2 becomes available for interaction with other proteins (Hoogenraad et al., 2001). These protein interactions seem to be determining for the cellular distribution of BICD2 and the fulfillment of its specific function (Figures 1, 2).
The Role of BICD2 in the Golgi Apparatus and Dynein-dynactin Complex
One of the most studied binding partners of BICD2 is the dynein-dynactin complex. Cytoplasmic dynein is the major responsible motor protein for transporting a large variety of cargos toward the minus ends of microtubules (Kardon and Vale, 2009). Dynactin is a protein complex that stimulates dynein processivity and participates in cargo binding (Holleran et al., 1998; Schroer, 2004). Dynein and dynactin directly bind to each other through the interaction of dynein intermediate chain and dynactin subunit p150 (Vaughan et al., 1995; Waterman-Storer et al., 1995; King et al., 2003). The N-terminal domain of BICD2 binds to the dynein-dynactin complex via interaction with the p50 subunit of the dynactin complex (Hoogenraad et al., 2001). Studies in vivo and in vitro suggested, that the N-terminus of BICD2 promotes a stable interaction between dynein and dynactin (Splinter et al., 2012). In this context it is particularly interesting to mention that mutations in cytoplasmic dynein DYNC1H1 causes SMALED1 (Harms et al., 2012; Tsurusaki et al., 2012; Peeters et al., 2015). See the Review Jaarsma and Hoogenraad (2015).
The C-terminal segment of BICD2 shows the highest degree of evolutionary conservation and is described as the cargo-binding domain (Hoogenraad et al., 2001; Terenzio and Schiavo, 2010). BICD2 is considered a linker protein that acts between a cargo bound to its C-terminal segment and the dynein-dynactin complex associated to its N-terminal segment. The C-terminal segment of BICD2 binds strongly to the active form of the RAB6A GTPase and is responsible for Golgi targeting (Hoogenraad et al., 2001, 2003).
RAB6A is part of a big RAB family of more than 60 members (Zerial and McBride, 2001). RAB proteins shift dynamically between an active membrane-associated GTP-bound form and an inactive cytosolic GDP-bound form (Pfeffer, 2001; Zerial and McBride, 2001). RAB proteins are associated with membranes via geranylgeranyl groups that are attached to cysteine residues at the C-terminus (Stenmark, 2009). Guanine nucleotide exchange factors (GEFs) are responsible for the activation of RAB proteins by catalyzing the exchange of GDP for GTP (Delprato et al., 2004; Stenmark, 2009). Experimental evidence suggested that GEFs are responsible for targeting the RAB proteins to specific membranes (Gerondopoulos et al., 2012; Blümer et al., 2013). The active, membrane-bound RAB proteins are involved in membrane traffic, by binding with specific proteins (Grosshans et al., 2006).
The small GTPase RAB6A is involved in intra-Golgi transport. Several studies support that RAB6A coordinates the retrograde COPI-independent Golgi-ER pathway, which is considered a recycling route for Golgi-resident glycosylation enzymes (Martinez et al., 1997; Girod et al., 1999; White et al., 1999; Storrie et al., 2000). The active form of RAB6A (GTP-bound) recruits BICD2 to the trans-Golgi membrane via direct interaction with the C-terminal domain of BICD2 (Hoogenraad et al., 2001; Matanis et al., 2002; Short et al., 2002; Bergbrede et al., 2009; Matsuto et al., 2015). Recent reports suggest that BICD2 may stabilize the active RAB6A, by inhibiting its GTPase activity and thus increasing the GTP-bound membrane-associated RAB6A (Matsuto et al., 2015). Once the C-terminal segment of BICD2 binds to RAB6A, the N-terminal segment of BICD2 becomes available and recruits the dynein-dynactin complex. This recruitment is considered a critical step for the microtubule retrograde traffic. In this manner, a coordinated action between RAB6A, BICD2 and the dynein-dynactin complex controls COPI-independent Golgi-ER transport (Matanis et al., 2002).
Hence BICD2 has an abundant CC structure, localizes at the Golgi complex, and interacts with a member of the RAB family of GTPases (RAB6A), it is considered to be a golgin (Barr and Short, 2003; Short et al., 2005; Goud and Gleeson, 2010).
Golgins are proteins associated with the Golgi apparatus and help to maintain the organized architecture of this dynamic organelle. One type of golgins consists of resident transmembrane-Golgi proteins which are necessary for tethering membranes together (Barr and Short, 2003). Another type of golgins corresponds to proteins that are recruited to the Golgi apparatus, and are associated with components of the trafficking machinery. BICD2 is included in this last class (Barr and Short, 2003).
Mutations in BICD2 cause Golgi Fragmentation
The effects of the BICD2 domains on the integrity of the Golgi apparatus have been studied in detail (Hoogenraad et al., 2001, 2003; Splinter et al., 2012). Although the C-terminal domain targets the BICD2 to the Golgi apparatus, the overexpression of the N-terminal segment perturbs the Golgi organization (Hoogenraad et al., 2001). Fragmentation of the Golgi apparatus has been associated with inhibition of dynein function (Burkhardt et al., 1997; Harada et al., 1998; Quintyne et al., 1999).
Based on the association of BICD2 with RAB6A and Golgi targeting, we performed immunofluorescent staining of the Golgi apparatus of fibroblast cells derived from the patients carrying heterozygous mutations in BICD2.
We observed a prominent fragmentation of the Golgi apparatus in primary fibroblast cells harboring the p.Thr703Leu substitution (C-terminal) and a milder fragmentation in cells with the p.Asn188Thr substitution (N-terminal) (Neveling et al., 2013) (Figure 3). The individuals carrying the mutation p.Thr703Leu show a more severe phenotype including congenital contractures, while the individuals carrying the mutation p.Asn188Thr do not present contractures. Thus, in the case of these two cell lines showing Golgi fragmentation, the grade of fragmentation seems to correlate with the severity of the disease (Neveling et al., 2013). Although, Golgi fragmentation is not a common feature in all the cell lines carrying mutations in BICD2 (Oates et al., 2013; Peeters et al., 2013).
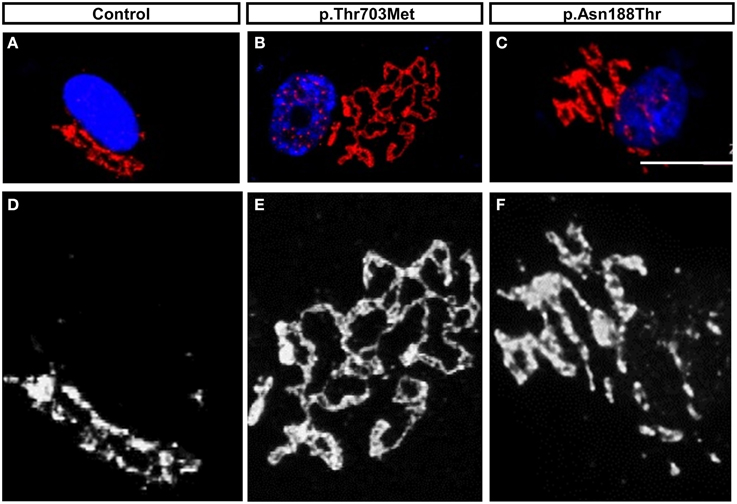
Figure 3. The mutations in BICD2, p.Thr703Leu and p.Asn188Thr, triggered Golgi-fragmentation in Primary Fibroblast Cells. (A–C) Modified data as published previously (Neveling et al., 2013), showing the Golgi apparatus of control fibroblasts and patients fibroblasts. The Golgi apparatus was immunostained with antibody against the Golgi marker ML160. (D–F) Magnification in monochrome of (A–C) for better visualization and comparison. Please note the prominent unfolded and disperse structure of the Golgi apparatus in cells carrying the mutation p.Thr703Leu and the intermedium or mild dispersion in the cells carrying the mutations p.Asn188Thr. For detail information and quantification, please read (Neveling et al., 2013).
The BICD2 mutations p.Thr703Leu and p.Asn188Thr may cause fragmentation of the Golgi apparatus due to conformational changes of BICD2 that alter protein function and affect the structure of the Golgi. How amino acid substitution at the positions Thr703 and Asn188 impact intramolecular interactions and probably protein folding remains elusive, as there is no crystalloid structure of BICD2 or the region harboring these mutations available. The alteration in the Golgi structure could also be a secondary effect of alteration in the microtubule organization. The integrity and positioning of the Golgi apparatus seems to have an important role in neuronal polarization and development (de Anda et al., 2005).
Potential Consequences of Golgi Fragmentation on Neuronal Development and Maintenance
Proper development of neurons is vital to fulfill and maintain the precise connectivity of the nervous system. Neurons are among the most polarized cell types and consist of a cell body from which specialized structures emerge, the neurites. In a very coordinated process, only one of the neurites grows long becoming an axon, while the other neurites acquire dendritic identity (Goslin et al., 1989; Craig and Banker, 1994; Polleux and Snider, 2010).
Previous studies have shown that in completely undifferentiated neurons, the centrosomes, Golgi apparatus and late/recycling endosomes cluster together to the area where the first neurite will form, which is in turn opposite from the plane of the last mitotic division (de Anda et al., 2005). The coordinated activity of these organelles is necessary for the polarization of the neuron.
Besides axon development, it has been suggested that the Golgi apparatus has an important role in dendrite maintenance and shape (Horton and Ehlers, 2003; Lewis and Polleux, 2012; Ori-McKenney et al., 2012). Non-neuronal cells contain one Golgi apparatus while many neurons contain several Golgi, the somatic Golgi and the dendritic Golgi centers called “Golgi outposts.” These dendritic Golgi outposts function similarly to the somatic Golgi, providing dendrites with substantial secretory capacity. The Golgi outposts appear during early neuronal differentiation, and may form as fragments derived from somatic Golgi that expand into dendrites, as the dendrites grow (Horton and Ehlers, 2003). The Golgi structure, dynamics and localization seem to have an important effect on neuronal development and maintenance, and an alteration in any of them could trigger degeneration or failure in correct maturation.
Additionally to SMALED2, other motor neurodegenerative diseases like ALS, have been associated with Golgi fragmentation (Gonatas et al., 1992; Mourelatos et al., 1994). However, it is still not clear why motor neurons but not other neuronal cells are affected. This effect might be attributable, to specific requirements of motor neurons during development that are different from other neuronal populations.
In conclusion, pathogenic substitutions in BICD2 causes SMALED2 with an autosomal dominant inheritance. Patients with SMALED2 show slowly to non-progressive muscle weakness of proximal and distal mainly of lower extremities, with a quite broad clinical spectrum. The majority of patients show congenital or early onset with foot deformities and joint contractures. However, some patients present a late onset and have very mild and non-progressive appearance. Commonly seems to be a wide aperture of the upper body (body-building shape). BICD2 is a golgin adaptor protein. The various BICD2 substitutions found in patients with SMALED2 impair interaction to other binding partners such as dynein-dynactin, RAB6A etc. and thus impair most likely cellular processes such as axonal transport, vesicular transport, Golgi integrity etc, which however needs further intensive functional studies. It is not understood why these mutations cause specifically loss of spinal motor neurons, since BICD2 is expressed in all neurons, is essential in neuronal migration and in positioning of the nucleus in the cell. There is also a quite broad phenotypic variability even in patients with the same mutation, suggesting the influence of modifying factors, which also need to be investigated.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The research leading to these results has received funding from the European Community's Seventh Framework Program FP7/2007-2013 under grant agreement no 2012-305121 (Project acronym NeurOmics), Deutsche Forschungsgemeinsschaft (Wi-945/14-2; Wi-945/16-1 and RTG 1960 to BW) and Center for Molecular Medicine Cologne (C11) to BW.
References
Adams, C., Suchowersky, O., and Lowry, R. B. (1998). Congenital autosomal dominant distal spinal muscular atrophy. Neuromuscul. Disord. 8, 405–408. doi: 10.1016/S0960-8966(98)00042-X
Bansagi, B., Griffin, H., Ramesh, V., Duff, J., Pyle, A., Chinnery, P. F., et al. (2015). The p.Ser107Leu in BICD2 is a mutation ‘hot spot’ causing distal spinal muscular atrophy. Brain. doi: 10.1093/brain/awv159. [Epub ahead of print].
Barr, F. A., and Short, B. (2003). Golgins in the structure and dynamics of the Golgi apparatus. Curr. Opin. Cell Biol. 15, 405–413. doi: 10.1016/S0955-0674(03)00054-1
Bergbrede, T., Chuky, N., Schoebel, S., Blankenfeldt, W., Geyer, M., Fuchs, E., et al. (2009). Biophysical analysis of the interaction of Rab6a GTPase with its effector domains. J. Biol. Chem. 284, 2628–2635. doi: 10.1074/jbc.M806003200
Blümer, J., Rey, J., Dehmelt, L., Mazel, T., Wu, Y. W., Bastiaens, P., et al. (2013). RabGEFs are a major determinant for specific Rab membrane targeting. J. Cell Biol. 200, 287–300. doi: 10.1083/jcb.201209113
Bruccoleri, R. E., Novotny, J., Keck, P., and Cohen, C. (1986). Two-stranded alpha-helical coiled-coils of fibrous proteins: theoretical analysis of supercoil formation. Biophys. J. 49, 79–81. doi: 10.1016/S0006-3495(86)83601-3
Bullock, S. L., and Ish-Horowicz, D. (2001). Conserved signals and machinery for RNA transport in Drosophila oogenesis and embryogenesis. Nature 414, 611–616. doi: 10.1038/414611a
Burkhardt, J. K., Echeverri, C. J., Nilsson, T., and Vallee, R. B. (1997). Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J. Cell Biol. 139, 469–484. doi: 10.1083/jcb.139.2.469
Craig, A. M., and Banker, G. (1994). Neuronal polarity. Annu. Rev. Neurosci. 17, 267–310. doi: 10.1146/annurev.ne.17.030194.001411
de Anda, F. C., Pollarolo, G., Da Silva, J. S., Camoletto, P. G., Feiguin, F., and Dotti, C. G. (2005). Centrosome localization determines neuronal polarity. Nature 436, 704–708. doi: 10.1038/nature03811
Delprato, A., Merithew, E., and Lambright, D. G. (2004). Structure, exchange determinants, and family-wide rab specificity of the tandem helical bundle and Vps9 domains of Rabex-5. Cell 118, 607–617. doi: 10.1016/j.cell.2004.08.009
Frijns, C. J., Van Deutekom, J., Frants, R. R., and Jennekens, F. G. (1994). Dominant congenital benign spinal muscular atrophy. Muscle Nerve 17, 192–197. doi: 10.1002/mus.880170210
Gerondopoulos, A., Langemeyer, L., Liang, J. R., Linford, A., and Barr, F. A. (2012). BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr. Biol. 22, 2135–2139. doi: 10.1016/j.cub.2012.09.020
Girod, A., Storrie, B., Simpson, J. C., Johannes, L., Goud, B., Roberts, L. M., et al. (1999). Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat. Cell Biol. 1, 423–430. doi: 10.1038/15658
Gonatas, N. K., Stieber, A., Mourelatos, Z., Chen, Y., Gonatas, J. O., Appel, S. H., et al. (1992). Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Am. J. Pathol. 140, 731–737.
Goslin, K., Birgbauer, E., Banker, G., and Solomon, F. (1989). The role of cytoskeleton in organizing growth cones: a microfilament-associated growth cone component depends upon microtubules for its localization. J. Cell Biol. 109, 1621–1631. doi: 10.1083/jcb.109.4.1621
Goud, B., and Gleeson, P. A. (2010). TGN golgins, Rabs and cytoskeleton: regulating the Golgi trafficking highways. Trends Cell Biol. 20, 329–336. doi: 10.1016/j.tcb.2010.02.006
Grosshans, B. L., Ortiz, D., and Novick, P. (2006). Rabs and their effectors: achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. U.S.A. 103, 11821–11827. doi: 10.1073/pnas.0601617103
Harada, A., Takei, Y., Kanai, Y., Tanaka, Y., Nonaka, S., and Hirokawa, N. (1998). Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J. Cell Biol. 141, 51–59. doi: 10.1083/jcb.141.1.51
Harms, M. B., Ori-McKenney, K. M., Scoto, M., Tuck, E. P., Bell, S., Ma, D., et al. (2012). Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 78, 1714–1720. doi: 10.1212/WNL.0b013e3182556c05
Holleran, E. A., Karki, S., and Holzbaur, E. L. (1998). The role of the dynactin complex in intracellular motility. Int. Rev. Cytol. 182, 69–109. doi: 10.1016/S0074-7696(08)62168-3
Hoogenraad, C. C., Akhmanova, A., Howell, S. A., Dortland, B. R., De Zeeuw, C. I., Willemsen, R., et al. (2001). Mammalian Golgi-associated Bicaudal-D2 functions in the dynein-dynactin pathway by interacting with these complexes. EMBO J. 20, 4041–4054. doi: 10.1093/emboj/20.15.4041
Hoogenraad, C. C., Wulf, P., Schiefermeier, N., Stepanova, T., Galjart, N., Small, J. V., et al. (2003). Bicaudal D induces selective dynein-mediated microtubule minus end-directed transport. EMBO J. 22, 6004–6015. doi: 10.1093/emboj/cdg592
Horton, A. C., and Ehlers, M. D. (2003). Dual modes of endoplasmic reticulum-to-Golgi transport in dendrites revealed by live-cell imaging. J. Neurosci. 23, 6188–6199.
Iannaccone, S. T., Browne, R. H., Samaha, F. J., and Buncher, C. R. (1993). Prospective study of spinal muscular atrophy before age 6 years. DCN/SMA Group. Pediatr. Neurol. 9, 187–193. doi: 10.1016/0887-8994(93)90082-N
Jaarsma, D., and Hoogenraad, C. (2015). Cytoplasmic dynein and its regulatory proteins in Golgi pathology in nervous system disorders. Front. Neurosci. 9:397. doi: 10.3389/fnins.2015.00397
Jaarsma, D., Van den Berg, R., Wulf, P. S., Van Erp, S., Keijzer, N., Schlager, M. A., et al. (2014). A role for Bicaudal-D2 in radial cerebellar granule cell migration. Nat. Commun. 5, 3411. doi: 10.1038/ncomms4411
Kardon, J. R., and Vale, R. D. (2009). Regulators of the cytoplasmic dynein motor. Nat. Rev. Mol. Cell Biol. 10, 854–865. doi: 10.1038/nrm2804
King, S. J., Brown, C. L., Maier, K. C., Quintyne, N. J., and Schroer, T. A. (2003). Analysis of the dynein-dynactin interaction in vitro and in vivo. Mol. Biol. Cell 14, 5089–5097. doi: 10.1091/mbc.E03-01-0025
Larsen, K. S., Xu, J., Cermelli, S., Shu, Z., and Gross, S. P. (2008). BicaudalD actively regulates microtubule motor activity in lipid droplet transport. PLoS ONE 3:e3763. doi: 10.1371/journal.pone.0003763
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. doi: 10.1016/0092-8674(95)90460-3
Lewis, T. L. Jr., and Polleux, F. (2012). Neuronal morphogenesis: golgi outposts, acentrosomal microtubule nucleation, and dendritic branching. Neuron 76, 862–864. doi: 10.1016/j.neuron.2012.11.019
Martinez, O., Antony, C., Pehau-Arnaudet, G., Berger, E. G., Salamero, J., and Goud, B. (1997). GTP-bound forms of rab6 induce the redistribution of Golgi proteins into the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 94, 1828–1833. doi: 10.1073/pnas.94.5.1828
Matanis, T., Akhmanova, A., Wulf, P., Del Nery, E., Weide, T., Stepanova, T., et al. (2002). Bicaudal-D regulates COPI-independent Golgi-ER transport by recruiting the dynein-dynactin motor complex. Nat. Cell Biol. 4, 986–992. doi: 10.1038/ncb891
Matsuto, M., Kano, F., and Murata, M. (2015). Reconstitution of the targeting of Rab6A to the Golgi apparatus in semi-intact HeLa cells: a role of BICD2 in stabilizing Rab6A on Golgi membranes and a concerted role of Rab6A/BICD2 interactions in Golgi-to-ER retrograde transport. Biochim. Biophys. Acta 1853, 2592–2609. doi: 10.1016/j.bbamcr.2015.05.005
McLachlan, A. D., and Karn, J. (1983). Periodic features in the amino acid sequence of nematode myosin rod. J. Mol. Biol. 164, 605–626. doi: 10.1016/0022-2836(83)90053-0
Mohler, J., and Wieschaus, E. F. (1986). Dominant maternal-effect mutations of Drosophila melanogaster causing the production of double-abdomen embryos. Genetics 112, 803–822.
Mourelatos, Z., Hirano, A., Rosenquist, A. C., and Gonatas, N. K. (1994). Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis (ALS). Clinical studies in ALS of Guam and experimental studies in deafferented neurons and in beta, beta'-iminodipropionitrile axonopathy. Am. J. Pathol. 144, 1288–1300.
Neveling, K., Martinez-Carrera, L. A., Hölker, I., Heister, A., Verrips, A., Hosseini-Barkooie, S. M., et al. (2013). Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am. J. Hum. Genet. 92, 946–954. doi: 10.1016/j.ajhg.2013.04.011
Novarino, G., Fenstermaker, A. G., Zaki, M. S., Hofree, M., Silhavy, J. L., Heiberg, A. D., et al. (2014). Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343, 506–511. doi: 10.1126/science.1247363
Oates, E. C., Reddel, S., Rodriguez, M. L., Gandolfo, L. C., Bahlo, M., Hawke, S. H., et al. (2012). Autosomal dominant congenital spinal muscular atrophy: a true form of spinal muscular atrophy caused by early loss of anterior horn cells. Brain 135, 1714–1723. doi: 10.1093/brain/aws108
Oates, E. C., Rossor, A. M., Hafezparast, M., Gonzalez, M., Speziani, F., MacArthur, D. G., et al. (2013). Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am. J. Hum. Genet. 92, 965–973. doi: 10.1016/j.ajhg.2013.04.018
Oh, J., Baksa, K., and Steward, R. (2000). Functional domains of the Drosophila bicaudal-D protein. Genetics 154, 713–724.
Ori-McKenney, K. M., Jan, L. Y., and Jan, Y. N. (2012). Golgi outposts shape dendrite morphology by functioning as sites of acentrosomal microtubule nucleation in neurons. Neuron 76, 921–930. doi: 10.1016/j.neuron.2012.10.008
Peeters, K., Bervoets, S., Chamova, T., Litvinenko, I., De Vriendt, E., Bichev, S., et al. (2015). Novel mutations in the DYNC1H1 tail domain refine the genetic and clinical spectrum of dyneinopathies. Hum. Mutat. 36, 287–291. doi: 10.1002/humu.22744
Peeters, K., Litvinenko, I., Asselbergh, B., Almeida-Souza, L., Chamova, T., Geuens, T., et al. (2013). Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance. Am. J. Hum. Genet. 92, 955–964. doi: 10.1016/j.ajhg.2013.04.013
Pfeffer, S. R. (2001). Rab GTPases: specifying and deciphering organelle identity and function. Trends Cell Biol. 11, 487–491. doi: 10.1016/S0962-8924(01)02147-X
Polleux, F., and Snider, W. (2010). Initiating and growing an axon. Cold Spring Harb. Perspect. Biol. 2:a001925. doi: 10.1101/cshperspect.a001925
Quintyne, N. J., Gill, S. R., Eckley, D. M., Crego, C. L., Compton, D. A., and Schroer, T. A. (1999). Dynactin is required for microtubule anchoring at centrosomes. J. Cell Biol. 147, 321–334. doi: 10.1083/jcb.147.2.321
Ran, B., Bopp, R., and Suter, B. (1994). Null alleles reveal novel requirements for Bic-D during Drosophila oogenesis and zygotic development. Development 120, 1233–1242.
Rossor, A. M., Oates, E. C., Salter, H. K., Liu, Y., Murphy, S. M., Schule, R., et al. (2015). Phenotypic and molecular insights into spinal muscular atrophy due to mutations in BICD2. Brain 138, 293–310. doi: 10.1093/brain/awu356
Rudnik-Schöneborn, S., Forkert, R., Hahnen, E., Wirth, B., and Zerres, K. (1996). Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: further delineation on the basis of SMN gene deletion findings. Neuropediatrics 27, 8–15. doi: 10.1055/s-2007-973741
Schroer, T. A. (2004). Dynactin. Annu. Rev. Cell Dev. Biol. 20, 759–779. doi: 10.1146/annurev.cellbio.20.012103.094623
Schüpbach, T., and Wieschaus, E. (1991). Female sterile mutations on the second chromosome of Drosophila melanogaster. II. Mutations blocking oogenesis or altering egg morphology. Genetics 129, 1119–1136.
Shen, J. C., Rideout, W. M. III, and Jones, P. A. (1994). The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 22, 972–976. doi: 10.1093/nar/22.6.972
Short, B., Haas, A., and Barr, F. A. (2005). Golgins and GTPases, giving identity and structure to the Golgi apparatus. Biochim. Biophys. Acta 1744, 383–395. doi: 10.1016/j.bbamcr.2005.02.001
Short, B., Preisinger, C., Schaletzky, J., Kopajtich, R., and Barr, F. A. (2002). The Rab6 GTPase regulates recruitment of the dynactin complex to Golgi membranes. Curr. Biol. 12, 1792–1795. doi: 10.1016/S0960-9822(02)01221-6
Splinter, D., Razafsky, D. S., Schlager, M. A., Serra-Marques, A., Grigoriev, I., Demmers, J., et al. (2012). BICD2, dynactin, and LIS1 cooperate in regulating dynein recruitment to cellular structures. Mol. Biol. Cell 23, 4226–4241. doi: 10.1091/mbc.E12-03-0210
Stenmark, H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525. doi: 10.1038/nrm2728
Steward, R. (1987). Dorsal, an embryonic polarity gene in Drosophila, is homologous to the vertebrate proto-oncogene, c-rel. Science 238, 692–694. doi: 10.1126/science.3118464
Storrie, B., Pepperkok, R., and Nilsson, T. (2000). Breaking the COPI monopoly on Golgi recycling. Trends Cell Biol. 10, 385–391. doi: 10.1016/S0962-8924(00)01818-3
Synofzik, M., Martinez-Carrera, L. A., Lindig, T., Schöls, L., and Wirth, B. (2014). Dominant spinal muscular atrophy due to BICD2: a novel mutation refines the phenotype. J. Neurol. Neurosurg. Psychiatr. 85, 590–592. doi: 10.1136/jnnp-2013-306777
Terenzio, M., and Schiavo, G. (2010). The more, the better: the BICD family gets bigger. EMBO J. 29, 1625–1626. doi: 10.1038/emboj.2010.78
Tsurusaki, Y., Saitoh, S., Tomizawa, K., Sudo, A., Asahina, N., Shiraishi, H., et al. (2012). A DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics 13, 327–332. doi: 10.1007/s10048-012-0337-6
Vaughan, K. T., Holzbaur, E. L., and Vallee, R. B. (1995). Subcellular targeting of the retrograde motor cytoplasmic dynein. Biochem. Soc. Trans. 23, 50–54. doi: 10.1042/bst0230050
Vazquez-Pianzola, P., Adam, J., Haldemann, D., Hain, D., Urlaub, H., and Suter, B. (2014). Clathrin heavy chain plays multiple roles in polarizing the Drosophila oocyte downstream of Bic-D. Development 141, 1915–1926. doi: 10.1242/dev.099432
Waterman-Storer, C. M., Karki, S., and Holzbaur, E. L. (1995). The p150Glued component of the dynactin complex binds to both microtubules and the actin-related protein centractin (Arp-1). Proc. Natl. Acad. Sci. U.S.A. 92, 1634–1638. doi: 10.1073/pnas.92.5.1634
Wee, C. D., Kong, L., and Sumner, C. J. (2010). The genetics of spinal muscular atrophies. Curr. Opin. Neurol. 23, 450–458. doi: 10.1097/WCO.0b013e32833e1765
White, J., Johannes, L., Mallard, F., Girod, A., Grill, S., Reinsch, S., et al. (1999). Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J. Cell Biol. 147, 743–760. doi: 10.1083/jcb.147.4.743
Wirth, B. (2000). An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 15, 228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9
Zerial, M., and McBride, H. (2001). Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117. doi: 10.1038/35052055
Zerres, K., and Rudnik-Schöneborn, S. (1995). Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch. Neurol. 52, 518–523. doi: 10.1001/archneur.1995.00540290108025
Keywords: BICD2, SMALED2, SMA, Golgi fragmentation, endocytosis, RAB6A, dynein, dynactin
Citation: Martinez-Carrera LA and Wirth B (2015) Dominant spinal muscular atrophy is caused by mutations in BICD2, an important golgin protein. Front. Neurosci. 9:401. doi: 10.3389/fnins.2015.00401
Received: 31 July 2015; Accepted: 09 October 2015;
Published: 05 November 2015.
Edited by:
Georg Haase, Centre National de la Recherche Scientifique, FranceReviewed by:
Albena Jordanova, University of Antwerp and Vlaams Instituut voor Biotechnologie, BelgiumOdile Boespflug-Tanguy, Paris Diderot University, France
Copyright © 2015 Martinez-Carrera and Wirth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brunhilde Wirth, brunhilde.wirth@uk-koeln.de