Article Text

Abstract
Frontotemporal dementia (FTD) is the second most common young-onset dementia and is clinically characterised by progressive behavioural change, executive dysfunction and language difficulties. Three clinical syndromes, behavioural variant FTD, semantic dementia and progressive non-fluent aphasia, form part of a clinicopathological spectrum named frontotemporal lobar degeneration (FTLD). The classical neuropsychological phenotype of FTD has been enriched by tests exploring Theory of Mind, social cognition and emotional processing. Imaging studies have detailed the patterns of atrophy associated with different clinical and pathological subtypes. These patterns offer some diagnostic utility, while measures of progression of atrophy may be of use in future trials. 30–50% of FTD is familial, and mutations in two genes, microtubule associated protein tau and Progranulin (GRN), account for about half of these cases. Rare defects in VCP, CHMP2B, TARDP and FUS genes have been found in a small number of families. Linkage to chromosome 9p13.2–21.3 has been established in familial FTD with motor neuron disease, although the causative gene is yet to be identified. Recent developments in the immunohistochemistry of FTLD, and also in amyotrophic lateral sclerosis (ALS), have led to a new pathological nomenclature. The two major groups are those with tau-positive inclusions (FTLD-tau) and those with ubiquitin-positive and TAR DNA-binding protein of 43 kDa (TDP-43) positive inclusions (FTLD-TDP). Recently, a new protein involved in familial ALS, fused in sarcoma (FUS), has been found in FTLD patients with ubiquitin-positive and TDP-43-negative inclusions. In this review, the authors discuss recent clinical, neuropsychological, imaging, genetic and pathological developments that have changed our understanding of FTD, its classification and criteria. The potential to establish an early diagnosis, predict underlying pathology during life and quantify disease progression will all be required for disease-specific therapeutic trials in the future.
- Frontotemporal dementia
- frontotemporal lobar degeneration
- tau
- TDP-43
- fus
- MAPT
- progranulin
- behavioural disorder
- dementia
- frontal lobe
- neurogenetics
- neuropathology
Statistics from Altmetric.com
- Frontotemporal dementia
- frontotemporal lobar degeneration
- tau
- TDP-43
- fus
- MAPT
- progranulin
- behavioural disorder
- dementia
- frontal lobe
- neurogenetics
- neuropathology
Introduction
Frontotemporal dementia (FTD) is the second most common early-onset dementia and is characterised clinically by progressive behavioural changes and frontal executive deficits and/or selective language difficulties. Although recognised over a century ago, the last few years have seen rapid advances in our understanding of FTD, its genetic causes and pathological substrates.1–5 In 1892, Arnold Pick described a patient with progressive aphasia and lobar atrophy,6 and in 1911, the presence of argyrophilic neuronal inclusions at neuropathological examination, later known as ‘Pick bodies’, was reported by Alois Alzheimer.7
The selective involvement of the frontal and/or temporal cortices with relative preservation of more posterior cerebral regions determines the presentation, and gives rise to the terms FTD as a clinical syndrome with distinct subtypes, and the term frontotemporal lobar degeneration (FTLD) to describe the pathological syndrome. The disease progresses from an insidious onset of behavioural change or language impairment and cognitive decline to a severe and more generalised dementia, accompanied by progressive cerebral hypometabolism and atrophy of frontal and temporal lobes preferentially.
The clinical spectrum of FTD encompasses distinct canonical syndromes: the behavioural variant of FTD (bvFTD) and the language variants, semantic dementia (SD) and progressive non-fluent aphasia (PNFA). There is also overlap of FTD with motor neuron disease (FTD-MND or FTD-ALS), as well as the parkinsonian syndromes, progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS).8 Recent advances in FTD have identified novel genetic defects and a chromosomal locus in hereditary forms of FTLD,9–14 as well as novel neuropathological associations, for example the proteins TAR DNA-binding protein of 43 kDa (TDP-43) and fused in sarcoma (FUS) are now recognised in the pathological classification of FTLD.15–19
In this review, we will discuss the different clinical variants, neuropsychological aspects, neuroimaging, hereditary forms, pathological subtypes and clinicopathological associations of FTD with the focus on recent developments.
Epidemiology
The exact prevalence of FTD remains uncertain, as there have been only a few studies, and these have produced a wide range of estimates. The highest prevalences have been reported from two independent studies in the UK and one Italian study with an estimated prevalence of FTD of 15–22 per 100 000 inhabitants aged 45–64 years,4 5 20 which was almost half the prevalence of AD in this age group.5 However, a study from The Netherlands estimated the prevalence of FTD to be significantly lower (9.4 per 100 000 in the age group of 60–69 years).21 The lower prevalence relative to AD in that series is consistent with some pathological series.22 23 The estimated prevalence in a Swedish population-based sample of 85-year-olds was 3.1 per 100 inhabitants.24
Two reported incidence studies of FTD were remarkably consistent: 3.5 and 4.1 cases per 100 000 person-years in the age-group of 45–64 years.25 26 There do not appear to be any clear gender differences in susceptibility.21 23 26 27
The average age at onset is around 50–60 years, although approximately 10% have an age at onset of over 70 years (up to 89 years).9 There is a wide range in durations of illness (2–20 years) partly reflecting different underlying pathologies. Patients with FTD-MND have the shortest survival with a mean of 3 years.9 28
Clinical presentation
BvFTD, SD and PNFA all share an insidious onset and inexorably progressive but variable decline. Each clinical syndrome is associated with topographically distinct cerebral involvement: with bvFTD associated with symmetrical (or right-sided) frontal and anterior temporal dysfunction, PNFA left frontotemporal dysfunction and SD anterior temporal (typically left more than right) deficits. BvFTD is the most common of these subtypes and accounts for about half of all cases9 29 without any clear differences in presentation between sporadic and familial bvFTD, or between late- and early-onset bvFTD.30 While all of the subtypes can occur in conjunction with motor neuron disease (FTD-MND), it is most commonly seen with bvFTD, occasionally with PNFA and only very rarely with SD.
Emotional blunting, loss of empathy, apathy, selfishness and neglect of personal hygiene are typical of bvFTD but may be seen in all subtypes.31 Other frequently reported symptoms are disinhibition, irritability, gluttony, altered preference for foods (particularly for sweets), wandering, pacing, motor and verbal stereotypies, and hoarding.31 It has been suggested that bvFTD can be subdivided into apathetic and disinhibited variants depending on initial presentation32; however, these symptoms frequently co-occur, and the usefulness of this distinction is questionable.30 A stereotyped-compulsive syndrome has been recognised as a third variant.33
A significant correlation with specific topographic patterns of atrophy, hypoperfusion or hypometabolism has been found for several of these symptoms. Apathy is associated with atrophy and dysfunction of the right anterior cingulate cortex and superior frontal gyrus,34 disinhibition with the right subgenual cingulate cortex and orbitofrontal cortex,34–36 overeating with an orbitofrontal–striatal circuit,37 and executive dysfunction with the dorsolateral and prefrontal cortex.38
The core features of current clinical criteria for bvFTD encompass an insidious onset and gradually progressive course, early disruption of social and personal conduct, early emotional blunting and lack of insight.2 3 Stereotypic behaviour, alterations in eating behaviour and loss of social awareness particularly support a diagnosis of FTD, while more posterior symptoms such as difficulty with spatial orientation and locating objects suggest Alzheimer's disease (AD).31 39 40 Apathy, mood changes and dysexecutive symptoms occur in both and have not been found to be effective discriminators of FTD from AD.39 41 42
The clinical criteria focus on behavioural changes rather than cognitive disturbances, and therefore might apply equally to a number of psychiatric syndromes, including (late-onset) depression and schizophrenia. However, virtually no studies have focused on the differentiation of bvFTD from psychiatric disorders. With this in mind, an interesting group of patients is the ‘non-progressive’, ‘benign’ or ‘slow’ bvFTD, who do not (or only slowly) progress over time, and do not show definite structural atrophy or hypometabolism many years from symptom onset.43–45 Behavioural symptoms may appear to progress according to carer description but without any measurable cognitive change.45 As these patients with a non-progressive bvFTD appear to have a normal life expectancy and seldom come to autopsy, the underlying pathology is still unknown.44 45 Possible diagnoses that have been suggested include decompensated Asperger syndrome or personality disorder, mild bipolar syndrome, or an otherwise previously undescribed neuropsychiatric syndrome with functional disruption of the same orbitofrontal–amygdala–polar network.44
Autopsy-proven studies have shown that the current clinical criteria correctly classify approximately 80–90% of bvFTD cases, whereas 3–17% are pathologically proven AD.46–49 However, the criteria lack sensitivity (37%) in the early phase of bvFTD.50 Therefore, revised criteria for bvFTD have been proposed in light of recent advances.51 The most important revisions are the incorporation of neuroimaging and genetic findings within the criteria and expansion of the role of supportive behavioural features for the diagnosis of bvFTD.51
The nosology of the language variants of FTD remains controversial. PNFA and SD are the canonical subtypes of what is collectively often termed primary progressive aphasia (PPA). Fluent speech, progressive impairment of single-word comprehension, preserved articulatory abilities and a multimodal breakdown of semantic memory are the characteristic features of SD.52 Patients with SD may show behavioural changes in the course of the disease similar to bvFTD.53 In particular, they may become egocentric and develop fixed daily routines.53 PNFA patients present with apraxia of speech and/or expressive agrammatism: single-word comprehension and object knowledge are relatively preserved, and behavioural symptoms are less common. However, there are patients with progressive language impairment who do not fit into the SD and PNFA: a third, more recently defined, subtype of PPA is the logopenic or phonological variant (LPA) which is characterised by a slow rate of speech output, word-finding difficulties, deficits in sentence repetition and occasional phonemic errors in spontaneous speech and naming, whereas motor speech, expressive grammar and single-word comprehension are relatively spared.52 54 It remains unclear whether there are further subtypes of PPA, although there is some evidence that patients with GRN mutations have a non-fluent PPA syndrome distinct to either PNFA or LPA.55
PPA subtypes have an association with different types of underlying pathology. SD is associated most commonly with FTLD-TDP type 117 pathology and only rarely with FTLD-tau or AD pathology.49 56 57 PNFA is commonly associated with FTLD-tau,56 58 59 although AD and, to a lesser extent, FTLD-TDP pathology have also been described.49 57 58 Finally, LPA is predominantly associated with AD pathology.58 59 Although these are relatively strong associations, they are not absolute, and it is currently not possible to predict with certainty the underlying pathology of specific PPA syndromes.57 However, using multimodal predictors including qualitative clinical features, neuropsychological test scores and atrophy on MRI improve the non-invasive prediction of the underlying pathology in non-fluent PPA (table 1).60
Motor neuron disease (MND) may occur early or late in the disease course in a subset of FTD patients.65 66 Muscle atrophy, weakness and fasciculations are often most prominent in the upper extremities and the tongue. The disease has a rapidly progressive course with a mean survival of 3 years. It is now accepted that FTD and MND are part of the same clinicopathological spectrum. A third of all FTD-MND cases have a positive family history for dementia, FTD, MND or FTD-MND. The causative gene defect in FTD-MND has yet to be discovered.
Some patients with predominantly right temporal lobe atrophy (RTLA) present with prominent behavioural changes, episodic memory disturbances, topographical disorientation and prosopagnosia.67 68 Patients with RTLA are usually diagnosed clinically with either bvFTD or SD.68 It has been suggested that patients with bvFTD and RTLA have FTLD-tau pathology, whereas patients with SD and RTLA have FTLD-TDP pathology.68
Neuropsychology and social cognition
Impairment of executive function including planning, organisation, judgement, problem solving and mental flexibility is characteristic of FTD,69 whereas memory, visual perception and spatial skills are usually relatively well preserved.50 70–74 However, executive dysfunction may be absent or overshadowed by pronounced behavioural changes in early disease and may also be seen in AD.75 Verbal fluency (letter and categorical) is usually impaired in bvFTD and PNFA, but to a lesser degree also in AD.76–78 In SD patients, semantic fluency is more impaired than letter fluency.76 78
Early episodic memory impairment, a characteristic feature of AD, has also been reported in pathologically proven bvFTD cases, and in patients with GRN gene mutations.79 80 Though orientation in time and place, delayed free recall and delayed recognition are more often impaired in AD than in FTD at initial assessment, it still remains difficult to differentiate FTD from AD in the early phase using standard neuropsychological tests.81
As standard neuropsychological tests cannot reliably differentiate bvFTD from AD,82–84 several investigators have explored the utility of emotional processing and social recognition tasks in the clinical diagnosis of FTD over recent years. Social dysfunction and emotional blunting commonly occur in FTD.44 75 85–91 Theory of Mind (ToM) tests require the interpretation of social situations and ascribing mental state to oneself and others, and may reveal subtle deficits not detected with standard neuropsychological testing.75 Recent reports suggest that the neural basis for ToM tasks, social cognition and empathy lies within the medio- and/or orbitofrontal cortex, which is affected early in bvFTD.92 Patients with bvFTD have impaired scores on these tests of social judgements and cognitive flexibility, and express concrete, literal interpretations.75 93 Performances in ToM tests do not correlate with executive functioning on standard neuropsychological testing in the early phase.85 94 95 However, in a more advanced stage, when atrophy spreads to dorsolateral frontal structures, the ToM ability and executive functioning become strongly related.75 93 94 Empathy as rated by care givers is clearly impaired in bvFTD and SD patients, and correlates with ToM tasks.85 96
In line with these observations, recognition of facial emotions is impaired in patients with bvFTD, in particular for negative emotions (anger, fear, sadness and disgust) (figure 1D).85 87–91 97 The same applies for the recognition of vocal emotions, in particular for anger and sad voices.89 Interpretation of sarcastic statements is impaired in FTD, and is correlated with the ability to recognise negative emotions.44 Self-conscious emotions, such as embarrassment and amusement, are another important aspect of emotion functioning which may be disrupted in FTD.98 99 Social cognition tests also seem to help to differentiate bvFTD patients with imaging abnormalities from the non-progressive bvFTD with normal neuroimaging.85 96

Imaging of frontotemporal dementia (FTD) subtypes. (A) Frontal atrophy on axial fluid attenuated inversion recovery MRI of a patient with behavioural variant of FTD (bvFTD). (B) Axial T1-weighted image with left temporal lobe atrophy in a patient with semantic dementia (SD). (C) Coronal T1-weighted MR image of a patient with progressive non-fluent aphasia (PNFA) and left inferior frontal and superior temporal atrophy. (D) Axial T1-weighted MR image in a patient with predominant right temporal lobe atrophy.
It will be interesting to investigate further whether impaired social cognition is a very early feature of familial FTD as has already been described in a single case study of a presymptomatic mutation carrier; studying presymptomatic mutation carriers may allow identification of sensitive (even preclinical) cognitive predictors of decline and its neural substrate.100
Neuroimaging
Patients with FTD classically have frontal and temporal atrophy, and hypometabolism which is often asymmetrical. In the clinical setting this is often best seen using volumetric structural MRI scans (with coronal T1 images being particularly useful for assessing asymmetry) or with functional imaging using either FDG-PET or, less commonly, HMPAO-SPECT. More recently, however, neuroimaging studies have aimed to refine these initial findings, mostly in clinical cohorts, but also in pathologically and genetically confirmed FTD.
Studies of mild bvFTD in clinically defined cohorts show involvement particularly of frontal and paralimbic areas,101 namely the anterior cingulate cortex and frontal insula as well as medial frontal and orbitofrontal cortices, hippocampus, striatum and thalamus, more in the right than in the left hemisphere (figure 1A). With increasing disease severity, more diffuse atrophy in similar areas is seen with involvement of more lateral frontal areas and subsequently more posterior temporal and anterior parietal atrophy.102 It remains unclear whether this pattern of atrophy is a feature of bvFTD independent of the underlying pathology (which is heterogeneous) or whether different pathologies have distinct patterns of atrophy. Unfortunately, there are currently no studies which directly compare all of the pathological subtypes. Patients with FUS pathology seem to have a similar pattern of frontal paralimbic atrophy to that described above but in addition have severe caudate involvement compared with FTLD-tau or FTLD-TDP.103–105 Studies comparing genetically defined FTD patients with either GRN or microtubule associated protein tau (MAPT) mutations 106 107 have described different patterns between the two groups: GRN mutations are associated with asymmetrical frontal, temporal and inferior parietal lobe atrophy, whereas MAPT mutations are associated with relatively symmetrical anteromedial temporal lobe and orbitofrontal grey matter atrophy.106 107 The presence of early parietal lobe atrophy in GRN mutations, a feature which may distinguish such cases from other FTD patients, has been shown in studies of presymptomatic mutation carriers.100 108 Whether patients with different mutations in the same gene have distinct patterns of atrophy is unclear, although one small study suggests there may be a difference between patients with MAPT mutations that affect splicing of exon 10 (more medial temporal lobe atrophy) compared with mutations that do not affect splicing of exon 10 (more lateral temporal lobe atrophy).109 Bringing these findings together, one study that used a cluster analysis to investigate bvFTD suggested there may be four types of bvFTD anatomically: a temporal-dominant subtype associated with MAPT mutations; a temporofrontoparietal subtype that can be associated with GRN mutations but also with other pathologies such as corticobasal degeneration (CBD); as well as frontal-dominant and frontotemporal subtypes.110 Larger studies of pathologically proven patients are needed to confirm these findings.
Early voxel-based morphometry (VBM) studies of SD showed asymmetrical atrophy of the anterior and inferior temporal lobes,111 112 usually affecting the left more than the right hemisphere (figure 1B). These findings were supported by subsequent region-of-interest (ROI) studies of the temporal lobe, which identified involvement particularly of the temporal pole and anterior parts of the entorhinal cortex, fusiform gyrus, inferior temporal gyrus, amygdala and hippocampus with relative sparing of the superior temporal gyrus.113 114 Most studies have used clinically defined cohorts, but one study looking at measurement of cortical thickness in patients with left greater than right temporal lobe atrophy showed a similar pattern of involvement in a pathologically confirmed cohort of patients, all with FTLD-TDP.115 This study also showed that areas within the left hemisphere outside the temporal lobe are involved with increasing disease severity, namely orbitofrontal, inferior frontal, insular and anterior cingulate cortices.115 Increasing involvement of the temporal lobe in the right hemisphere is seen with disease progression.115 116 A mirror-image pattern of initial atrophy and disease progression seems to occur in those with right greater than left temporal lobe involvement (RTLA).117 Although SD is characteristically FTLD-TDP pathologically, in a small number of cases, Pick's disease (FTLD-tau) and occasionally AD pathology can be seen. One small study showed similar patterns of atrophy in the FTLD-TDP and FTLD-tau but with a qualitatively different pattern in those with AD who had mostly hippocampal involvement, lack of the knife-edge anterior temporal atrophy seen in the other groups and without the sparing of the superior temporal gyrus.118
Studies of PNFA are fewer and more heterogeneous, which reflects the clinical heterogeneity of this group. As with SD, there is asymmetrical involvement with more atrophy in the left hemisphere and most significant involvement of the inferior frontal lobe and anterior insula (figure 1C).52 116 119 120 With increasing severity, there is involvement of left superior temporal, middle and superior frontal and anterior parietal lobes.116 ROI studies have also shown involvement of the caudate in PNFA.121 There are few pathologically confirmed studies of PNFA, and these are usually in mixed pathological groups, but they show similar findings to the clinical cohort studies.116 122 Some small studies suggest there may be different patterns of atrophy in PNFA patients with PSP clinically (and therefore likely pathologically) compared with those without PSP,123 and also in those with GRN mutations (FTLD-TDP pathologically) compared with those without.63 More detailed studies of PNFA subgroups will be needed to confirm these findings (figure 1).
Being able to distinguish FTD from AD is important clinically, and recent studies have suggested that atrophy or cortical thinning of precuneus, posterior cingulate, posterior temporal and parietal areas is characteristic of AD pathology independent of clinical diagnosis and is therefore helpful in distinguishing those with atypical AD presentations (which may include bvFTD or a progressive aphasia) from those with FTLD pathology.124–126 Clinically, however, VBM or cortical thickness studies are unlikely to be available, and simpler techniques such as visual rating scales have been developed which can help to differentiate FTD from AD.127 More sophisticated methods using techniques such as support vector machines are being developed which allow automatic classification of patients into FTD or AD groups with little user input necessary, although currently these are computationally demanding.128 Another possibility is to use support vector machine-based MRI analyses that integrate grey matter and diffuse tensor imaging (DTI), which has shown accurate pathological or CSF-defined categorisation of FTLD and AD.129
A different neuroimaging tool that accurately differentiates FTLD from AD is arterial spin labelling (ASL) perfusion MRI, which reveals non-invasive quantification of cerebral blood flow, without the use of ionising radiation as in SPECT or PET.130 Patients with AD pathologically can also be defined using amyloid molecular imaging (eg, PIB-PET)131 but the availability of such scans is currently limited to a few large research centres.
One of the more novel concepts to emerge from recent neuroimaging studies of FTD using the technique of resting-state fMRI is the idea that FTD is caused by degeneration within specific intrinsic functional connectivity networks that are selectively vulnerable to FTLD pathologies.132 Consistent with earlier VBM findings in structural MRI studies of bvFTD, resting-state fMRI studies show attenuated connectivity within an anterior ‘salience’ network of dorsal anterior cingulate and frontoinsular cortices which has connectivity to subcortical and limbic structures.133 In contrast, there appears to be enhanced connectivity in the more posterior ‘default’ network which has been shown to be affected in AD.133 These findings have been linked to specific neuropathological findings (early involvement of von Economo neurons in FTD),134 and further work will be needed to look at whether specific pathological subtypes can be linked to specific and distinct neural network degeneration.
Cerebrospinal fluid and plasma biomarkers
Currently cerebrospinal fluid (CSF) biomarkers have limited ability to identify FTD reliably. This might be explained by both the pathological heterogeneity and the large variation in neurodegenerative severity. Levels of CSF tau in FTD are normal, increased or even decreased.135 Levels of CSF phosphorylated tau are essentially normal in FTD, in contrast with AD. Levels of CSF amyloid β1–42 have been found to be either decreased or in the normal range. An indication of lower amyloid β1–40 levels in FTD compared with AD and control subjects, might be particularly useful to distinguish FTD patients from subjects without a neurodegenerative disorder.135
Decreased levels of progranulin protein are found in plasma, serum and cerebrospinal fluid (CSF) by ELISA, and may reliably differentiate GRN mutations carriers from non-carriers.136–140
It remains to be investigated if measurement of plasma or CSF levels of TDP-43 is useful diagnostically, as plasma phosphorylated TDP-43 levels have been found to be correlated with the extent of TDP-43 pathology in FTLD.141 142
Recent biomarker studies on CSF are using multianalyte profiling to derive novel biomarkers for neurodegenerative disorders and have delivered some promising neuropeptides (agouti-related peptide (AgRP), adrenocortotrophic hormone (ACTH), IL-17 and IL-23 and Fas) which are useful in distinguishing FTLD-TDP from FTLD-tau patients.143 144
Genetics
A positive family history has been found in 30–50% of patients with bvFTD, whereas patients with SD or PNFA have a much lower frequency.9–11 21 145 146 The heritability in FTD-MND differs between studies from 10 to 60%.9 11 146 An autosomal dominant mode of inheritance is found in 10–27% of all FTD patients.9–11 21 145 146
Genetic heterogeneity of FTLD is reflected by the identification of mutations in the MAPT and progranulin (GRN) genes in approximately 50% of the familial cases, whereas mutations in the valosin containing protein (VCP), charged multivesicular body protein 2B (CHMP2B), TAR-DNA binding protein (TARDP) and fused in sarcoma (FUS) genes are found in less than 5%. Familial FTD-MND has been linked to chromosome 9, but the causative gene defect has yet to be discovered.
Microtubule associated protein tau (MAPT)
More than 40 mutations in the MAPT gene have been identified in families with FTD and parkinsonism linked to chromosome 17q (FTDP-17) with accumulation of hyperphosphorylated tau protein in neurons and/or glial cells (http://www.molgen.ua.ac.be/FTDmutations).12 Alternative splicing of exons 2, 3 and 10 of the MAPT gene gives rise to six isoforms: three isoforms containing three amino-acid repeats (3R), and three isoforms with four repeats (4R).147 Mutations can be distinguished into missense mutations in exon 9–13 affecting the normal function of the tau protein to stabilise microtubules, and intronic and some coding mutations affecting the splicing of exon 10 at the mRNA level, resulting in a change in ratio of 3R to 4R tau isoforms.148
The mean age at onset is 55 years and usually shows a small intrafamilial variation between 45 and 65 years, although the disease may present before the age of 40 years or after 70 years in a few mutations.149 The mean duration of illness is approximately 9 years, but varies between 5 and 20 years. There exists a dementia-dominant phenotype with prominent behavioural changes including disinhibition and obsessive–compulsive behaviour149 and a parkinsonism-dominant phenotype with CBS or PSP-like syndromes.80 Patients may develop language problems—for example, mild semantic impairment during the illness.150
Progranulin
More than 60 mutations in the GRN gene on chromosome 17 (1.7 Mb centromeric to the MAPT gene) have been identified to date, and account for approximately 5–10% of all FTD patients, and up to 22% in familial FTD.9 13 14 151–153 Its frequency is similar to that of MAPT gene mutations in hereditary FTD.9 146 GRN gene mutations are occasionally reported in sporadic cases.9 151–153 Whether this is due to a low penetrance of the GRN mutation in one of the parents or to a spontaneous mutation in the patient is unknown.
GRN encodes the progranulin protein, which is a growth factor implicated in wound healing and tumour growth inflammation, and is abundantly expressed in specific neuronal subsets.154 The neuropathology of patients with GRN mutations is characterised by tau-negative, and ubiquitin- and TDP-43-positive inclusions.155
The mean age at onset is around 60 years, ranging from 35 to 89 years, with a penetrance of 90% by the age of 70 years.80 151 Within families, the onset age shows considerable variation with a difference of up to 20 years between consecutive generations.9 80 The mean duration is 8 years (range 3–22 years).
Apathy and social withdrawal are the most common behavioural changes. Twenty-five per cent of patients present with early isolated language dysfunction, suggestive of an anomic non-fluent type (without motor speech impairment) and with relatively early single word comprehension impairment.156 Hallucinations and delusions are frequently reported.157–159 Episodic memory deficits occur in 10–30%, and may lead to the clinical diagnosis of amnestic variant of MCI or together with parietal deficits, such as dyscalculia, visuospatial dysfunction and limb apraxia to AD.157 158 160–162
Extrapyramidal features are frequently seen and include CBS with limb apraxia, asymmetrical parkinsonism and dystonia.157 158 163–165 ALS is only a very rare part of the clinical spectrum within GRN families9 151 161 163 166—for example, it has been reported in a single patient of a large Canadian family.13 167
Other hereditary forms
The genetic heterogeneity of FTD is further emphasised by the rare occurrence of mutations in the VCP, CHMP2B, TARDP and FUS genes.168–171 VCP gene mutations are associated with inclusion body myopathy (90%), Paget's disease of the bone (45%) and FTD (38%) (IBMPFD), presenting between the age of 40 and 60 years.168 172
The clinical presentation of CHMP2B gene mutations consists of a frontal lobe syndrome and a more global cognitive impairment, with parkinsonism, dystonia, pyramidal signs and myoclonus later in the course of the disease.173 ALS has been reported in only two patients.174
TARDBP gene mutations on chromosome 1 are found in 5% of familial ALS,170 175–182 and occasionally in FTD or FTD-MND cases.170 171 Also, FUS gene mutations are found in 5% of the familial ALS cases,183–186 and in one clinical bvFTD patient, but not in FTD-MND.187 However, for the majority of the FTD-MND families, the genetic defect has yet to be identified. A number of these families have been linked to a locus on chromosome 9p13.2–21.3, but at time of writing an exhaustive sequencing of all genes in this region has not revealed any coding or splice-donor site mutations.188–193
There still remains a subgroup of FTD patients with a positive family history without known gene mutations. These patients usually have bvFTD and memory problems with or without MND, TDP-43 pathology and neuronal loss and gliosis of the cornu ammonis 1 and subiculum of the hippocampus (hippocampal sclerosis) at neuropathological examination.9
Genetic risk factors
Several investigators have tried to identify genetic risk factors for FTD. Homozygosity for the T allele of the SNP rs5848 was found to have a 3.2-fold increased risk for developing FTLD-TDP,194 but this observation could not be replicated in other studies.195 196 The same is true for three other SNPs of the GRN gene, which were initially found to be associated with younger onset age or shorter survival in FTLD or ALS.9 194 Also, an association of the Cystatin C gene (CST3) haplotype B, the ε4 allele of the apolipoprotein E gene (APOE), and heterozygosity of the codon 129 polymorphism of the prion protein gene (PNRP) could not be confirmed in further studies.197–201 Finally, SNPs in the Ubiquitin associated protein 1 (UBAP1) gene have been associated with FTD,202 which was supported by a reduced mRNA expression from the disease-associated haplotype in a quantitative analysis.202
Recently, an international genome-wide association study (GWAS) with pathologically proven FTLD-TDP patients has demonstrated a significant association with three SNPs within the TMEM106B gene on chromosome 7p21.203 TMEM106B variants also contribute to genetic risk for FTLD-TDP in individuals with GRN gene mutations.203 TMEM106B encodes an uncharacterised transmembrane protein of 274 amino acids.203 Expression data showed increased TMEM106B expression in the frontal cortex of FTLD-TDP than in controls, suggesting that increased TMEM106B expression in the brain might be linked to mechanisms of disease in FTLD-TDP and that risk alleles at TMEM106B confer genetic susceptibility by increasing gene expression.203
Genetic screening in clinical practice
The benefit of genetic screening in FTD depends on the strength of the family history and the clinical subtype. Genetic defects, either MAPT or GRN, are most often found in patients with an autosomal dominant form of bvFTD.9 146 Genetic screening in SD is unlikely to be useful, although patients who develop semantic impairment later in the illness may have a MAPT gene mutation,9 146 whereas a GRN mutation can be found in a familial form of PNFA.9 146 156 Current gene defects are very rare in FTD-MND, and genetic screening is therefore likely not to be useful at present.9 146 Screening in sporadic patients will be of little value, as a very few mutations have been found in sporadic patients, except for those with a concealed or incomplete family history. In this latter group, careful consideration is necessary before embarking on genetic screening.146
Pathology
FTLD is the common underlying pathology of clinical FTD subtypes, and also includes ALS, PSP and CBD. The major pathological hallmark of FTLD is selective atrophy of the frontal and temporal cortex, with neuronal loss, gliosis and spongiosis of the superficial layers, especially of layer II. The nomenclature has been changed several times since it was first described by Arnold Pick over a 100 years ago.6 The term Pick's disease is now reserved for cases of FTLD with intraneuronal argyrophilic inclusions, the so-called Pick bodies, which consist of abnormal three-repeat tau protein (FTLD-tau).
FTLD is a neuropathologically heterogeneous disorder, which can be divided into two major subtypes; FTLD with tau-positive inclusions (FTLD-tau), and FTLD with ubiquitin-positive and TDP-43-positive, but tau-negative inclusions (FTLD-TDP).18 FTLD-tau includes patients with MAPT mutations, Pick's disease, PSP, CBD, argyrophilic grain disease (AGD) and multiple system tauopathy with dementia (MSTD).18 MAPT mutations are associated with different types of tau inclusions (Pick bodies, neurofibrillar tangles and pretangles) in the frontal and temporal cortex, hippocampus and subcortical nuclei, and sometimes in midbrain, brainstem, cerebellum and spinal cord.149 Glial tangles and coiled bodies in white matter are found in a few MAPT mutations and consist predominantly of four-repeat tau isoforms.149
FTLD-TDP is the second major subtype of FTLD, with ubiquitin-positive inclusions, which have the TDP-43 protein as major constituent.204 The further classification into four different FTLD-TDP subtypes according to the morphology and distribution of the inclusions15 16 can be predicted to some extent by the clinical picture: SD is strongly associated with abundant dystrophic neurites (type 1), FTD-MND with numerous neuronal cytoplasmatic inclusions in both superficial and deep cortical laminae (type 2), GRN mutations are characterised by numerous cytoplasmatic inclusions, dystrophic neurites and neuronal intranuclear inclusions (type 3), and VCP mutations are characterised by numerous intranuclear and infrequent number of neuronal cytoplasmatic inclusions and dystrophic neurites (type 4).15 17 It remains unclear what differences in underlying pathophysiology determine the distinction between these TDP-43 subtypes.
A small number of FTLD cases with ubiquitin-positive, TDP-43 negative pathology,205–208 have recently shown immunoreactivity with the FUS antibody.19 103 None of these FTLD-FUS cases had FUS gene mutations.208 FTLD-FUS cases are characterised by a young age at onset, bvFTD, negative family history and caudate atrophy on MRI.103 104 FUS-positive inclusions are also found in patients with neuronal filament inclusion disease (NIFID).209 NIFID patients mostly present with bvFTD symptoms, a negative family history and pyramidal and/or extrapyramidal movement disorder.209
The FUS protein contains 526 amino acids and is as a nuclear protein involved in DNA repair and the regulation of RNA splicing.183 184 Mutations in the FUS gene on chromosome 16 have emphasised its pathogenetic role in the clinicopathological spectrum of FTD and ALS.187
Finally, the pathological heterogeneity of FTLD has been further emphasised by cases with ubiquitin-positive, TDP-43 and FUS-negative inclusions, termed FTLD-UPS. Most of the FTLD-UPS cases carry a CHMP2B mutation,210 but there remain a few without CHMP2B mutations.208 Further research on FTLD-UPS is necessary to elucidate the full complement of FTLD pathologies.208
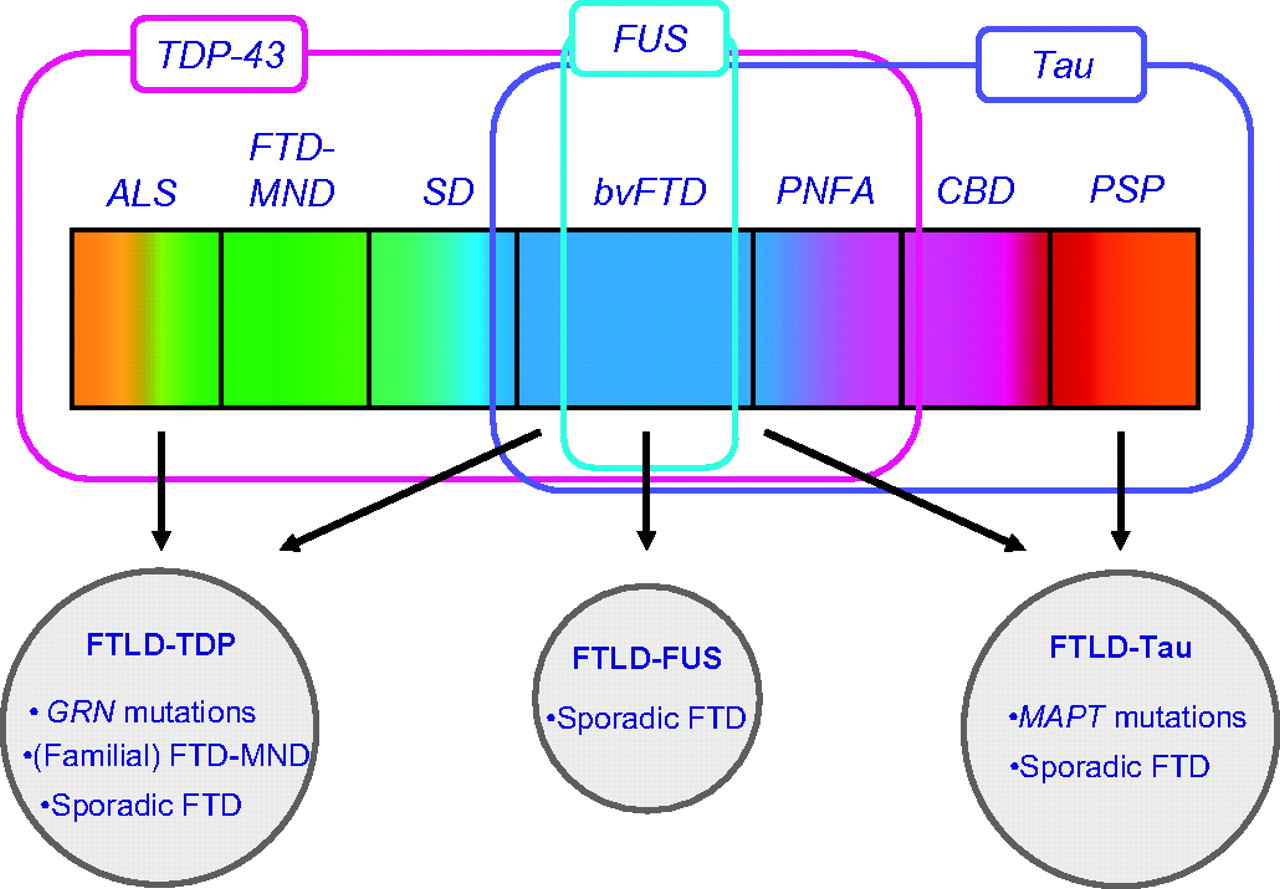
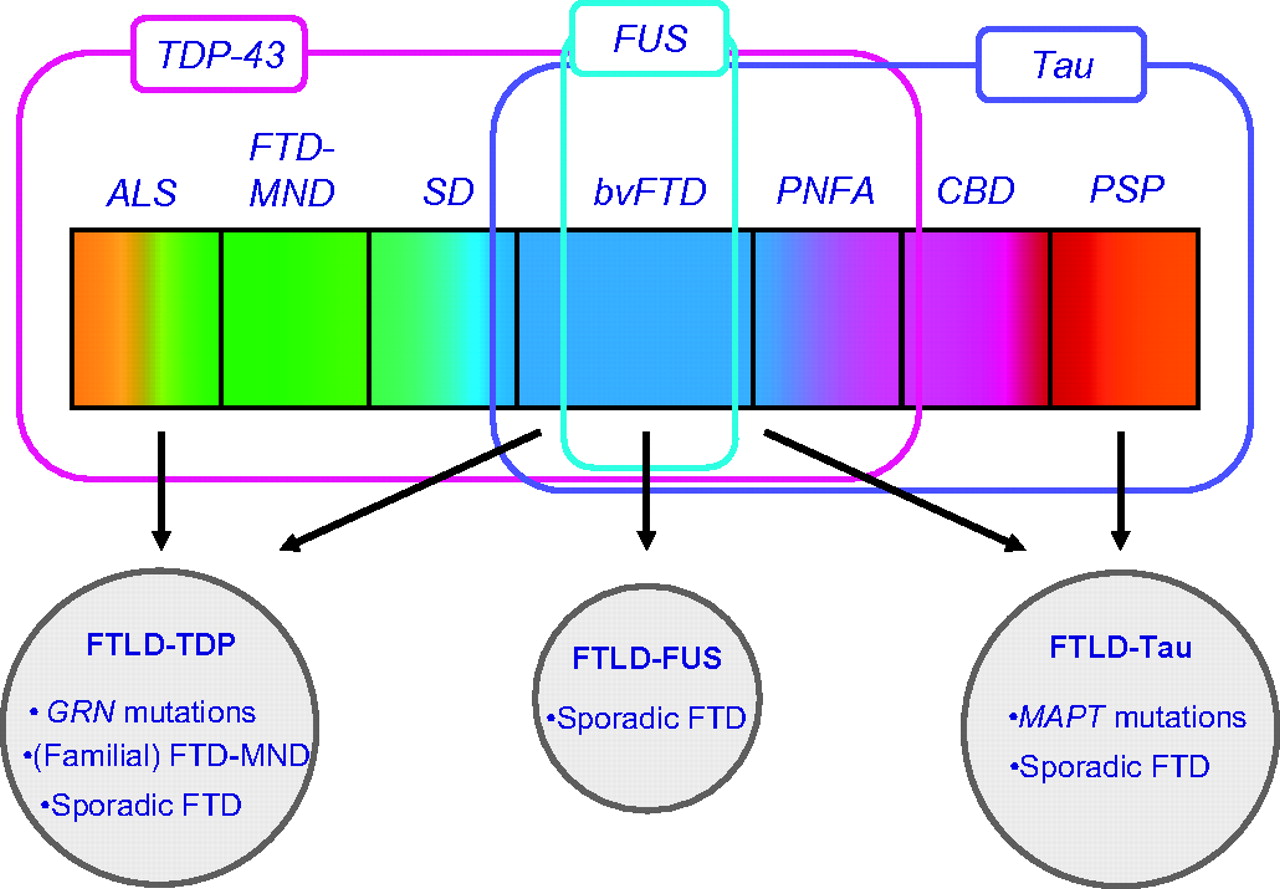
Future clinicopathological studies, including neuroimaging and genetics, are necessary to improve the prediction of the underlying pathology. In particular, the prediction of the underlying pathology in (sporadic) bvFTD will be important, as tau-, TDP-43- or FUS pathology could be the disease-modifying protein in these patients (figure 2).
{kind=link}
{kind=link}
Clinical, genetic and pathological spectrum of frontotemporal lobar degeneration.
Future directions
Important advances in the field research on FTD over the last decade have led to an impressive change in the clinical recognition of this disease. Future scientific efforts should focus on three major lines of research: (1) to improve the early detection of the disease; (2) to develop reliable markers in predicting the underlying pathology; and (3) to unravel its pathophysiology in order to develop therapeutic strategies preventing or delaying the disease process.
Concerning the clinical diagnosis, an international study has been initiated to revise the clinical criteria based on a large sample of pathologically proven cases. The aim is that neuroimaging and genetic data, and the most salient clinical features should be incorporated in a revised set of simplified criteria of bvFTD. A second clinical issue will be to monitor the progression of the disease in individual patients, which has now become available by the recent introduction of FTD rating scale (FRS)211 characterising the features of different severity stages. Finally, the use of social cognition tasks will help in the early detection of bvFTD and discrimination from non-progressive bvFTD. Their use offers us the opportunity to investigate the relative contributions of individual brain regions to social cognition in FTD.
Although relatively specific atrophy patterns have been found in clinical FTD subtypes, neuroimaging features as biomarkers for underlying pathology in bvFTD have yet to be determined. Support vector machines and arterial spin labelling are new neuroimaging tools to accurately differentiate FTD from AD. Another novel and promising neuroimaging technique is resting-state fMRI, which has shown changes in the salience network in FTD. An interesting question will be whether the early (or even presymptomatic) MAPT or GRN mutation carriers can be detected using this technique.
The differentiation of PPA into SD, PNFA and LPA has proven to be an important step in predicting underlying pathology in these groups: TDP-43 pathology is most commonly found in SD, tau-pathology in PNFA and AD pathology in LPA. Multimodal predictors, including clinical parameters, neuropsychological test scores and atrophy patterns, will improve the prediction of the underlying pathology in clinical PPA syndromes. However, radioactive compounds to detect tau or TDP-43 pathology in the brain with PET scanning would be of great help to differentiate bvFTD into its two major pathological subtypes during life. The recent recognition of the FUS protein as a pathological component of neuronal inclusions in a specific subtype of FTLD emphasises the existence of different pathways and will also contribute to further understanding of the underlying pathophysiology. Another strategy would be the development of new CSF biomarkers, which could be derived by large-scale proteomics analysis.
Several common (MAPT, GRN) and rare (VCP, CHMP2B, TARDBP, FUS) genetic factors have been found in hereditary FTD over recent years. However, we still have to identify one or more gene defects in familial FTD with and without MND. Whole exome sequencing as innovative genetic technique might reveal new genetic defects in small families with FTD and for pathologically well-characterised FTLD subtypes (such as FTLD-FUS). Identification of novel genetic defect(s) will help to understand the pathophysiology of TDP-43 in hereditary and probably also of the sporadic FTLD-TDP. A large genome-wide association study of more than 3000 DNA samples is currently under way and may hopefully reveal additional genetic factors with a small effect on the disease process.
All these small steps in unravelling the pathophysiology should finally lead to the development of therapeutic interventions for FTD.
References
Footnotes
Funding HS and JCvS are funded by Stichting Dioraphte and Hersenstichting. The Dementia Research Centre is an Alzheimer's Research Trust Co-ordinating centre and receives support from the NIHR Biomedical Research Centres scheme. NCF is funded by the Medical Research Council and is an NIHR Senior Investigator.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.