


Abstract
Bovine adrenal zona fasciculata (AZF) cells express bTREK-1 background K+ channels that set the resting membrane potential. Whole-cell and single-channel patch-clamp recording were used to compare five Ca2+ channel antagonists with respect to their potency as inhibitors of native bTREK-1 K+ channels. The dihydropyridine (DHP) Ca2+ channel antagonists amlodipine and niguldipine potently and specifically inhibited bTREK-1 with IC50 values of 0.43 and 0.75 μM, respectively. The other Ca2+ channel antagonists, including the DHP nifedipine, the diphenyldiperazine flunarizine, and the cannabinoid anandamide were less potent, with IC50 values of 8.18, 2.48, and 5.07 μM, respectively. Additional studies with the highly prescribed antihypertensive amlodipine showed that inhibition of bTREK-1 by this agent was voltage-independent and specific. At concentrations that produced near complete block of bTREK-1, amlodipine inhibited voltage-gated Kv1.4 K+ and T-type Ca2+ currents in AZF cells by less than 10%. At the single-channel level, amlodipine reduced bTREK-1 open probability without altering the unitary conductance. The results demonstrate that selected DHP L-type Ca2+ channel antagonists potently inhibit native bTREK-1 K+ channels, whereas other Ca2+ channel antagonists also inhibit bTREK-1 at higher concentrations. Collectively, organic Ca2+ channel antagonists make up the most potent class of TREK-1 inhibitors yet described. Because TREK-1 K+ channels are widely expressed in the central nervous and cardiovascular systems, it is possible that some of the therapeutic or toxic effects of frequently prescribed drugs such as amlodipine may be due to their interaction with TREK-1 K+ rather L-type Ca2+ channels.
TREK-1 belongs to the mechanogated, thermosensitive phospholipid- and fatty acid-activated family of two pore/four transmembrane segment, leak-type K+ channels (Lesage and Lazdunski, 2000). In humans, TREK-1 channels are widely expressed in CNS neurons, the small intestine, and ovaries (Lesage and Lazdunski, 2000). In bovine adrenocortical cells, where TREK-1 was first identified, these channels set the resting membrane potential, and they function pivotally in the physiology of adrenocorticotropic hormone- and angiotensin II-stimulated cortisol and aldosterone secretion (Enyeart et al., 2005).
In neurons, TREK-1 channels may mediate pain and thermosensitivity, confer neuroprotection in global ischemia, and serve as a target for general anesthetics (Franks and Honoré, 2004). Recently, a role for TREK-1 K+ channels in clinical depression has emerged when it was observed that TREK-1 knockout mice displayed a remarkable depression-resistant phenotype (Heurteaux et al., 2006). This finding suggests TREK-1 as a potential target for a new generation of antidepressants that would function by selective inhibition of these channels in CNS neurons.
TREK-1 K+ channels display a unique pharmacological profile. Significantly, they are relatively insensitive to standard antagonists of voltage-gated K+ channels, including tetraethylammonium (TEA) and 4-amino-pyridine (4-AP) (Gomora and Enyeart, 1999a). In contrast, we discovered that several agents that potently and preferentially inhibit low-voltage-activated T-type Ca2+ channels are also potent TREK-1 antagonists. Specifically, in studies on bovine AZF cells, diphenylbutylpiperidine (DPBP) antipsychotics, including penfluridol, fluspirilene, and pimozide, inhibit native bTREK-1 channels with IC50 values ranging from 187 to 354 nM (Gomora and Enyeart, 1999a). Penfluridol inhibited cloned bTREK-1 channels expressed in human embryonic kidney 293 cells with similar potency (Enyeart et al., 2002a). Mibefradil, a novel Ca2+ antagonist that preferentially inhibits T- over L-type Ca2+ channels, blocked native bTREK-1 channels with an IC50 value of 0.5 μM (Gomora and Enyeart, 1999b; Alloui et al., 2006).
The potent inhibition of bTREK-1 channels by these Ca2+ antagonists was unexpected, and it suggested that perhaps other organic Ca2+ antagonists might also inhibit bTREK-1. In this regard, DHP Ca2+ antagonists were of particular interest. DHPs are widely prescribed for the treatment of hypertension and angina (Hoffman, 2005; Michel, 2005). Although the DHPs uniformly and potently block L-type Ca2+ channels, it has been reported that several of these agents also inhibit T-type Ca2+ channels (Heady et al., 2001).
In patch-clamp recordings from bovine AZF cells, we have explored the inhibition of native bTREK-1 channels by several DHP Ca2+ antagonists, including one antagonist that potently inhibits T-type Ca2+ channels, and by two additional T-channel blockers (Fig. 1). We discovered that two DHP Ca2+ antagonists, amlodipine and niguldipine, inhibited bTREK-1 K+ channels at submicromolar concentrations. Amlodipine, the most potent bTREK-1 antagonist, did not inhibit T-type Ca2+ channels in these cells.
Materials and Methods
Tissue culture media, antibiotics, fibronectin, and fetal bovine sera (FBS) were obtained from Invitrogen (Carlsbad, CA). Coverslips were from Bellco (Vineland, NJ). Phosphate-buffered saline, enzymes, BAPTA, MgATP, and nifedipine (C17H18N2O6) were from Sigma-Aldrich (St. Louis, MO). Amlodipine (C20H25ClN2O5), niguldipine (C36H39N3O6), and flunarizine (C26H26F2N2) were obtained from BIOMOL Research Laboratories (Plymouth Meeting, PA), and anandamide (C22H37NO2) was purchased from Tocris Cookson Inc. (Ellisville, MO).

Chemical structure of Ca2+ channel and bTREK-1 K+ channel antagonists.
Isolation and Culture of AZF Cells. Bovine adrenal glands were obtained from steers (age 2–3 years) at a local slaughterhouse. Isolated AZF cells were obtained and prepared as described previously (Enyeart et al., 1996). After isolation, cells were either resuspended in DMEM/Ham's F-12 (1:1) with 10% FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and the antioxidants 1 μM tocopherol, 20 nM selenite, and 100 μM ascorbic acid (DMEM/Ham's F-12+) and plated for immediate use, or they were resuspended in FBS/5% dimethyl sulfoxide, divided into 1-ml aliquots, and stored in liquid nitrogen for future use. For patch-clamp experiments, cells were plated in DMEM/Ham's F-12+ in 35-mm dishes containing 9-mm2 glass coverslips. Coverslips were treated with 10 μg/ml fibronectin at 37°C for 30 min, and then they were rinsed with warm, sterile phosphate-buffered saline immediately before plating cells. Cells were maintained at 37°C in a humidified atmosphere of 95% air, 5% CO2.
Patch-Clamp Experiments. Patch-clamp recordings of K+ channel currents were made in the whole-cell and outside-out patch configuration. For whole-cell recordings, the standard pipette solution consisted of 120 mM KCl, 1 mM CaCl2, 2 mM MgCl2, 11 mM BAPTA, 10 mM HEPES, 1 mM ATP, and 200 μM GTP, with pH titrated to 7.1 using KOH. Pipette solution of this composition yielded a free Ca2+ concentration of 2.2 × 10–8 M, as determined by the Bound and Determined software program (Brooks and Storey, 1992). The external solution consisted of 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, and 5 mM glucose, with pH adjusted to 7.4 using NaOH. The standard external and pipette solutions used for single-channel recording from outside-out patches were identical to those used for whole-cell recordings.

Concentration-dependent inhibition of bTREK-1 by amlodipine. Whole-cell K+ currents were recorded from AZF cells at 30-s intervals in response to voltage steps to +20 mV applied from a holding potential of –80 mV with or without 10-s prepulses to –20 mV. After bTREK-1 reached a maximal amplitude, cells were superfused with amlodipine at concentrations ranging from 0.2 to 10 μM. A, K+ current records with (right traces) or without (left traces) 10-s prepulses to –20 mV. Numbers on traces correspond to currents recorded at times indicated in B. B, bTREK-1 amplitudes recorded with (○) or without (•) depolarizing prepulses are plotted against time. Amlodipine was superfused as shown at the concentrations indicated. Numbers on plot at left correspond to currents in A. C, inhibition curve: fraction of unblocked bTREK-1 current is plotted against amlodipine concentration. Data were fit with an equation of the form I/IMAX = 1/[1 + (B/IC50)X], where B is the amlodipine concentration. IC50 is the concentration that reduces bTREK-1 by 50%, and X is the Hill coefficient. Values are mean ± S.E.M. of indicated number of determinations.
Patch-clamp recordings of T-type Ca2+ currents were made in the whole-cell configuration. The standard pipette solution was 120 mM CsCl, 1 mM CaCl2, 2 mM MgCl2, 11 mM BAPTA, 10 mM HEPES, and 1 mM MgATP, with pH titrated to 7.2 with CsOH. The external solution consisted of 117 mM TEA-Cl, 5 mM CsCl, 10 mM CaCl2, 2 mM MgCl2, 5 mM HEPES, and 5 mM glucose, with pH adjusted to 7.4 with TEA-OH. All solutions were filtered through 0.22-μm cellulose acetate filters.
Recording Conditions and Electronics. AZF cells were used for patch-clamp experiments 2 to 12 h after plating. Typically, cells with diameters <15 μm and capacitances of 10 to 15 pF were selected. Coverslips were transferred from 35-mm culture dishes to the recording chamber (volume, 1.5 ml) that was continuously perfused by gravity at a rate of 3 to 5 ml/min. For whole-cell recordings, patch electrodes with resistances of 1.0 to 2.0 MΩ were fabricated from Corning 0010 glass (WPI, Sarasota, FL). These electrodes routinely yielded access resistances of 1.5 to 4.0 MΩ and voltage-clamp time constants of <100 μs. For single-channel recordings, patch electrodes with higher resistances (3–5 MΩ) were used. K+ currents were recorded at room temperature (22–25°C) according to the procedure of Hamill et al. (1981) using an EPC-7 patch-clamp amplifier (List Electronics, Darmstadt, Germany).
Pulse generation and data acquisition were done using a personal computer and pCLAMP software with Digidata 1200 interface (Molecular Devices, Sunnyvale, CA). Currents were digitized at 2 to 10 KHz after filtering with an eight-pole Bessel filter (Frequency Devices, Haverhill, MA). Linear leak and capacity currents were subtracted from current records using summed scaled hyperpolarizing steps of one-half to one-fourth pulse amplitude. Data were analyzed using pCLAMP (CLAMPFIT 9.2, FETCHAN 6.04, and PSAT 6.04) and SigmaPlot, version 10.0, software (Systat Software, Inc., Point Richmond, CA). Drugs were applied by bath perfusion, controlled manually by a six-way rotary valve. p values were calculated using Student's t test.

Amlodipine inhibition of bTREK-1 is voltage-independent and specific. A, voltage-independent inhibition. K+ currents were activated at 30-s intervals by voltage steps of varying size from a holding potential of –80 mV, before and after superfusing 10 μM amlodipine. bTREK-1 current amplitudes in the absence and presence of amlodipine are plotted against test potential. B, specificity: K+ currents were recorded in response to voltage steps to +20 mV applied at 30-s intervals from a holding potential of –80 mV. After bTREK-1 reached a maximal amplitude, cell was superfused with 10 μM amlodipine. Current traces are recorded at indicated times in the absence and presence of amlodipine. C, summary of experiments as described in B. Bars indicate percentage of Kv1.4 or bTREK-1 K+ current inhibited by 2 or 10 μM amlodipine as indicated. Values are mean ± S.E.M. of indicated number of separate determinations.
Results
Bovine AZF cells express voltage-gated, rapidly inactivating Kv1.4 K+ channels and bTREK-1 background K+ channels (Mlinar and Enyeart, 1993; Mlinar et al., 1993; Enyeart et al., 2002a). In whole-cell patch-clamp recordings, bTREK-1 amplitude increases over a period of minutes before reaching a stable maximum. Expression of bTREK-1 is enhanced when pipette solutions contain nucleotide triphosphates at millimolar concentrations (Enyeart et al., 2002a). The absence of time- and voltage-dependent bTREK-1 inactivation allows this current to be isolated in whole-cell recording using either of two voltage-clamp protocols. When voltage steps of 300-ms duration are applied from a holding potential of –80 mV, bTREK-1 can be measured near the end of a voltage step when the transient Kv1.4 current has inactivated (Fig. 2A, left traces). Alternatively, bTREK-1 can be selectively activated by an identical voltage step after a 10-s prepulse to –20 mV has completely inactivated Kv1.4 channels (Fig. 2A, right traces). Measurement of bTREK-1 by either method provided nearly identical results.
Inhibition of bTREK-1 by DHP Ca2+Channel Antagonists. Three DHP Ca2+ antagonists, amlodipine, niguldipine, and nifedipine, were compared with respect to their potency as inhibitors of bTREK-1 K+ channels in bovine AZF cells. Of these three antagonists, amlodipine and niguldipine inhibited bTREK-1 at submicromolar concentrations, whereas nifedipine was significantly less potent. In the experiments illustrated in Fig. 2, A and B, bTREK-1 grew to a stable maximal amplitude over a 15-min period before the cell was superfused with amlodipine at concentrations between 0.2 and 10 μM. Amlodipine specifically inhibited bTREK-1 in a concentration-dependent manner with an IC50 of 0.43 μM (Fig. 2C). Amlodipine is charged, but highly lipid soluble at physiological pH (Mason et al., 1989). Inhibition of bTREK-1 by amlodipine was slowly reversible with washing (Fig. 2B, right).
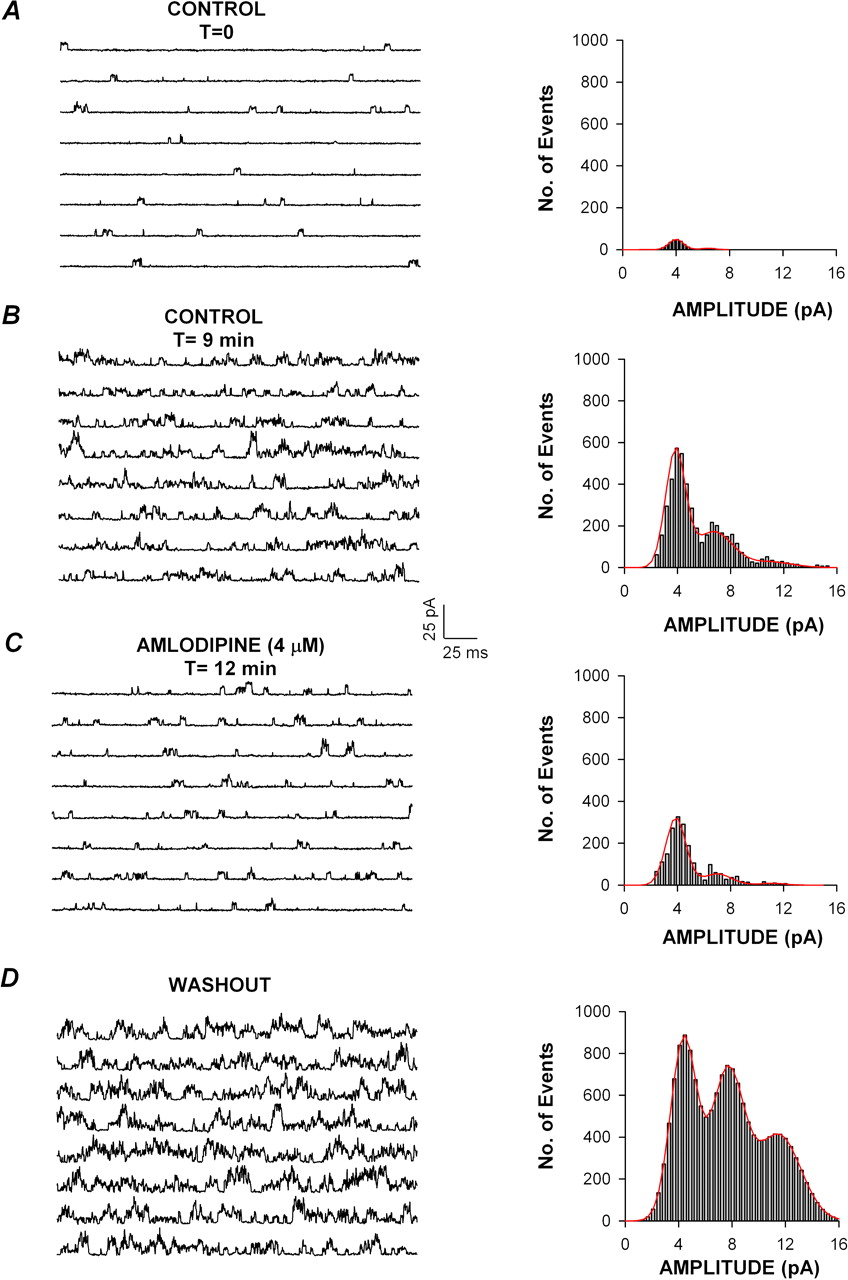
Effect of amlodipine on unitary bTREK-1 channel activity. Unitary bTREK-1 currents were recorded from excised patches in the outside-out configuration in response to voltage steps to +30 mV, applied from a holding potential of –40 mV. Each amplitude histogram was constructed from idealized channel opening obtained from unitary currents recorded during 80 to 96 separate voltage steps of 300-ms duration. Unitary current amplitudes were distributed into bins of 0.18-pA width. Currents were filtered at cut-off frequency of 2 kHz and sampled at 5 kHz. Similar results were obtained in each of four outside-out patches. Traces and corresponding amplitude histograms in control saline (A), in control saline after 9 min of recording to show run-up of current (B), in the presence of 4 μM amlodipine (C), and after washing with control saline (D). The continuous line in the histograms represents the first-order (A), second-order (B and C), or third-order Gaussian fit (D) of the data. Similar results were obtained in each of four outside-out patches.
Although it is a background or leak-type K+ channel, bTREK-1 activation is weakly voltage-dependent with open probability enhanced at more positive potentials (Enyeart et al., 1997). Amlodipine inhibited bTREK-1 with equal effectiveness over a wide range of test potentials. The experiment illustrated in Fig. 3A shows current-voltage relationships recorded before and after superfusing a cell with 10 μM amlodipine. In the presence of amlodipine, bTREK-1 was completely inhibited at all test potentials between –60 and +40 mV. In contrast, the rapidly inactivated Kv1.4 current was prominently expressed in the presence of amlodipine (Fig. 3A, right traces).
The selectivity of amlodipine as an inhibitor of bTREK-1 compared with Kv1.4 was quantitated by comparing Kv1.4 current amplitudes before and after inhibition of TREK-1 currents by amlodipine (Fig. 3B). At concentrations of 2 and 10 μM, amlodipine inhibited Kv1.4 K+ currents by only 4.3 ± 1.1% (n = 6) and 12.7 ± 6.9% (n = 4), respectively. By comparison, in the same experiments, bTREK-1 was inhibited by 76.4 ± 3.8% (n = 8) and 96.1 ± 1.9% (n = 5), respectively (Fig. 3C).

Niguldipine inhibition of bTREK-1. A and C, concentration-dependent inhibition of bTREK-1. Whole-cell K+ currents were recorded at 30-s intervals in response to voltage steps to +20 mV, applied from –80 mV with or without 10-s prepulses to –20 mV. After bTREK-1 reached a maximal amplitude, cells were superfused with niguldipine at concentrations from 0.2 to 10 μM. A, K+ current records with (right traces) or without (left traces) 10-s prepulses to –20 mV. Numbers on traces correspond to those on plot of bTREK-1 amplitudes at right. B, voltage-independent inhibition. K+ currents were activated by voltage steps of varying size from –80 mV before and after superfusing 10 μM niguldipine. bTREK-1 current amplitudes in the absence and presence of niguldipine are plotted against test potential. C, inhibition curve: fraction of unblocked bTREK-1 current is plotted against niguldipine concentration. Data were fit with an equation of the form I/IMAX = 1/[1 + (B/IC50)X], where B is niguldipine concentration, IC50 is the concentration that reduces bTREK-1 by 50%, and X is the Hill coefficient. Values are mean ± S.E.M. of indicated number of determinations. D, specificity: K+ currents were recorded in response to voltage steps to +20 mV from a holding potential of –80 mV. After bTREK-1 reached a maximal amplitude, cells were superfused with 2 or 10 μM niguldipine. Bars indicate percentage of Kv1.4 or bTREK-1 K+ current inhibited by 2 or 10 μM niguldipine as indicated. Values are mean ± S.E.M. of indicated number of separate determinations.
Amlodipine Reduces bTREK-1 Open Probability but Not Unitary Conductance. In single-channel recordings from excised outside-out patches, amlodipine reduced the activity of bTREK-1 channels without changing the amplitude of the unitary currents. Figure 4 shows unitary bTREK-1 currents, recorded from an excised outside-out patch in response to voltage steps to +30 mV. Under these conditions, a single type of K+ channel was typically active in the membrane patch. Histogram analysis of unitary current amplitudes showed a major peak with a mean of 4.10 ± 0.55 pA (Fig. 4A).
As in whole-cell recordings, bTREK-1 channel activity increased spontaneously with time in outside-out patches. As channel activity increased, histogram analysis showed several peaks, each with a mean amplitude a multiple of the first (Fig. 4B). However, in contrast to whole-cell recordings, bTREK-1 activity did not reach a stable maximal value in excised patches. In spite of continuous channel run-up, bTREK-1 channel activity was markedly inhibited upon superfusing 4 μM amlodipine (Fig. 4C). Although channel open probability was reduced by amlodipine, the unitary current amplitude remained essentially constant at 4.04 ± 0.81 pA. The inhibition of bTREK-1 activity was reversible with washing, revealing the continuous time-dependent increase in bTREK-1 activity (Fig. 4D). Overall, these results clearly demonstrate that amlodipine affects bTREK-1 gating, rather than permeation.
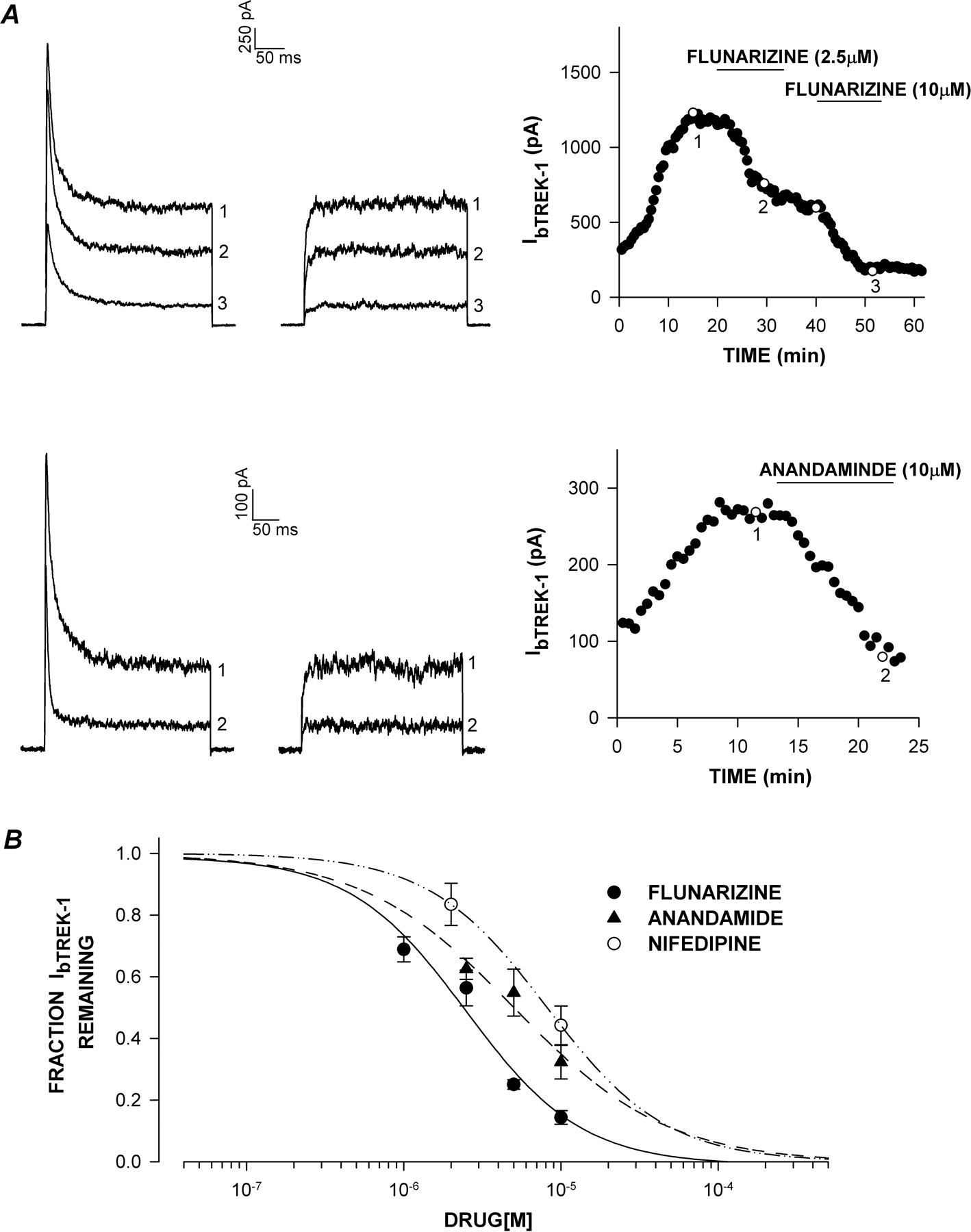
bTREK-1 inhibition by flunarizine, anandamide, and nifedipine. Whole-cell K+ currents were recorded from AZF cells at 30-s intervals in response to voltage steps to +20 mV applied from a holding potential of –80 mV with or without 10-s prepulses to –20 mV. After bTREK-1 reached maximal amplitude, cells were superfused with one of the three Ca2+ antagonists at concentrations from 0.5 to 10 μM. A, flunarizine and anandamide. K+ current records with (right traces) and without (left traces) 10-s prepulses to –20 mV in the presence of flunarizine (top) or anandamide (bottom). bTREK-1 amplitudes recorded with (○) or without (•) depolarizing prepulses are plotted against time at right. Numbers on plot correspond to those on current traces. B, inhibition curves. fraction of unblocked bTREK-1 current is plotted against niguldipine, flunarizine, or nifedipine concentration as indicated. Data were fit with an equation of the form I/IMAX = 1/[1 + (B/IC50)X], where B is the drug concentration, IC50 is the concentration that reduces bTREK-1 by 50%, and X is the Hill coefficient. Values are mean ± S.E.M. of from three to nine separate determinations.
Dwell time analysis of unitary bTREK-1 currents showed that under control conditions, channel kinetics could be described by single open (τo) and closed (τc) time constants. Amlodipine increased τc with little effect on the mean open time. In the experiment illustrated in Fig. 4, amlodipine increased τc from the control value of 10.9 ms to 18.4 ms. By comparison, τo in control saline (0.773) did not differ significantly from that determined in the presence of amlodipine (0.680 ms). The increase in τc observed in the presence of amlodipine occurred despite the spontaneous increase in single-channel activity. Accordingly, in the experiment shown, τc decreased markedly upon washing from 18.4 to 2.62 ms.
Effects of Other Ca2+Channel Antagonists on bTREK-1. Two other DHP Ca2+ antagonists, niguldipine and nifedipine, were compared with respect to their potency as inhibitors of bTREK-1. Although both of these agents inhibit L-type Ca2+ channels, only niguldipine has been reported to also block T-type channels with similar potency (Romanin et al., 1992). We found that niguldipine also potently inhibited bTREK-1 K+ channels with an IC50 value of 0.75 μM (Fig. 5, A and C). In contrast, nifedipine inhibited bTREK-1 only at much higher concentrations (IC50 value of 8.18 μM) (Fig. 6B).
The inhibition of bTREK-1 by niguldipine was voltage-independent, specific, and not easily reversed with washing. As illustrated in Fig. 5B, 10 μM niguldipine inhibited bTREK-1 with equal effectiveness at test potentials between –60 and +40 mV.
With regard to selectivity, niguldipine at concentrations of 2 and 10 μM inhibited Kv1.4 K+ currents by only 0.61 ± 0.39% (n = 5) and 3.79 ± 2.69% (n = 4), respectively. In contrast, in the same experiments, bTREK-1 was inhibited by 79.2 ± 2.92% (n = 7) and 94.7 ± 1.58% (n = 5), respectively (Fig. 5D).

Inhibition of T-type Ca2+ current by amlodipine. T-type Ca2+ currents were activated by short (10-ms) or long (300-ms) voltage steps to 0 or –5 mV, applied at 30-s intervals from a holding potential of –80 mV. After recording Ca2+ currents in standard saline, cells were superfused sequentially with amlodipine at 10 and 20 μM. A, T-type Ca2+ current records in response to 300-ms (left) or 10-ms (right) depolarizing steps, showing steady-state block of T-type current by amlodipine. Ca2+ current amplitudes are plotted against time in the presence of 10 and 20 μM amlodipine. C, bars indicate percentage of IT-Ca inhibited by 10 and 20 μM amlodipine. Values are mean ± S.E.M. of indicated number of separate determinations.
Flunarizine is a diphenyldiperazine that is used clinically to treat migraine and epilepsy. Flunarizine blocks L-, T-, and N-type Ca2+ channels (Heady et al., 2001; Santi et al., 2002). Two of the three subtypes of T-type Ca2+ channels are blocked by flunarizine at submicromolar concentrations (Santi et al., 2002). Flunarizine also inhibited bTREK-1 K+ channels with an IC50 value of 2.48 μM (Fig. 6, A and B).
Anandamide is an endogenous cannabinoid found in the brain that directly blocks two T-type Ca2+ channel subtypes, with IC50 values of approximately 1 μM (Chemin et al., 2001). Anandamide also inhibited bTREK-1 channels but less potently (IC50 value of 5.10 μM) (Fig. 6, A and B).
Amlodipine Inhibition of T-type Ca2+Current. Bovine adrenocortical cells express α1H (Cav3.2) T-type Ca2+ channels that are inhibited by submicromolar concentrations of DPBPs and mibefradil (Enyeart et al., 1993; Gomora et al., 2000; Schrier et al., 2001). Previous studies on native and cloned bTREK-1 demonstrated that these K+ channels were potently inhibited by the same organic Ca2+ channel antagonists that potently block T-type Ca2+ channels in bovine AZF cells (Gomora and Enyeart, 1999a,b; Enyeart et al., 2002a).
Experiments were done to determine whether amlodipine would also inhibit T-type Ca2+ current in AZF cells. The rapidly inactivating and slowly deactivating T-type Ca2+ current was recorded in response to either long or short depolarizing steps (Fig. 7A). At concentrations that produced near complete inhibition of bTREK-1, amlodipine was ineffective at blocking T-type current in these cells. Even at higher concentrations of 10 and 20 μM, amlodipine inhibited T currents by only 15.6 ± 2.0% (n = 5) and 28.8 ± 4.5% (n = 4), respectively (Fig. 7, B and C).
Discussion
In this study, it was discovered that selected DHP Ca2+ channel antagonists potently inhibit native bTREK-1 K+ channels. The most potent of these antagonists, amlodipine, inhibits bTREK-1 half-maximally at a concentration of 0.43 μM, ranking it among the most potent TREK-1 antagonists yet identified (Table 1). In this regard, it is interesting to note that among the 29 drugs listed in Table 1, the six most potent drugs are organic Ca2+ antagonists. Each of these antagonists inhibits TREK-1, with an IC50 value less than 1 μM. These Ca2+ antagonists inhibit TREK-1 at concentrations more than 10,000-fold lower than traditional antagonists of voltage-gated K+ channels, including 4-AP and TEA. The potent inhibition of bTREK-1 by amlodipine is of particular significance, because amlodipine is used clinically in the treatment of hypertension, and it is among the most frequently prescribed drugs in cardiovascular pharmacology.
Pharmacology of TREK-1 K+ channel inhibition
Comparison of T-Type Ca2+Channel and bTREK-1 K+Channel Inhibition. Previous studies suggested that TREK-1 was inhibited by Ca2+ antagonists that potently inhibit T-type Ca2+ channels (Gomora and Enyeart, 1999a,b; Enyeart et al., 2002a). The findings of this present study indicate that the previously noted correlation was coincidental. Specifically, amlodipine is at least 100-fold less potent than mibefradil and DPBPs as an inhibitor of T-type Ca2+ channels in AZF and other cells (Enyeart et al., 1993; Gomora et al., 2000; Heady et al., 2001; Santi et al., 2002). In contrast, amlodipine inhibits bTREK-1 with an IC50 value comparable with that of the DPBPs and mibefradil (Gomora and Enyeart, 1999a,b; Enyeart et al., 2002a). Although niguldipine is 50 to 100 times more potent than amlodipine at inhibiting T-type Ca2+ channels in various cells, it was slightly less potent than amlodipine as an inhibitor of bTREK-1 (Romanin et al., 1992; Stengel et al., 1998; Heady et al., 2001). In bovine AZF cells, 20 μM amlodipine inhibited T-type Ca2+ channels by only 29.0%, indicating that it was approximately 100-fold more potent as an inhibitor of bTREK-1.
Flunarizine is a relatively potent T-channel antagonist, and it inhibits α1G and α1I channels, with IC50 values less than one micromolar (Santi et al., 2002). However, flunarizine was 5-fold less potent than amlodipine as an inhibitor of native bTREK-1 channels. Likewise, anandamide, which inhibits α1H and α1I T-type currents, with IC50 values of approximately 1 μM, was 10-fold less potent than amlodipine as a bTREK-1 inhibitor (Chemin et al., 2001). Previously, 3 μM anandamide was reported not to significantly inhibit rat TREK-1 K+ channels expressed in COS cells (Maingret et al., 2001). In our experience, continuous run-up of TREK-1 activity in transfected cells makes it difficult to measure block at low antagonist concentrations.
Mechanism of bTREK-1 Inhibition by Amlodipine. cAMP inhibits hippocampal TREK-1 K+ channels by a mechanism that reportedly involves the interconversion between leak-type and voltage-dependent phenotypes (Bockenhauer et al., 2001). Consequently, cAMP was much less effective at inhibiting these neuronal K+ channels at increasingly positive test potentials. In contrast, amlodipine inhibited bTREK-1 with equal effectiveness over a wide range of test voltages. Although amlodipine did not alter the voltage-dependent activation of bTREK-1, single-channel recording suggested that inhibition occurred through an effect on channel gating rather than permeation. Specifically, although TREK-1 channel open probability was markedly reduced, the unitary conductance remained unchanged. Nevertheless, our results do not exclude the possibility that this charged DHP inhibits bTREK-1 by pore occlusion.
Amlodipine and TREK-1 in the Cardiovascular System. Amlodipine is one of the most effective Ca2+ antagonists prescribed in the treatment of hypertension (Fleckenstein et al., 1989). It is not known whether any of the actions of amlodipine on the cardiovascular system could be due to inhibition of TREK-1 channels. In humans, TREK-1 channels are primarily expressed in the brain, ovaries, and the small intestine (Lesage and Lazdunski, 2000). But TREK-1-like K+ channels have also been identified in the mammalian heart, including atria and ventricles, where they are modulated by β adrenergic agonists and ATP (Aimond et al., 2000; Terrenoire et al., 2001; Tan et al., 2002; Li et al., 2006). The role of TREK-1 channels in cardiovascular function has not been determined. At therapeutic concentrations, amlodipine and other Ca2+ antagonists may interact with TREK-1 K+ channels and L-type Ca2+ channels in the cardiovascular system.
TREK-1 Antagonists in the CNS and Depression. TREK-1 K+ channels are also widely distributed throughout the brain and spinal cord, with particularly high expression in the basal ganglia, hippocampus, and parts of the cerebral cortex (Hervieu et al., 2001; Talley et al., 2001). At the cellular level, TREK-1 channels are distributed over the entire neuronal membrane, including the cell body and processes. Although amlodipine is charged at physiological pH, it has an extremely high lipid-to-water partition coefficient (Mason et al., 1989). The possibility that amlodipine could produce CNS effects through interaction with neuronal TREK-1 channels cannot be excluded.
In this regard, the recent finding that deletion of TREK-1 in knockout mice produced a depression-resistant phenotype raises the possibility that drugs that potently block TREK-1 would have therapeutic value as antidepressants (Heurteaux et al., 2006). Interestingly, fluoxetine, a widely used antidepressant that acts primarily as a serotonin uptake inhibitor, also inhibits TREK-1 K+ channels (Kennard et al., 2005) (Table 1).
Several DHP Ca2+ channel antagonists, including nifedipine have been reported to possess antidepressant activity in tests on mice and rats (Czyrak et al., 1989; Cohen et al., 1997). However, amlodipine lacked activity in these animal models of depression (Cohen et al., 1997). Since amlodipine is more than 10 times more potent than nifedipine as a TREK-1 antagonist, its lack of antidepressant activity argues against a role of this or other TREK-1 blockers as antidepressants. Accordingly, other drugs that potently inhibit bTREK-1, including the DPBPs niguldipine and mibefradil, have not been reported to possess antidepressant properties.
Thus, although some overlap has been reported between inhibition of TREK-1 K+ channels and antidepressant activity for several drugs, including fluoxetine and selected DHPs, the correlation seems to be coincidental. It remains to be seen whether TREK-1 channel antagonists like amlodipine will modulate other TREK-1-related functions including nociception, neuroprotection, and anesthesia.
Footnotes
-
This work was supported by National Institutes of Health Grant R01-DK47875 (to J.J.E.).
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.125245.
-
ABBREVIATIONS: CNS, central nervous system; TEA, tetraethylammonium; 4-AP, 4-amino-pyridine; AZF, adrenal zona fasciculata; DPBP, diphenylbutylpiperidine; DHP, dihydropyridine; FBS, fetal bovine serum; BAPTA, 11,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; DMEM, Dulbecco's modified Eagle's medium.
- Received May 3, 2007.
- Accepted July 9, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}