


Abstract
This study examined the ability of the endocannabinoids 2-arachidonoyl glycerol (2-AG) and noladin ether as well as the synthetic cannabinoid CP-55,940 [(-)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl) cyclohexanol] to regulate three intracellular effectors via CB2 receptors in transfected Chinese hamster ovary cells. Although the three agonists regulate all effectors with equivalent efficacy, the rank order of potencies differs depending on which effector is evaluated. Noladin ether and CP-55,940 most potently inhibit adenylyl cyclase, requiring higher concentrations to stimulate the extracellular signal-regulated kinase subgroup of the mitogen-activated protein kinases (extracellular signal-regulated kinase-mitogen-activated protein kinase; ERK-MAPK) and Ca2+-transients. In contrast, 2-AG most potently activates ERK-MAPK, necessitating greater concentrations to inhibit adenylyl cyclase and even higher amounts to stimulate Ca2+-transients. Endocannabinoids also seem to be more “efficient” agonists at CB2 receptors relative to synthetic agonists. 2-AG and noladin ether require occupancy of less than one-half the number of receptors to produce comparable regulation of adenylyl cyclase and ERK-MAPK, relative to the synthetic cannabinoid CP-55,940. The CB2 antagonist 6-iodo-2-methyl-1-[2-(4-morpholinyl)-ethyl]-1H-indol-3-yl](4-methoxyphenyl)-methanone (AM630) reverses the actions of all agonists except Ca2+-transient stimulation by 2-AG. However, the effect of 2-AG on Ca2+-transients is attenuated by a second CB2 antagonist N-[(1S)-endo-1,3,3-trimethylbicyclo[2.2.1]heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-1-pyrazole-3-carboxamide (SR144528). This suggests that 2-AG stimulates Ca2+-transients by binding to sites on CB2 receptors distinct from those occupied by AM630 and the other cannabinoids examined. Agonists produce no effects in pertussis toxin-treated cells. In summary, cannabinoid agonists distinctly bind to CB2 receptors and display different rank order of potencies and fractional receptor occupancies for regulation of intracellular effectors. These data provide direct evidence for agonist-directed trafficking of response by endocannabinoids acting at CB2 receptors.
Δ9-Tetrahydrocannabinol is the main psychoactive constituent in the plant Cannabis sativa (marijuana) and produces its effects by activation of CB1 (Matsuda et al., 1990) and CB2 (Munro et al., 1993) cannabinoid receptors. CB1 receptors are expressed throughout the central nervous system (Herkenham et al., 1990), whereas CB2 receptors are expressed predominantly in immune cells and non-neuronal tissues (Galiegue et al., 1995). Both CB1 and CB2 receptors inhibit the activity of the intracellular effector adenylyl cyclase via pertussis toxin (PTX)-sensitive Gi/Goα-subunits (Howlett, 1985; Mechoulam et al., 1995) and stimulate the activity of the extracellular signal-regulated kinase subgroup of the mitogen-activated protein kinases (ERK-MAPK) through Gβγ subunits (Bouaboula et al., 1995, 1996). Stimulation of CB1 (Sugiura et al., 1997) and CB2 (Sugiura et al., 2000) receptors also results in transient increases intracellular free Ca2+ concentrations via a phospholipase-Cβ (PLCβ)-mediated mechanism in cellular models expressing endogenous cannabinoid receptors. Unlike CB1 receptors, CB2 receptors seem to lack the ability to regulate the activity of either voltage-gated Ca2+ or inward rectifying K+ channels (Felder et al., 1995).
Endocannabinoids are lipid-derived native ligands that activate CB1 and CB2 receptors (Di Marzo et al., 2004). To date, four have been recognized: anandamide, 2-AG, noladin ether, and virodhamine (Howlett et al., 2002). 2-AG is a full agonist at CB1 and CB2 receptors. Anadamide and virodhamine seem to act as partial agonists at both receptors. Noladin ether, once classified as a selective CB1 partial agonist (Hanus et al., 2001), has been shown recently to also fully activate CB2 receptors at nanomolar concentrations (Shoemaker et al., 2005).
Recent studies suggest that different endocannabinoids, acting through CB2 receptors, have distinct effects in regulating specific functions of immune and inflammatory cells. In particular, 2-AG induces pronounced migration and proliferation of these cells types, whereas other endogenously occurring or synthetically derived cannabinoids produce only modest or no effects at all (Jorda et al., 2002; Kishimoto et al., 2003; Walter et al., 2003; Carrier et al., 2004; Oka et al., 2004). The synthetic cannabinoid agonist CP-55,940 actually blocks 2-AG-induced migration of myeloid precursor cells (Alberich Jorda et al., 2004). In all studies, the actions of 2-AG are mediated specifically through activation of CB2 receptors, and, in cases where tested, involve stimulation of the intracellular effector ERK-MAPK (Kishimoto et al., 2003; Walter et al., 2003; Alberich Jorda et al., 2004; Carrier et al., 2004). Although presently unknown, the differential actions of these agonists at CB2 receptors could be due to their ability to selectively activate distinct signaling pathways (e.g., agonist-directed trafficking of response).
Evidence suggests that G protein-coupled receptors (GPCRs) exist in multiple active receptor conformations (Kenakin, 2002). It has been predicted that binding of a particular agonist to a GPCR results in enrichment of a unique set of receptor conformations based on the microaffinity of the agonist for each conformation. Because distinct conformations could presumably couple receptors differently to specific G proteins and intracellular effectors, individual agonists could ultimately produce distinct effects. Numerous studies provide support that individual agonists acting at several different classes of GPCRs (Figini et al., 1997; Berg et al., 1998; Wiens et al., 1998), including CB1 receptors (Bonhaus et al., 1998), are able to traffic intracellular responses in a ligand-dependent manner. Furthermore, using plasmon-waveguide resonance spectroscopy, Alves et al. (2003) have recently provided direct evidence for the existence of distinct topographical configurations of human δ opioid receptors with discrete affinities between individual G protein subclasses and different ligand-induced states.
The purpose of this study was to determine whether agonists acting at CB2 receptors selectively direct the trafficking of intracellular responses. This was accomplished by examining the ability of the endocannabinoids 2-AG and noladin ether as well as the synthetic cannabinoid CP-55,940 to regulate three intracellular effectors via CB2 receptors in transfected Chinese hamster ovary (CHO) cells. The results reported here provide evidence that endocannabinoids acting at CB2 receptors selectively direct intracellular signaling.
Materials and Methods
Materials. Penicillin/streptomycin (10,000 IU/ml and 10,000 μg/ml), geneticin (G418), fetal calf serum, and Dulbecco's modified Eagle's medium (DMEM) were purchased from Mediatech (Herndon, VA). The transfection agent lipofectin and serum-free medium Opti-MEM were obtained from Invitrogen (Carlsbad, CA). CP-55,940, noladin ether, and AM630 were procured from Tocris Cookson Inc. (Ellisville, MO). SR144528 was obtained from the National Institute on Drub Abuse drug inventory supply and control system (Bethesda, MD). [3H]CP-55,940 (168 Ci/mmol) was obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). [3H]Adenine (26 Ci/mmol) was purchased from Vitrax (Placenia, CA). Pertussis toxin was acquired from List Biological Laboratories Inc. (Campbell, CA). All other reagents were purchased from Fisher Scientific (Pittsburgh, PA).
Cell Culture and Stable Transfection. CHO cells were stably transfected with human CB2 receptor cDNA (Guthrie Research Institute, Sayre, PA) and were cultured in DMEM with 10% (v/v) fetal calf serum, 0.05 IU/ml penicillin, 50 μg/ml streptomycin, and 250 μg/ml selection antibiotic G418. CHO-CB2 cells were maintained in a humidified atmosphere of 5% CO2, 95% O2 at 37°C. Experiments were conducted with cells maintained between passages 4 and 18. In cases where PTX or chronic drug treatments were examined, drugs were added to the medium for 24 h before the assays, and flasks were washed twice with warmed DMEM to remove residual drug or toxin before beginning an assay. Stable cells lines expressing CB2 receptors were created using the cationic-lipid lipofectin as described previously (Shoemaker et al., 2005). CHO cells were cultured to 80% confluence (3 × 106 cells in 100-mm dishes) and incubated for 6 h with 5 μg of pcDNA3.1 plasmids (Invitrogen, Carlsbad, CA) containing the cDNA encoding for the CB2 receptor, and 15 μg of lipofectin reagent in serum-free Opti-MEM. Selective antibiotic (1 mg/ml geneticin) was added to the cell culture medium 48 h after transfection, and surviving colonies were picked 14 days after beginning selection. To confirm CB2 receptor expression, competition binding using whole cells obtained from each colony was performed using [3H]CP-55,940 (0.2 nM) displaced by nonradioactive CP-55,940 (1 μM) as described below. The clone expressing the highest level of CB2 receptor binding (e.g., CHO-CB2) was selected for future studies. For all studies, CHO-CB2 cells were maintained in DMEM containing 250 μg/ml geneticin.
Membrane Preparation. Pellets of frozen/thawed cells were resuspended in a homogenization buffer containing 50 mM HEPES, pH 7.4, 3 mM MgCl2, and 1 mM EGTA. Using a 40-ml Dounce glass homogenizer (Wheaton, Philadelphia, PA), samples were subjected to 10 complete strokes and centrifuged at 18,000 rpm for 10 min at 4°C. After repeating the homogenization procedure twice more, the samples were resuspended in HEPES buffer (50 mM, pH 7.4) and subjected to 10 strokes using a 7-ml glass homogenizer. Membranes were stored in aliquots of approximately 1 mg/ml at -80°C.
Competition Binding. Increasing concentrations of various nonradioactive cannabinoid ligands were incubated with 0.1 nM [3H]CP-55,940 in a final volume of 1 ml of binding buffer as described previously (Shoemaker et al., 2005). Each binding assay contained 50 μg of membrane protein, and reactions were incubated for 90 min at room temperature with mild agitation. Nonspecific binding was defined as binding observed in the presence of 10 μM of nonradioactive CP-55,940. Reactions were terminated by rapid vacuum filtration through Whatman GF/B glass fiber filters followed by two washes with ice-cold binding buffer. Analysis of the binding data were performed using the nonlinear regression (Curve Fit) function of GraphPad Prism version 4.0b (GraphPad Software Inc., San Diego, CA) to determine the concentration of the drug that displaced 50% of [3H]CP-55,940 (IC50). A measure of affinity (Ki) was derived from the IC50 values using the Cheng-Prusoff equation.
Measurement of cAMP Levels. The conversion of [3H]adenine labeled ATP pools to cyclic AMP was used as a functional measure of cannabinoid agonist activity (Shoemaker et al., 2005). CHO-CB2 cells were seeded into 17-mm (24-well) plates (4 × 106 cells/plate) and cultured to confluence. The day of the assay, an incubation mixture of DMEM containing 0.9% NaCl, 500 μM 3-isobutyl-1-methylxanthine, and 2 μCi/well [3H]adenine was added to the cells for 2 h at 37°C. The [3H]adenine-containing incubation mixture was removed, and the cannabinoid agonists were added to the cells for 15 min at 37°C in a Krebs-Ringer-HEPES buffer (110 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 25 mM glucose, 55 mM sucrose, and 10 mM HEPES, pH 7.4) containing 500 μM 3-isobutyl-1-methylxanthine and 10 μM forskolin. The reaction was terminated with the addition of 50 μl of 2.2 N hydrochloric acid. [3H]cAMP was separated by column chromatography. Ten milliliters of liquid scintillation cocktail was added to the final eluate before counting on a Packard Tri-Carb 2100TR liquid scintillation counter.
ERK-MAPK Assay: Western Blot. CHO-CB2 cells were seeded into 17-mm (24-well) plates 24 h before the assay in serum-free DMEM at a density required to achieve 90% confluence at the time of the experiment. Before the assay, cells were rinsed once with warmed (37°C) serum-free DMEM. The indicated concentrations of drugs were diluted in serum-free DMEM and exposed to the plated cells floating on a 37°C water bath for times ranging from 1 to 15 min. The plates were immediately placed on ice after the incubation was complete and subsequently rinsed three times with ice-cold phosphate-buffered saline (PBS) (0.1 M NaCl and 0.01 M NaH2PO4). After removal of the last wash, 100 μl of loading buffer (0.0625 M Tris, 2% SDS, 10% glycerol, and 5% 2-mercaptoethanol, pH 6.8) was added to each well, and plates were floated for 10 min on a 37°C water bath. Protein concentration of all samples was determined and 50 μg was separated by SDS-polyacrylamide gel electrophoresis on gels containing 10% acrylamide. The ECL method of immunoblotting was used (GE Healthcare, Piscataway, NJ). Gels were transferred to Hybond-ECL nitrocellulose membranes and incubated for 4 h at 4°C while shaking with 10% milk in blotting buffer (TBS-0.1%) (25 mM Tris-HCl, pH 7.6, 0.09% NaCl, and 0.1% Tween 20). Blots were then rinsed three times for 5 min each with TBS-0.1% and incubated overnight at 4°C while shaking with a primary antibody (1:1000) recognizing only the phosphorylated form of ERK-MAPK (e.g., active p44/42 ERK-MAPK) (Cell Signaling Technology Inc., Beverly, MA). Blots were rinsed as described previously and incubated with the secondary antibody (donkey anti-rabbit immunoglobin horseradish peroxidase; 1:10,000) for 2 h at 4°C while shaking in blotting buffer containing 10% milk. The secondary antibody was removed and blots were rinsed three times for 5 min each with TBS-0.3%, followed by three rinses for 5 min each with TBS-0.1%. Blots were then incubated for 1 min with equal volumes of ECL detection reagents 1 and 2, wrapped in Saran plastic wrap, and exposed to Hybond-ECL X-ray film for periods varying between 30 s and 10 min. After scanning, the blot was stripped and reprobed as described previously using a primary antibody (1:1000) recognizing both phosphorylated and non-phosphorylated forms ERK-MAPK (e.g., total ERK-MAPK) (Cell Signaling Technology Inc.). Immunopositive bands were quantified by densitometry using the NIH Image software program (version 1.56). To determine the relative amount of the target protein recognized, the area of each band was traced and multiplied by its mean optical density.
ERK-MAPK Assay: Modified Fast-Activated Cell-Based Enzyme-Linked Immunosorbent Assay (FACE) Assay. Adapted from the Active Motif (Carlsbad, CA) FACE assay, wells of a 96-well plate were coated with 10 μg/ml poly-l-lysine (Sigma-Aldrich, St. Louis, MO) before seeding of cells. CHO-CB2 cells were seeded in serum-free DMEM 24 h before the assay at a density required to achieve 90% confluence at the time of the experiment. Before the assay, cells were rinsed once with warmed (37°C) serum-free DMEM. The indicated concentrations of drugs were diluted in serum-free DMEM and exposed to the plated cells floating on a 37°C water bath for times ranging from 1 to 15 min. The plates were immediately placed on ice after the incubation was complete and subsequently rinsed three times with ice-cold PBS. After removal of the last rinse, cells were fixed with a 4% formaldehyde solution (Fisher Scientific) in PBS. Fixed cells were then rinsed three times for 5 min each with wash buffer (0.1% Triton X-100 in PBS) and quenched for 20 min with a 1% hydrogen peroxide and 1% sodium azide solution in wash buffer. After three rinses of 5 min each with wash buffer, nonspecific binding was blocked by a 1-h incubation of cells with 5% milk in wash buffer at room temperature while shaking. Cells were then rinsed twice for 5 min with wash buffer and incubated overnight at 4°C while shaking with a primary antibody (1:500) recognizing only the phosphorylated form of ERK-MAPK (e.g., active p44/42 ERK-MAPK) (Cell Signaling Technology Inc.). In some experiments, separate plates of cells treated in a parallel fashion were incubated with a primary antibody (1:500) recognizing both phosphorylated and non-phosphorylated forms of ERK-MAPK (e.g., total ERK-MAPK) (Cell Signaling Technology Inc.). Cells were rinsed three times for 5 min each with wash buffer and incubated with the secondary antibody (donkey anti-rabbit immunoglobin horseradish peroxidase; 1:2000) for 1 h at room temperature in wash buffer while shaking. Cells were rinsed three times for 5 min each with wash buffer followed by two rinses of 5 min each with PBS. SuperSignal West Femto reagent (50 μl) (Pierce Chemical, Rockford, IL) was added to each well while plates were floating on an ice bath. Plates were warmed to room temperature and immediately scanned using a SPECTRAFluor Plus luminometer (Tecan U.S., Durham, NC). Plates were rinsed twice for 5 min each with wash buffer and twice for 5 min each with PBS. Protein concentration was determined by adding 100 μl of crystal violet (Fisher Diagnostics, Middletown, VA) to the each well and incubating plates at room temperature for 30 min. Cells were then rinsed three times for 5 min with PBS and incubated for 1 h with 100 μl of 1% SDS in PBS. Plates were scanned using a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA) set to detect fluorescent emission at 595 nm.
Measurement of Calcium Transients. To measure intracellular Ca2+, CHO-CB2 cells were suspended in a modified Geys buffer (145 mM NaCl, 5 mM KCl, 1 mM Na2HPO4, 0.5 mM MgSO4, 1 mM CaCl2, 5 mM glucose, 10 mM HEPES, and 1 mM probenecid, pH 7.4). Cells were loaded with 5 μM of the intracellular dye Fura-2 acetoxymethyl ester (Calbiochem, San Diego, CA) for 20 min at 25°C. After two washes with modified Geys buffer, cells were resuspended in the same buffer to a concentration of ∼1.5 × 106 cells/ml. Using a Hitachi F2000 spectrofluorometer (Hitachi Instruments, Danbury, CT) equipped with a thermostatic cell holder and magnetic stirrer, 1.5-ml aliquots of cells inside a 1-cm2 cuvette were warmed to 37°C for 3 min before measurement of Fura-2 fluorescence. Samples were continuously stirred throughout the experiment with a 1-cm grooved magnetic stir disc. A 30-s baseline reading was taken before the addition of the indicated concentrations of cannabinoid agonists. Measurements of the 340:380-nm emission ratio were recorded every 0.5 s using an excitation wavelength of 510 nm. Maximum and minimum fluorescence were determined by the addition of 0.2% Triton X-100 and 10 mM EGTA (final concentrations), respectively. Each set of experiments was completed within 30 to 60 min after loading the Fura-2 acetoxymethyl ester dye. Intracellular free calcium was calculated with the following formula (eq. 1):  where R is the 340:380 fluorescence ratio, and Kd = 224 nM.
where R is the 340:380 fluorescence ratio, and Kd = 224 nM.
Statistics. All data are expressed as mean ± S.E.M. For parameters estimated from the log-concentration axis (e.g., ED50 and Ki), the averages were calculated as the geometric mean of the log ED50 or log Ki values (Kenakin, 1977). As such, the S.E.M. is presented as an asymmetrical range around the means (e.g., not centered around the mean values). Unless otherwise stated in the figure legends, data are represented by a minimum of three separate experiments, each performed in triplicate. All curve-fitting and statistical analysis was conducted by employing the computer program GraphPad Prism version 4.0b (GraphPad Software Inc.). To compare three or more groups, statistical significance of the data was determined by a one-way ANOVA, followed by a post hoc comparison using a Tukey's or Dunnett's test. To compare two groups, the nonpaired Student's t test was used.
Results
CB2 Receptors Regulate Three Intracellular Effectors in Transfected CHO Cells. In contrast to an initial study suggesting that the putative endocannabinoid noladin ether is a CB1-selective partial agonist (Hanus et al., 2001), we recently reported (Shoemaker et al., 2005) that this ligand binds to CB2 receptors expressed in CHO-CB2 cells with nanomolar affinity, activates G proteins, and inhibits adenylyl cyclase activity with equivalent efficacy relative to the full agonists 2-AG and CP-55,940. Furthermore, it was demonstrated that chronic exposure to noladin ether produces down-regulation and desensitization of CB2 receptors, similar to that observed after prolonged treatment of cells with CP-55,940. These findings indicate that CB2 receptors in CHO-CB2 cells are negatively coupled to adenylyl cyclase.
CB2 receptors have previously been shown to also activate ERK-MAPK in transfected CHO cells (Bouaboula et al., 1996). As such, we examined the ability of cannabinoids to regulate ERK-MAPK activity in our cellular model (Fig. 1). A standard Western blot technique was used for initial studies in which cells are exposed to cannabinoids, immediately solubilized, and cellular proteins separated by SDS-polyacrylamide gel electrophoresis. The relative amount of active (phosphorylated) and total forms of ERK-MAPK in a given sample are then quantified by immunoblots performed with selective antibodies (Fig. 1A). Optimization of this technique demonstrated that 2-AG produces rapid activation of ERK-MAPK (within 1 min), with maximal stimulation observed by 5 min and activation decreasing by 15 min (data not shown). Additional experiments established that both CP-55,940 and noladin ether also produce maximal ERK-MAPK stimulation at 5 min (data not shown). As such, all subsequent experiments were conducted using an optimal drug exposure time of 5 min. 2-AG also produces a concentration-dependent increase in the amount of active (Fig. 1B, open squares) but not total ERK-MAPK (data not shown). To develop a more quantifiable, sensitive, and high-throughput technique to measure ERK-MAPK activity, we modified the FACE assay developed by Active Motif. In this technique, cells seeded into 96-well plates are exposed to agonist and immediately fixed, washed, and incubated with antibodies selective for the active and total forms of ERK-MAPK. After incubation with appropriate secondary antibodies, the amount of chemiluminescence is then quantified after scanning using a luminometer. Initial experiments with this technique also show that 2-AG produces a time- (data not shown) and concentration-dependent (Fig. 1B, filled squares) activation of ERK-MAPK identical to that determined by the Western blot method. 2-AG does not alter the levels of total ERK-MAPK in cells treated in a parallel manner (data not shown). Confident that the modified FACE technique produces accurate results, this method was used for the remainder of the study. Results obtained from both methods indicate that in addition to regulation of adenylyl cyclase, CB2 receptors in CHO-CB2 cells are positively coupled to a second intracellular effector, ERK-MAPK.

Activation of ERK-MAPK in CHO-CB2 cells by the endocannabinoid 2-AG: comparison of Western blot and modified FACE techniques. The level of activated ERK-MAPK was monitored in whole CHO-CB2 cells by using an antibody that selectively recognizes the phosphorylated (active) state of ERK-MAPK as described under Materials and Methods. Quantification of active ERK-MAPK levels was accomplished by Western blotting and a modified FACE assay. A representative Western blot is presented in A. Concerning the Western blot technique, immunopositive bands were quantified by scanning densitometry using NIH Image software (B, □). Concerning the FACE assay, levels of chemiluminescence were quantified by scanning of 96-well plates using a SPECTRAFluor Plus luminometer (B, ▪). Data are presented graphically for both assays as the percentage of the maximal response occurring in the presence of increasing concentrations of the endocannabinoid agonist 2-AG. Concentration-effect curves are representative of experiments repeated four (Western blot) or 11 (FACE) times.
Stimulation of CB2 receptors produces transient increases in intracellular free Ca2+ concentrations via a PLCβ-mediated mechanism in cellular models expressing endogenous cannabinoid receptors (Sugiura et al., 2000). CB2-mediated signaling through this pathway was examined in our cellular model (Fig. 2). Intracellular [Ca2+]i was measured in stirred suspensions of CHO-CB2 cells maintained at 37°C. Basal (unstimulated) [Ca2+]i averages 85 ± 3 nM (N = 12 flasks). Addition of CB2 agonists CP-55,940, noladin ether, or 2-AG produces a rapid, transient (Fig. 2), and concentration-dependent (Figs. 3, 4, 5) rise in [Ca2+]i. The rise of [Ca2+]i typically peaks within 30 s and returns to basal levels by 90 s. These results indicate that CB2 receptors in CHO-CB2 cells are positively coupled to a third intracellular effector, PLCβ, resulting in transient increases in intracellular Ca2+ levels.
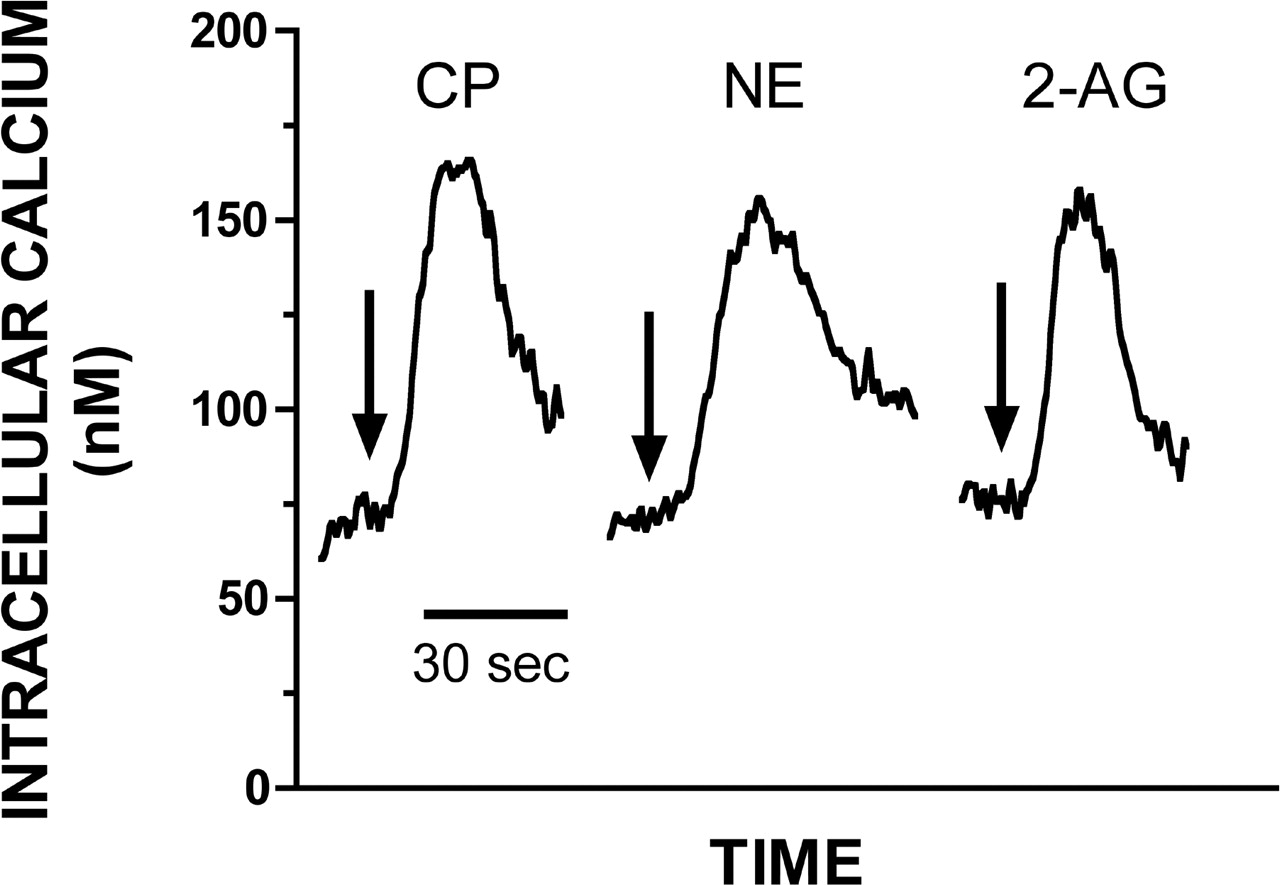
Representative responses for activation of Ca2+-transients by cannabinoid agonists CP-55,940 (CP), noladin ether (NE), and 2-AG in CHO-CB2 cells. Presented are individual responses to maximally effective concentrations of CP-55,940 (1 μM), noladin ether (10 μM), and 2-AG (100 μM). The arrow indicates the time that each agonist was added. Responses presented are representative of experiments repeated at least four times.
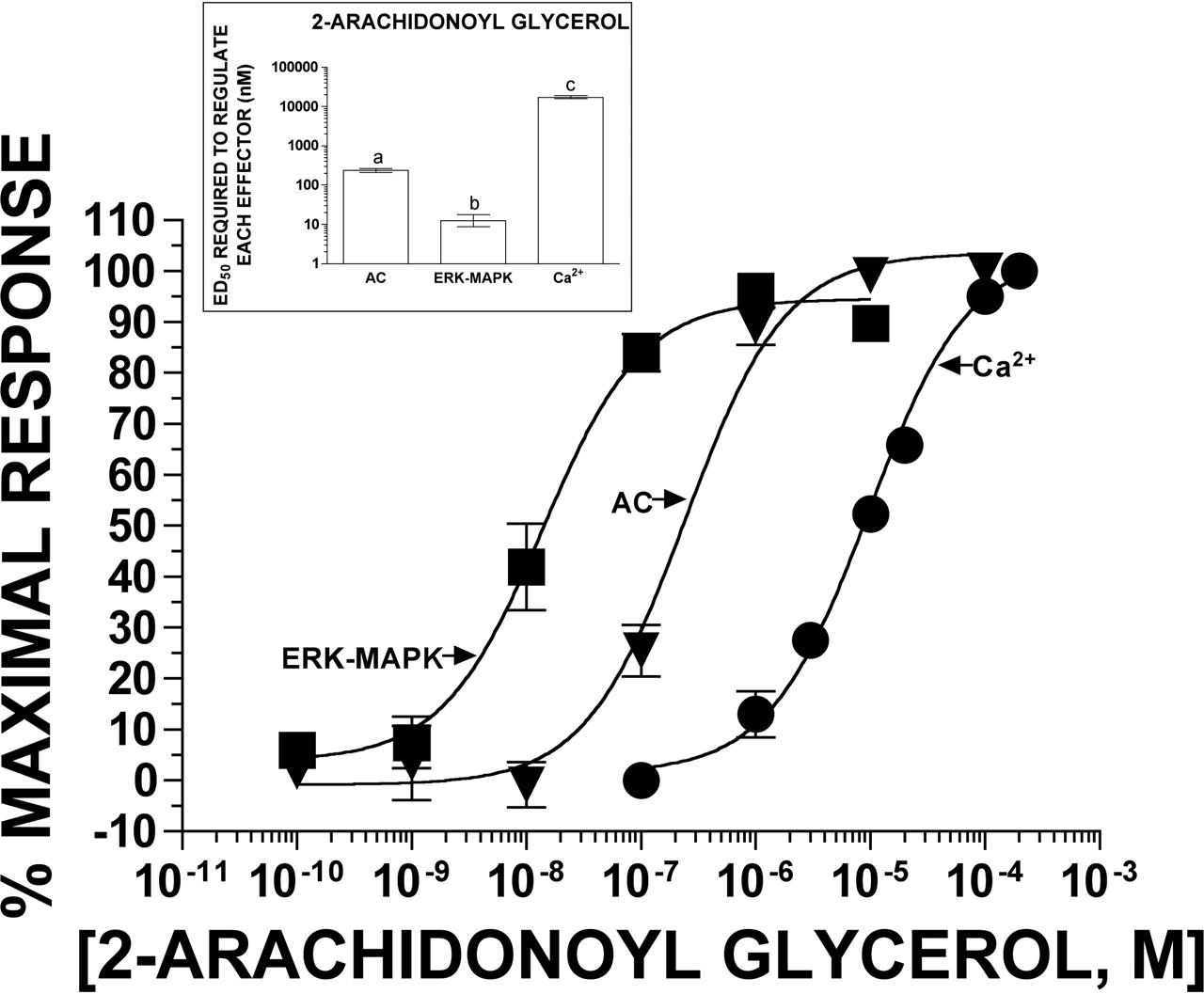
CP-55,940 and Noladin Ether Most Potently Regulate Adenylyl Cyclase, Whereas 2-AG Most Potently Regulates ERK-MAPK. To determine the potency and efficacy of individual cannabinoid agonists to regulate each of the three effectors, concentration-effect experiments were conducted (Figs. 3, 4, 5; Table 1). To visually aid in the graphical comparison of ED50 values, data for all assays are presented as the percentage of the maximal response. Specifically, all responses were normalized for each ligand in each assay (e.g., each ligand defined its own 100%). Results from the concentration-dependent inhibition of adenylyl cyclase activity by CP-55,940, noladin ether, and 2-AG presented here were published in a previous study (Shoemaker et al., 2005). The synthetic, nonselective cannabinoid agonist CP-55,940 regulates adenylyl cyclase, ERK-MAPK, and Ca2+-transients in a concentration-dependent manner (Fig. 3; Table 1). CP-55,940 most potently inhibits the activity of adenylyl cyclase with an ED50 value of 0.39 nM. The concentration of CP-55,940 required to achieve half-maximal activation of ERK-MAPK (2.61 nM) or Ca2+-transients (5.27 nM) is significantly greater (P < 0.01) than that needed to inhibit adenylyl cyclase. The endocannabinoid noladin ether regulates the three effectors with the same rank order of relative potency as CP-55,940 (Fig. 4; Table 1): adenylyl cyclase (ED50 = 34.9 nM) > ERK-MAPK (ED50 = 243 nM) = Ca2+-transients (ED50 = 555 nM). In marked contrast to the initial two cannabinoids examined, 2-AG most potently regulates ERK-MAPK (ED50 = 12.4 nM), requiring significantly greater (P < 0.01) concentrations to inhibit adenylyl cyclase (ED50 = 238 nM) and even higher amounts to stimulate Ca2+-transients (ED50 = 17,400 nM) (Fig. 5; Table 1). We have previously shown that the potency of 2-AG to regulate adenylyl cyclase in CHO-CB2 cells is unaltered by addition of 100 μM phenylmethylsulfonyl fluoride, a commonly used nonspecific enzymatic inhibitor to prevent degradation (Shoemaker et al., 2005). Therefore, it is also unlikely that the ED50 of 2-AG required to regulate ERK-MAPK or Ca2+-transients is altered by 2-AG metabolism in this cellular model.
Receptor affinity and regulation of adenylyl cyclase, ERK-MAPK, and Ca2+-transients by cannabinoid agonists in CHO-CB2 cells Competition binding was performed in the presence of 0.1 nM [3H]CP-55,940 and increasing concentrations of the competing drugs. The ED50 and EMAX values for regulation of all intracellular effectors were derived from nonlinear regression of full concentration-effect curves (Figs. 3, 4, 5). Data are presented as the mean ± S.E.M. determined from 3 to 11 separate experiments, each performed in triplicate.

Concentration-dependent regulation of adenylyl cyclase, ERK-MAPK, and Ca2+-transients by the synthetic cannabinoid agonist CP-55,940 in CHO-CB2 cells. The ability of increasing concentrations of CP-55940 to regulate adenylyl cyclase (▾), ERK-MAPK (▪), and Ca2+-transients (•) was examined in whole CHO-CB2 cells as described under Materials and Methods. CP-55940 produces concentration-dependent regulation of all three effectors with a relative rank order potency of adenylyl cyclase > ERK-MAPK = Ca2+-transients (inset). To visually aid in the graphical comparison between inhibition of adenylyl cyclase and activation of ERK-MAPK and Ca2+-transients, all data are presented as the percentage of maximal response. Data represent the mean ± S.E.M. of four (adenylyl cyclase), three (ERK-MAPK), or five (Ca2+-transients) experiments, each performed in triplicate. The ED50 values are presented graphically in the inset and numerically in Table 1. The EMAX values are presented graphically in Fig. 6 and numerically in Table 1. ED50 values that are designated with different letters (a and b) are significantly different, P < 0.01 (one-way ANOVA followed by Tukey's post hoc comparison).

Concentration-dependent regulation of adenylyl cyclase, ERK-MAPK, and Ca2+-transients by the endocannabinoid agonist noladin ether in CHO-CB2 cells. The ability of increasing concentrations of noladin ether to regulate adenylyl cyclase (▾), ERK-MAPK (▪), and Ca2+-transients (•) was examined in whole CHO-CB2 cells as described under Materials and Methods. Noladin ether produces concentration-dependent regulation of all three effectors with a relative rank order potency of adenylyl cyclase > ERK-MAPK = Ca2+-transients (inset). To visually aid in the graphical comparison between inhibition of adenylyl cyclase and activation of ERK-MAPK and Ca2+-transients, all data are presented as the percentage of maximal response. Data represent the mean ± S.E.M. of nine (adenylyl cyclase), four (ERK-MAPK), or six (Ca2+-transients) experiments, each performed in triplicate. The ED50 values are presented graphically in the inset and numerically in Table 1. The EMAX values are presented graphically in Fig. 6 and numerically in Table 1. ED50 values that are designated with different letters (a and b) are significantly different, P < 0.01 (one-way ANOVA followed by Tukey's post hoc comparison).
Contrary to the differences observed for the rank order of potencies, all three cannabinoids produce equivalent maximal activation/inhibition of the three effectors (Table 1). Agonists produce from 47 to 57% inhibition of adenylyl cyclase, 52 to 66% stimulation of ERK-MAPK, and 96 to 108% activation of Ca2+-transients. These results demonstrate that although all cannabinoid agonists regulate the three effectors examined with equivalent efficacy, the rank order or potency for regulation differs depending on which effector is evaluated.
The ED50 values for regulation of adenylyl cyclase and ERK-MAPK by CP-55,940 and 2-AG presented here are similar to those reported previously (Howlett et al., 2002). Since noladin ether has been classified as a selective CB1 partial agonist (Hanus et al., 2001) until our recent publication (Shoemaker et al., 2005), no functional studies have been reported concerning its action at CB2 receptors. Interesting, in the only report (Bouaboula et al., 1996) in which the regulation of both adenylyl cyclase and ERK-MAPK were concurrently evaluated in CHO-CB2 cells, CP-55,940 and WIN 55,212-2 similarly produce more potent regulation of adenylyl cyclase (2 and 3 nM, respectively) relative to ERK-MAPK (8 and 12 nM, respectively). To our knowledge, the ability of 2-AG or noladin ether to regulate these effectors concurrently in the same cell line via CB2 receptors has never been evaluated.
Although it has been demonstrated that cannabinoid agonists stimulate Ca2+-transients via endogenously expressed CB2 receptors in HL-60 cells (Sugiura et al., 2000), other studies report that activation of transfected CB2 receptors in CHO cells is unable to elevate intracellular calcium concentrations (Felder et al., 1995). Although the reasons for the differences between these past studies and the present investigation are not known, one potential explanation for the stimulatory effects in CHO-CB2 cells reported here might be due to the choice of agonists used. Whereas the previous studies observed no effect of WIN 55,212-2, anadamide, and HU-210 on intracellular calcium concentrations, none of the agonists tested in the present study were evaluated.

Concentration-dependent regulation of adenylyl cyclase, ERK-MAPK, and Ca2+-transients by the endocannabinoid agonist 2-AG in CHO-CB2 cells. The ability of increasing concentrations of 2-AG to regulate adenylyl cyclase (▾), ERK-MAPK (▪), and Ca2+-transients (•) was examined in whole CHO-CB2 cells as described under Materials and Methods. 2-AG produces concentration-dependent regulation of all three effectors with a relative rank order potency of ERK-MAPK > adenylyl cyclase > Ca2+-transients (inset). To visually aid in the graphical comparison between inhibition of adenylyl cyclase and activation of ERK-MAPK and Ca2+-transients, all data are presented as the percentage of maximal response. Data represent the mean ± S.E.M. of six (adenylyl cyclase), 11 (ERK-MAPK), or three (Ca2+-transients) experiments, each performed in triplicate. The ED50 values are presented graphically in the inset and numerically in Table 1. The EMAX values are presented graphically in Fig. 6 and numerically in Table 1. ED50 values that are designated with different letters (a–c) are significantly different, P < 0.01 (one-way ANOVA followed by Tukey's post hoc comparison).
Cannabinoid Agonists Regulate Effectors Specifically by Activation of CB2 Receptors That Couple to Gi/Go-Type G Proteins. CB2 receptors are known to inhibit adenylyl cyclase (Felder et al., 1995), activate ERK-MAPK (Bouaboula et al., 1996), and stimulate Ca2+-transients (Sugiura et al., 2000) by coupling to Gi/Go proteins. To determine whether such coupling occurs in our cellular model, the ability of cannabinoid agonists to regulate all three effectors was examined in CHO-CB2 cells treated overnight with PTX (200 ng/ml) to selectively inactivate Gi/Go proteins (Fig. 6, right). This pretreatment eliminates (P < 0.01) the ability of CP-55,940, noladin ether, and 2-AG to regulate adenylyl cyclase (Fig. 6A), ERK-MAPK (Fig. 6B), and Ca2+-transients (Fig. 6C).
To verify that effector regulation was mediated specifically through CB2 receptors, a selective CB2 receptor antagonist/ inverse agonist, AM630, was used (Fig. 6, left). We have previously demonstrated that the affinity of AM630 for CB2 receptors expressed in CHO-CB2 cells is 22.5 (21.1–24.0) nM (Shoemaker et al., 2005). AM630 (10 μM) administered alone produces no effect on basal ERK-MAPK activity or intracellular Ca2+ concentrations (data not shown). However, as might be expected of an inverse agonist acting at constitutively active CB2 receptors, AM630 (10 μM) given alone results in a slight, but significant (P < 0.05), 5 to 10% increase in basal cAMP levels. AM630 (10 μM) administered 5 min before the addition of CP-55,940 and noladin ether blocks (P < 0.01) the ability both agonists to inhibit adenylyl cyclase (Fig. 6A), activate ERK-MAPK (Fig. 6B), or stimulate Ca2+-transients (Fig. 6C). AM630 produces a similar blockade (P < 0.01) of the ability of 2-AG to regulate adenylyl cyclase and ERK-MAPK but surprisingly does not attenuate the stimulation of Ca2+-transients by 2-AG.

The effect of pretreatment with a CB2 antagonist (left) or PTX (right) on cannabinoid agonist regulation of adenylyl cyclase, ERK-MAPK, and Ca2+-transients in CHO-CB2 cells. Left, adenylyl cyclase, ERK-MAPK, and Ca2+-transient assays were conducted in whole cells as described under Materials and Methods. CHO-CB2 cells were incubated with the CB2-selective antagonist AM630 (10 μM) or vehicle for 5 min before the addition of the indicated agonist. For adenylyl cyclase experiments, concentrations of 1 nM CP-55,940, 1 μM noladin ether, and 1 μM 2-AG were used. For ERK-MAPK experiments, concentrations of 10 nM CP-55,940, 1 μM noladin ether, and 100 nM 2-AG were used. For Ca2+-transients experiments, concentrations of 30 nM CP-55,940, 1 μM noladin ether, and 10 μM 2-AG were used. Pretreatment with AM630 significantly reverses the regulation of all effectors by all three agonists except Ca2+-transient stimulation by 2-AG. Data are presented as the mean ± S.E.M. for three to six independent experiments performed in triplicate. Right, CHO-CB2 cells were exposed to PTX overnight (200 ng/ml) and washed twice before all effector assays. For adenylyl cyclase experiments, concentrations of 10 nM CP-55,940, 10 μM noladin ether, and 10 μM 2-AG were used. For ERK-MAPK experiments, concentrations of 10 nM CP-55,940, 1 μM noladin ether, and 100 nM 2-AG were used. For Ca2+-transient experiments, concentrations of 30 nM CP-55,940, 1 μM noladin ether, and 10 μM 2-AG were used. Pretreatment with PTX completely blocks the regulation of all effectors by all cannabinoid agonists examined. Data are presented as the mean ± S.E.M. for three to six independent experiments performed in triplicate. *, **, significantly different from the percentage of inhibition produced by the indicated agonist in cells not pretreated with AM630 or pertussis toxin (unpaired Student's t test, P < 0.05, 0.01).
These results indicate that noladin ether and CP-55,940 produce their effects through action at CB2 receptors that couple to Gi/Go proteins. Although 2-AG also inhibits adenylyl cyclase and stimulates ERK-MAPK through Gi/Go-coupled CB2 receptors, the failure of AM630 to reverse the stimulation of Ca2+-transients required further analysis.
2-AG Stimulates Ca2+-Transients by Binding to Sites on CB2 Receptors Distinct from Those Occupied by AM630. It is possible that the regulation of Ca2+-transients by 2-AG is due to action at a native receptor in CHO cells, other than CB2 receptors. To examine this question, the ability of 2-AG to regulate Ca2+-transients in CHO cells not expressing CB2 receptors was evaluated (Fig. 7A). CHO cells stably transfected with FLAG-tagged δ opioid receptors (CHO-FLAG-DOR) were used for these experiments. Since δ opioid agonists produce effects on intracellular Ca2+ concentrations similar to that observed for cannabinoid receptors (Jin et al., 1992), use of these cells allows the testing of an important positive control for these studies. The cannabinoid agonists CP-55,940, noladin ether, and 2-AG are unable to produce measurable stimulation of Ca2+-transients in the CHO-FLAG-DOR cells. In marked contrast, activation of δ-opioid receptors by the full agonist DPDPE increases intracellular calcium levels by 101 ± 16.5%.
Although AM630 was not examined, a previous report demonstrated that another selective CB2 antagonist, SR144528, was able to completely antagonize 2-AG-mediated stimulation of Ca2+-transients via endogenous CB2 receptors expressed in HL-60 cells (Sugiura et al., 2000). Therefore, the effect of SR144528 on 2-AG-mediated stimulation of Ca2+-transients in CHO-CB2 cells was examined (Fig. 7B). SR144528 (10 μM) administered alone did not alter basal Ca2+-concentrations (data not shown). As observed previously (Fig. 6C), AM630 (10 μM) had no effect on the level of Ca2+-transient stimulation by 2-AG (77 versus 79%). In contrast, SR144528 (10 μM) significantly (P < 0.05) reduced 2-AG-mediated stimulation of Ca2+-transients (77 versus 48%). Although SR144528 only partially blocked the effect of 2-AG, these results suggest that stimulation of Ca2+-transients by 2-AG in CHO-CB2 cells is mediated, at least in part, by CB2 receptor activation.
To provide additional evidence that the action of 2-AG on Ca2+-transients was indeed mediated via CB2 receptors, CHO-CB2 cells were treated chronically (12 h) with a receptor saturating concentration of CP-55,940 (1 μM) (Fig. 7C, left). We have previously shown that these conditions produce both CB2 receptor down-regulation and a dramatic reduction in the ability of cannabinoid agonists to inhibit adenylyl cyclase upon subsequent challenge (e.g., desensitization) (Shoemaker et al., 2005). Chronic exposure of CHO-CB2 cells to CP-55,940 results in a 70 to 85% reduction in ability of all three cannabinoids (including 2-AG) to induce intracellular calcium release. Such homologous desensitization, coupled with a total absence of effect in cells lacking CB2 receptors, strongly indicate that the stimulation of Ca2+-transients by 2-AG is mediated by CB2 receptor activation in CHO-CB2 cells.

Characterization of specific CB2 receptor involvement for activation of Ca2+-transients by 2-AG in CHO-CB2 cells. A, ability of cannabinoid agonists CP-55,940 (1 μM), noladin ether (10 μM), and 2-AG (10 μM) and δ opioid agonist DPDPE (1 μM) to stimulate Ca2+-transients in CHO cells lacking CB2 receptors (CHO-FLAG-DOR) was evaluated as described under Materials and Methods. Cannabinoid agonists produce no measurable effect, whereas the δ opioid agonist DPDPE produces an increase in intracellular calcium level of over 100%. Data are presented as the mean ± S.E.M. for three to six independent experiments performed in triplicate. B, effect of selective CB2 cannabinoid antagonists AM630 (10 μM) and SR144528 (10 μM) on Ca2+-transient stimulation by 2-AG (10 μM). CHO-CB2 cells were incubated with the CB2-selective antagonist or vehicle for 5 min before the addition of 2-AG. SR144528, but not AM630, produced a significant reduction in 2-AG-induced stimulation of Ca2+-transients. Data are presented as the mean ± S.E.M. for six to eight independent experiments performed in triplicate. C, ability of cannabinoid agonists to stimulate Ca2+-transients in CHO-CB2 cells was evaluated before and after pretreatment with CP-55,940 (1 μM, 12 h), U73122 (10 μM, 5 min), or U73347 (10 μM, 5 min). Chronic exposure to CP-55,940 (left) or inhibition of PLCβ by the active U73122 compound (middle), but not the inactive U73347 compound (right) blocks Ca2+-transient stimulation by all cannabinoid agonists examined. Data are presented as the mean ± S.E.M. for three to seven independent experiments performed in triplicate. +, Significantly different from percentage of stimulation produced by all cannabinoid agonists (one-way ANOVA followed by Tukey's post hoc comparison, P < 0.01). *, significantly different from the respective percentage of stimulation produced by 2-AG or 2-AG + AM630 (one-way ANOVA followed by Tukey's post hoc comparison, P < 0.05). **, significantly different from the respective percentage of stimulation produced by the indicated agonist in cells not pretreated with CP-55,940, U73122, or U73347 (unpaired Student's t test, P < 0.01).
The next set of experiments was conducted to determine whether 2-AG and the other cannabinoid agonists produce Ca2+-transients by similar postreceptor mechanisms (Fig. 7C, middle and right). The kinetics of intracellular Ca2+ release and results obtained from previous studies (Sugiura et al., 2000) indicates that CB2 agonists activate Ca2+-transients by a signaling pathway involving PLCβ. To examine this possibility, CHO-CB2 cells were preincubated for 5 min with 10 μM selective PLCβ inhibitor U73122 or inactive analog U73343. U73122 inhibited the rise in [Ca2+]i elicited by CP-55,940, noladin ether, or 2-AG by 90 ± 10% (Fig. 7C, middle), indicating that activation of PLCβ is involved in CB2-mediated production of Ca2+-transients in transfected CHO cells. Importantly, as predicted, the inactive analog U73343 was ineffective in blocking Ca2+-transient stimulation (Fig. 7C, right). Neither U73122 nor U73343 altered basal [Ca2+]i (data not shown). These results suggest that 2-AG, CP-55,940, and noladin ether all activate Ca2+-transients via a similar mechanism involving PLCβ.
Collectively, these data indicate that 2-AG stimulates Ca2+-transients by binding to sites on CB2 receptors distinct from those occupied by AM630 and the other cannabinoid agonists examined. Therefore, although the CB2 antagonist AM630 is unable to attenuate this response, experiments using CHO-FLAG-DOR cells, chronic agonist treatment of CHO-CB2 cell, and results with a second CB2 antagonist SR144528 strongly indicate that the stimulation of Ca2+-transients by 2-AG is nevertheless mediated specifically through action at CB2 receptors.
Relationship of Fractional Receptor Occupancy (FRO) to Agonist-Directed Trafficking of Response. To provide additional evidence that cannabinoid agonists selectively traffic intracellular responses, the FRO required for agonists to regulate each effector was compared. The law of mass action predicts that, at equilibrium, the FRO is the proportion of all receptors that are bound by drug at a given concentration (Tallarida, 1990). FRO is calculated using the following formula (eq. 2): 
Based on these assumptions, it is predicted that one-half of the receptors are occupied when the concentration of a ligand used is equal to the Ki of the drug being investigated. Extending this relationship to a range of different concentrations, the theoretical proportion of receptors occupied with respect to increasing concentrations of CP-55,940 (A), noladin ether (B), or 2-AG (C) were calculated and are represented by the sigmoidal curves presented in Fig. 8. Similarly, the FRO of the three agonists required to produce half-maximal regulation of each effector (e.g., the FRO at ED50) were determined and are presented graphically as the filled symbols superimposed on the receptor occupancy curve for each agonist. As would be anticipated based on ED50 values alone, CP-55,940 and noladin ether require the least receptor occupancy to regulate adenylyl cyclase, whereas 2-AG requires occupancy of the fewest receptors to activate ERK-MAPK. Although not obvious when considering only ED50 values, noladin ether requires occupancy of considerably fewer receptors than CP-55,940 to produce equivalent regulation of all effectors examined (AC, 7 versus 40%; ERK-MAPK, 34 versus 82%; and Ca2+, 54 versus 90%). This indicates that even though both agonists share the same rank order of potency to regulate effectors, the endocannabinoid noladin ether seems to be considerably more “efficient” than CP-55,940 in transducing intracellular signals through CB2 receptors in this cellular model. The endocannabinoid 2-AG is also a very efficient agonist, requiring only 1% receptor occupancy to half-maximally activate ERK-MAPK, relative to 19 and 94% FRO to produce similar regulation of adenylyl cyclase and Ca2+-transients, respectively. These results indicate that in addition to comparing rank orders of potency, other aspects of agonist-directed trafficking might be revealed by also examining the FRO required to regulate effectors.
Discussion
This study is the first to provide evidence that two endogenous cannabinoid agonists, 2-AG and noladin ether, selectively traffic intracellular responses through CB2 receptors. This is indicated by a reversal of potency for individual agonists regulating distinct effectors via CB2 receptors. Specifically, 2-AG most potently activates ERK-MAPK, requiring greater concentrations to inhibit adenylyl cyclase, and even higher amounts to stimulate Ca2+-transients. In contrast, noladin ether and the synthetic cannabinoid CP-55,940 most potently inhibit adenylyl cyclase, necessitating higher concentrations to stimulate ERK-MAPK and Ca2+-transients. 2-AG also seems to stimulate Ca2+-transients by binding to CB2 receptors in a unique manner, relative to noladin ether or CP-55,940, that cannot be reversed by the antagonist AM630.
CHO-CB2 cells used here express a moderately high density of CB2 receptors of 1.44 pmol/mg protein (Shoemaker et al., 2005). Several cell lines and tissues possess comparable levels of CB2 receptors. For example, CB2 receptors are expressed at a density of 0.525 pmol/mg in myelomonocytic U937 cells (Bouaboula et al., 1993) and 0.441 pmol/mg in invertebrate macrophages (Stefano et al., 1996). Since partial agonists often display efficacy similar to full agonists in cellular models overexpressing GPCRs (Kenakin, 2002), it is not surprising that all agonists demonstrate similar efficacy in this study. As such, it is likely that this model is not suitable for detection of agonist-selective differences in efficacy. It is also unfortunate that a lack of commercially available irreversible antagonists for CB2 receptors precludes depletion of spare cannabinoid receptors that could benefit efficacy comparisons. In spite of this, analysis of the relative rank order of potencies required for different agonists to regulate distinct effectors as a means to detect ADTR in transfected cell lines is appropriate and has been used successfully for this purpose (Gurwitz et al., 1994; Sagan et al., 1999).
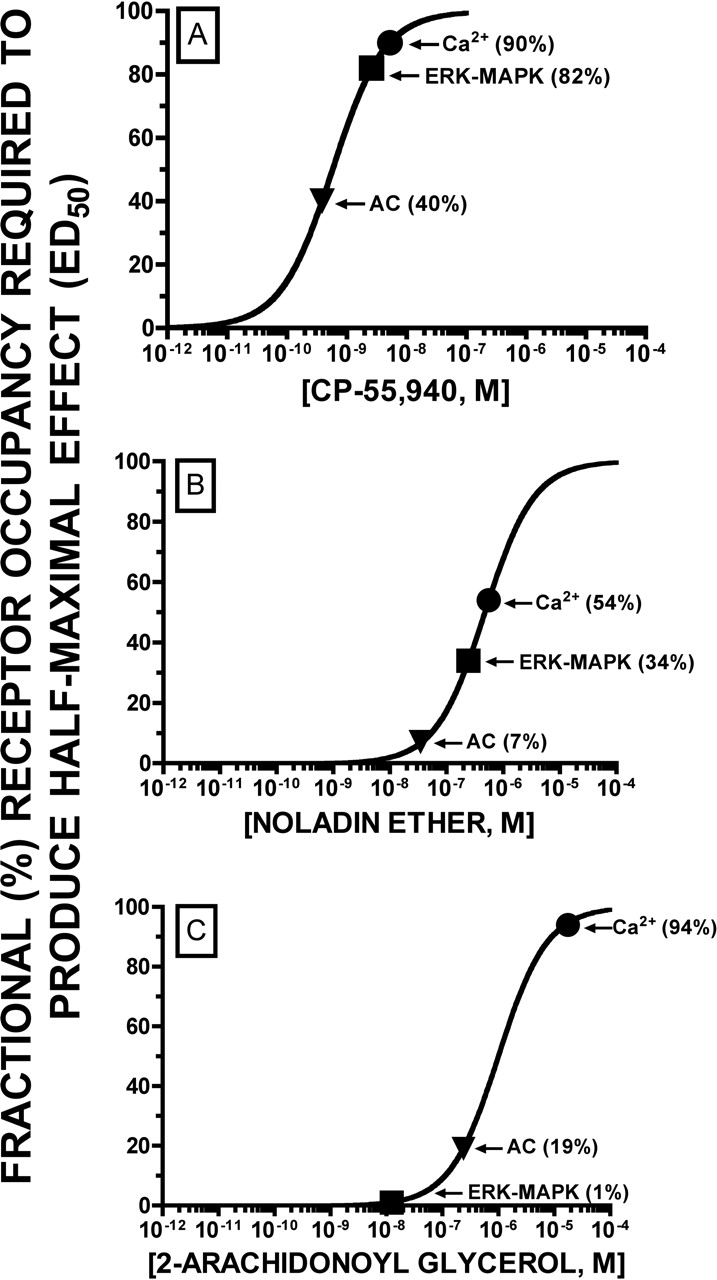
FRO required for cannabinoid agonists to produce half-maximal regulation of adenylyl cyclase, ERK-MAPK, and Ca2+-transients in CHO-CB2 cells. The theoretical proportion of receptors occupied with respect to increasing concentrations of CP-55,940 (A), noladin ether (B), or 2-AG (C) were calculated and are represented by the sigmoidal curves presented in each graph. The FRO of each agonist required to produce half-maximal regulation of adenylyl cyclase (▾), ERK-MAPK (▪), and Ca2+-transients (•) were also calculated and are presented as the filled symbols superimposed on the receptor occupancy curve for each agonist. The number in parentheses beside each symbol is the calculated FRO for regulation of each effector (e.g., the FRO at ED50). CP-55,940 and noladin ether require the least receptor occupancy to regulate adenylyl cyclase, whereas 2-AG requires occupancy of the fewest receptors to activate ERK-MAPK. Also demonstrated is the observation that noladin ether requires occupancy of considerably fewer receptors than CP-55,940 to produce equivalent regulation of all effectors examined.
Many studies suggest that individual agonists acting at several types of GPCRs selectively traffic responses by activating distinctive blends of intracellular effectors. Berg et al. (1998) show that in CHO cells transfected with 5-HT2A and 5-HT2C receptors, agonists differ in their efficacies to regulate two intracellular effectors; phospholipase-C-mediated inositol phosphate accumulation and phospholipase A2-mediated arachidonic acid release (Berg et al., 1998). Moreover, the “best-fit” of concentration-response curves for 5-HT2C agonists indicate a three-state model of receptor activation, suggesting that the pathway-dependent efficacies might be explained by two active states of the receptor. In CHO cells expressing M1 muscarinic receptors, potency differences of carbachol, pilocarpine, and AF102B to regulate several intracellular effectors also indicate that selective activation is associated with ligand-specific receptor recognition (Gurwitz et al., 1994). Interestingly, mutation of a cluster of serine residues in the fifth transmembrane domain of the D2S dopamine receptor results in an inability of dopamine, but not several other D2S agonists, to inhibit adenylyl cyclase activity in transfected C6 glioma cells or to activate inwardly rectifying K+ channels in Xenopus laevis oocytes (Wiens et al., 1998). The authors suggest the existence of several conformations of the receptor that are differentially sensitive to serine point mutations. In guinea pig tracheal tube preparation, there are two groups of tachykinin NK1 receptor agonists: one group activates endogenous NK1 receptors to produce nitric oxide release, whereas a second group does not (Figini et al., 1997). Last, in CHO cells transfected with CB1 receptors, the cannabinoid agonist WIN 55,212-2 is equally efficacious in producing inhibition or stimulation of adenylyl cyclase activity (Bonhaus et al., 1998). In contrast, two other cannabinoid agonists, anandamide and CP-55,940, are much less efficacious in stimulating cAMP accumulation relative to inhibiting its formation.
This study is one of the first to provide direct evidence that agonists acting at CB2 receptors also selectively direct signaling. More significant is the observation that two structurally related endocannabinoids produce ADTR. Studies suggest that different intracellular effectors might be responsible for producing distinct physiological effects. For example, the respiratory depressive effects of μ opioid agonists seem to be selectively related to inhibition of adenylyl cyclase activity in respiratory neurons because concurrent administration of 5-HT4A receptor agonists that act by increasing cAMP levels, totally reverse fentanyl-induced respiratory depression, whereas having no effect on analgesia (Manzke et al., 2003). If individual cannabinoid ligands produce selective regulation of effectors by CB2 receptors, agonists might be developed that at optimal concentrations preferentially activate pathways responsible for the therapeutic effects of cannabinoids, while avoiding activation of other pathways potentially mediating undesirable actions.
In addition to providing evidence that ADTR occurs at CB2 receptors, the preferential activation of the ERK-MAPK pathway by 2-AG, relative to noladin ether and CP-55,940, might provide insight into the cellular basis for previously reported agonist selective actions of cannabinoids. For example, in HL-60 cells differentiated into a macrophage-like state, 2-AG produces marked migration through a CB2- and ERK-MAPK-dependent pathway (Kishimoto et al., 2003). In contrast, noladin ether only weakly stimulates migration, whereas anadamide, CP-55,940, WIN 55,212-2, and several other cannabinoids have no effect. 2-AG also results in pronounced ERK-MAPK-dependent migration of myeloid precursor cells via overexpressed CB2 receptors, whereas anadamide produces near negligible effects, and other cannabinoids are devoid of activity (Jorda et al., 2002). Microglial cell migration, a neuroinflammatory response to dying neurons, is initiated in response to CB2 receptor activation by 2-AG, but not by two other putative endocannabinoids, and is dependent on ERK-MAPK activation (Walter et al., 2003). Although involvement of ERK-MAPK was not tested, activation of CB2 receptors by 2-AG induces the migration of EoL-1 human eosinophilic leukemia cells, noladin ether is only weakly effective, and anadamide does not induce migration (Oka et al., 2004). In all the cited studies, 2-AG induces pronounced migration of cells, whereas other endogenously occurring or synthetically derived cannabinoids produce only modest or no effects at all. In addition, migration induced by 2-AG was shown to occur through activation of CB2 receptors and ERK-MAPK. As such, it is tempting to speculate that this rather selective, robust ability of 2-AG to induce migration of variety of cell types might be due to the ability of 2-AG to preferentially regulate ERK-MAPK via CB2 receptors relative to other cannabinoids.
Evidence indicates that individual ligands subtly differing in structure bind cannabinoid receptors in distinct manners (Shim et al., 1998). Such overlapping binding sites for individual cannabinoids may explain the ability of the selective CB2 antagonist SR144528, but not AM630, to attenuate the regulation of Ca2+-transients by 2-AG. Although AM630 was not examined, a previous report similarly demonstrated that SR144528 completely antagonized 2-AG-mediated stimulation of Ca2+-transients via endogenous CB2 receptors expressed in HL-60 cells (Sugiura et al., 2000). Interestingly, similar to our findings with 2-AG, a recent study reported that the reduction in coronary perfusion pressure produced by anadamide is reversed by SR144528 but not by AM630 (Ford et al., 2002). Selective antagonism has also been observed for other lipid-signaling molecules. For example, oleamide produces a concentration-dependent increase in cAMP accumulation in HeLa cells expressing 5-HT7 receptors that is attenuated by the 5-HT antagonist ketanserin but not by clozapine (Thomas et al., 1997). It is proposed that oleamide produces functional effects through binding to an allosteric site on 5-HT7 receptors. Relevant to the present study, distinct binding properties of cannabinoids could potentially contribute to the selective signaling characteristics of 2-AG relative to noladin ether and CP-55,940 reported here.
A final potentially significant finding of this study is that endocannabinoids seem to be more “efficient” agonists at CB2 receptors relative to synthetic agonists. The two endocannabinoids examined, 2-AG and noladin ether, require occupancy of less than one-half the number of receptors to produce comparable regulation of adenylyl cyclase and ERK-MAPK, compared with the synthetic cannabinoid CP-55,940. Indeed, 2-AG and noladin ether require only 1 or 7% receptor occupancy to regulate ERK-MAPK or adenylyl cyclase, respectively. Similar to CP-55,940, we have observed a relatively large receptor occupancy requirement for regulation of adenylyl cyclase by the synthetic cannabinoid WIN 55,212-2 compared with that needed by the endocannabinoids (data not shown). Although speculative, endocannabinoids might improve the efficiency of CB2 signaling by enhancing receptor/G protein interaction and subsequent signal amplification relative to synthetic agonists. Importantly, a large receptor reserve for cannabinoids acting at CB1 receptors in the central nervous system to release acetylcholine has been observed (Gifford et al., 1999). The nonselective cannabinoid agonist WIN 55,212-2 requires only 0.13 and 7.5% receptor occupancy to produce half-maximal and maximal acetylcholine release from hippocampal slices, respectively. If such large receptor reserves for cannabinoid-mediated effects are common, even low levels of endogenously released agonists might be sufficient to activate appreciable second messenger systems and maintain an endocannabinoid “tone” as proposed by others (Steffens et al., 2003).
In summary, this study indicates that endocannabinoids acting at CB2 receptors selectively direct intracellular signaling. It will be important to determine whether similar ADTR by CB2 ligands occurs in cells expressing lower levels of receptors or in phenotypic cells that express native CB2 receptors. Given potentially important roles for CB2 receptors during development of nociceptive and inflammatory states (Di Marzo et al., 2004), understanding ADTR by cannabinoids acting at CB2 receptors could significantly impact future drug development for these diseases.
Footnotes
-
This work was supported in part by National Institute of on Drug Abuse Grant DA13660 (to P.L.P.).
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.105.089474.
-
ABBREVIATIONS: CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2; PTX, pertussis toxin; ERK-MAPK, extracellular signal-regulated kinase subgroup of the mitogen-activated protein kinases; PLCβ, phospholipase-Cβ; 2-AG, 2-arachidonoyl glycerol [(5Z,8Z,11Z,14Z)-5,8,11,14-eicosatetraenoic acid, 2-hydroxy-1-(hydroxymethyl) ethyl ester]; CP-55,940, (-)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl) cyclohexanol; GPCR, G protein-coupled receptor; CHO, Chinese hamster ovary; DMEM, Dulbecco's modified Eagle's medium; AM630 [6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)-methanone; SR144528, N-[(1S)-endo-1,3,3-trimethylbicyclo[2.2.1]heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-1-pyrazole-3-carboxamide; PBS, phosphate-buffered saline; ECL, enhanced chemiluminescence; FACE, fast-activated cell-based enzyme-linked immunosorbent assay; TBS, Tris-buffered saline; ANOVA, analysis of variance; WIN 55,212-2, (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone; HU-210, (6aR)-trans-3-(1,1-dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol; DOR, δ opioid receptor; DPDPE, [d-Pen2,d-Pen5]-enkephalin; U73122, 1-[6-[[17-β-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione; U73343, 1-[6-[[17-β-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-2,5-pyrrolidinedione; FRO, fractional receptor occupancy; AF102B, (±)-cis-2-methyl-spiro(1,3-oxathiolane-5,3′)quinuclidine; AC, adenylyl cyclase; ADTR, agonist-directed trafficking of response; 5-HT, 5-hydroxytryptamine.
- Received May 12, 2005.
- Accepted July 26, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}