Article Text

Abstract
Objective To assess the safety and efficacy of the B lymphocyte (anti-CD20) antibody, rituximab, in the treatment of steroid-resistant moderately active ulcerative colitis (UC).
Methods A double-blinded, randomised controlled trial with a 2:1 ratio of treatment:placebo (phase II) was carried out in the setting of a University teaching hospital. The subjects comprised 24 patients with moderately active UC who have either failed to respond to conventional corticosteroid therapy or who have relapsed during corticosteroid withdrawal. Five of 8 placebo-treated patients and 12 of 16 rituximab-treated patients were receiving azathioprine, 6-mercaptopurine or methotrexate. Two infusions of rituximab 1 g in 500 ml of 0.9% saline intravenously over 4 h (n=16) or saline placebo (n=8) were given at 0 and 2 weeks. Patients still receiving corticosteroids on entry (placebo group 7/8; rituximab group 14/16) continued a standard steroid tapering regimen. The primary end point was remission (Mayo score ≤2) at 4 weeks. Secondary end points included response (Mayo score reduced ≥3) at 4 and 12 weeks.
Results Mayo score at entry was higher in rituximab-treated patients (mean 9.19; 95% CI 8.31 to 10.06) than for placebo patients (7.63; 6.63 to 8.62, p=0.03). At week 4 only 1/8 placebo-treated patients and 3/16 rituximab-treated patients were in remission (p=1.0), but 8/16 rituximab-treated patients had responded compared with 2/8 placebo-treated patients, with a median reduction in Mayo score of 2.5 (rituximab) compared with 0 (placebo; p=0.07). This response was only maintained to week 12 in 4/16. Mucosal healing was seen at week 4 in 5/16 rituximab-treated patients and 2/8 placebo-teated patients (non-significant). Rituximab was well tolerated, with one chest infection, three mild infusion reactions plus one case of (probably unrelated) non-fatal pulmonary embolism.
Conclusions Rituximab has no significant effect on inducing remission in moderately active UC not responding to oral steroids. There was a possible short-term response that was not sustained. Rituximab is well tolerated in UC.
Clinical trial number NCT00261118.
- Colitis
- ulcerative/therapy
- colitis ulcerative/pathology
- randomised controlled trial
- rituximab
- human
- adult
- B cell
- clinical trials
- inflammatory bowel disease
- ulcerative colitis
Statistics from Altmetric.com
- Colitis
- ulcerative/therapy
- colitis ulcerative/pathology
- randomised controlled trial
- rituximab
- human
- adult
- B cell
- clinical trials
- inflammatory bowel disease
- ulcerative colitis
Significance of this study
What is already known about this subject?
There is evidence that ulcerative colitis (UC) is, in part, an autoimmune disease.
Rituximab (an anti-CD20 antibody) has been shown to be effective therapy in other autoimmune diseases.
Medical treatment options for steroid-refractory UC are limited.
What are the new findings?
In this first in human trial, rituximab is not effective in gaining remission in steroid-refractory UC.
There may be a modest short-term effect but this is not maintained.
Rituximab is well tolerated.
How might it impact on clinical practice in the foreseeable future?
B cells may play a limited role in the pathophysiology of active UC.
If rituximab is indicated for another reason, it can be given safely to people with UC.
Introduction
Although the pathogenesis of ulcerative colitis (UC) is incompletely understood, there is evidence that it may be an autoimmune disease, whereas in Crohn's disease evidence of autoimmunity is modest.1 Early studies in children reported that 80% had antibodies against colonic epithelial cells,2 and later studies confirmed the frequent presence of circulating antibodies against colon epithelial goblet cells.3 4 About two-thirds of patients are positive for pANCA (perinuclear anticytoplasmic neutrophil antibody),5 directed against components of neutrophil leucocytes.6 7
The presence of pANCA in UC sera has been regarded as an epiphenomenon, but the myeloperoxidase-directed pANCA that occurs in some forms of vasculitis is itself the cause of vasculitis in animal models.8 pANCA cross-links myeloperoxidase expressed on the neutrophil outer membrane, thus triggering neutrophil activation, leading to glomerulonephritis and vasculitis in mice infused with pANCA. Although pANCA in UC is not directed against myeloperoxidase, various neutrophil granule components, including possibly the UC pANCA epitope(s), may become expressed on the outer membrane of the activated neutrophil. Separately, antibodies to tropomyosin that also occur in UC may induce complement deposition and destruction of colonic epithelial cells.9
For most autoimmune diseases, the MHC (major histocompatiblity complex) region on the short arm of chromosome 6 is the strongest genetic component. UC susceptibility has been linked to a narrow MHC-related genomic window (IBD3) which contains the human leucoyte antigen (HLA)-DRB1 gene, and recent evidence suggests that the HLA-DRB1*1101 allele plays a primary role in UC susceptibility.10 Pooled analysis also substantiates the association of UC with HLA-DRB1*0103 and HLA-DRB1*1502.11 Class II HLA associations would implicate CD4 helper T cells and, indirectly, B cells whose activity they are largely responsible for inducing, in UC pathogenesis.12
Traditionally the profile of proinflammatory cytokine production in UC was thought to be in keeping with a B lymphocyte-predominant, T helper 2 (Th2) type of response.13 However, more recent evidence suggests the involvement of more complex cytokine pathways with contribution from Th17 cells driven by the production of interleukin 23.14 Further evidence of autoimmunity in UC is the association with other autoimmune diseases such as thyroiditis, autoimmune haemolytic anaemia and autoimmune hepatitis.15
Rituximab is a humanised antibody directed against CD20, a cell surface molecule expressed by mature B lymphocytes but not by their precursor cells, and its intravenous infusion produces an effective but reversible depletion in B cell numbers. Because of the pivotal role of B lymphocytes in the development and perpetuation of autoimmune diseases, rituximab has been used successfully in several autoimmune diseases including rheumatoid arthritis (RA), systemic lupus erythematosus, Sjogren syndrome, dermatomyositis and autoimmune haemolytic anaemia.16 The mechanism of action of rituximab is mediated through antibody-dependent cell-mediated immunity, complement-dependent cell-mediated cytotoxicity and promotion of CD20-positive cell apoptosis. Rituximab has been particularly extensively investigated in RA, a disease with well-described autoreactive B cells producing autoantibodies such as rheumatoid factor and anticitrullinated peptide. Trials reported to date are largely in patients non-responsive to anti-tumour necrosis factor α (TNFα) treatment. Meta-analyses of three studies including 823 patients comparing rituximab plus a disease-modifying antirheumatic drug (DMARD) versus placebo plus a DMARD showed a relative effect of 2.92 (95% CI 1.76 to 4.83).17 Pooled analysis of the long-term safety of rituximab in clinical trials in patients with RA (n=5013 patient-years of rituximab exposure) is reassuring. The most frequent adverse events were infusion-related reactions (25% during the first infusion, <1% of which were considered serious). The overall serious infection rate was 4.31/100 patient-years (95% CI 3.77 to 4.92).18 Two recent trials have demonstrated the efficacy of rituximab (given weekly for 4 weeks) in inducing remission in ANCA-associated diseases: ANCA-associated vasculitis19 and ANCA-associated renal vasculitis.20
In contrast, three cases of UC have been reported in which it has been suggested that rituximab might have caused exacerbation or new onset of disease.21–23 One case had pre-existing UC and had received multiple treatments, and relatively short follow-up was presented, one developed new-onset typical UC and a third developed a UC-like colitis which resolved with steroid therapy and did not recur. We had previously noted a case of azathioprine-treated UC complicated by rectal Epstein–Barr virus-associated B cell lymphoma in whom chemotherapy that included rituximab had led not only to cure of the lymphoma but also to long-term remission of UC despite withdrawal of azathioprine.
We therefore hypothesised that B cell depletion by rituximab in active steroid non-responsive UC could be effective in inducing remission.
Methods
Trial design
This randomised double-blind placebo-controlled pilot trial compared the efficacy of two infusions of 1 g of rituximab (Roche, Basel, Switzerland) versus placebo in inducing remission in active steroid-resistant UC. Randomisation was 2:1 for active therapy compared with placebo. The trial was conducted in a single centre (Clinical trial number NCT00261118).
Ethical considerations
The trial was approved by the Liverpool Research Ethics Committee.
Participants
Eligible participants were patients with an established diagnosis of UC by conventional clinical, endoscopic and histological criteria, aged >18 years at trial entry, and capable of giving written consent. Active steroid-resistant UC was defined as a Mayo score24 (also known as a Disease Activity Index) of 6–12 inclusive and failure of response to at least 2 weeks of oral prednisolone (40 mg/day) or relapse of disease on tapering the prednisolone dose. Exclusion criteria were: first attack of UC, severe UC (presence of any of: temperature >37.5°C, pulse rate >100 bpm, focal severe or rebound abdominal tenderness, haemoglobin <10.0 g/dl, serum albumin <3.5 g/dl, transverse colon diameter >5.0 cm on plain abdominal x-ray), patients with a stoma, positive stool culture for pathogens or test for Clostridium difficile at screening within 7 days prior to trial entry, those for whom a baseline Mayo score could not be reliably calculated (frequent use of laxatives; eg, for proximal constipation or antimotility agents for control of diarrhoea) and disease extending <20 cm from the anal margin. Women were excluded if they were at risk of pregnancy and not willing to use a reliable form of contraception (oral contraceptive and barrier or barrier plus spermicide), were pregnant, postpartum (<3 months) or breast feeding.
Concomitant medication
Immunosuppressive medication (azathioprine, 6-mercaptopurine or methotrexate) had to be taken at a continuous stable dose for the previous 3 months and 5-aminosalicylates for the previous month. Patients were excluded if there had been any change to rectal therapy for colitis within the previous 2 weeks. All patients were naive to anti-TNFα therapy.
Intervention
Eligible patients received either rituximab 1 g in 500 ml of 0.9% saline infused into a peripheral vein over 2 h or 500 ml of 0.9% saline infused into a peripheral vein over 2 h as placebo. Infusions were identical in appearance. This regimen was repeated at 2 weeks. Patients who were receiving oral prednisolone 40 mg/day on entry continued to receive oral prednisolone 40 mg/day for 2 weeks then 30 mg for 2 weeks, then 20 mg/day for 2 weeks, then reduced by 5 mg/day every 7 days until off prednisolone. Patients entering the trial because of relapse of UC on tapering steroids did not have the dose of steroids increased but were kept stable for 2 weeks and then reduced as per the protocol above.
Evaluation schedule
At screening, routine bloods were evaluated (urea and electrolytes, albumin, full blood count and C-reactive protein (CRP)) and peripheral blood B lymphocyte counts measured by flow cytometric analysis for CD19-positive cells (Coulter EPICS XL-MCL, Beckman Coulter, Miami, Florida, USA). CD19 was selected as it is a specific pan-B cell marker distinct from CD20 against which rituximab is directed. All patients underwent flexible sigmoidoscopy and biopsies (20 cm from the anal margin) within a week prior to randomisation. Patients were reviewed at weeks 2, 4, 8 and 12, and safety follow-up was undertaken at week 24. At each visit, blood urea and electrolytes, albumin, full blood count, CRP and CD19 count (week 0, 4 and 8 only) were performed. Flexible sigmoidoscopy and biopsies (20 cm from the anal margin) were performed at weeks 4 and 12. Full Mayo score was calculated at weeks 0, 4 and 12, and partial Mayo score (without endoscopic scores) was calculated at weeks 2 and 8. Inflammatory bowel disease (IBD) quality of life questionnaires (IBDQs)25 were completed at weeks 0, 4 and 12.
Withdrawal
Patients were withdrawn from the trial if their Mayo score increased by ≥3 points at week 4 and/or clinical deterioration requiring alternate therapy and/or an adverse event sufficient to preclude further dosing with rituximab occurred. For withdrawn patients the last value was carried forward.
Definition of outcomes
The primary outcome measure was remission defined as a decrease in Mayo score to ≤2 points at week 4. Secondary outcome measures were clinical response (defined as a decrease in Mayo score by ≥ 3 points at weeks 4, 8 (partial Mayo score) and 12), remission at weeks 8 and 12, endoscopic mucosal healing at weeks 4 and 12 (Mayo endoscopic score of 0 or 1), and improvement in the IBD-specific Quality of Life Index at weeks 4 and 12. Histological improvement of disease activity at 4 and 12 weeks was compared with baseline and scored blinded as follows: 0=no polymorphs, 1=small numbers of polymorphs in the lamina propria with minimal infiltration of crypts; 2=prominent polymorphs in the lamina propria with infiltration of ≥50% of crypts; 3=florid polymorph infiltrate with crypt abscesses; 4=florid acute inflammation with ulceration. Post hoc analysis of CD20 expression in colonic biopsies in patients who had received rituximab was performed using anti-CD20cy (B cell clone L26, Dako Ltd, Ely, UK) immunohistochemistry. Blinded assessment (to the timing on biopsies) was performed for CD20 as follows: 0=no staining, 1=staining of scattered cells, 2=staining of cells and aggregates, 3=staining of numerous cells and aggregates.
Sample size and statistical analysis
Sample size calculations were based on 80% power for excluding, at p<0.05, an 80% remission rate with active treatment compared with a predicted 25% placebo remission rate with 2:1 randomisation of rituximab compared with placebo. These assumptions resulted in a sample size of 16 patients receiving rituximab and 8 patients receiving placebo.
All analyses used two-sided tests. Formal hypothesis testing of the primary outcome was by the Fisher exact test. Wilcoxon signed rank test was used for comparisons against baseline for changes in secondary quantitative end points. Blinded and unblinded data were reviewed by two independent reviewers.
Randomisation
Randomisation was 2:1 rituximab:placebo and allocated in blocks of five by the pharmacy department of the hospital. The pharmacist randomising the patients had no other participation in the trial. Randomisation allocation was concealed from participants and investigators until after the final patient completed the trial. Assessment of response or remission was made before unblinding of treatment allocation.
Results
Baseline characteristics
A total of 24 patients were enrolled (n=16 rituximab and n=8 placebo) from 2004 to 2009 in a single centre. All patients received active drug or placebo. The baseline characteristics were mostly similar in both groups (table 1). However, the baseline mean Mayo score was significantly higher in the rituximab group (9.2 (1.7)) compared with placebo (7.6 (1.2)) (mean (SD)) p=0.03, and all the steroid non-responders (n=7) were in this group. There was a non-significant trend towards more extensive disease in the placebo group. The flow of patients in the trial is shown in figure 1.
Baseline characteristics
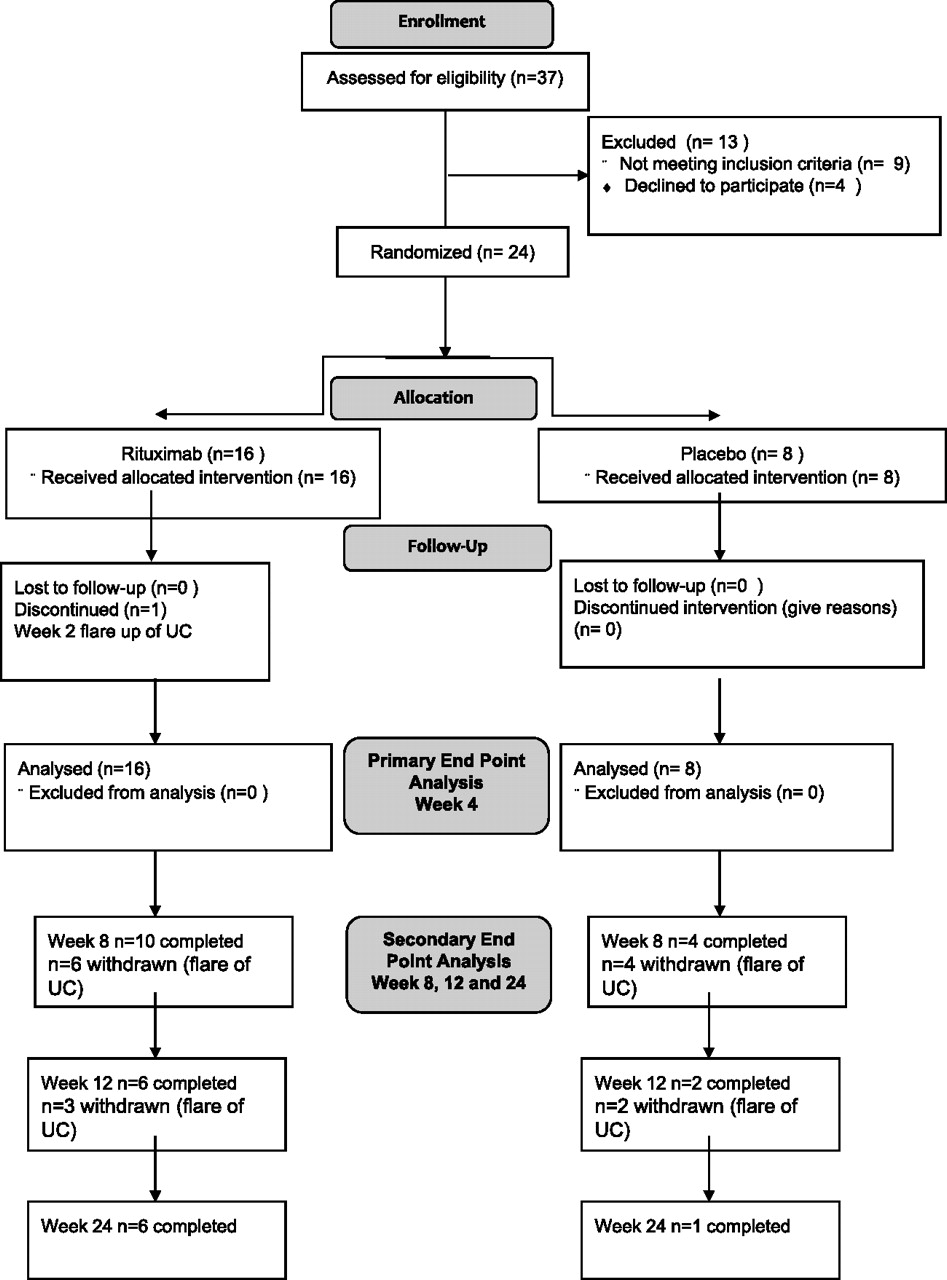
CONSORT study flow diagram. UC, ulcerative colitis.
Mayo score
At week 4, only 1/8 placebo-treated patients (12.5%) and 3/16 rituximab-treated patients (19%) were in remission (p=1.0, Fisher exact test). Likewise, there were no significant differences in remission at week 8 (1/16 rituximab vs 1/8 placebo, p=1.0) or week 12 (0/16 rituximab vs 1/8 placebo, p=0.33) (figures 2 and 3). However at week 4, 8/16 (50%) rituximab-treated patients had shown a response (decrease in Mayo score by ≥3) compared with 2/8 (25%) of the placebo group, with a median reduction in Mayo score of 2.5 (rituximab) compared with 0 (placebo; p=0.07). By week 8 this difference in response was lost (6/16 rituximab vs 1/8 placebo, p=0.36) (week 12 (4/16 rituximab vs 1/8, p=0.67)) and there were no significant differences in reduction of Mayo score at week 8 (median reduction 0.5 rituximab vs 0 placebo, p=0.25) or week 12 (median reduction 0 points for both groups. p=0.25). Because of the significant difference in baseline Mayo score, post hoc analysis was performed with a definition of response as a fall of ≥30%. Using this definition, there were no significant differences in response rate at week 4 (9/16 rituximab vs 3/8 placebo, p=0.67) or week 12 (5/16 rituximab vs 1/8 placebo, p=0.63).

Full Mayo score at baseline, week 4 and week 12. At baseline, mean (SD) Mayo score was significantly higher in the rituximab group (9.2 (1.7)) compared with the placebo group (7.6 (1.2)), p=0.03. There were no significant differences in the proportion of patients achieving remission (decrease in Mayo score to ≤2 points) at week 4 (primary outcome measure) or week 12 (secondary outcome measure). At week 4, 8/16 (50%) rituximab-treated patients had shown a response (decrease in Mayo score score by ≥3) compared with 2/8 (25%) placebo-treated patients, with a median reduction in Mayo score of 2.5 (rituximab) compared with 0 (placebo; p=0.07), but this effect was lost by week 12.

Partial Mayo score (without endoscopic scores) at weeks 2, 8 and 24. There were no significant differences in remission at any time point, nor any significant differences in reduction of Mayo score at any time point.
Mucosal healing
At baseline all patients had a Mayo endoscopic score ≥2. Endoscopic mucosal healing (score 0 or 1) was seen at week 4 in 5/16 rituximab-treated patients and 2/8 placebo-treated patients (p=1.0, Fisher exact test) and at week 12 in 2/16 rituximab-treated patients and 1/8 placebo-treated patients (p=1.0, Fisher exact test). Histological healing (score of 0) was seen at week 4 and 12 in 1/16 rituximab-treated patients and 0/8 placebo-treated patients (p=1.0, Fisher exact test).
IBD quality of life questionnaires
Mean (SD) IBDQ score at entry was 112 (31) for rituximab and 116 (36) for placebo. At week 4 the mean improvement of IBDQ score was 27 (40) points for rituximab and 1 (26) point for placebo (p=0.13), and at week 12 mean improvement was 17 (45) points for rituximab and 2 (29) points for placebo (p=0.40) (figure 4).

Mean (SD) inflammatory bowel disease quality of life questionnaire (IBDQ) score at entry was 112 (31) for rituximab and 116 (36) for placebo. At week 4 the mean improvement in the IBDQ was 27 (40) points for rituximab and 1 (26) point for placebo (p=0.13), and at week 12 mean improvement was 17 (45) points for rituximab and 2 (29) points for placebo (p=0.40).
CRP/white blood cell (WBC)/lymphocyte counts/ANCA
There were no significant changes in the values for CRP (Supplementary figure 1), haemoglobin, total WBC count or albumin at any time point (data not shown). Due to assay problems early in the study only 7/16 patients in the rituximab group (6/7 positive) and 2/8 in the placebo group had pANCA testing (2/2 positive). Therefore, further subgroup analysis in relation to pANCA status was not performed.
Blood CD19 and colonic tissue CD20 expression
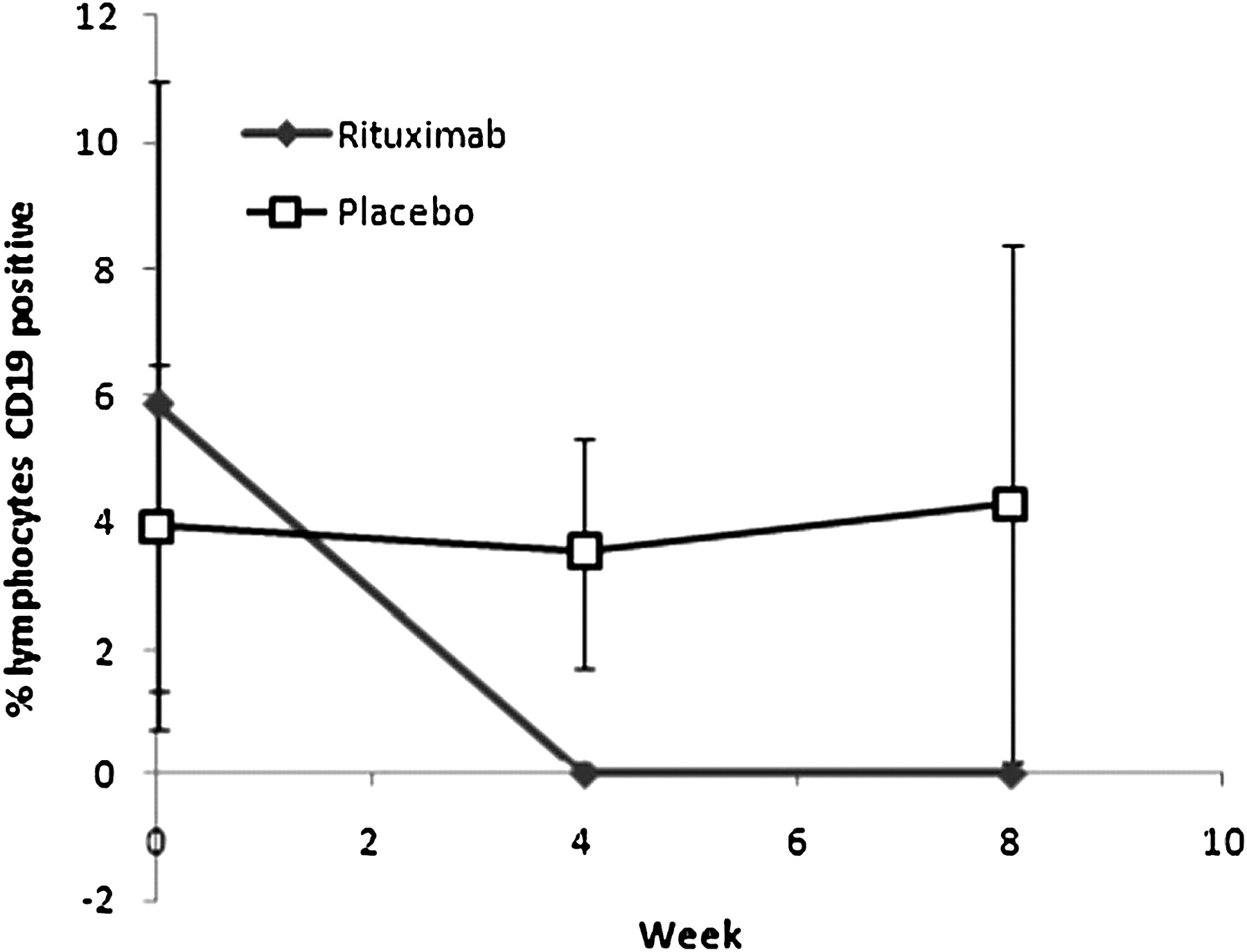
The mean (SD) percentage of peripheral blood lymphocytes which were CD19-positive B cells at baseline was 5.86% (5.13%) for rituximab patients and 3.93% (2.59%) for placebo patients (p=0.34). CD19 B cell counts fell significantly in the rituximab group to 0.02 (0.03) at week 4 and 0.01 (0.03) at week 8 (p=0.005), with no fall in the placebo group: 3.54 (1.8) at week 4 and 4.29 (4.11) at week 8 (figure 5). In all patients who received rituximab, at week 4 CD19 counts fell to <0.08% of total peripheral blood lymphocytes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean (SD) percentage lymphocytes which were CD19-positive B cells at baseline, week 4 and week 8. CD19 B cell counts fell significantly in the rituximab group to 0.02 (0.03) at week 4 and 0.01 (0.03) at week 8 (p=0.005), with no fall in the placebo group: 3.54 (1.8) at week 4 and 4.29 (4.11) at week 8.
Post hoc analysis of CD20 expression in colonic mucosal biopsies of patients who received rituximab showed that before treatment all patients had either score 2 or 3 (either staining of cells and aggregates or staining of numerous cells and aggregates). All but one patient receiving rituximab had lost CD20 expression at week 4 (score 0) (Supplementary figure 2). The single patient with residual CD20 expression had score 3 at baseline and score 2 at week 4. Biopsies were available from three rituximab-treated patients at week 12, all of whom were negative at week 4, and all these continued to be negative for CD20.
Safety
All patients completed follow-up at the primary end point at week 4. Only 1/8 placebo patients completed 24 weeks in the trial, with all withdrawals because of worsening or continuing UC; four patients before week 8, two before week 12 and one before week 24. In the rituximab group, 6/16 completed 24 weeks follow-up with all withdrawals because of worsening or continuing UC; six before week 8, two before week 12 and two before week 24 (figure 1). There were no significant differences in the adverse event profile of the two groups (table 2). A total of 15/16 rituximab and 8/8 placebo patients reported adverse effects. Four serious adverse events occurred; three for rituximab (one hospitalisation for flare up of UC, one for hospitalisation with pulmonary embolism and one pregnancy) and one for placebo (hospitalisation for flare up of UC). There was no difference in the incidence of infection in the two groups. The patient who became pregnant following rituximab had a negative pregnancy test at screening and was aware of the contraceptive advice. She had a positive pregnancy test at week 4, the pregnancy was uneventful and the baby had a normal lymphocyte count at birth and has remained well since.
Adverse events (AEs)
Discussion
In this randomised, placebo-controlled phase II trial, rituximab had no effect in achieving remission in steroid-non-responsive UC. Although there was a non-significant increase in response rate at week 4 this was not sustained. Rituximab showed a biological effect by a profound reduction in CD19 levels and loss of CD20 expression in colonic tissue. We conclude that knocking out mature B cells with rituximab is not effective treatment for steroid-non-responsive UC.
The trial is limited by the small sample size. However, this was a pilot/proof of concept trial with placebo blinding and it was therefore only powered for a large difference in effect between rituximab and placebo (80% vs 25% remission rates). There is, however, no sign of any difference in remission rates between rituximab and placebo, and the fact that only one of 16 rituximab-treated patients was in remission by week 8 suggests that there is insufficient efficacy to justify further study of what is an expensive and relatively potent immunological therapy.
The dose of rituximab was the same as that used in large trials in RA and it is unlikely that a different dosing regime would be more effective, particularly as a biological response was achieved with this regime, as reflected in the marked reduction in CD19, maintained at least until week 8, and absence of CD20 expression in colonic tissue in nearly all patients at week 8. The same dosing regime in RA achieved a 50% improvement in American College of Rheumatology (ACR) criteria in 43% (rituximab and methotrexate) compared with 13% for methotrexate alone (p=0.005).26
The group of patients enrolled in this study had refractory UC and many had long-term refractory disease on immunosuppression. It remains possible that rituximab might have an effect at an earlier stage in UC.
Three previous case reports have suggested that rituximab may have adverse effects in UC.21–23 One describes the onset of UC after rituximab therapy for Graves disease.21 The UC responded to conventional therapy. The second was a paediatric patient with nephrotic syndrome refractory to conventional therapy who developed a new-onset severe UC-like colitis 42 days after treatment with rituximab.22 The colitis may have been triggered by torovirus and the colitis responded to corticosteroids. No relapse of colitis occurred in 2 years of follow-up. The third describes the use of rituximab in a patient with chronically active medication-refractory UC, in which the UC worsened after rituximab administration.23 However, due to the multiple medications used it is difficult to ascribe this directly to rituximab. There are some animal data suggesting that B cells may be protective in IBD by regulation of T cell activity and production of interleukin 10.27 28 In contrast, in a murine model of Crohn's ileitis in SCID mice, co-transfer of B cells together with CD4 T cells caused more intestinal inflammation than transfer of CD4 T cells alone, suggesting that B cells worsen colitis.29
One patient became pregnant during the trial after receiving rituximab. There were no adverse events during the pregnancy and the baby had normal B cell counts at delivery, and at follow-up has suffered no adverse effects and has normal development. There are limited data on safety of rituximab in pregnancy but the few case reports have not shown adverse effects on the baby.30 It is likely that the lack of adverse effect on fetal B cell development in our patient is due to the fact that rituximab is an immunoglobulin G antibody and does not cross the placenta until after week 16 and she received rituximab before conception. One report describes depletion of neonatal B cells with third trimester administration of rituximab with subsequent recovery at month 6.31
Rituximab has no significant effect on inducing remission in moderately active UC unresponsive to oral steroids. There may be a modest therapeutic effect in the short term but no sustained effect. Rituximab is well tolerated with no increase in side effects compared with placebo. In contrast to previous case reports, rituximab was not associated with worsening of UC and may be considered for use in patients with UC who have other indications for rituximab.
Acknowledgments
We thank Professor Mark Pritchard and Dr Melita Gordon, Univerity of Liverpool, for independent blinded and unblinded review of data.
References
Supplementary materials
Online only data supplement
Files in this Data Supplement:
Footnotes
Funding The costs of the rituximab and placebo were provided by an unrestricted grant from Roche, UK. The company had no other involvement in the trial. The trial was designed, conducted and results analysed by the authors.
Competing interests None.
Ethics approval This study was conducted with the approval of the Liverpool medical research ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.