Article Text

Abstract
Background: In hereditary non-polyposis colorectal cancer, over 90% of the identified mutations are in two genes, hMSH2 and hMLH1. A large proportion of the mutations detected in these genes are of the missense type which may be either deleterious mutations or harmless polymorphisms.
Aim: To investigate whether nine missense and one splice site mutation of hMLH1 and hMSH2, in 10 kindreds with a familial history of colorectal cancer or young age of onset, could be interpreted as pathogenic.
Methods: Clinical and genetic characteristics were collected: (i) evolutionary conservation of the codon involved; (ii) type of amino acid change; (iii) occurrence of mutation in healthy controls; (iv) cosegregation of mutation with disease phenotype; (v) functional consequences of gene variant; and (vi) microssatellite instability and immunoexpression of hMSH2 and hMLH1 analysis.
Results: Seven different missense and one splice site mutation were identified. Only 1/8 was found in the control group, 2/7 occurred in conserved residues, and 5/7 resulted in non-conservative changes. Functional studies were available for only 2/8 mutations. Segregation of the missense variant with disease phenotype was observed in three kindreds.
Conclusion: In the majority of families included, there was no definitive evidence that the missense or splice site alterations were causally associated with an increased risk of developing colorectal cancer. Until further evidence is available, these mutational events should be regarded and interpreted carefully and genetic diagnosis should not be offered to these kindreds.
- hereditary non-polyposis colorectal cancer
- genetic diagnosis
- mismatch repair genes
- missense and splice site mutations
- HNPCC, hereditary non-polyposis colorectal cancer
- MMR, mismatch repair
- DGGE, denaturing gradient-gel electrophoresis
- PCR, polymerase chain reaction
- MSI, microsatellite instability
Statistics from Altmetric.com
- hereditary non-polyposis colorectal cancer
- genetic diagnosis
- mismatch repair genes
- missense and splice site mutations
- HNPCC, hereditary non-polyposis colorectal cancer
- MMR, mismatch repair
- DGGE, denaturing gradient-gel electrophoresis
- PCR, polymerase chain reaction
- MSI, microsatellite instability
Hereditary non-polyposis colorectal cancer (HNPCC; OMIM120435-6) is a cancer susceptibility syndrome linked to inherited defects in human mismatch repair (MMR) genes.1, 2 It has been associated with an increased lifetime risk of colorectal as well as a number of extracolonic cancers, such as those of the endometrium or small bowel.3, 4 Genetic testing for patients complying with the Amsterdam criteria for HNPCC diagnosis5 is currently performed in the hope of identifying asymptomatic gene carriers in whom application of appropriate screening and intervention programmes will result in decreased morbidity and mortality.6–,8 Over 90% of the identified mutations are in two genes, hMSH2 and hMLH1, located on chromosomes 2p and 3p, respectively.9–,11 The nature of the mutations differs between the hMSH2 and hMLH1 genes. Whereas most mutations affecting the hMSH2 gene predict the premature termination of the reading frame, 50% of the mutations occurring in the hMLH1 gene are of the missense type.11 The pathogenic significance of these amino acid substitutions is often difficult to evaluate and may present an obstacle to genetic testing in HNPCC.12–,14
Functional impairment of proteins resulting from these gene alterations is probably the critical factor in deciding on the pathogenicity of a given mutation. Two functional assays have been established to help identify and evaluate whether mutations in the MMR genes are pathogenic.15, 16 The first is a “dominant negative” mutator assay in yeast15 while the second functional test is a protein-protein assay measuring the degree of interaction between hMLH1 and hPMS2.16 However, as stated by previous authors,12 the lack of validation of these assays in controlled human studies makes it premature to use them as the sole basis for making clinical recommendations.
Other criteria used to predict the pathogenicity of these missense mutations are: (i) the codon involved is evolutionary conserved, (ii) the alteration leads to a non-conservative amino acid change, (iii) the missense mutation does not occur in the general population, and (iv) the mutation cosegregates with the disease phenotype in the family.
In the present study, we report on nine kindreds with a familial or personal history suggestive of HNPCC in whom we detected missense mutations in either the hMSH2 or hMLH1 MMR genes. Additionally, one young patient carrying a splice site mutation of doubtful pathogenic effect was also included. Using some of the above mentioned criteria, as well as microsatellite instability analysis and immunoexpression of MMR genes in tumour tissue, we tried to determine whether these variants could be interpreted as disease causing mutations or only rare polymorphisms.
MATERIAL AND METHODS
Subjects
Forty one subjects from 10 Portuguese families with a personal or familial history of colorectal cancer were included in the present study.17 Patients were identified by self referral or health care provider referral to our cancer genetics clinic. Four of these families complied with the Amsterdam criteria for HNPCC diagnosis, two did not meet all of the criteria, and the remaining four were young patients with colorectal cancer (less than 35 years of age). Personal and family cancer histories and demographic data were obtained from the proband and participating relatives, and cancer diagnoses and deaths were confirmed by review of the medical records, pathology reports, or death certificates. All index cases included in this study gave written informed consent to participate in the study. The prevalence of the variants found in patients was evaluated in 50 healthy blood donors.
Molecular methods
Genetic analysis was performed on a blood specimen from one affected member of each family. DNA was extracted from venous blood using the guanidine/HCl method. Mutation analysis in the hMLH1 and hMSH2 genes was performed using GC clamped denaturing gradient-gel electrophoresis (DGGE), as described previously.18 Primers used for amplification of the hMSH2 and hMLH1 genes have been published previously.19, 20 For optimal DGGE conditions, DNA melting behaviour simulations were performed with the MELT87 program which was developed by Dr L Lerman.21 DNA fragments that displayed an abnormal DGGE pattern were sequenced using the same primers as for DGGE but without the GC clamp. The amplified fragment was purified using QIAquick polymerase chain reaction (PCR) purification kit protocol (Qiagen, Ontario, Canada). [γ-32P] dATP end labelled primer was used for cycle sequencing using the fmol DNA sequencing system kit (Promega, Madison, Wisconsin, USA). The products were resolved in 7% Long Ranger (JT Baker, Deventer, the Netherlands) acrylamide gels containing 7 M urea.
Microsatellite instability analysis
Tumours arising in the context of HNPCC typically exhibit the so-called microsatellite instability (MSI) phenomenon, characterised by expansion or contraction of short repeat sequences of DNA at multiple loci.22, 23 MSI was analysed in colorectal tumours from nine individuals/families included in the study. DNA was extracted from fresh frozen tissue from colorectal tumours and their corresponding normal mucosa using a previously described method and PCR amplified at five loci containing di or mononucleotide repeated sequences (CA)n : BAT-26, BAT-40, D2S123, D5S346, and D17S783, using primers specific for each locus and obtained from Research Genetics (Huntsville, Alabama, USA). The PCR reaction was performed in a total volume of 12.5 μl using 200 mM concentration of each deoxynucleotide triphosphate, 10 pmol of each primer, 75 ng of DNA, 0.5 U of Taq polymerase (BRL, Eggenstein, Germany), and 1 μCi of 32P-dCTP. Samples were processed through 35 cycles of amplification with annealing temperatures which ranged from 55 to 60°C. PCR products were diluted 1:1 with loading buffer containing 95% formamide, denatured, and electrophoresed on polyacrylamide gels containing 6.9 M urea and 32.5% formamide. Gels were fixed in 10% acetic acid, dried, and exposed overnight to MP film (Amersham Corp., UK) at −70°C. Tumour MSI was defined as changes in size bands observed in neoplastic DNA but not visible in the corresponding non-neoplastic DNA.
Immunoexpression of hMSH2 and hMLH1 genes
Formalin fixed paraffin embedded tissue samples of tumour and normal tissue were sectioned. Deparaffinisation and rehydration were performed using xylene and alcohol. Optimal antigen retrieval was obtained using a domestic pressure cooker for one minute at maximum pressure with 0.01 M citrate buffer, pH 6.0.24 Two monoclonal mouse antibodies reacting against hMLH1 (clone G168-728-Pharmigen) and hMSH2 (clone FE11-Oncogene) protein gene products were used in the study.25 Optimal dilutions of the antibodies in Tris buffered saline were 1:150 and 1:300, respectively, for hMSH2 and hMLH1. Bound antibody was detected using biotinylated rabbit F (ab)`2 antibody to mouse immunoglobulin (Dako, Glostrup, Denmark) diluted in Tris buffered saline (1:250) with normal serum (1:25). An avidin-biotin complex linked to horseradish peroxidase (Vector, Burlingame, USA) diluted 1:50 in Tris buffered saline was then used. All incubations were carried out at room temperature and the primary antibody was incubated overnight at 4°C. A solution of diaminobenzidine was used as chromogenic substrate for peroxidase. A positive reaction was recognised whenever there was unequivocal nuclear staining of neoplastic cells. Tumour cells without nuclear staining in the presence of normally stained non-neoplastic stromal cells were considered to exhibit an abnormal pattern of expression.
RESULTS
Characterisation of the eight variants found in the 10 families/individuals included in the study are presented in table 1⇓.
Evaluation of potential pathogenicity of missense variants found in the hMLH1 and hMSH2 genes
In family No 1, the germline mutation was found to affect exon 17, codon 659 of the hMLH1 gene CGA to CTA (arg to leu). This mutation has previously been reported to result in aberrant splicing with deletion of exon 17, thereby leading to a non-functional protein.26 Segregation analysis also supported a pathogenic role for this mutation as affected members (II:2 and III:1; fig 1⇓) were shown to carry this same mutation while II:5 did not. MSI was positive in 2/5 markers and nuclear expression of the hMLH1 gene was absent in tumour tissue from one affected patient.

Pedigree of family No 1.
Family No 2 presented with a germline mutation affecting exon 16, codon 607 of the hMLH1 gene (table 1⇑). Although occurring in a non-conserved codon, this variant results in a non-conservative amino acid substitution (table 1⇑), and it was not observed in any of the 50 healthy controls included in the study. Segregation analysis showed that individuals II:8 and III:9 did not carry this mutation while affected member III:1 who presented with colon cancer at the age of 41 presented with this missense variant (fig 2⇓). MSI analysis was negative for all five markers analysed and immunoexpression of MMR genes was normal for both the hMSH2 and hMLH1 genes. No functional studies are available, but except for the absence of MSI, the remaining criteria support the hypothesis that L607H could determine an increased risk of colonic cancer in this family.

Pedigree of family No 2.
Family No 3 (fig 3⇓) was found to carry a missense mutation in exon 8, codon 213 of the hMLH1 gene. In contrast with previous missense mutations, this is a conservative substitution in the sense that both amino acids are similar (val to met) (table 1⇑). Segregation analysis in this family was carried out in five relatives. Three unaffected members were negative for this mutation (IV:2, IV:8, and IV:20), one healthy family member aged 47 was found to carry this variant (IV:18), and more surprisingly, one affected member who presented with colorectal cancer at the age of 44 years (IV:17) was found to be negative for this mutation. Immunoexpression of both MMR genes was normal and MSI was negative for all five markers analysed. Thus in the present kindred, although functional studies are not yet available and this variant was not found in the general population, the type of amino acid change, absence of MSI, as well as results of segregation analysis suggest that V213M might be a rare polymorphism rather that a pathogenic mutation. For these reasons, this kindred was not offered genetic diagnosis.

Pedigree of family No 3.
Family No 4 (fig 4⇓) was found to carry a mutation in exon 16, codon 618 (AAG to ACG) of the hMLH1 gene. This alteration was not present in any of the 50 healthy controls analysed and previous studies described it as being pathogenic.13, 27 Also, both the dominant negative mutator assay and the protein-protein assay measuring the degree of interaction between hMLH1 and hPMS2 showed that the K618T mutation resulted in substantial loss of protein function.15, 16 However, segregation analysis showed that family member V:5, who had colorectal cancer at the age of 53 years, did not carry this same mutation, similar to the unaffected individual VI:2, whereas individual VI:1, presently healthy at age 36 years, was found to carry this missense mutation. MSI was not observed in tumour tissue and nuclear expression of both MMR genes was normal. Thus, and in contrast with most criteria analysed, segregation analysis does not support a role for K618T in cancer development in this family. Until further clarification, genetic diagnosis is not being offered to this family.

Pedigree of family No 4.
Family No 5 (fig 5⇓) was found to carry two mutations in the hMSH2 gene. One was a nonsense mutation on exon 10, codon 518, CAG to TAG (stop codon) and the other was a missense mutation on exon 6, codon 322 GGC to GAC. Although we may assume that the truncating mutation is the disease causing event, we tried to investigate whether the missense variant played any role in determining cancer susceptibility in this family. Previous studies first interpreted G322D as being pathogenic based on location in an evolutionary conserved amino acid.28, 29 Further support for this hypothesis, and although functional studies are not yet available, is that this variant leads to a non-conservative amino acid change and it was not found in any of the 50 healthy controls included in the present study. However, studies including a higher number of controls report a frequency of 0–3% in the reference population20, 30–,32 which reduces the probability that it is functionally significant. Consistent with the hypothesis that G322D might solely be a bystander mutation are the results from segregation analysis. Thus none of these mutations were detected in a healthy subject (IV:7) whereas family members IV:2 and IV:4 who presented with colonic tumours at ages 48 and 43 years, respectively, were found to carry the truncating mutation but not the missense variant (fig 5⇓). Taken together these data further support the view that G322D should probably be regarded as a rare polymorphism, at least in the present kindred. MSI analysis was positive in 5/5 markers and immunohistochemistry for the hMSH2 gene was negative in tumour tissue.

Pedigree of family No 5.
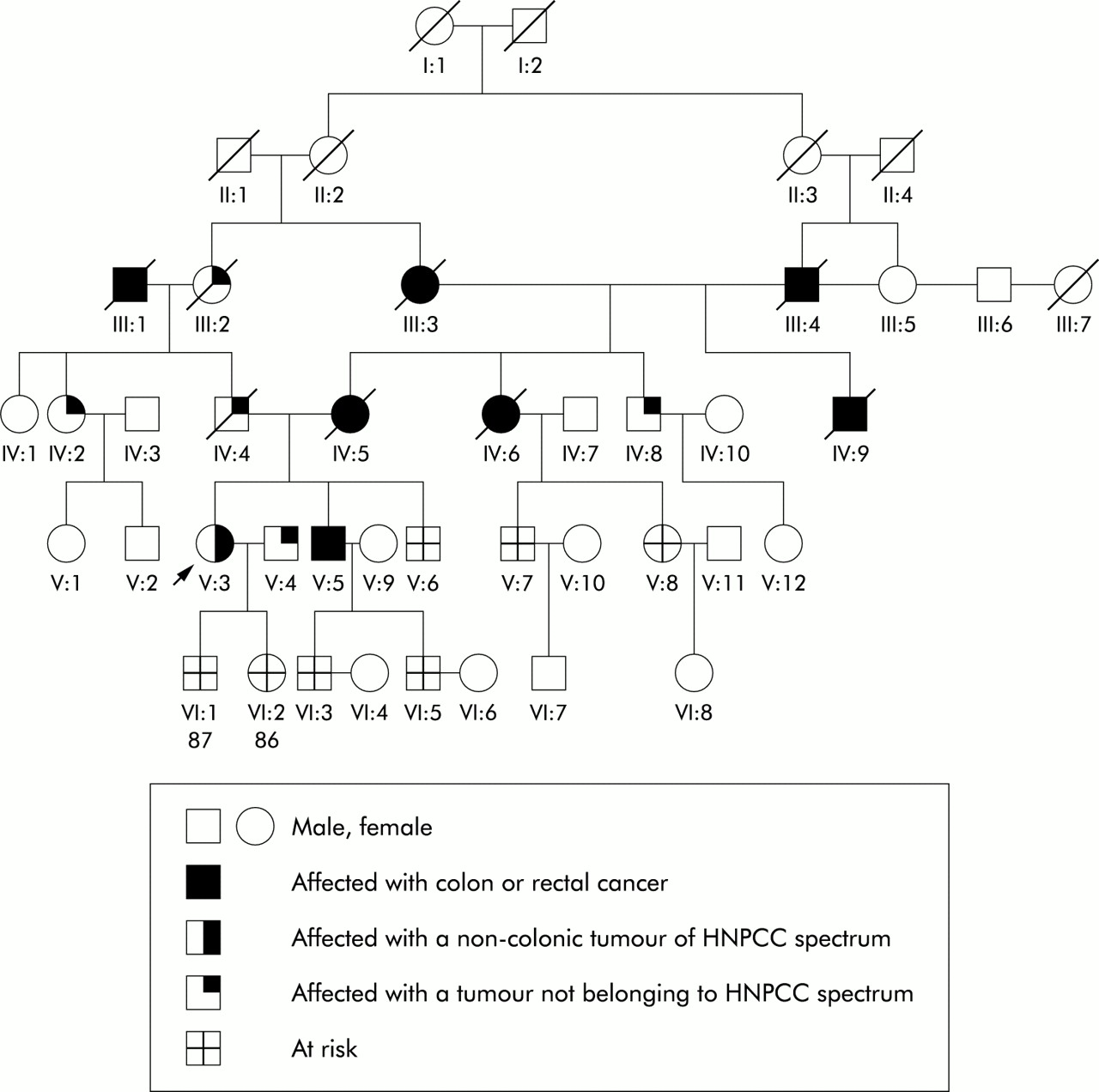
Family No 6 was found to carry a missense variant on exon 12A of the hMLH1 gene (table 1⇑) which occurs in a non-conserved codon and which leads to a non-conservative amino acid alteration. It was not in any of the 50 healthy blood donors included in the present study and no functional studies of this alteration are available in the literature. As shown in fig 6⇓, segregation analysis showed that this variant was segregated with disease phenotype in most instances. Except for family members III:9 and IV:8 who are healthy and aged 62 and 30 years, respectively, all of the remaining family members who presented with colon cancer were found to carry this missense variant. No tumour tissue was available to analyse MSI or immunoexpression of MMR genes in this family.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pedigree of family No 6.
Patient No 1 was found to carry the same missense mutation found in family No 4, which despite occurring in a non-conserved codon, leads to a non-conservative amino acid change, and previously performed functional studies indicate significant loss of function for this gene variant.15, 16 This patient is a 35 year old woman who presented with a mucinous carcinoma of the right colon. Her mother was reported as having died at age 62 years of a colon cancer and her father, presently healthy, was not shown to carry the K618T variant. Immunoexpresssion was normal for both genes, and tumour samples from this patient did not show MSI in any of the five markers analysed. From the data available it is hard to draw any conclusions as to whether this missense mutation plays a role in determining increased cancer risk.
Patients Nos 2 and 3 were found to carry missense mutations in the hMLH1 (exon 17, codon 648 CCC to TCC) and hMSH2 genes (exon 6, codon 322, GGC to GAC), respectively (table 1⇑). The latter alteration has been discussed above as being either a low penetrance mutation or low frequency polymorphism with normal function.28, 33, 34 Analysis of tumour tissue from this patient showed normal expression of both MMR genes and lack of MSI in any marker analysed. The P648S mutation found in patient No 2 leads to a non-conservative change but occurs in a non-conserved codon and, more importantly, it was found in 3/50 healthy donors which is highly suggestive that it is most probably a frequent polymorphism. MSI was positive in 2/5 markers and nuclear expression of hMLH1 was negative in tumour tissue. Because no other relatives were affected, segregation analysis could not be carried out in these kindreds.
Patient No 4 is a 28 year old male who presented with a cancer in the transverse colon which exhibited typical pathological features of the mutator phenotype (poorly differentiated mucinous carcinoma with Crohn-like infiltrate). Microsatellite analysis was positive for 4/5 markers and MMR gene mutation analysis showed a splice site mutation in intron 7, codon 1277 of the hMSH2 gene. Because it was a splice site mutation, it was interpreted as being most probably pathogenic. Also, immunoexpression of hMSH2 was negative in tumour tissue. To clarify whether this germline alteration was inherited or whether it appeared as a de novo mutation, the patient's mother, aged 64 years, and two siblings were tested for this same mutation. All three family members tested positive for this same mutation. We are now performing cDNA sequencing which might help us to understand whether this is truly a disease causing mutation with low penetrance.
DISCUSSION
HNPCC is an hereditary form of colorectal cancer associated with germline mutations in MMR genes. Genetic testing is being performed in these high risk families in the hope of identifying healthy carriers who can be enrolled in surveillance programmes or offered the possibility of prophylactic surgery.7, 35 Ninety per cent of the identifiable mutations are located in the hMLH1 or hMSH2 genes.9–,11 Whereas most of the mutations found in the hMSH2 gene are nonsense, frameshift, and splice site mutations which can be easily interpreted as pathogenic,36, 37 missense mutations in the hMLH1 gene constitute half of the reported mutations.11 To be able to counsel patients in these families regarding the risk of colorectal cancer, and because gene testing is evolving from a research to a clinical service model,12 it is necessary to fully understand the role played by these missense variants in increasing cancer susceptibility. Recently, two groups reported functional tests for MMR missense mutations and tested the mismatch repair functionality of most of the missense mutations in the hMLH1 gene described so far.15, 16 Interestingly, the majority of the investigated missense mutations had impaired repair function which led the authors to conclude that these missense variants were likely to be pathogenic.15, 16 Unfortunately, most of the variants detected in our study have not been tested in any of these previous studies. Of the two cases for which there were functional test available, in one (K618T—family No 4), segregation analysis was strongly against a pathogenic mutation. This clearly demonstrates that at this point, clinical recommendations cannot be based solely on these tests. Also, these functional assays are labour intensive, time consuming, and not readily available in most laboratories. This situation leads to a difficult scenario in which the clinician is frequently left with a genetic diagnosis difficult to interpret and dangerous to propose to other family members.
To illustrate these difficulties, in the present study we reported on 10 families with colorectal cancer who were found to harbour mutations in either the hMSH2 or hMLH1 genes. Whenever possible, we used the five previously defined criteria to help us clarify whether these alterations could be interpreted as disease causing mutations or as simple polymorphisms.
Occurrence of these amino acid substitutions in the general population is usually one of the four criteria used to distinguish a pathogenic mutation from a silent polymorphism. Because there are no studies of this kind in the Portuguese population, we thought it would be important to include a group of healthy controls. Although 50 controls is clearly a small number, only one of the eight mutations observed in these high risk individuals was found in the control group, which could suggest that in most instances we were in fact dealing with disease causing mutations.
Further support for this hypothesis comes from the fact that most of these mutations were non-conservative (5/7), 3/8 occurred in conserved residues, and some were already described as potentially pathogenic (3/8).
Segregation analysis is another criteria which may help in clarifying whether or not a missense mutation is involved in cancer pathogenesis. However, as shown in the present study, in the absence of large kindreds with numerous affected subjects, this analysis is often not possible or inconclusive. Thus although in 3/10 families we observed that the missense variant cosegregated with the disease phenotype, in most families analysed the segregation analysis was rather difficult to interpret. In some instances, affected individuals did not carry the mutation found in the proband whereas in other cases healthy individuals, sometimes in their forties, were shown to carry the mutation found in the affected proband. Thus in family No 4, a K618T mutation was found. Both functional tests for MMR genes published so far15, 16 concluded that this mutation interfered substantially with protein function. However, segregation analysis in this kindred was highly suspicious of a non-pathogenic mutation as an affected member who presented with colon cancer at the age of 53 years did not carry this variant. Similarly, in family No 5, segregation analysis clearly showed that the missense variant did not segregate with disease phenotype although previous studies suggested that this variant could be associated with an increased susceptibility of developing hereditary cancer.28, 29, 33, 34 In this family it is also worth noting that two mutations were found in the hMSH2 gene which further supports the concept that mutational analysis should always be as complete as possible before genetic diagnosis is offered.
In patient No 4 a splice site mutation on the hMSH2 gene was found, which was readily interpreted as being pathogenic. However, this variant was shared by two siblings as well as by his mother aged 64 years and apparently healthy. As the risk of colon or endometrial cancer in a female carrier varies from 30% to 70%,3, 38, 39 it is possible that the healthy mother demonstrates non-penetrance of this predisposing mutation. However, clinical recommendations based on these results cannot be made until further evidence confirming a pathogenic role for this mutation is available.
Thus due to the fact that penetrance of the syndrome is far less than 100% with not all HNPCC patients developing tumours and some of these presenting late in life, segregation analysis may not be very helpful in deciding on the pathogenicity of a given missense alteration.
To indirectly address the issue of whether these missense mutations had any functional consequences in MMR gene expression, MSI and immunoexpression of both hMLH1 and hMSH2 genes in colorectal tumours from affected individuals were analysed. Four of nine tumours analysed exhibited MSI in two or more of the five loci analysed. However, three cases in whom no MSI was observed occurred in patients harbouring mutations in exon 16 of the hMLH1 gene. As recently reported by Liu and colleagues,13 tumours from patients with exon 16 missense alterations often lack MSI. In these cases, the mechanism involved in carcinogenesis is probably different from that generally hypothesised. As these authors state, this is important from a clinical point of view when we think of the MSI test as a criterion for MMR mutation screening.13
With respect to immunohistochemical analysis of MMR genes, as these antibodies recognise a specific epitope located at the C terminus of the protein, it would be expected that mutations which result in a splicing abnormality with premature truncation of the gene product would not express the corresponding protein in neoplastic cells. In fact, colonic tumours from patients belonging to family No 1, patient No 2, and from patient No 4 had no detectable hMLH1 and hMSH2 proteins, respectively. However, to our knowledge, the effect of missense mutations on protein expression is still not known. We could hypothesise that amino acid substitutions which interfere with normal protein structure affect the stability of the gene product and thereby result in reduced expression of the protein. This could strongly suggest that these non-truncating mutations are pathogenic. However, in the present series, only one case which did not originate splicing abnormalities displayed negative staining for either the hMLH1 or hMSH2 MMR gene. Thus our results do not seem to indicate that immunohistochemistry plays any valuable role in evaluating pathogenicity of missense variants in HNPCC kindreds.
In conclusion, it is important that we are able to identify patients with HNPCC accurately for genetic counselling, screening, and prevention, as well as for predictive testing of unaffected family members. However, our results together with previously published data show that in the present series, only 1/10 variants in the hMLH1 and hMSH2 genes met all of the criteria required to be considered conclusively pathogenic. Even the use of multiple criteria did not enable us to give a definitive answer in the large majority of patients/families studied.
Clearly, further work is necessary to resolve this issue, including follow up studies of these families and subjects who carry these missense variants. However, at the present moment, the prevalence of missense mutations, genetic heterogeneity of the syndrome, and limited availability of validated functional assays present a challenge in the interpretation of genetic test results of HNPCC families. Our data clearly demonstrate that, as opposed to what happens with familial adenomatous polyposis coli, another syndrome of hereditary colorectal cancer, genetic diagnosis in HNPCC cannot be simply given as positive, negative, or non-informative. It needs to be carefully interpreted and, for that reason, should remain in specialised centres with geneticists and clinicians working in close collaboration before a recommendation is made to the patient.
Acknowledgments
This work was supported by a research grant from Ministério da Saúde, 1999.