Article Text

Abstract
Genome-wide association studies have identified over 200 loci associated with IBD. We and others have recently shown that, in addition to variants in protein-coding genes, the majority of the associated loci are related to DNA regulatory elements (DREs). These findings add a dimension to the already complex genetic background of IBD. In this review we summarise the existing evidence on the role of DREs in IBD. We discuss how epigenetic research can be used in candidate gene approaches that take non-coding variants into account and can help to pinpoint the essential pathways and cell types in the pathogenesis of IBD. Despite the increased level of genetic complexity, these findings can contribute to novel therapeutic options that target transcription factor binding and enhancer activity. Finally, we summarise the future directions and challenges of this emerging field.
- Ibd - genetics
- gene regulation
- genetic polymorphisms
- gastrointestinal pathology
- gene targeting
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Inflammatory bowel disease
IBD is a group of disorders of the GI tract that are characterised by intermittent, chronic or progressive inflammation. There are two main groups of IBD: Crohn’s disease (CD) in which transmural inflammation of the intestinal wall occurs in patches throughout the whole intestine, and UC in which inflammation is limited to the mucosa of the colon and rectum.1 The pathogenesis of IBD is multifactorial and includes genetic susceptibility as a major contributor. Alongside the growing number of identified genetic risk variants for IBD, there is growing knowledge on the role and functions of many different elements that often reside outside of the protein-coding regions of the human genome. This impacts the interpretation of the role that these variants play in IBD pathogenesis. In this review, we will introduce the concept of DNA regulatory regions, elucidate the possible roles of non-coding regulatory DNA in IBD pathogenesis and discuss how this creates novel therapeutic possibilities.
Genetic susceptibility in IBD pathogenesis
It has been known for decades that both CD and UC show familial clustering, although in the absence of a clear Mendelian inheritance pattern. Therefore, both diseases are generally considered polygenic disorders with variable phenotypic penetrance. Reported risk ratios for siblings of patients with CD and UC compared with the general population vary between 15–42 and 7–17, respectively.2 Three nationwide twin studies performed in Sweden, Denmark and the UK revealed increased concordance of IBD in monozygotic twins when compared with dizygotic twins, thereby elucidating that familial clustering is not solely based on shared environmental factors but rather on a shared genetic background.3–5 In CD, the difference in concordance is more pronounced than it is in UC, implying a greater genetic influence on the pathogenesis of CD than UC. The Swedish study further revealed significantly higher co-occurrence rates of phenotypic characteristics in monozygotic twins when compared with dizygotic twins, especially for age of onset and location of disease, suggesting that the disease and the phenotypic manifestations of IBD are heritable.15 Both disease phenotype and clinical course have been shown to be influenced by IBD-associated genetic variants.6 7
Based on these observations, a substantial effort was made to identify genetic elements involved in IBD pathogenesis. In this respect, multiple genome-wide association studies (GWAS) were performed over the past years.8–13 These studies assayed common genetic variants (single nucleotide polymorphisms [SNPs]) spanning the whole genome in search of SNPs that are significantly over-represented in patients when compared with healthy controls. SNPs that occurred more frequently in patients are thus called disease-associated variants.
Many genetic loci and variants that are located side by side are inherited together during meiosis (ie, there is a very small change of the occurrence of recombination sites between them). Due to this phenomenon, associated SNPs are generally considered to be markers for other variants located in the coding region of nearby genes. This approach has led to the identification of numerous crucial genes and pathways involved in IBD pathogenesis and has allowed for the development of novel therapeutic approaches.14 GWAS meta-analyses have identified over 200 loci that are associated with IBD and account for the increased risk of development of the disease.8 15 16 Because the search for candidate genes at the associated loci has proven difficult, most loci are not yet functionally linked to IBD. There are indications that pathogenic processes between CD and UC are convergent and share the majority of their associated loci (eg, the IL23R locus).8 17
Overall, IBD consists of a range of inflammatory disorders in the GI tract with distinctive onsets, severities, localisations and complications. Although a clear role for genetic susceptibility in IBD pathogenesis has been identified through multiple GWAS, the translation of these findings into patient benefits has been limited. The majority of SNPs that are associated with IBD were found to be located in non-coding DNA. Therefore, these SNPs cannot be causative in the sense that they directly lead to amino acid changes at the protein level.8–12 18–21 Notably, knowledge on the functional non-coding elements in the human genome has tremendously increased. This knowledge can now be used to better interpret GWAS findings. Here we review new approaches in which the focus shifts from coding sequences to other functional parts of the human genome and how this further enhances our knowledge of causative IBD genetic mutations and the awaited therapeutic perspectives.
DNA regulatory elements
Genomic DNA functions as a carrier of information that dictates the primary sequences of genes. In addition to this important function, non-coding DNA contains elements that are involved in transcriptional regulation.22 According to the 2013 University of California Santa Cruz (UCSC) genome build (GRCh38.p12, December 2013, last updated January 2018), the human genome consists of 3 609 003 417 base pairs and 20 376 coding genes.23 The coding sequences of genes make up only 2% of the whole genome, which designates 98% of the three billion base pairs as non-coding DNA.24 Over the last years, some functions of non-coding DNA have been discovered, and the role of regulatory sequences in transcriptional regulation, development, disease and determination of cell type specificity is now widely appreciated.25–28 Multiple distinct elements have been identified, each of which displays specific characteristics and plays different roles in transcriptional regulation (figure 1).
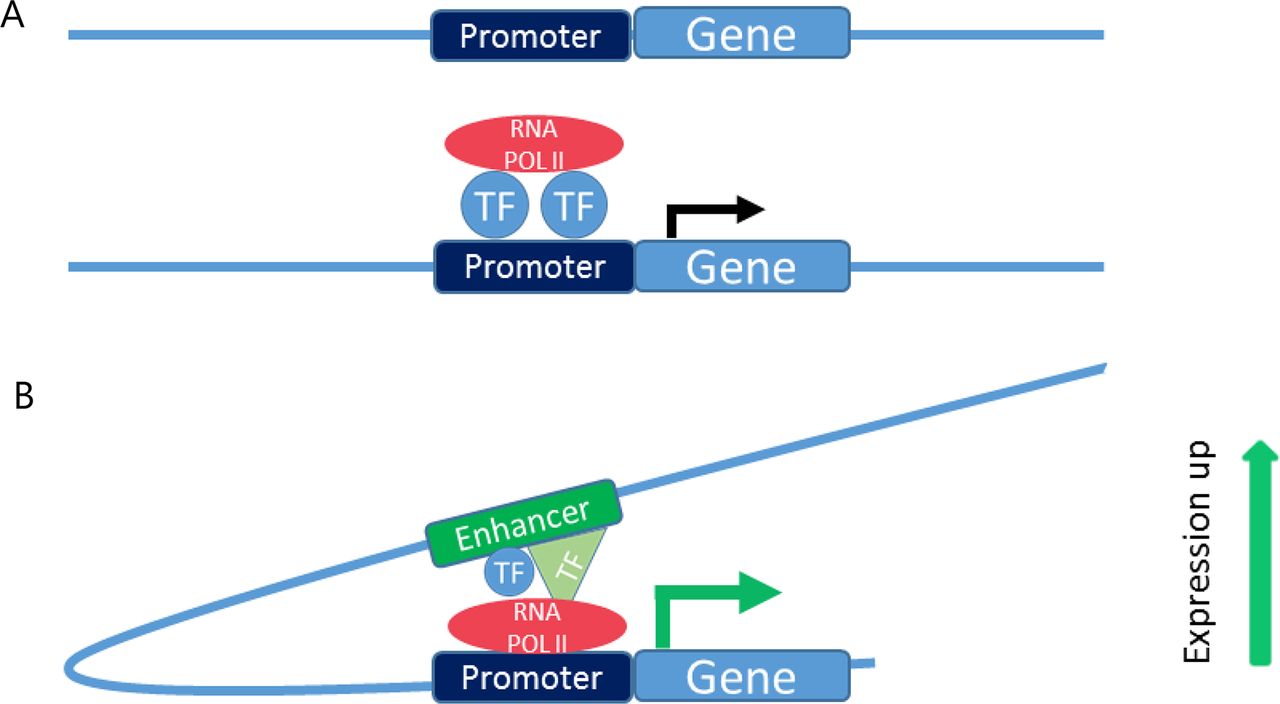
DNA regulating elements. (A) Promoter elements are located directly upstream of genes. By binding transcription factors and recruiting RNA polymerase II, promoters mediate the transcription. (B) Enhancer elements are located distally from the gene(s) they regulate(s). Active enhancer elements increase transcription levels, which is mediated through transcription factor binding. RNA POL II, RNA polymerase II; TF, transcription factor.
Enhancers
Enhancers are located distal from the genes they regulate and can be found up to several megabases away from the transcription start site.29 They are involved in enhancing the transcription of a regulated gene (figure 1A,B). These distal regulatory elements can regulate multiple genes and one gene can be regulated by multiple enhancers. The mechanisms of enhancer function have been studied at both the molecular (enhancement of transcription) and cellular (cell differentiation and development) levels. The mechanism through which enhancers regulate the transcription from promoters has been widely discussed.30 Multiple models have been proposed, and the ‘looping’ model received support after the development of a method known as chromosome conformation capture (3C). The looping model states that the action of enhancers relies on a physical interaction between the enhancer and the promoter of a reporter gene. Various 3C-based methods have been developed to enable the identification of genomic regions that are in physical contact with each other. By using these techniques, long-range interactions between enhancers and promoters have been discovered.31 In studies of the β-globin locus and its distant locus control region, specific interactions between enhancer-bound and promoter-bound transcription factors (TFs) have been discovered.32 33 Through these and other studies it is now generally accepted that enhancer function is established by TFs that bind to transcription factor binding sites (TFBS) and subsequently by the interaction between the TFs that are bound to promoters at one site and the TFs bound to enhancers at other sites.31
Enhancers play a key role in the establishment of complex expression patterns that determine the diversity of spatial and temporal gene expression within an organism.22 Enhancer activity is highly cell type-specific and can be correlated with gene expression patterns. Some enhancers can be found in clusters that drive the expression of cell type-specific genes. These clusters of enhancers are called super-enhancers and have been shown to be enriched in disease-associated variants.34 35 The combination of the histone modifications histone 3 lysine 4 monomethylation (H3K4me1) and histone 3 lysine 27 acetylation (H3K27ac) predominantly marks active enhancers, whereas H3K27 acetylation or H3K4me1 alone is predominantly found at ‘poised’ or inactive genes.36–38 Interestingly, many regulatory elements can have both enhancer and promoter characteristics and can therefore show different histone marks in time, in cell types and cell states.39–43 By investigating H3K4me1 distribution, between 24 000 and 36 000 enhancers were identified per cell line. Surprisingly, only a minority of these locations (approximately 5000) overlapped between the two cell types examined, showing that enhancers exhibit cell type-specific patterns. The contribution of epigenetic signatures on development is further established by the finding that fetal gut shows distinct DNA methylation patterns and dynamics from paediatric and adult intestinal tissue.44 By identifying the divergent patterns of enhancer activity in different cell types within one organism, enhancer activity was shown to play an important role during differentiation and development.45 46
Promoters
Promoters are regulatory elements that can be found at the 5’-end of a gene and are involved in transcription initiation. Promoter sequences contain TFBS that recruit (TFs) and assemble the transcription preinitiation complex that guides RNA polymerase II to the transcription start site.47 The location of promoters relative to genes, usually located just upstream of a gene, has facilitated their identification and annotation (figure 1A).48 Although active promoters are classically marked by histone 3 lysine 4 trimethylation (H3K4me3) histone modifications,37 recent studies have shown that some elements can exhibit promoter activity for one gene and enhancer activity for another gene.39 41 43 As described above, one DNA regulatory element (DRE) can be marked by different histone modifications, which reflects the potential of elements to execute multiple regulatory functions.40 Promoters and enhancers might therefore no longer be seen as individual entities, but rather as a spectrum of DRE activity.41
Next to enhancers and promoters, multiple other non-coding elements are involved in the regulation of gene expression. For example silencer and insulator elements are mainly involved in negative regulation of gene expression. As such, expression levels are subject to the composition of all sorts of elements along the chromosomes. In contrast to the cell type-specific activity of enhancer elements, the activity of most other elements is consistent throughout different cell types, as is reflected by their stable chromatin states.49
Mechanisms through which variants in DRE cause human genetics disease
Since the knowledge on DREs has increased, it has become clear that DREs are involved in determining cell types and differentiation. Sequence variants can be located in the whole genome, including DREs, and causative variants in both coding and non-coding elements are important players in the pathogenesis of complex genetic diseases such as IBD.
There are multiple ways through which sequence variants in DREs can contribute to the development of a genetic disease. One of the proposed mechanisms is through the alteration of TFBS. In this model, a sequence variant in the TFBS changes the affinity of a TF for this specific sequence. This can subsequently change the regulatory activity of DREs. Figure 2 shows an example of an altered nuclear factor kappa B (NFKB)-binding site in an enhancer that regulates the expression of the PTGER4 gene. The disruption of this binding site results in less binding of NFKB and subsequently a decreased expression of an IBD-associated gene (PTGER4).50 Another mechanism through which variants can contribute to pathogenic processes is by their influence on DRE activity and corresponding histone modifications and DNA methylation profiles51 52–55. Recently, these theoretical models were supported by the finding that allelic differences lead to allele-specific activity of many regulatory layers. Sequence variants were shown to affect transcription levels, binding of TFs, DNA methylation, histone modifications and histone positioning at the locus of the variant.56–58 Furthermore, this sequence-based variation in regulatory modules can be transmitted from parent to child, indicating that altered transcription regulation might be a basis for the heritable pathogenic processes.59 60

Influence of sequence variants on protein expression levels. The presence of an SNP in the TFBS of an active enhancer enables the enhancer to increase or decrease the transcription of genes. (A) Example of an SNP (rs4495224) that is associated with Crohn’s disease and is located in a TFBS in an enhancer element that regulated the expression of PTGER4. In silico analysis demonstrated that the C-allele of the rs4495224 polymorphism enables the binding of NFKB.50 (B) The A-allele of the rs4495224 polymorphism is thought to alter the TFBS in an enhancer, which will cause decreased affinity of NFKB. This could result in less enhancer activity and a subsequent decrease in PTGER4 expression levels. NFKB, nuclear factor kappa B; RNA POL II, RNA polymerase II; SNP, single nucleotide polymorphism; TF, transcription factor; TFBS, transcription factor binding site.
Besides variants in DREs, there are variants in non-protein-coding DNA that result in a phenotype through different mechanisms. Variants that cause alternative splicing and thereby affect the protein structure have been described.61 Furthermore, variants in genes that code for microRNAs or long non-coding RNAs can affect the function of these non-coding RNAs and can thereby contribute to IBD pathogenesis.62 63
Several pathogenic mechanisms through which non-coding DNA can result in disease phenotypes have been delineated in monogenic diseases. For example, preaxial polydactyly was found to be caused by a single mutation in a regulatory element of the Sonic hedgehog gene that is found 1 Mb upstream of the gene.29 This study and other studies64 demonstrate the possibility that even a single deleterious sequence variant in a regulatory region can cause disease. The mechanisms through which less common genetic variants (ie, deletions, translocations and so on) alter enhancer activity and contribute to genetic disease have been extensively reviewed.65
Taken together, these data establish that sequence variants in non-coding DNA can influence the activity of regulatory elements and that such sequence variants are often associated with human genetic diseases.
The majority of IBD-associated variants map to DREs
To understand the pathogenesis of IBD, the major contributors should be identified on the level of proteins, RNA transcripts and consequently genes. The genetic background of IBD has been successfully studied through GWAS, but unfortunately extracting the major players and causative genes is not straightforward. Initially, the translation of GWAS data into candidate genes focused on variants that could be linked to genes via their localisation in close proximity to the associated variant (figure 3A,B).11 19 20 However, the association of many variants that lie outside gene bodies cannot be linked to missense mutations.

Candidate gene approaches for disease-associated variants. (A) In genome-wide association studies, sequence variants at multiple genomic loci are studied. In this example, studying sequence variants at an IBD-associated locus reveals a C-allele was found more frequently in the patient population; therefore, this allele is determined to be a disease-associated variant. (B) Model of classical candidate gene approaches. A disease-associated variant is considered to be a marker for a causative coding variant in a gene that is transmitted to offspring on the same stretch of DNA, that is, inherited on the same haploblock. This gene is considered to be a possible candidate gene. For example, the IBD-associated SNP rs630923 (A/C) is located in the vicinity of CXCR5. CXCR5 is therefore considered to be an IBD candidate gene.8 (C) Novel model of candidate gene approaches. In this model, the disease-associated variant lies within a DNA regulating element. The SNP in this element results in changes in the expression of the target gene. The target gene can be located outside the haploblock and will therefore not be found by using the classical model. For example, the IBD-associated SNP rs630923 is located in an enhancer that was found to regulate the IL10RA gene that is found in another haploblock. Therefore, IL10RA is considered as a candidate gene as well.87 SNP, single nucleotide polymorphism.
As over 80% of the human genome consists of functional regulatory elements, it was hypothesised that the association of non-coding SNPs to IBD could be due to their effect on regulatory elements. Maurano et al determined the large overlap between GWAS-associated SNPs for multiple diseases and open chromatin (defined by DNase hypersensitivity sites [DHS]). DHS are a proxy for chromatin that is accessible for proteins such as TFs, and therefore active regulatory elements are generally found to map to DHS. This study revealed a significant enrichment of disease-associated SNPs to DHS and showed that these putative regulatory elements can interact with promoters over long genomic distances in vivo.52 Figure 3C shows how novel approaches can result in the identification of novel IBD candidate genes (eg, IL10RA) at previously associated loci. Disease-associated SNPs were also found to be enriched in a combination of DNase footprints, TFBS and histone modifications.66 The role of the chromatin landscape in IBD was further established by the finding that the colon tissue of patients with CD can be subdivided into two clinically relevant subtypes based on chromatin accessibility profiles.67
To address whether loci specific for IBD localise to DRE, we profiled active regulatory elements in cell types that are relevant for IBD. We found that 56% of IBD-associated SNPs can be linked to either immune or intestinal epithelial cell-specific regulatory regions (as determined based on the presence of histone modifications).18 This colocalisation occurred approximately three times more frequently in IBD-associated SNPs when compared with randomised sets of SNPs. Furthermore, the enrichment of the localisation of IBD-associated SNPs in enhancers and promoters that are activated on active UC and CD has been revealed.68 These data suggest that a large part of the IBD-associated loci can be explained by sequence variants within DREs.
Individual non-coding IBD variants alter DNA regulatory functions
As the vast majority of associated SNPs lies in non-coding DNA, much effort has gone into identifying candidate genes by linking specific variants to genomic regulatory functions. Delineating individual loci has led to the identification of pathogenic regulatory mechanisms and the genes that are affected by non-coding variants.
IRGM locus
The association of the IRGM gene locus to IBD has been established through multiple GWAS.69–71 IRGM is of great interest as it is involved in the early phases of autophagy, a process implicated in IBD pathogenesis.69 72 Indepth sequencing of the coding sequence of this gene could not reveal any non-synonymous mutations.69 This implies that the causal variant must be located in non-coding sequences. Indeed, a common 20 kb deletion polymorphism and two small insertions were found upstream of the IRGM gene body and turned out to be in strong linkage disequilibrium (LD) with the most strongly CD-associated SNP. Subsequently, the effect of the deletion and insertions was studied.69 71 The risk alleles were found to perturb IRGM expression levels, and subsequently manipulated IRGM expression affected cellular autophagy.71 Finally, a family of miRNAs are involved in the regulation of IRGM expression and were found to downregulate the protective allele, but not the risk allele.73
PTGER4 locus
A GWAS by Libioulle et al identified a novel IBD-risk locus on chromosome 5p13.1. The associated risk variants map to a 1.25 Mb gene desert, which complicated candidate gene identification. To test the regulatory potential of the locus, the effect of the SNPs on expression levels of the neighbouring genes was profiled. This approach revealed two SNPs that significantly increase the expression of PTGER4.12 In silico analysis showed that this is likely caused by the alteration of two TFBS (NFKB and XBP1 [X-box binding protein 1], respectively). The increased affinity of these TFs for the associated locus could explain the increased expression of PTGER4.50 The role of this locus is supported by the increased susceptibility of Ptger4 mutant mice to dextran sulfate sodium (DSS)-induced colitis and the established roles of NFKB and XBP1 in IBD pathogenesis.74 Furthermore, variants at the associated locus were systematically screened for aberrant regulatory capacity in lymphoblastoid cells. This revealed a single SNP in a distal enhancer that affects the expression of PTGER4 in vitro and could be rescued by genome editing with CRISPR/Cas9.75
TRIB1 locus
There are two SNPs associated with CD that are located upstream of the coding sequence are found at open and therefore likely active chromatin.52 These two SNPs were found to alter T-Bet binding motifs and result in reduced T-bet binding in vivo. This is relevant because T-Bet is a TF that is involved in T cell differentiation and plays a key role in T cell-mediated colitis.76 Furthermore, a direct link between T-bet and TRIB1 was established by showing that T-Bet−/− mice show decreased TRIB1 expression.77 These data show that these CD-associated SNPs alter transcription regulatory processes that correlate with TRIB1 expression.
IL18RAP locus
In a GWAS from 2010 an IBD-associated variant on chromosome 2q12 was identified. This variant was studied through mRNA profiling in whole blood samples. This revealed that the minor allele specifically correlated with altered expression of the IL18RAP gene.78 Other associated SNPs at the same locus were found to alter the T-bet TFBS that results in differential expression of IL18RAP.
TPL2 locus
TPL2 (MAP3K8) maps to a locus that is associated to CD.8 Although no variants that caused amino acid changes were found, TPL2 was an obvious IBD candidate gene because it is an important player in T cell and innate responses, and it induces cytokine production in a pattern recognition receptors-signalling cascade.79 In mice, TPL2 was shown to contribute to the development of colitis.80 81 Hedl and Abraham revealed the putative role of associated variants on DNA regulation, by showing that the disease-associated allele has an increased expression of TPL2 in monocyte-derived macrophages.82 Furthermore, this study elucidated that carriers of the risk allele demonstrate increased NOD2 and NFKB signalling that subsequently results in increased cytokine production.
The role of regulatory elements in IBD studied through systematic approaches
The studies described above concern examples of the association of single IBD-associated loci with a putative DNA regulatory function. However, there is a strong need for novel approaches to systematically identify the genes that are regulated through elements that carry IBD variants (figure 4A).

Candidate gene identification through eQTLs and 4C. (A) Identification of candidate genes based on the proximity of a gene to the enhancer that carries the IBD variant is a biased approach. From the linear composition of DNA, it is not possible to identify the genes that are regulated by associated enhancers. (B) eQTLs are used to identify differential expression between alleles that carry different IBD-associated variants. The differentially expressed genes are considered to be candidate genes. (C) 4C is used to study the 3dimensional conformation of the chromatin and identify candidate genes based on their physical interaction with IBD-associated enhancers. 4C, circular chromatin conformation capture; eQTLs, expression quantitative trait loci.
IBD-associated variants overlap eQTLs and influence transcription levels
A common feature of regulatory elements is their influence on RNA expression. Therefore, SNPs that contribute to a disease phenotype are expected to affect allele-specific RNA expression levels. This mechanism is used to profile eQTLs (expression quantitative trait loci), that is, loci that contain sequence variants that influence the expression of specific genes, thereby identifying SNPs that are linked to transcription regulation (figure 4B). As regulatory activity is cell type-specific, the influence of sequence variants is limited to cells in which the overlapping DRE is active. Therefore, eQTL databases of many cell types have been developed.78 83–85 eQTL profiles of cell types that are likely involved in the IBD pathogenesis are used to assign candidate genes to IBD-associated SNPs that influence expression levels. This approach is now commonly used to complement candidate gene approaches that are based on genomic distances between SNPs and genes.8 9 13 15 Using eQTLs and regulatory information enables the identification of candidate genes for many loci that could not have been done previously. This is exemplified by a GWAS in which only 3 of 38 novel SNPs were in LD with known missense mutations, whereas 14 SNPs showed eQTL effects.13
Besides allele-specific influence on RNA expression, SNPs can also influence chromatin landscapes and TF binding. IBD-associated SNPs were found to influence the accessibility of the DNA and thereby likely influence the extent to which DREs can execute their function in individuals that carry risk variants.52 Furthermore, IBD-associated SNPs were found to be enriched for localisation to TFBS16 18 52 68 and to result in allele-specific affinity of TFs.77 86 Finally, DNA methylation that influences DNA accessibility was confirmed to show allele-specific patterns.53 As such, various layers and mechanisms have been discovered that enable a better understanding of the complex role that genetic variants can play, ultimately through (defected) regulation of DNA transcription of relevant IBD-related genes.
Candidate genes physically interact with DNA regulatory regions
Many studies specifically analyse whether associated SNPs affect the expression of genes that are located in the vicinity of the SNP. To overcome this biased approach for the identification of candidate genes, we developed a novel, unbiased method that complements classical approaches to candidate gene identification (figure 4C).87 This approach relies on the physical interaction between enhancer elements and the genes they regulate. Using 4C-seq (circular chromatin conformation capture sequencing), we studied each gene that interacts with one of the 92 active enhancer elements that carry an IBD-associated SNP. We applied this technique to intestinal epithelial cells (IECs), monocytes and lymphocytes and identified 902 putative candidate genes with an average distance of 300 kilo base pairs per interaction. These results emphasise that genes that contribute to the IBD pathogenesis can be located further from the associated SNP than is assumed in classical candidate gene approaches and has recently been applied to other complex genetic diseases.88 89 This approach identified noteworthy genes including ATG9A,90 a gene involved in autophagy that has been implicated in IBD pathogenesis, and IL10RA, a gene that has been shown to play a role in monogenic forms of IBD.91
Gene prioritisation
With increasing knowledge on the regulatory functions of the majority of IBD-associated loci, the complexity of the genetic background of IBD seems to be ever expanding. Therefore, in silico methods are being developed that integrate all regulatory, chromatin, coding and non-coding information to identify the key regulators in IBD.92 93 Peters et al applied a predictive model to identify gene networks that play a role in IBD and subsequently validated 12 key drivers of IBD pathogenesis. Our approach, through systematic analysis of chromatin interactions, helped identify common upstream regulators of the IBD candidate genes.87 As such, our results suggest an important role for HNF4A (a TF that belongs to the nuclear hormone receptor super-family) in both intestinal epithelium and immune cells. These in silico approaches are crucial to translate the extending number of IBD-associated genes to key regulators of the main pathogenic pathways and finally to identify novel therapeutic targets.
Regulatory information creates novel insight in cell types and disease states
On the one hand, the cell type specificity of DRE activity can complicate the studies of genetic variants in regulatory elements, as the functional impact of each associated SNP may be present only in specific cells. Reciprocally, this phenomenon can help us to pinpoint cell types that are involved in IBD pathogenesis as the active regulatory elements from these cells are enriched for IBD-associated variants. Therefore, a characteristic of DRE that seemed complicating at first has paved the way for the identification of cell types that play a role in the IBD pathogenesis. The first study to apply this strategy showed that T helper 17 and T helper 1 cells have the highest accumulation of SNPs in accessible chromatin.52 Studying the enrichment of IBD-associated SNPs in cell type-specific active DRE revealed marked differences between UC and CD.
We and others have shown that in UC both IECs and immune cells seem to be important players, whereas in CD IECs are found to play a less important role than immune cells.16 18 68 94 This implies that there are distinct pathological processes underlying CD and UC and that these are limited to immune cells in CD, although in UC the intestinal tissue itself is a major player.
DREs are differentially active among cell types and among cell states. Studying the activation of monocytes through stimulation with lipopolysaccharides (LPS) or interferon gamma (IFNγ) revealed many context-specific eQTLs. This means that some SNPs will only affect pathogenicity on, for example, inflammation or during infection. These context-specific eQTLs are now used in search of causative genes in GWAS.15 A recent study has identified eQTLs in ileal biopsies from patients in different stages of CD (healthy, complication-free disease and disease progression with stricturing or penetrating disease). Through this approach, disease stage-specific eQTLs have been identified. The IBD-associated SNPs that form these eQTLs are therefore likely stage-specific and may be used to predict disease progression.
DREs as therapeutic targets
The majority of IBD-associated SNPs have been shown to affect transcription regulation by altering the sequence of DRE. This creates possibilities for novel therapeutic strategies. Based on the common features of the SNPs that are involved in DNA regulation, druggable targets may be defined.
The presence of SNP in a disease-associated locus can result in changes in TFBS and in the chromatin landscape. Although there is an interplay between the deposition of histone modifications and the affinity of transcription factors for TFBS, they can be targeted through different mechanisms. Here we will discuss how the chromatin landscape and key IBD TFs can be targeted and we will review the progress that has been made on this score.
Targeting the chromatin landscape
The activity of DREs strongly correlates with the co-occurrence of histone modifications, DNA methylation and TF binding.95 Histone modifications are covalent post-translational modifications of one of the four histone tails including acetylation and methylation (figure 5). The tails of histones H3 and H4 are important for transcriptional regulation of numerous genes. Modifications of histones by acetylation are known to weaken the chemical attractions between nucleosome components, enabling the DNA to uncoil from nucleosomes and allowing access to proteins important for transcription, such as RNA polymerase II and TFs. Acetylation of histones by ‘writer’ enzymes called acetyltransferases is known to increase the expression of the genes that are regulated by the acetylated enhancers.96 The addition of a methyl group is mediated by the writer enzyme called histone methyltransferases (HMT).97 This enzyme can either activate or further repress transcription, depending on the histone tail, and subsequently the amino acid that is being methylated and the presence of other methyl or acetyl groups in the vicinity.96 Acetyl and methyl groups can be removed by ‘eraser’ enzymes called histone deacetylases (HDACs) and histone demethylases, respectively.97 Moreover, ‘reader’ enzymes such as bromodomains can recognise histone modifications and direct a specific transcriptional outcome by modifying chromatin structure or recruiting machinery involved in gene expression.98 Due to their influence on chromatin structure and transcription, drugs targeting enzymes capable of adding (writers), removing (erasers) and recognising (readers) major enhancer-associated histone modifications could be a promising therapy for reversing aberrant DRE activity seen in the context of disease. One such histone modification which has shown great promise in the treatment of IBD is histone acetylation. Table 1 presents the available compounds with clinical potential.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histone modifications. Histone tails that belong to one of the four subgroups of histones can be post-translationally modified by covalent attachment of (among others) acetyl and methyl groups. These modifications are deposited and removed by specific enzymes. Each modification has a different effect on transcriptional activity. (A) Repressive histone modifications cause the DNA to be densely packed and be inaccessible for transcription factors. (B) Activating histone modifications are found at accessible DNA, which can result in transcription activation of genes found in this genomic region. HAT, histone acetyltransferase; HDAC, histone deacetylase; HDMT, histone demethylase; HMT, histone methyltransferase.
Therapeutics that target chromatin landscape and key regulators: overview of putative and novel therapeutics for IBD based on their potential to target chromatin modifiers or key regulators
HDAC inhibitors
The first potential targets are HDACs. Studies in both murine and human immune cells and colonic mucosa have shown that a lack of DRE acetylation of immune genes is involved in the pathogenesis of IBD.99 Inhibitors of HDACs increase the levels of acetylation, and administration to mice resulted in the amelioration of colitis, a reduction of proinflammatory cytokines and a decrease of migratory inflammatory cells in the colonic mucosa.100 There are 11 isoforms of HDACs, each of which can be inhibited by specific compounds (table 1).101 HDAC inhibitors have shown to have multiple substrates that are not limited to HDACs. The effect of HDAC inhibitors can therefore not be automatically and solely ascribed to their effect on the chromatin landscape.102 103 Here, we will review HDAC inhibitors that affect the chromatin landscape and subsequently result in a putative beneficial effect in IBD. Many of these inhibitors are already being used in the clinic, mainly as anticancer treatment.104 One HDAC inhibitor, the short-chain fatty acid butyrate, is already used as treatment for IBD.105 The putative mechanism is inhibition of HDAC9, which enhances histone H3 acetylation in the promoter region of NOD2.106 107 In both an in vitro study on human IECs and in in vivo murine models of experimental colitis, this drug increased Nod2 expression that was associated with reduced nuclear TF NFKB signalling, reduction of inflammation and improved integrity of the intestinal epithelium.106 107 The effect of butyrate on IEC proliferation was shown to be due to its effect on HDAC activity and independent of its potential to target G protein-coupled receptors.108
Suberoylanilide hydroxamic acid and valproic acid are HDAC inhibitors that have passed to advanced clinical stages for anticancer treatment.109 These inhibitors cause a dose-dependent increase in H3 acetylation at the site of inflammation and are associated with macroscopic and histological reduction of disease severity, as well as marked suppression in proinflammatory cytokine expression in the colon.100 109 This anti-inflammatory effect could be explained by HDAC inhibition in dendritic cells that results in decreased expression of inflammatory cytokines.110 111 Furthermore, the HDAC inhibitor givinostat is being studied in clinical trials for systemic-onset juvenile idiopathic arthritis and has obtained a good safety profile.112 113
KAT2B is a lysine acetyltransferase that is downregulated in inflamed colonic tissue of patients with CD and UC. Inhibition of KAT2B by anacardic acid demonstrated reduced levels of histone H4 lysine 5 acetylation (H4K5ac) in the interleukin-10 (IL-10) promoter region which was associated with a dose-dependent decrease in the expression of IL-10.99 HDAC1-selective inhibitors such as tacedinaline and quisinostat promote H4K5 acetylation and restore IL-10 transcription.99 The reduction of IL-10 has been linked to IBD as patients with deleterious mutations in IL-10, IL10RA and IL10RB suffer from severe infantile-onset IBD.114
Overall, many studies have demonstrated that these small molecules can target important pathways in the pathogenesis of IBD, and their efficacy is currently being studied in clinical trials.
BET inhibitors
BET (bromodomain and extraterminal) proteins are ‘reader’ proteins which recognise acetylated lysine residues on regulatory elements and influence expression by recruiting TFs and chromatin remodelling factors such as the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex.115 BET proteins can be specifically found at super-enhancers that are characterised by extensive acetylation of histone H3 at lysine 27 and increased binding TFs. Super-enhancers are highly cell type-specific and cell state-specific and might therefore be a target in disease-specific processes116–119 After all, both healthy controls and patients with IBD may contain the same SNPs in an enhancer, but the enhancer may only be active due to tissue or cell context-like inflammation and the presence of cytokines. Therefore, the enhancers that only become active in certain contexts are more likely to have an influence on the pathogenesis of a disease, and thus it is beneficial that BET inhibitors are studied in the context of inflammatory disease. There is evidence that inhibiting BET proteins leads to a reduction in inflammation-associated gene expression and can modulate the proinflammatory activity of adaptive immune cells.120 For example, the BET inhibitors JQ1, I-BET762 and I-BET151 were shown to reduce the production of proinflammatory cytokines in monocytes and macrophages stimulated by LPS in vitro and in mice.120 Studies on human inflamed joint synovial fluid show that treatment with JQ1 on CD4+ memory/effector T cells preferentially inhibited the expression of genes involved in proinflammatory and cytokine-related processes regulated by super-enhancers.121 Although BET inhibitors have not been studied in the context of IBD, their immune-modulating potential points to promising effects for IBD treatment.
MT inhibitors
The methyltransferase EZH2 (enhancer of zeste homolog 2) is an important player in IBD. Patients show significantly reduced expression, and it has been shown in mice that inhibition of EZH2 leads to increased immune responsiveness that is associated with an increased sensitivity to DSS-induced and 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced experimental colitis.122 In addition, mice that lack EZH2 specifically in regulatory T cells (Tregs) develop spontaneous IBD. EZH2 functions as a cofactor of FOXP3 for the regulation of Treg-specific gene networks. As dysregulation of EZH2 plays a role in the development of IBD in both mice and humans, treatment that restores HMT activity in Tregs could be beneficial for treating IBD.122
Targeting key regulators
Inflammatory pathways that play a role in the pathogenesis of IBD can be targeted through cytokine inhibitors. This way, the cascade that results from cytokine-receptor binding can be blocked, after which inflammation is dampened. Several monoclonal antibodies have been developed and have become common therapeutics used in IBD. The majority of clinically approved antibodies target tumour necrosis factor α, a cytokine that can be produced by many cell types and is found to be upregulated in IBD.123 Other cytokines including IFNγ, IL-6, transforming growth factor beta (TGFB) and IL-12/p40 are being studied as therapeutic targets in IBD with varying degrees of success.124 125 However, these targets may not completely represent the common pathways that have been identified through integrating genetic and epigenetic data.
As described above, recently, many attempts have been done to identify key regulators in the pathogenesis of IBD. The goal of these approaches is to identify single players preferably affecting multiple IBD genes at the same time.87 92 Many of these key regulators turn out to be TFs. This can be explained by both the many associated loci that contain the same binding motifs for a limited number of TFs and by the involvement of pathways that are regulated by a limited number of TFs. Alterations in TF activity therefore can play a central role in the IBD pathogenesis, and targeting these factors seems a valid approach. However, TFs have proved to be challenging targets and have often been termed ‘undruggable’.97 Nevertheless, several strategies for TF targeting are being developed (table 1).
HNF4A
We and others have identified HNF4A as an important key regulator in IBD that interferes with pathways in multiple relevant cell types.87 92 HNF4A provides an interesting target, as there are multiple levels in which HNF4A contributes to IBD: enrichment of IBD-SNPs in HNF4A binding sites, altered DNA binding,93 regulation of expression of IBD candidate genes87 and differential expression of HNF4A itself. Notably, Hfn4a knockout mice are prone to develop DSS-induced colitis.126 There are currently no HNF4A agonists ready to be tested in clinical studies. Conversely, multiple HNF4A inhibitors are used in vitro.127 However, HNF4A seems to have a protective effect in IBD; therefore, agonists rather than antagonists are of interest as putative therapeutic compounds. Identification of HNF4A agonists has proven difficult and there are currently no compounds available that upregulate HNF4A. Nevertheless, studies on the in vivo ligands of HNF4A reveal that medium-chain and long-chain fatty acids are the natural ligands and activators of HNF4A.128–130
NFKB
As for agonists of HNF4A, finding specific antagonists for another IBD key regulator, NFKB, has been a challenge. For NFKB, an alternative TF targeting approach has shown success in this context. By using decoy oligonucleotides, NFKB-DNA binding is decreased through competitive inhibition. Decoy oligonucleotides are short, double-stranded DNA molecules containing the binding motif of a specific TF. When TFs bind to decoy oligonucleotides, their availability for binding to DREs decreases. A major limitation of this technique is that decoy oligonucleotides are easily degraded by nucleases. However, recent developments have increased their stability and made them more nuclease-resistant. Administration of an NFKB decoy oligonucleotide in rats limited the expression of proinflammatory pathways and showed increased survival rate on inducing DSS-induced colitis.131 Another NFKB inhibitor, dehydroxymethylepoxyquinomicin, blocks the nuclear translocation of NFKB and ameliorated experimental TNBS-induced and DSS-induced colitis in mice.132 Finally, the natural NFKB inhibitor curcumin has shown positive effects on the treatment of IBD in phase I, II and III trials.133
STAT3
STAT3 is a key TF in the pathogenesis of IBD. The nuclear translocation of STAT3 is mediated by Janus kinase (JAK).134 Therefore, inhibition of JAKs subsequently leads to the inhibition of STAT3 and thereby of the downstream JAK/STAT pathway. On the discovery of a role of STAT3 in IBD pathogenesis, JAK inhibitors have been developed and tested in clinical trials. To date, such drugs have not been proven efficacious in CD.135 136 However, JAK inhibitors are currently used for the treatment of UC.137 138
Conclusion and discussion
Although GWAS of IBD have revealed many associated sequence variants and some involved genes, these findings only explain a minor portion of the genetic background of IBD.8 19 Therefore, new insights into the genetic make-up of these complex genetic diseases are needed. We have reviewed the accumulating evidence regarding the contribution of sequence variants in DREs to the pathogenesis of IBD. This contribution is supported by many studies, and steps are being taken to translate these findings into new diagnostic, preventive and therapeutic measures.
Studying genetic variants in regulatory elements and their effect on the pathogenesis of complex genetic diseases remains challenging. The context-specific activity of DRE makes their identification and annotation of DRE difficult. The limited effect of SNPs in DRE compared with the effect of variants that alter gene coding sequences further complicates the identification of pathogenic regulatory variants. Over the last years, multiple high-throughput techniques have been developed that enable the efficient annotation of DRE and the evaluation of allele-specific effects. These techniques are built on epigenetic signatures, chromosome conformation, genome editing and self-transcribing activity of regulatory elements and have been thoroughly reviewed by Elkon and Agami.139 To assay the effects of genetic variants on the activity of regulatory region, the cell type-specific activity of enhancers needs to be taken into account as the phenotype that is caused by an SNP might only be present in a limited number of cell types. Assaying an SNP in the wrong cell type or developmental stage will not reveal a deleterious effect. This is further complicated by the finding that the phenotype of some genetic variants is dependent on the cell state (eg, activated cells vs non-activated cells).84 Similar to eQTL studies, the cell type, developmental stage and cell state-dependent effect of SNPs on DRE causes a high chance of false-negative detection. To increase detection of disease-associated SNPs, assays should be performed in a multitude of cell types and conditions. To address this, eQTL databases that contain a plethora of cell types and developmental stages are now being developed.140
New therapeutic measures for IBD include targeting histone writers, readers and erasers, as well as key regulators of IBD networks. A possible drawback of targeting histone modifications and key regulators is that the effect of the compounds will not be limited to the tissues that are affected by the disease. However, the predictive value of IBD-associated SNPs on the pathogenic cell types can be a lead to develop therapeutics that can be delivered to specific cells. This may be associated with adverse effects as is seen for many therapeutics that target general processes, including immune modulators and chemotherapeutics. The extent of the adverse effects and the efficacy of these putative new compounds are currently being studied in clinical trials. Although most trials concern malignant diseases, the outcome of the trials can be highly relevant for IBD (as for safety, dose ranging and side effects). In table 1 we reviewed multiple compounds that have shown to be effective in vitro and in vivo and are already being used in human diseases.
We believe that recent insights into the roles of DRE in IBD will result in the development of novel therapeutic strategies. With novel techniques like CRISPR/Cas9, which are now widely used for scientific purposes, genetic diseases may eventually be treated by restoring genetic defects. Although we reviewed the contribution of genetic variants to the pathogenesis of IBD, we have not discussed the therapeutic potential of genome editing. As patients carry multiple SNPs that contribute to the pathogenesis of IBD, treatment of patients by targeting these common variants through genome editing does not seem an efficient approach, even more so because the loci but not the disease-causing variants have been identified. However, profiling the disease-associated SNPs in individual patients could be useful in predicting response to therapeutics and thereby in defining a personalised therapeutic strategy. We therefore suggest including SNP profiles in the analyses of clinical trials to establish the variants that are predictive for treatment outcome.141 142
In this review, we have shown that sequence variants in DREs are involved in the pathogenesis of IBD. The majority of IBD-associated sequence variants colocalise with active DREs, and the mechanisms through which these SNPs lead to pathogenic processes have been intensively studied over the last decade. This resulted in the identification of novel therapeutic targets including regulatory layers such as the chromatin landscape and key regulatory TFs.
Acknowledgments
We want to thank Jorg van Loosdregt for reading the manuscript and providing valuable comments.
References
Footnotes
Contributors CAM performed the literature search and was involved in writing the manuscript. ACJvdL performed the literature search and was involved in writing the manuscript. EESN supervised the project and was involved in writing and editing the manuscript. MM supervised the project and was involved in writing and editing the manuscript.
Funding This work was supported by the Alexandre Suerman Stipendium (2014) through CAM and WKZ Funds (2012) through MM.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.