Article Text

Abstract
Colorectal cancer is a complex disease resulting from somatic genetic and epigenetic alterations, including locus-specific CpG island methylation and global DNA or LINE-1 hypomethylation. Global molecular characteristics such as microsatellite instability (MSI), CpG island methylator phenotype (CIMP), global DNA hypomethylation, and chromosomal instability cause alterations of gene function on a genome-wide scale. Activation of oncogenes including KRAS, BRAF and PIK3CA affects intracellular signalling pathways and has been associated with CIMP and MSI. Traditional epidemiology research has investigated various factors in relation to an overall risk of colon and/or rectal cancer. However, colorectal cancers comprise a heterogeneous group of diseases with different sets of genetic and epigenetic alterations. To better understand how a particular exposure influences the carcinogenic and pathologic process, somatic molecular changes and tumour biomarkers have been studied in relation to the exposure of interest. Moreover, an investigation of interactive effects of tumour molecular changes and the exposures of interest on tumour behaviour (prognosis or clinical outcome) can lead to a better understanding of tumour molecular changes, which may be prognostic or predictive tissue biomarkers. These new research efforts represent ‘molecular pathologic epidemiology’, which is a multidisciplinary field of investigations of the inter-relationship between exogenous and endogenous (eg, genetic) factors, tumoural molecular signatures and tumour progression. Furthermore, integrating genome-wide association studies (GWAS) with molecular pathological investigation is a promising area (GWAS-MPE approach). Examining the relationship between susceptibility alleles identified by GWAS and specific molecular alterations can help elucidate the function of these alleles and provide insights into whether susceptibility alleles are truly causal. Although there are challenges, molecular pathological epidemiology has unique strengths, and can provide insights into the pathogenic process and help optimise personalised prevention and therapy. In this review, we overview this relatively new field of research and discuss measures to overcome challenges and move this field forward.
- Colorectal carcinoma
- multistep carcinogenesis
- etiologic
- risk factor
- survival
- molecular change
- prevention
- cancer epidemiology
- cancer prevention
- cancer susceptibility
- carcinogenesis
- colorectal neoplasia
Statistics from Altmetric.com
- Colorectal carcinoma
- multistep carcinogenesis
- etiologic
- risk factor
- survival
- molecular change
- prevention
- cancer epidemiology
- cancer prevention
- cancer susceptibility
- carcinogenesis
- colorectal neoplasia
Introduction to molecular pathological epidemiology
Molecular pathological epidemiology, the concept of which has been consolidated by Ogino and Stampfer,1 is a relatively new field of epidemiology based on molecular classification of cancer. In molecular pathological epidemiology, a known or suspected aetiological factor is examined in relation to a specific somatic molecular change, in order to gain insights into the carcinogenic mechanism.1 In recent years, there has been a new direction of this field where we examine an interactive effect of tumorous molecular features and lifestyle or other exposure factor on tumour behaviour (prognosis or clinical outcome).2 In this review, we focus on colorectal neoplasia, overview the current status of molecular pathological epidemiology, describe various challenges in this field, and propose future directions.
Molecular classification of colorectal cancer
Colorectal cancer is a disease that is characterised by uncontrolled growth of colorectal epithelial cells. According to the theory of multistep carcinogenesis,3 4 colorectal epithelial cells accumulate a number of molecular changes and eventually become fully malignant cells. Genetic and epigenetic events during the carcinogenesis process differ considerably from tumour to tumour. Thus, colorectal cancer is not a single disease. Rather, colorectal cancer encompasses a heterogeneous complex of diseases with different sets of genetic and epigenetic alterations. Essentially, each tumour arises and behaves in a unique fashion that is unlikely to be exactly recapitulated by any other tumour.5
We typically classify colorectal cancers into categories according to a well-defined molecular feature (eg, microsatellite instability, MSI-high vs microsatellite stability, MSS), because substantial evidence suggests that tumours with similar characteristics (eg, MSI-high) have arisen by similar mechanisms and will behave in a similar fashion.5 Thus, the major purposes of molecular classification are: (1) to predict natural history (ie, prognosis); (2) to predict response or resistance to a certain treatment or intervention; and (3) to examine the relationship between a certain aetiological factor (ie, lifestyle, environmental or genetic) and a molecular subtype, so that we can provide evidence for causality and optimise preventive strategies.
For any marker for molecular classification, we need to consider two key points. The first question is whether a given molecular feature reflects genome-wide changes. For example, MSI, chromosomal instability (CIN), the CpG island methylator phenotype (CIMP), and global DNA hypomethylation reflect genome-wide or epigenome-wide aberrations. Because these molecular features often confound the relationship between a locus-specific change and an exposure or outcome of interest, it is important to consider potential confounding by these genome-wide features whenever one examines locus-specific changes. The second question is whether a given molecular change has by itself driven cancer initiation or progression, or is simply linked to other important molecular events. For example, loss of heterozygosity (LOH) events may not by itself cause tumour progression; rather, underlying genomic instability (ie, CIN) or functional loss of important genes within the lost chromosomal segment may cause tumour progression. Nevertheless, even if a given molecular change is consequential rather than causal, the change not only can be a good surrogate marker of a certain cancer pathway, but also may ultimately become a driver in later steps of tumour progression.
Emergence and evolution of molecular pathological epidemiology
Traditional epidemiology research has investigated lifestyle, environmental or genetic factors that might increase or decrease risk of developing colorectal cancer.6 7 The weight of the evidence, in conjunction with results from in vitro and animal models or human experimental trials, can lead to particular factors being widely considered to be aetiologically linked to cancer. Aetiological factors which have been implicated in colorectal carcinogenesis include red and processed meat, excess alcohol intake, deficiency of B and D vitamins, obesity, physical inactivity, diabetes mellitus, smoking, family history of colorectal cancer, inflammatory bowel diseases, among others.8 More recently, the field of molecular epidemiology has evolved since the 1990s, encompassing genome-wide association studies (GWAS) since 2000s.9 10 Molecular epidemiology refers to a specialised field of epidemiology where investigators examine genetic and molecular variation in a population and its interaction with dietary, lifestyle or environmental factors, to find clues to plausible causative links between aetiological factors and diseases. However, the mechanisms with which plausible aetiological factors influence the carcinogenic process remain largely speculative.
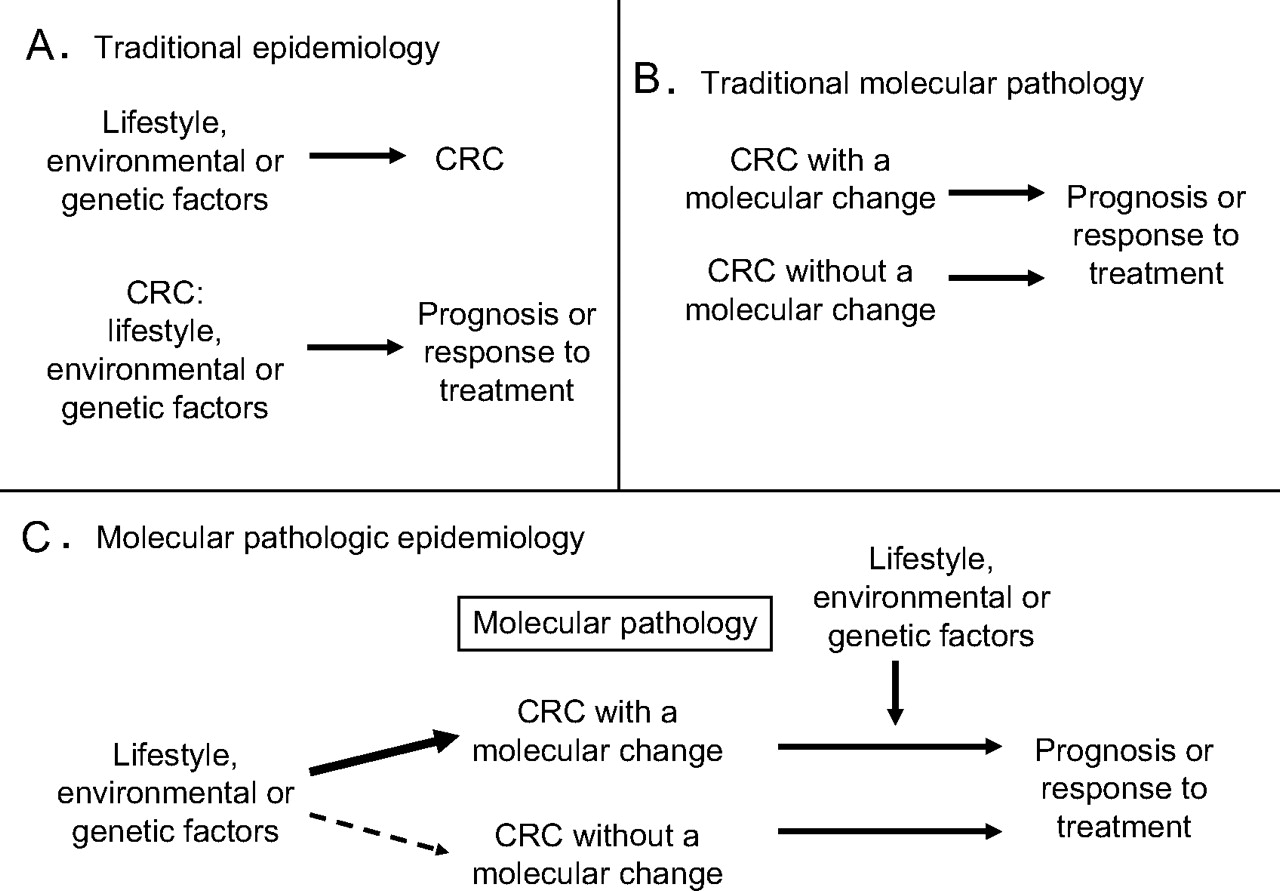
In traditional molecular pathology, investigators examine molecular characteristics in tumour cells to better understand carcinogenic processes and tumour cell behaviour. In the last two decades, our knowledge of somatic molecular alterations in the carcinogenic process has substantially improved.5 11–16 As illustrated in figure 1, these two approaches, epidemiology and molecular pathology, have converged to improve our understanding of how certain exposures influence carcinogenesis by examining molecular pathological marks of tumour initiation or progression, in relation to the exposures of interest.1 This represents a relatively new field of scientific investigation, which has been coined ‘molecular pathological epidemiology’.1 If a specific lifestyle or dietary factor can prevent the occurrence of a specific somatic molecular change, it would add considerable scientific basis to such a preventive strategy. Specificity of the association for a certain molecular change provides further evidence for a causal relationship. For an individual who has a susceptibility to a specific somatic molecular change, we may be able to develop a personalised preventive strategy, which targets specific molecules or pathways.

Illustration of traditional epidemiology (A), traditional molecular pathology (B), and molecular pathological epidemiology (C). Note that molecular pathology plays a central role in molecular pathological epidemiology. Molecular pathological epidemiology addresses the question of whether a particular exposure factor is associated with a specific molecular change in colorectal cancer (C, left side), as well as the question of whether a specific molecular change can interact with a particular exposure factor to affect tumour cell behaviour (C, right side). The latter represents a new direction of molecular pathological epidemiology where results can provide additional insights on the mechanism of how the tumoural molecular change and the exposure factor of interest influence tumour cell behaviour. CRC, colorectal cancer.
Table 1 (Available in online) is a comprehensive list of molecular pathological epidemiology studies on colorectal neoplasia.17–151 One challenge is that, despite a number of studies on some topics (eg, one-carbon metabolism gene polymorphisms and epigenetic changes), generalisable confirmed findings are uncommon. We discuss possible reasons and various challenges in a later section. Nonetheless, there have been observations confirmed by notable independent studies: a case–control study by Slattery et al120 and a prospective cohort study by the Iowa Women's Health Study147 have independently shown that cigarette smoking is associated with CIMP-positive tumour, and with BRAF-mutated tumour. As another example, the association between obesity and microsatellite stable (MSS) tumour has been demonstrated by three independent case–control studies, including Slattery et al,124 the North Carolina Colon Cancer Study,122 and the Colon Cancer Family Registry.93 With regard to germline genetic variants and molecular changes, MLH1 rs1800734 promoter SNP has been associated with MSI-high tumours in three independent case–case and case–control studies,29 117 121 and MGMT rs16906252 promoter SNP has been associated with MGMT promoter methylation and loss of expression in colorectal cancer57 and normal colorectal mucosa and peripheral blood cells in individuals without cancer.152 153 These consistent data across different studies increase validity of each other's findings and support aetiological roles of cigarette smoking, obesity and germline variants in specific pathways of colorectal carcinogenesis. Ultimately, our understanding of these specific neoplasia pathways will clarify areas for disease intervention.
Recently, GWAS have identified a number of candidate susceptibility loci for colorectal cancer.9 10 Currently, a significant limitation in interpreting GWAS results is our limited understanding of the functional relevance of risk alleles identified by GWAS. As a promising future direction, a molecular pathological epidemiology approach can be used to validate findings of GWAS in certain ways. First, if a candidate cancer susceptibility variant is hypothesised to regulate expression of a nearby gene, the relationship between the variant and gene expression in tumour tissue can be examined.36 Second, if a candidate variant is hypothesised to cause a genetic or epigenetic alteration in a critical pathway, the relationship between the variant and tumorous molecular alterations in the particular pathway can be examined.135 Specificity of the relationship between the variant and the tumour molecular alterations will provide additional evidence to support a causal effect of the putative cancer susceptibility allele.
Additional examples of studies and findings on three specific areas (energy balance, inflammation, and one-carbon metabolism) will be discussed in later sections because these have been particularly active areas of investigations.
Study design in molecular pathological epidemiology
Figure 2 illustrates three basic approaches to investigate the relationship between an exposure (eg, smoking) and a tumour molecular change (eg, KRAS mutation). A fourth approach, an interventional cohort study (not illustrated in figure 2) is a ‘gold standard’; however, to date no interventional molecular pathological epidemiology data have been published.

{kind=link}
{kind=link}
Comparison of a case–case study design (A), a case–control study design (B) and a prospective cohort study design (C). Smoking status is used as an example of an exposure variable, and KRAS mutation status in colorectal cancer as an outcome variable. See detailed explanations in text. CRC, colorectal cancer.
The first approach is a ‘case–case’ approach (figure 2A), where tumours are classified into subtypes according to a molecular feature, and then distributions of an exposure variable of interest among different subtypes are compared. For example, if it is hypothesised that smoking causes KRAS mutation, one may expect that a group of cancer patients showing a KRAS mutation would contain a higher fraction of smokers than a group of cancer patients showing KRAS wild-type. A limitation of this approach is that it is not possible to obtain information on the distribution of an exposure variable among the background population that has given rise to the cancer cases. Thus, the direction of any association cannot be determined; if there is a positive association between smoking and KRAS-mutated tumours (ie, a negative association between smoking and KRAS wild-type tumours), it is uncertain whether smoking protects against KRAS wild-type tumours, or smoking causes KRAS-mutated tumours.
The second approach is a case–control study (figure 2B), where non-cancer control subjects should ideally be randomly sampled from the background population that has given rise to the cancer cases. In traditional cancer epidemiology, distributions of an exposure of interest between cases and controls are compared. In molecular pathological epidemiology, distributions of a given exposure can be compared between cancer cases with a specific molecular alteration (eg, KRAS mutation), cancer cases without the alteration, and controls. If the exposure has caused the particular alteration, a higher fraction of exposed individuals would be expected among cancer cases with the alteration but not among cancer cases without the alteration, compared to controls. Nevertheless, case–control approaches in molecular pathological epidemiology face the same inherent limitations of traditional case–control studies. Such caveats include recall bias, differential selection bias between cases and controls, among others. One advantage of a case–control design over a prospective cohort design is its relative ease to recruit a large number of colorectal cancer cases. Important examples of case–control studies include the Colon Cancer Family Registry,26 47 92–94 103 105 109–111 115 116 154 a population-based case–control study of colorectal cancer by Slattery et al,95–99 119–121 124–140 155 156 and the Molecular Epidemiology of Colorectal Cancer Study in northern Israel.36 69 118 157–159
The third approach is a prospective cohort study (figure 2C), which is less prone to potential bias related to case–case and case–control designs. A nested case–control design, a case–case design within a prospective cohort study, and a case–cohort design160 are derivatives of prospective cohort studies. In molecular pathological epidemiology, investigators examine the incidence rates of cancer with a specific alteration (eg, KRAS mutation) in exposed versus unexposed individuals, as well as the incidence rates of cancer without the specific alteration in exposed versus unexposed individuals. If the exposure causes the particular alteration, one would expect to see a higher incidence rate of cancer with the alteration in exposed individuals than in unexposed individuals, and similar incidence rates of cancer without the alteration between the exposed and unexposed groups. In molecular pathological epidemiology of colorectal cancer, to date, seven prospective cohort studies have published substantial data: the European Prospective Investigation into Cancer and Nutrition,53 66 161–164 the Health Professionals Follow-up Study (HPFS),2 18–25 38 40 45 54–64 71 145 149 150 the Iowa Women's Health Study,147 165 166 the Melbourne Collaborative Cohort Study,146 167–169 the Netherlands Cohort Study,27 49 73 77–90 the Northern Sweden Health and Disease Study,144 170 171 and the Nurses' Health Study.2 18–25 38 40 45 54–64 71 145 148–150 172 Prospective cohort studies require substantial numbers of participants, substantial follow-up time and funding support, and substantial efforts by the researchers and other personnel. Therefore, judicious utilisation of the existing resource of prospective cohort studies is a cost effective approach.
Interactive effect of exposure and tumOUR feature on tumour aggressiveness: new direction of molecular pathological epidemiology
As a new direction of molecular pathological epidemiology, our group has started examining how lifestyle or genetic factors interact with tumour molecular features to influence tumour cell behaviour (prognosis or clinical outcome) (figure 1C). Table 2 (available online) lists studies on interactive prognostic effects of lifestyle or genetic factors and tumoural features in colorectal cancer.2 18–21 34 45 55 56 58–64 173–178 In traditional molecular pathology, investigators examine tumorous molecular characteristics to better predict prognosis and response to specific treatments.11 In addition to tumorous molecular features, lifestyle, environmental or genetic factors likely influence tumour cell behaviour through the tumour microenvironment. Lifestyle factors (eg, physical activity or smoking) or genetic factors (eg, SNPs or family history) have been shown to influence clinical outcome of colorectal cancer patients.168 179–185 To better understand how a certain lifestyle, environmental or genetic factor influences tumour cell behaviour, it is of interest to examine interactive prognostic effects of the lifestyle, environmental or genetic factor and tumour molecular features. If a particular exposure is associated with worse outcome only among patients with a specific tumour molecular change, but not among those without the molecular change, this provides evidence that the exposure factor might influence tumour aggressiveness through that molecular change or pathway. We will discuss specific examples in the following sections.
Interactive prognostic effects of obesity, physical activity and tumoUR changes
Studies have shown that obesity is associated with worse survival of colon cancer patients.168 186–189 However, how obesity affects clinical outcome of cancer patients remains largely unknown. In 2008, our group started a new direction of molecular pathological epidemiology, to examine an interactive prognostic effect of obesity (prediagnosis body mass index) and FASN (fatty acid synthase) expression in colon cancer.2 We found that the adverse prognostic effect of obesity was present in patients with FASN-positive colon cancers, but not in patients with FASN-negative colon cancers.2 These data suggest that excessive energy present in obese patients may contribute to growth and proliferation of tumour cells with FASN activation.2 This study has opened new opportunities for investigating how lifestyle factors affect tumour cell behaviour through cellular molecules. In traditional epidemiology, investigators examine the relationship between an exposure factor (eg, obesity) and survival of cancer patients regardless of tumour molecular subtype; thus, mechanistic hypotheses remain speculative. For example, it is hypothesised that obesity increases tumour aggressiveness potentially through a certain cellular molecule such as FASN. Without analysis of FASN in tumour, the hypothesis still remains speculative. In molecular pathological epidemiology, we can specifically test the hypothesis by examining the relations between obesity and patient survival in tumour FASN-positive cases and between obesity and patient survival in tumour FASN-negative cases.2 If the hypothesis is true, we expect to observe the significant obesity/survival relationship in FASN-positive cases, but not in FASN-negative cases.2
Our subsequent investigations have found that a number of other tumour molecular changes interact with prediagnosis body mass index to modify tumour aggressiveness.59 61 62 Those tumour changes include STMN1 expression,59 CDKN1A (p21) expression,62 and CDKN1B (p27) cellular localisation,61 all of which have been linked to energy balance and related signal transduction pathways.190–193 In addition, our analysis on interactive prognostic effects of physical activity and tumour markers have revealed that post-diagnosis physical activity is beneficial only in patients with CDKN1B nuclear-positive colon cancers, but not in patients with CDKN1B-altered or lost colon cancers.174 These results collectively provide evidence for tumour–host interactions (energy balance status and tumour molecular alterations) that influence tumour cell behaviour.
Inflammation and carcinogenesis
Epidemiological studies have shown that regular use of aspirin or non-steroidal anti-inflammatory drug is associated with decreased risks of colorectal cancer and adenomas.194–203 Randomised trials have confirmed that regular use of aspirin204–206 or other inhibitors of PTGS2 (prostaglandin endoperoxide synthase 2, cyclooxygenase-2, COX-2)207–209 decreases the risk of developing colorectal adenomas. Experimental evidence suggests an important role of PTGS2 in colorectal carcinogenesis.210–212 Thus, it is hypothesised that PTGS2 (COX-2) inhibitors may prevent colorectal tumour through inhibition of PTGS2. Molecular pathological epidemiology research has provided further insights on mechanisms of cancer preventive effect of PTGS2 inhibition. Utilising the Nurses' Health Study and the Health Professionals Follow-up Study (HPFS), we found that regular aspirin use decreases the risk of cancers with PTGS2 (COX-2) over-expression, but not that of cancers without PTGS2 over-expression.145 This specific inverse association between aspirin use and incidence of PTGS2-positive cancer provides further evidence for the carcinogenic role of PTGS2 (COX-2), and for the role of PTGS2 (COX-2) inhibitors in cancer prevention.
We have also shown that PTGS2 (COX-2) over-expression is associated with aggressive tumour behaviour,176 and that regular aspirin use after colorectal cancer diagnosis significantly decreases mortality in patients with PTGS2-positive cancers, but not in patients with PTGS2-negative cancers.173 This specificity of the relation between aspirin use and low mortality in PTGS2-expressing cases provides additional evidence for the role of PTGS2 inhibition in prevention of cancer progression.
One-carbon metabolism, germline variants, and somatic epigenetic changes
Colorectal cancer is a complex disease resulting from both genetic and epigenetic alterations, including abnormal DNA methylation patterns.213 214 DNA hypomethylation at LINE-1 repetitive elements has been associated with poor prognosis in colon cancer.177 LINE-1 hypomethylation may provide alternative promoter activation,215 and contribute to non-coding RNA expression that regulates expression of many genes.216 217 Retrotransposons activated by DNA hypomethylation may transpose themselves throughout the genome, leading to gene disruptions218 and chromosomal instability (CIN).219 220 In addition, there exists a specific tumour phenotype, the CpG island methylator phenotype (CIMP), characterised by a propensity for widespread CpG island hypermethylation.221 High degree of CIMP (CIMP-high) is a distinct phenotype,5 15 222–225 and the most common cause of microsatellite instability (MSI) in colorectal cancer through epigenetic inactivation of a mismatch repair gene MLH1.226–230 Independent of MSI, CIMP-high is associated with older age, female gender, proximal tumour location,228 231 232 high tumour grade, signet ring cells,233 BRAF mutation,228 231 232 wild-type TP53,228 234 inactive PTGS2 (COX-2),234 inactive CTNNB1 (β-catenin),235 loss of CDKN1B (p27),236 high-level LINE-1 methylation,231 237 stable chromosomes,238 239 and expression of DNMT3B,175 CDKN1A (p21)240 and SIRT1.55 Thus, CIMP status is a potential confounder for many locus-specific tumour variables.5 Moreover, the relationship between KRAS mutation and another type of CIMP (‘CIMP-low’,5 231 241–245 ‘CIMP2’,246 and ‘intermediate-methylation epigenotype’247) has been demonstrated. Importantly, different CIMP subtypes appear to show different locus-specific methylation patterns.231 244 246–248 Accumulating evidence suggests that CIMP-high colorectal cancers arise through the ‘serrated pathway’,249–259 which has substantial implications in studies on colorectal polyps and adenomas, because of potential differences in detection rates, removal rates and natural histories between conventional and serrated precursor lesions. The elucidation of mechanisms of epigenetic aberrations will improve our understanding of the carcinogenic process.
One-carbon metabolism is considered to play major roles in DNA synthesis and methylation reactions.260 In most epidemiological studies, low folate intake has been associated with higher risks of colorectal cancer261–266 and adenoma.266–269 However, results from randomised clinical trials of folic acid supplementation among individuals with a prior history of colorectal adenomas have been disappointing. A meta-analysis of these randomised trials270 has demonstrated that folic acid supplementation does not decrease adenoma recurrence risk after short-term follow-up. In fact, one randomised trial271 272 suggested a potential tumour-promoting effect of folic acid supplementation. Thus, there has been much controversy on dietary folate, folic acid fortification/supplementation and risks of colorectal neoplasia.270 272–274 Examining molecular changes in tumour cells in relation to folate intake may provide additional insights on the possible link between one-carbon metabolism and carcinogenesis.
Folate deficiency is associated with an increase in de novo DNA methyltransferase activity.275 276 Altered levels of folate metabolites and intermediates are associated with aberrant DNA methylation patterns.43 277 The MTHFR rs1801131 polymorphism (codon 429) has been associated with colon cancer with the CpG island methylator phenotype (CIMP) in case–control and case–case studies,38 95 although another case–cohort study has not confirmed this finding.84 Notably, the latter case–cohort study has shown that the MTR rs1805087 polymorphism is inversely associated with CIMP in men.84 Collectively, genetic variations in one-carbon metabolism pathways may play roles in epigenetic events during carcinogenesis.
With regard to global DNA methylation level, experimental data support a link between folate level and cellular DNA methylation level.278–280 In our prospective cohort studies, subjects reporting low folate intake experienced an increased risk of colon cancer with global DNA (LINE-1) hypomethylation, but folate intake had no influence on a risk of LINE-1 hypermethylated cancer.150 In a randomised, double-blinded, placebo-controlled trial, folic acid supplementation was inversely associated with global DNA hypomethylation in normal colon mucosa.281 However, in the Aspirin/Folate Polyp Prevention Trial, there was no significant influence on LINE-1 methylation in normal colon mucosa by folic acid supplementation.282
Besides influence of one-carbon nutrients, local DNA sequence context may influence assembly of a methylation reaction machinery and locus-specific DNA methylation. Studies have shown that cis-acting elements cause allele-specific methylation in the mammalian genome.283–286 Thus, germline variations in putative cis-acting elements may influence epigenetic status; such examples include MLH1 rs1800734 promoter SNP,29 117 121 and MGMT rs16906252 promoter SNP.57 152 153
Challenges in molecular pathological epidemiology
Although molecular pathological epidemiology is a very promising field, a number of challenges exist. Molecular pathological epidemiology research has the same set of inherent limitations as traditional epidemiology research and pathology research, including those related to bias (eg, selection bias, recall bias, measurement errors, and misclassification), confounding, generalisability and causal inference. In addition, there are other issues specific to molecular pathological epidemiology. Many of the issues have previously been discussed.287–289 In this section, we systematically discuss various issues specific to molecular pathological epidemiology and propose measures to overcome those issues.
Selection bias
Since we can analyse only a finite number of cases, controls or cohort participants, selection bias is a universal issue. The use of cancer cases in one or a few hospitals may be a source of selection bias since patients have selected the one or few hospitals based on referral or their own preference. To decrease bias due to differential hospital selection by patients, a large population-based investigation or multicentre investigation is desirable. To minimise selection bias, it is necessary to make the best effort to retrieve enough tissue materials from as many hospitals and pathology laboratories as possible.
In molecular pathological epidemiology, a tumour tissue retrieval rate is almost inevitably less than 100%.156 290 Patient and disease characteristics may influence the tissue retrieval rate. Specimen availability may be related to tumour size and patient outcome;291 this may be especially true in colorectal adenomas. A large epidemiological study has shown that tumour tissue retrieval rates in early-stage intramucosal cancer and advanced stage IV cancer are lower compared to stage I–III cancers.156 Nonetheless, both case–control and prospective cohort studies have shown that demographic features and dietary and other exposure factors are similar between cases with tumour tissue analysed and those without available tumour tissue.145 156
Another source of selection bias is treatment before surgical resection of tumour. While this has not been a major issue in colon cancer, treatment prior to surgical resection of rectal cancer is now common. First, selection of patients for treatment is likely non-random and influenced by many factors. Second, treatment before surgery can eliminate most or all tumour cells in resection specimens in some patients, while treatment is ineffective in other patients. Thus, availability of ample tumour cells is determined by treatment effect which is likely influenced by tumour molecular characteristics. Third, treatment itself can introduce molecular changes which may not naturally occur. Thus, if treatment is administered before surgical resection, it is recommended to collect tumour specimens that were taken prior to such treatment.
Sample size
In studies on tumour prognostic markers, a frequent problem is using inappropriate sample sizes that are too small to conduct robust statistical analysis and draw meaningful conclusions.292 Small sample sizes lead to a number of problems including a large variation of an effect estimate with wide confidence limits, random and non-random selection bias, and publication bias. Publication bias refers to a phenomenon that studies with null findings have a higher likelihood of being unwritten and unpublished compared to studies with ‘significant’ findings. In the published literature, small underpowered studies with ‘significant’ findings have been over-represented, relative to small underpowered studies with null findings. In a meta-analysis of TP53 alterations and head and neck cancer outcome,293 not only publication bias, but also selective presentations of data in many small studies appear to be a serious problem that can lead to biased and misleading conclusions.
In molecular pathological epidemiology, sample size is a substantial issue. Even when a parent study is large-scale, any given molecular pathological epidemiology study requires multiple exclusions based on availability of tumour tissue materials and valid assay results. In molecular pathological epidemiology, by definition, a subset analysis for different outcomes (a molecular change present vs absent) is performed. A sample size for a smaller subset may not be large enough to provide adequate statistical power. Population-based studies have shown that molecular subtyping is often skewed: BRAF mutation (10–15% mutated vs 85–90% wild-type),146 228 294 295 PIK3CA mutation (15–20% mutated vs 80–85% wild-type),296 297 NRAS mutation (2% mutated vs 98% wild-type),40 MSI (15% high vs 85% low/MSS),85 93 127 231 KRAS mutation (35–40% mutated vs 60–65% wild-type),27 242 298 or CIMP (10–20% high vs 80–90% low/negative;170 243 294 299 or 15–30% positive vs 70–85% negative85 146 228). Therefore, for any future cancer epidemiology research, one should design a study as large as possible, because tumour molecular subtyping is increasingly common in cancer epidemiology.
Measurement error and misclassification
In addition to measurement error and misclassification in exposure variables and covariates, non-trivial measurement error and misclassification may be present in an outcome variable (ie, tumour molecular subtyping). This particular combination (ie, measurement errors and misclassification in both exposure and outcome assessments) is a unique challenge in molecular pathological epidemiology.
Tumour molecular and immunohistochemical assays should be validated and monitored for their precision and accuracy. In immunohistochemical analysis, it is possible to observe a correlative error between two completely unrelated proteins because of the presence of poor quality tissue specimens, which fail to react with any specific antibody leading to false negative results.5 Thus, in such poor quality cases, negativity of one protein tends to coincide with negativity of another protein even with the absence of any true association. Since those cases with poor quality materials are inevitably present in large-scale epidemiological studies, one should be cautious when interpreting an apparent positive correlation between two proteins by immunohistochemistry assays.5 The presence of internal control in tumour tissue may solve this problem to some extent.
To decrease run-to-run variability in immunohistochemical assays, the use of tissue microarray (TMA) is recommended.289 All cases in the same TMA slide can be processed and treated in a similar manner during immunostaining. We recommend inclusion in TMA of normal tissue adjacent to tumour tissue from the same individual whenever normal tissue is available. Normal colon mucosa may serve as an internal control. Tissue cores can be separately taken from tumour edge and centre and labelled as such. Because TMA is cost efficient for a large-scale study, any epidemiological study or clinical trial should consider TMA for immunohistochemical evaluations of expression of multiple proteins.
Multiple hypothesis testing
Multiple hypothesis testing is a common issue in epidemiology, and is even more problematic in molecular pathological epidemiology. By definition, molecular pathological epidemiology involves subset analyses on tumour subtypes, which exacerbate the potential for false positive findings due to multiple hypothesis testing.1 If one crosses a wide range of lifestyle and other exposure variables with a variety of molecular changes, the likelihood for a nominally significant chance finding is high.1 In this post-genomic era, we can potentially generate a countless number of hypotheses as we have already experienced in GWAS.300–302 False positive findings can potentially confuse the literature, scientific field, and clinical practice.303 If a higher significance level is required, then we require a large sample size.
An important question is whether the molecular pathological epidemiology approach should be hypothesis-driven or exploratory as GWAS. If the former is the case, how can we prioritise various hypotheses to allocate our limited resources? If the latter is the case, how can we make formal rules of statistical significance and validation of findings? The border between hypothesis-driven research and exploratory research may not be distinct in molecular pathological epidemiology. For example, a proposed link between smoking and MSI-high, CIMP-high or BRAF-mutated colon cancers may be regarded as either exploratory or hypothesis-based. Where do we draw a line between hypothesis testing versus exploration? At the very least, an initial few studies examining the relationship between a certain exposure and a specific molecular change should be regarded as exploratory and hypothesis-generating. Any generated hypothesis needs to be validated by independent datasets.
We acknowledge that any novel hypothesis could at first result from a fortuitous discovery by multiple hypothesis testing. If we successfully implement proper measures, the pace of our new discovery can be much faster than before. To generate and test new hypotheses, validate new findings, solidify new knowledge, and implement new public health recommendations and measures, we should develop an optimal and standardised way of streamlining the sequence of discoveries and validation in molecular pathological epidemiology.
Generalisability
All issues mentioned above affect generalisability of study findings. Many findings by molecular pathological epidemiology studies (as shown in tables 1 and 2 available online) are yet to be validated in other independent datasets. It is challenging since there is a wide variety in study designs and populations, and differences in tumour molecular assays add further diversity between different studies. On the other hand, because of the presence of such enormous heterogeneity between different studies, consistent findings across different studies can be regarded as generalisable findings.
Direct causation of molecular changes versus selective advantage
Although molecular pathological epidemiology illuminates carcinogenic mechanisms, it still needs experimental data to confirm a causal relationship. There still remains a question whether an exposure of interest can either directly or indirectly cause a specific molecular change, or create a specific environment which can provide selective advantage for clonal expansion of a cell with a specific molecular change. Tumour molecular alterations may not only represent the interactions of carcinogens with DNA repair mechanisms or epigenetic machinery, but also reflect the tissue-specific selection of those alterations that provide pre-malignant and malignant cells with a clonal growth advantage.
How we can examine the process of tumour progression in observational molecular pathological epidemiology
Since some molecular changes have been known to occur early (eg, APC loss, KRAS mutation), aetiological factors which appear to cause those early events can be considered to contribute to tumour initiation/progression early in the carcinogenic process. Another way is to analyse colorectal polyp/adenoma and colorectal cancer within the same population, and investigate how an exposure of interest is related to somatic molecular events in cancer and precursor lesions.
Multidisciplinary research environment and cross-training and education
Molecular pathological epidemiology is transdisciplinary and interdisciplinary by nature (see Stokols et al304 for the definitions of transdisciplinarity and interdisciplinarity). It requires expertise of diverse fields including, at least, epidemiology, biostatistics, pathology and oncology. Therefore, a collaborative environment is essential, and cross-training and education are extremely useful to advance this interdisciplinary area of science. In particular, training in epidemiology and biostatistics during pathology training is very beneficial.305 Increasing needs and a trend for team science rather than solo science have been well documented.304 306 307
Future direction and concluding remarks
‘Molecular pathological epidemiology’ is a relatively new, evolving field of epidemiology which is designed to elucidate how various exposures affect initiation, transformation and progression of neoplasia.1 A new direction of molecular pathological epidemiology is to investigate interactive effects of dietary or lifestyle exposures and tumour molecular features on tumour behaviour (prognosis or clinical outcome), so that one can attribute the effects of dietary or lifestyle variables to a specific molecular subtype of cancer.2 A number of hurdles must be overcome because of unique and new challenges which we have not faced in traditional epidemiology research. To overcome those issues, it is necessary to coordinate research efforts around the world and to possibly formulate a system where new findings can be discovered and validated. As a result, molecular pathological epidemiology research will continue to provide profound insights on carcinogenic processes and help us to optimise prevention and treatment strategies.
Acknowledgments
We thank all investigators who have contributed to this emerging multidisciplinary field of science.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
Supplementary materials
Online only appendix
Files in this Data Supplement:
Footnotes
Funding This work was supported by U.S. National Institute of Health (P01 CA87969, to S.E. Hankinson; P01 CA55075, to W.C. Willett; P50 CA127003, to CSF; R01 CA137178, to ATC; K07 CA122826, to SO; and R01 CA151993, to SO).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.