Article Text

Abstract
Objective The transcription factor Kruppel-like factor 2 (KLF2) modulates the expression of multiple endothelial vasoprotective genes. In the absence of KLF2, the endothelial phenotype becomes dysfunctional. To date, blood-derived shear stress is the main physiological stimulus identified to trigger and sustain endothelial KLF2 expression. Portal hypertension is a common complication of cirrhosis. Sinusoidal distortion and endothelial dysfunction play a significant role in its pathogenesis. This study aimed to assess whether abnormal intrahepatic haemodynamics in cirrhosis could modify KLF2 expression and consequently its downstream transcriptional programmes.
Design Rats received carbon tetrachloride or vehicle for two (acute injury), six (early cirrhosis) and twelve weeks (advanced cirrhosis). Systemic and hepatic haemodynamic parameters were measured in vivo. Hepatic expression of KLF2 and its vasoprotective targets were determined. Additionally, KLF2 expression was determined in liver sections, in freshly-isolated hepatic endothelial cells, and in livers from simvastatin-treated cirrhotic animals.
Results Cirrhotic livers have increased endothelial KLF2 expression compared with controls. KLF2 elevation, observed at six weeks of cirrhosis induction, was accompanied by a parallel increase in portal pressure and an increase in the expression of its target genes eNOS, thrombomodulin and CNP. Simvastatin administration further increased hepatic KLF2 and target genes expression.
Conclusions This study shows an increase in the expression of the vasoprotective transcription factor KLF2 in the cirrhotic liver, accompanied by an activation of its downstream transcriptional programmes. These data suggest that the marked increase in KLF2 expression may represent an endothelial compensatory mechanism to improve the ongoing vascular dysfunction in the cirrhotic liver.
- Endothelial cells
- endothelial dysfunction
- endothelium
- eNOS
- gene expression
- KLF2
- nitric oxide
- portal hypertension
- statins
Statistics from Altmetric.com
- Endothelial cells
- endothelial dysfunction
- endothelium
- eNOS
- gene expression
- KLF2
- nitric oxide
- portal hypertension
- statins
Significance of this study
What is already known about this subject?
The transcription factor KLF2 protects the endothelium inducing the expression of a variety of vasoprotector genes/proteins.
Shear stress is the most potent inducer of KLF2 expression.
Most of these findings were described in cultured endothelial cells isolated from large vessels. Characterisation of KLF2 in microvascular vessels or specialised endothelium such as the liver SEC has never been reported.
What are the new findings?
SEC express KLF2 in a shear stress-dependent manner.
Cirrhotic livers exhibit an upregulation in KLF2 and its vasoprotective target genes mRNA expression (including eNOS and thrombomodulin).
The determined increment in KLF2 mRNA expression results in an increase in KLF2 and thrombomodulin protein expression; however, eNOS protein synthesis is misregulated in cirrhotic livers.
Simvastatin upregulates hepatic KLF2 and its derived vasoprotective transcriptional programmes.
How might it impact on clinical practice in the foreseeable future?
Our data suggest that upregulation in KLF2 expression may represent a compensatory mechanism to improve the sinusoidal endothelial dysfunction characteristic of the cirrhotic liver.
Our data open the rationale to investigate whether modulating KLF2 expression/activity may be a potential target in the management of cirrhosis and in liver preservation.
Kruppel-like factors (KLF) are a subclass of the zinc finger family of transcription factors that regulate cellular growth and tissue development.1 KLF typically bind to GC-rich or CACCC sequences in the promoter region of target genes to regulate their transcriptional activity. One member of the KLF family, KLF2, is highly expressed in the vascular endothelium and it is required for normal vessel development.2 3 It has been demonstrated that KLF2 expression confers endothelial protection against inflammation, thrombosis and vasoconstriction (figure 1). In fact, KLF2 expression attenuates cytokine-mediated induction of pro-inflammatory targets such as E-selectin,4 inhibits the expression of vascular destabilisation molecules such as angiopoietin 2,5 induces gene expression of antithrombotic agents such as the blood coagulation inhibitor thrombomodulin,6 7 and potently activates vasodilatory pathways such as the endothelial-derived hyperpolarising factor c-type natriuretic peptide (CNP) or the endothelial nitric oxide synthase (eNOS).5

Scheme of Kruppel-like factor 2 (KLF2) induction and regulation in endothelial cells. Mitogen-activated protein kinase 5 (MEK5), mitogen-activated protein kinase 7 (ERK5), myocyte enhancing factor 2 (MEF2), KLF2, endothelial nitric oxide synthase (eNOS) and thrombomodulin (TM).
Blood flow-derived shear stress is one of the most important biomechanical stimuli that induces KLF2 expression, both in vivo and in cultured endothelial cells.5 8 9 In addition, endothelial cells exposed to disturbed shear stress do not express KLF2, providing evidence that KLF2 expression is selectively induced by distinct types of flow.5 8 9
Portal hypertension syndrome is defined by a pathological increase in the portal venous pressure derived from increments in intrahepatic resistance (IHR) and portal blood flow (PBF) and represents a major complication of liver cirrhosis.10 This increased liver resistance to PBF is partly the result of an injured and therefore dysfunctional hepatic endothelium, which synthesises large amounts of vasoconstrictor prostanoids11 12 and reduced quantities of vasodilators such as nitric oxide (NO).13
The main hypothesis of the present study was that hepatic haemodynamic variations occurring during cirrhosis development could modify the hepatic endothelial KLF2 expression, thus altering the vasoprotective KLF2 target gene expression. In the present study we thus characterised the expression of KLF2 and its main target genes at three different phases of the induction of experimental cirrhosis.
Methods
Induction of cirrhosis by CCl4
Eighteen male Wistar rats weighing 50–75 g underwent inhalation exposure to carbon tetrachloride (CCl4) for 2, 6 or 12 weeks (n=6 per group). Phenobarbital (0.3 g/l) was added to the drinking water as previously described.11 After these periods of CCl4 administration, treatment was stopped and the subsequent experiments were performed 1 week later. Age-matched control animals (n=6 per group) received only phenobarbital. The animals were kept in environmentally controlled animal facilities at the IDIBAPS. All experiments were approved by the Laboratory Animal Care and Use Committee of the University of Barcelona, and were conducted in accordance with the ‘Guide for the Care and Use of Laboratory Animals’ (National Institutes of Health, NIH publication 86-23, revised 1985).
In-vivo haemodynamic studies
Under anaesthesia with intraperitoneal ketamine hydrochloride (Ketalar, 100 mg/kg bodyweight; Parke-Davis SL, El Prat de Llobregat, Barcelona, Spain) and midazolam (5 mg/kg bodyweight; Reig Jofre SA, Sant Joan d'Espi, Barcelona, Spain) a tracheotomy was performed and a polyethylene PE-240 tubing was inserted into the trachea to ensure a patent airway. PE-50 catheters were introduced into the femoral artery, for arterial pressure recording (mm Hg), and into the portal vein through an ileocolic vein, to measure portal pressure (mm Hg). Then, the portal vein was carefully dissected free from connective tissue, and a non-constrictive perivascular transit-time ultrasonic flow probe (2PR, 2-mm diameter; Transonic Systems, Ithaca, New York, USA) was placed around this vessel. The flow probe was connected to a flow metre, to measure the portal vein blood flow (ml/min 100 g/bodyweight). Intrahepatic resistance (mm Hg/ml per min/g) was calculated as: portal pressure/(portal vein blood flow/liver weight). Blood pressures and flows were registered on a multichannel computer-based recorder (PowerLab; ADInstruments, Colorado Springs, Colorado, USA). The external zero reference point was placed at the midportion of the animal. Haemodynamic data were collected after a 30-min stabilisation period.
Liver histology
Liver tissue blocks from animals receiving CCl4 were fixed in 10% buffered paraformaldehyde and embedded in paraffin. Sections (10 μm) were cut and stained with haematoxylin and eosin (H&E) and with Masson's trichromic staining specifically to stain fibrous tissue components. The degree of fibrosis was assessed using image analysis techniques. Briefly, 10 Masson's staining representative sections per animal were obtained and the positive area was measured with freeware NIH Image J 1.38 (National Institute of Health, Bethesda, Maryland, USA). The results were expressed as a fibrosis ratio (%), calculated as the ratio of the Masson's positive area to the total area examined.
SEC isolation and exposure to shear stress
Sinusoidal endothelial cells (SEC) were isolated from control and cirrhotic rat livers (n=3 per group) and cultured as previously described.12 14 Briefly, after liver collagenase perfusion and isopycnic sedimentation of the resulting dispersed cells through a two-step density gradient of Percoll, pure monolayer cultures of SEC were established by selective attachment on a substrate of collagen I. SEC monolayers were cultured in the presence of vascular endothelial growth factor (VEGF; 40 ng/ml) and exposed to static conditions or to unidirectional laminar flow for 12 h using a cell culture flow chamber (IBIDI, Munich, Germany). Applied shear stress (14.1 dyn/cm2) was previously characterised for SEC in-vitro flow studies.15 All experiments were performed on high purity (>90%) and viability (>95%) cells on the first passage. To preserve its typical phenotype shear stress stimulus was initiated 12 h after SEC isolation.
In-vivo KLF2 induction
A subgroup of cirrhotic animals (12 weeks of CCl4 exposure, n=6 per group) received the KLF2-inducer simvastatin (25 mg/kg per day, by mouth), or its vehicle, for 3 days.16 Afterwards, the expression of KLF2 and its vasoprotective target genes was determined as described below.
RNA isolation and reverse transcription
Total RNA was isolated from frozen control and CCl4-treated rat livers and from fresh SEC using the Trizol method (Invitrogen, El Prat de Llobregat, Barcelona, Spain). RNA was treated with DNAse (Ambion, Austin, Texas, USA) to eliminate contaminating DNA. For complementary DNA synthesis, 1 μg of total RNA was retrotranscribed using Moloney-murine leukemia virus (MLV) reverse transcriptase and random hexamers, as described by the manufacturer (Invitrogen).
Real-time quantitative PCR of KLF2 and its target genes
cDNA templates were amplified by real-time quantitative (RT)–PCR using the fluorescent TaqMan technology (Applied Biosystems, Foster City, California, USA) on an ABI Prism 7900 sequence Detection System (Applied Biosystems). The quantification of rat KLF2, its target genes thrombomodulin, eNOS, CNP, VEGF, angiopoietin 2 and the endogenous control 18S RNA was performed using predesigned gene expression assays obtained from Applied Biosystems according to the manufacturer's protocol.
Each PCR reaction was carried out with 2 μl of the hepatic cDNA sample, 1×TaqMan Universal PCR Master Mix (Applied Biosystems), and primers and probe in a final volume of 20 μl, as recommended by the manufacturer. After an initial denaturation step at 95°C for 10 min, 40 cycles were performed as follows: 95°C for 15 s and 60°C for 1 min.
All experiments were performed in duplicate and several negative controls were included. Gene expression was related to a standard curve derived from serial dilutions (10−1–10−4) of a random sample cDNA. Standard curves were constructed by plotting the log of standard dilutions versus the threshold cycle (CT) values, CT being the fractional cycle number at which the fluorescence passes a fixed threshold. The messenger RNA concentration of each gene in hepatic samples was calculated referring the sample CT to the standard curve, and normalised with the corresponding value of endogenous control CT as recommended in the TaqMan user's manual. Values were expressed as relative units.
Western blot analysis of KLF2 and its targets
Protein expression for KLF2, thrombomodulin, eNOS, phosphorylated eNOS at Ser1176 (P-eNOS), VEGF and CNP in rat livers from CCl4-treated and control rat livers was assessed by western blot. Livers were collected, snap frozen in liquid nitrogen and homogenised in triton-lysis buffer as previously described.17 Aliquots from each sample containing equal amounts of protein (100 μg) were run on a 10% sodium dodecylsulphate polyacrylamide gel, and transferred to a nitrocellulose membrane. After the transfer, the blots were subsequently blocked for 1 h with Tris-buffered saline containing 0.05% (vol/vol) Tween 20 and 5% (wt/vol) non-fat dry milk and subsequently incubated with primary antibodies overnight at 4°C. Then membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature.
Protein expression was determined by densitometric analysis using the Science Lab Image Gauge (Fuji Photo Film GMBH, Düsseldorf, Germany). After stripping, blots were assayed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology, Santa Cruz, California, USA) expression as standardisation of sample loading. Quantitative densitometric values of all proteins were normalised to GAPDH.
Immunohistochemistry
KLF2 immunostaining was performed in paraffin-embedded liver sections from control and cirrhotic livers using a goat-anti-KLF2 antibody (N-13; Santa Cruz Biotechnology)18 or phosphate-buffered saline, as negative control, and diaminobenzidine as chromogen. Slides were counterstained with H&E, and images were acquired using a microscope equipped with a digital camera.
Drugs and reagents
Collagenase was from Roche Diagnostics (Mannheim, Germany). Percoll was from Amersham Biosciences (Uppsala, Sweden). Reagents for cell culture were provided by Biological Industries Ltd. (Kibbutz Beit Haemek, Israel). Gey's balanced salt solution (GBSS), acrylamide and other chemical reagents were purchased from Sigma (Tres Cantos, Madrid, Spain).
Statistical analysis
Statistical analysis was performed using the SPSS 14.0 for Windows statistical package. All results are expressed as mean±SEM. Comparisons between groups were performed with analysis of variance followed by Tukey's test or with Student's t test or the Mann–Whitney t test when adequate. Differences were considered significant at a p value less than 0.05.
Results
Haemodynamic changes during induction of cirrhosis by CCl4 administration
Two weeks of cirrhosis induction did not modify any of the systemic or the hepatic haemodynamic parameters compared with control animals (figure 2).
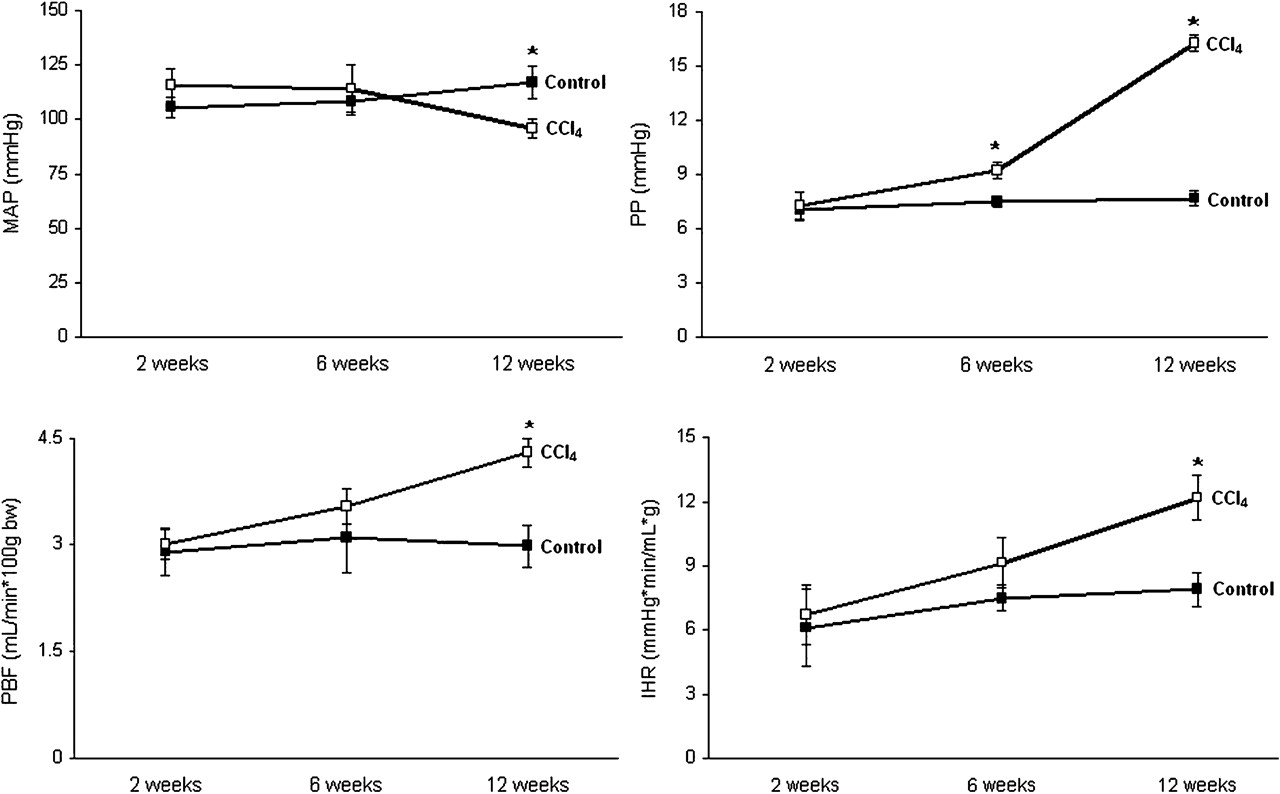
Haemodynamic studies. Mean arterial pressure (MAP), portal pressure (PP), portal blood flow (PBF) and intrahepatic resistance (IHR) variations along cirrhosis induction in rats by carbon tetrachloride (CCl4) inhalation (CCl4 group) compared with age-matched vehicle-receiving animals (control group). Results are shown as mean±SEM. (*p<0.05 vs control).
At 6 weeks of CCl4 treatment rats showed a significant increase in the portal pressure compared with those animals receiving vehicle. This was associated with non-significant increases in IHR and PBF (figure 2).
After 12 weeks of CCl4 administration, animals presented with ascites, hypotension and a further significant increase in portal pressure, due to both a rise in PBF and in intrahepatic vascular resistance compared with animals receiving vehicle (figure 2).
Liver fibrosis
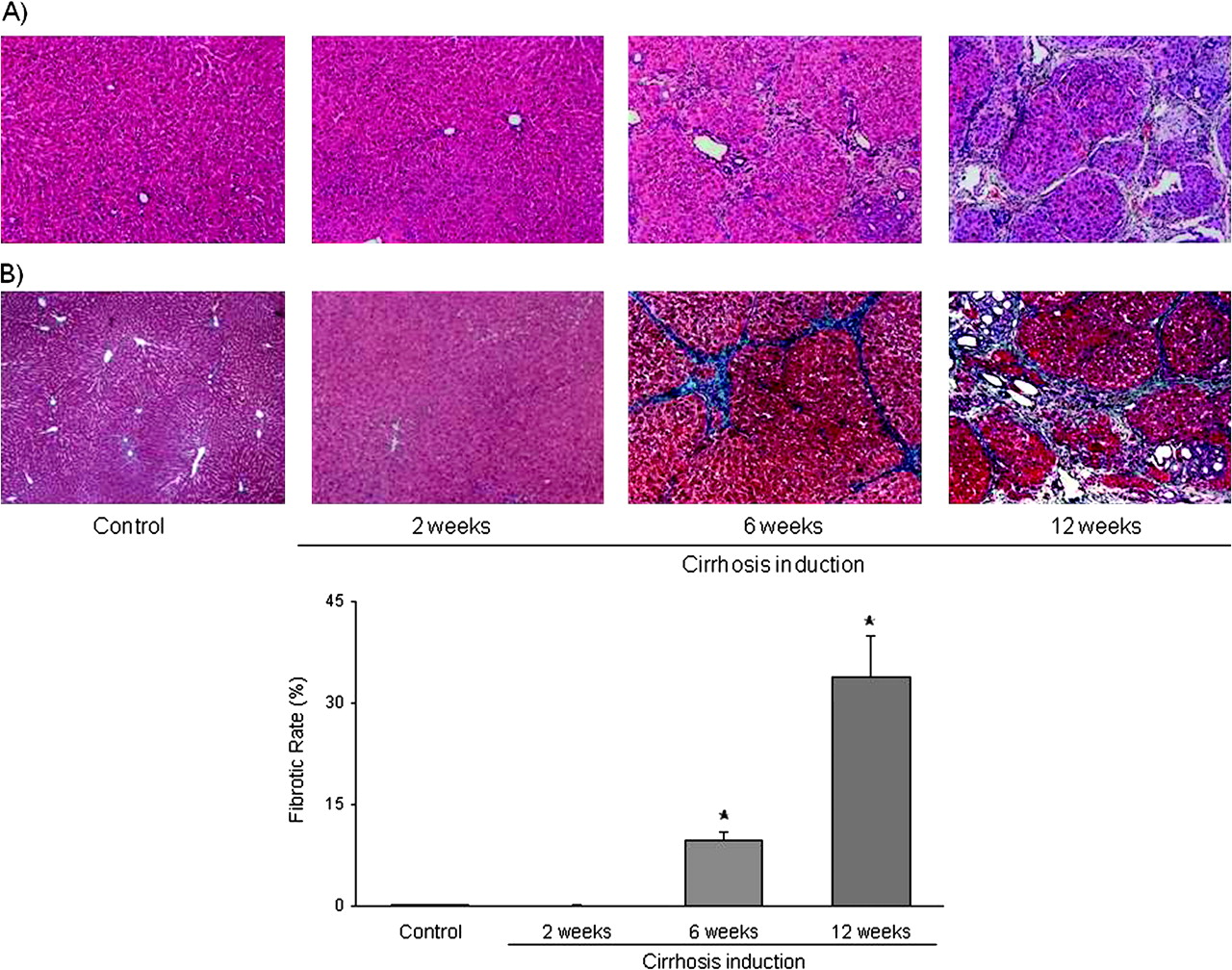
CCl4 administration led to progressive hepatic fibrosis and architectural damage (figure 3). Whereas no significant histological changes were observed after 2 weeks of CCl4 administration, at 6 weeks of starting the cirrhosis induction programme rats receiving CCl4 presented with periportal fibrosis and the formation of thin septa, with complete nodule formation in half of the animals. These changes were approximately threefold greater at 12 weeks, when rats had already developed advanced cirrhosis and ascites.

Histopathology analysis. (A) Representative images of haematoxylin and eosin-stained liver sections from control and carbon tetrachloride (CCl4)-treated rats. (B) Top: Representative images of Masson's trichromic stained liver sections from control and CCl4-treated rats. Images visualised and collected with light microscopy. Original magnification 20×. Bottom: Fibrosis quantification, values represent the ratio of the Masson's positive area to the total examined area (n=10 images per group).
Hepatic KLF2 expression
Control and CCl4-treated rat livers expressed KLF2 mRNA and protein.
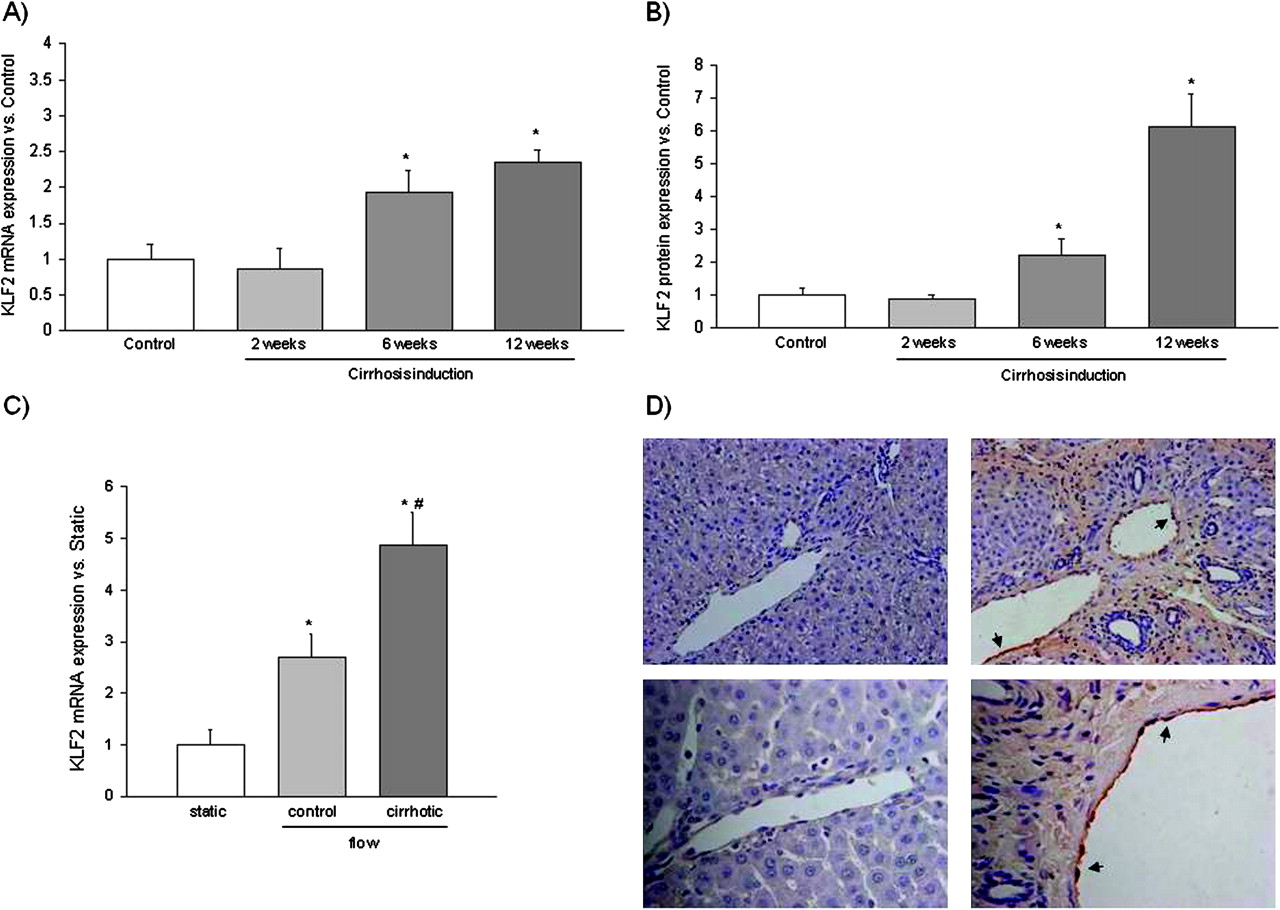
No differences in KLF2 mRNA or protein expression were observed after 2 weeks of vehicle or CCl4 administration (figure 4A,B). However, after 6 weeks CCl4-treated animals showed a significant increase in the hepatic KLF2 mRNA and protein expression compared with the matched control animals. Differences between both groups increased at 12 weeks of CCl4 administration, when animals had developed advanced cirrhosis (figure 4A,B).

Kruppel-like factor 2 (KLF2) expression. (A) Hepatic KLF2 mRNA expression levels in rats receiving carbon tetrachloride (CCl4) for 2, 6 and 12 weeks (n=6 per group). Values for KLF2 amplification from liver cDNA have been normalised to an endogenous reference gene (RNA 18S). Values (mean±SEM) are normalised to age-matched control liver expression (*p<0.05 vs control). (B) Hepatic KLF2 protein expression in rats receiving CCl4 (n=6 per group). Densitometry analysis of western blots were normalised to glyceraldehyde-3-phosphate dehydrogenase and referred to controls (n=6). Values represent mean±SEM. (*p<0.05 vs control). (C) KLF2 mRNA expression determined in sinusoidal endothelial cells, freshly isolated from control and cirrhotic rat livers, cultured under static conditions or exposed for 12 h to laminar shear stress (n=3 per group). Values (mean±SEM) are normalised to endogenous reference gene (*p<0.05 vs its corresponding static; #p<0.05 vs control). (D) KLF2 protein expression (dark brown areas, indicated with arrowheads) detected by immunohistochemistry in control (left) and cirrhotic (right) rat liver sections. Representative images were visualised with a light microscope and are showed with 20× (top) and 40× (bottom) magnifications.
To confirm the hepatic subcellular source of KLF2 expression and to know whether hepatic KLF2 expression responds to variations in flow stimulus, KLF2 expression was analysed in freshly isolated SEC exposed to static or physiological flow conditions and in liver sections. Figure 4C shows that SEC, freshly isolated from control or cirrhotic livers, express KLF2 in a flow-dependent manner; indeed SEC exposed for 12 h to flow-derived shear stress exhibited a significant upregulation in KLF2 expression compared with cells cultured under static conditions. KLF2 increment was significantly higher in cirrhotic SEC than in controls.
Localisation of KLF2 using immunohistochemistry revealed that hepatic KLF2 protein expression, at least in cirrhotic livers, is almost solely endothelial. In fact, KLF2 protein expression was specifically detected in the endothelium of both portal and arterial beds. In contrast, intrahepatic KLF2 was barely detected in control rats using immunostaining, confirming a lower expression of this transcription factor in control livers (figure 4D). Unspecific background staining detected in fibrotic tissue from cirrhotic slides is shown in supplementary figure 1 (available online only).
KLF2 target gene expression
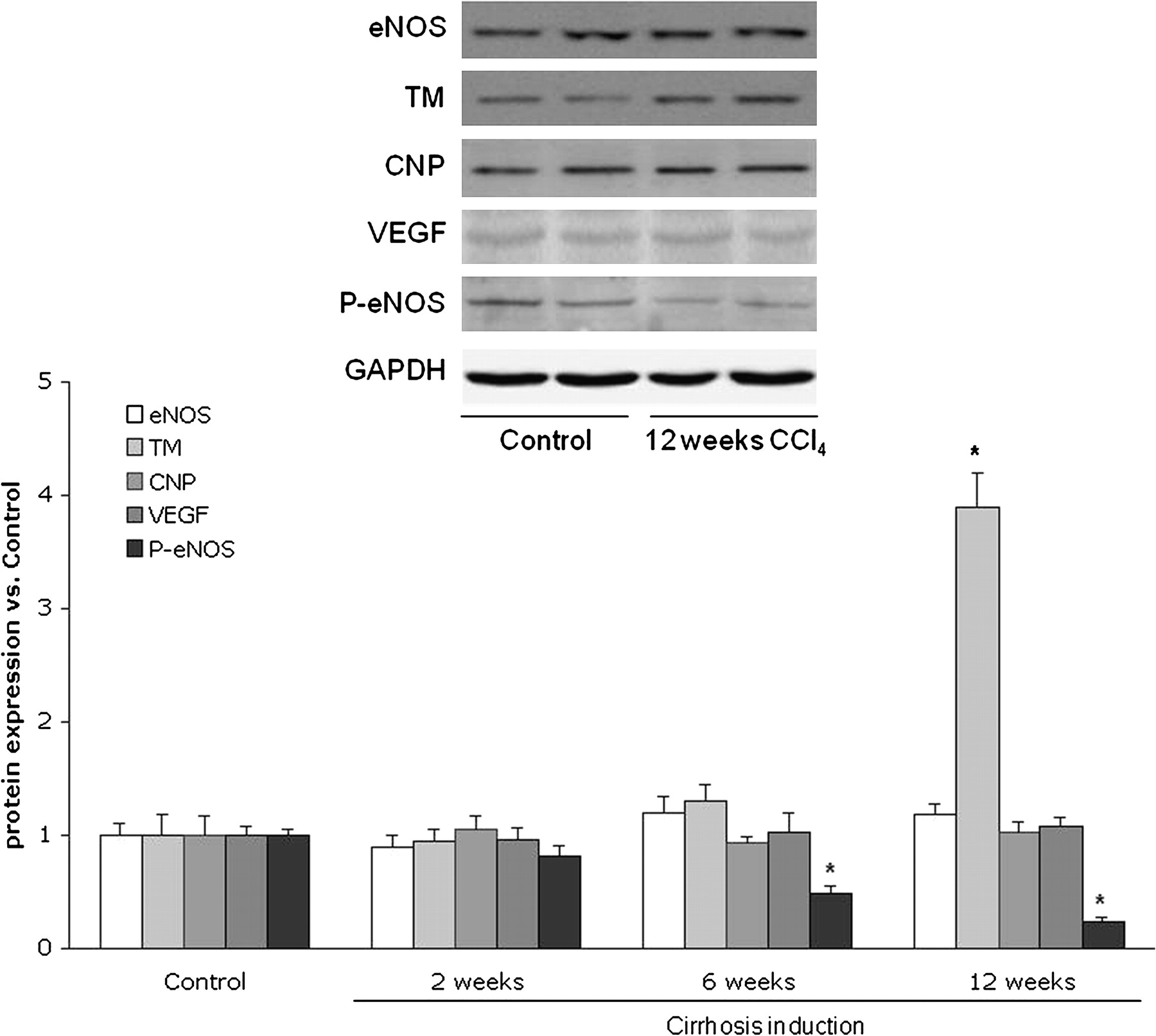
Analysis of KLF2-targeted genes and proteins involved in vascular physiology showed no differences in their hepatic expression comparing animals that received CCl4 or vehicle for 2 weeks (figures 5 and 6). However, in accordance with the observed increases in KLF2 expression, animals receiving CCl4 for 6 weeks showed a marked increase in hepatic eNOS, thrombomodulin and CNP mRNA expression compared with those receiving vehicle (figure 5), whereas no differences were observed in angiopoietin 2 and VEGF mRNA expression. In contrast, at this point of cirrhosis development no differences in any of the evaluated KLF2-targeted hepatic proteins were observed compared with control animals (figure 6); however, a significant reduction in eNOS phosphorylation was observed in CCl4-treated animal livers compared with controls (figure 6).
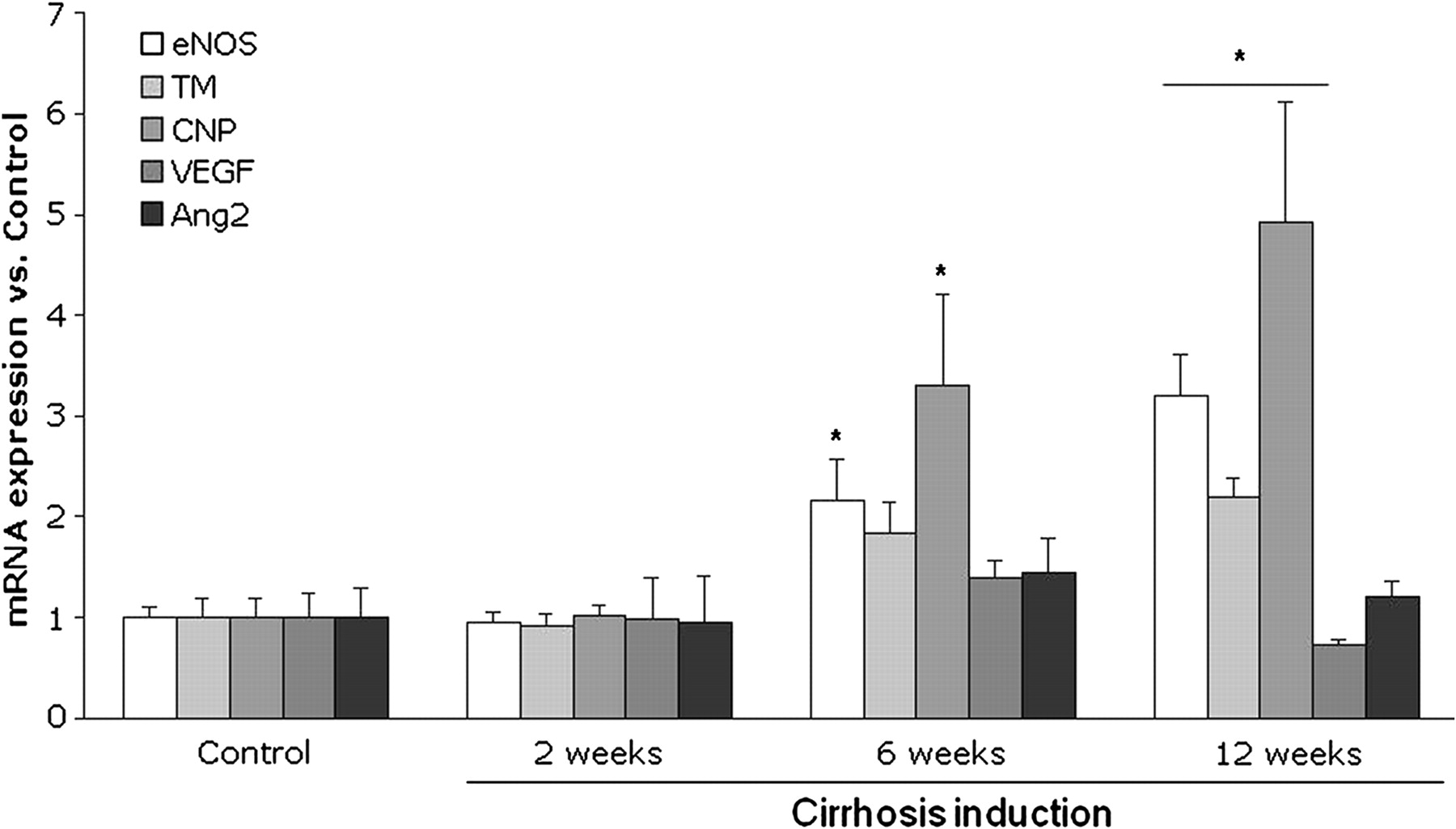
Hepatic Kruppel-like factor 2 (KLF2) target genes mRNA expression levels in rats receiving carbon tetrachloride for 2, 6 and 12 weeks (n=6 per group). Values for endothelial nitric oxide synthase (eNOS), thrombomodulin (TM), c-type natriuretic peptide (CNP), vascular endothelial growth factor (VEGF) and angiopoietin 2 (Ang2) amplification from liver cDNA have been normalised to an endogenous reference gene (RNA 18S). Values (mean±SEM) are normalised to age-matched control liver expression (*p<0.05 vs control).

Hepatic endothelial nitric oxide synthase (eNOS), thrombomodulin (TM), vascular endothelial growth factor (VEGF), phosphorylated-eNOS (P-eNOS) and c-type natriuretic peptide (CNP) protein expression in rats receiving carbon tetrachloride (CCl4). Top, representative western blots of indicated proteins. Bottom, densitometry analysis of western blots normalised to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and referred to age-matched controls (n=6 per group). Values represent mean±SEM (*p<0.01 vs control).
Animals with advanced cirrhosis (12 weeks of CCl4) showed a clear and significant increase in hepatic eNOS, thrombomodulin and CNP mRNA expression, whereas no differences in angiopoietin 2 and VEGF gene expressions were observed (figure 5). Thrombomodulin protein expression was increased, there were no differences in the protein expression of eNOS, VEGF and CNP and there was a significant reduction in p-eNOS in cirrhotic rat livers compared with controls (figure 6).
Effects of simvastatin on the expression of KLF2 and vasoprotective genes in cirrhotic rat livers
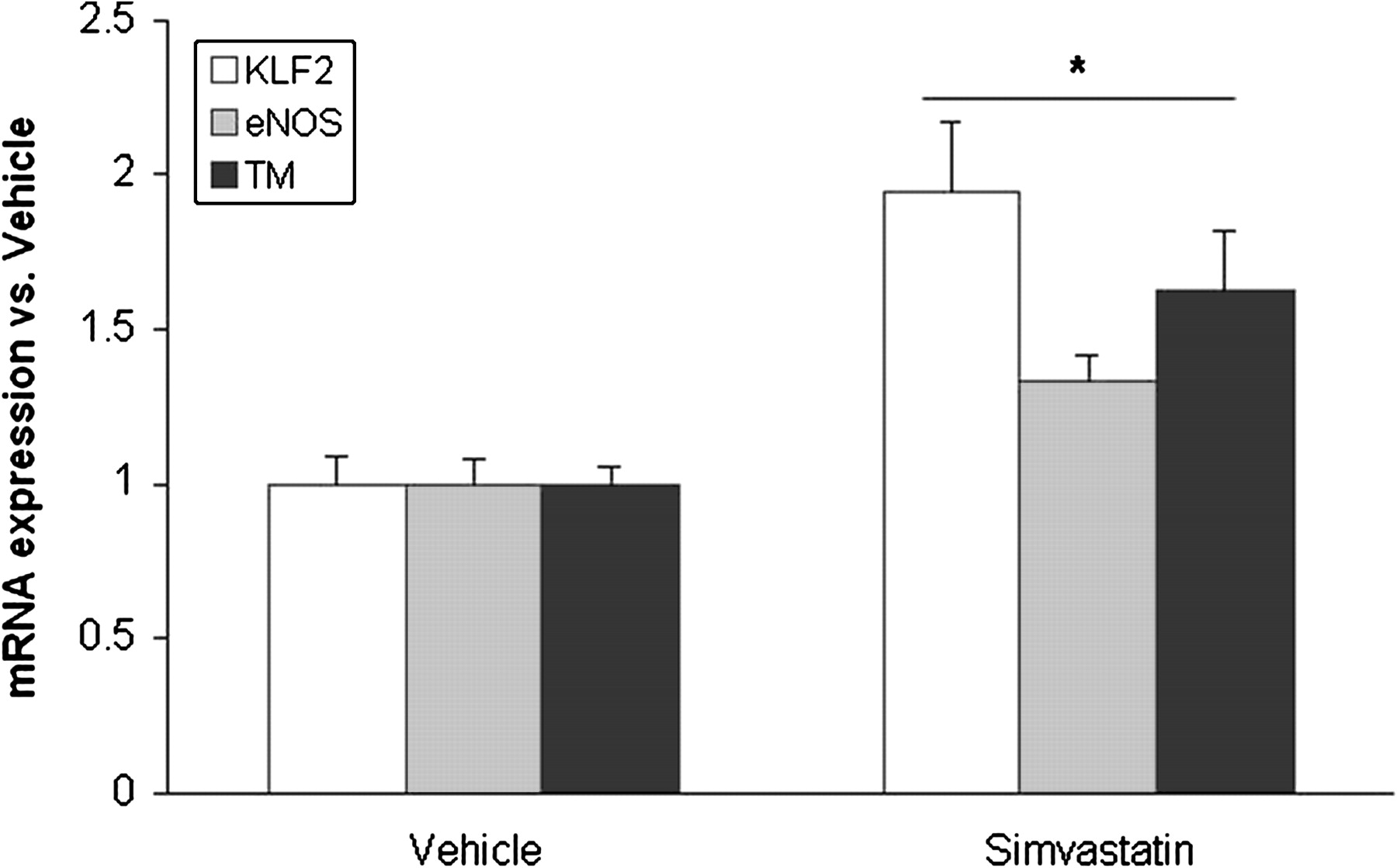
Cirrhotic animals treated with simvastatin showed a significant increase in hepatic KLF2 and its target gene expression compared with cirrhotic animals receiving vehicle (figure 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hepatic Kruppel-like factor 2 (KLF2), endothelial nitric oxide synthase (eNOS) and thrombomodulin (TM) mRNA expression in cirrhotic rats (12 weeks of carbon tetrachloride administration) treated for 3 days with simvastatin or its vehicle (n=6 per group). Values for gene amplification from cDNA have been normalised to an endogenous reference gene (RNA 18S). Values (mean±SEM) are normalised to vehicle-receiving animals (*p<0.05 vs vehicle).
Discussion
The vascular endothelium is an essential organ implicated in the regulation of vasomotor tone, vascular homeostasis and inflammatory processes.19 In a healthy liver, sinusoidal endothelium plays an important role regulating hepatic vascular tone by releasing vasoactive substances that diffuse to the hepatic stellate cells inducing their constriction or relaxation, thereby changing sinusoidal diameter and modulating intrahepatic haemodynamics.20 21 However, cirrhotic liver endothelium loses its vasodilatory, antithrombotic, and anti-inflammatory properties to acquire a vasoconstrictor, prothrombotic and inflammatory phenotype in the context of hepatic sinusoidal dysfunction.13 22–24 In fact, it is well known that cirrhotic hepatic endothelium produces large amounts of vasoconstrictor substances (eg, thromboxane A2)12 and reduced quantities of vasodilators (principally NO)13 22 contributing to increase IHR and consequently aggravating portal hypertension syndrome. However, the molecular mechanisms responsible for endothelium phenotype modulation during cirrhosis development remain poorly understood.23 25
In other vascular pathologies, different noxious stimuli such as turbulent flow can render the endothelium dysfunctional. However, healthy laminar flow modulates the phenotype of endothelial cells, conferring strong antithrombotic, anti-inflammatory and vasodilator properties to the endothelium.19 In addition, it has been shown that this vasoprotective phenotype is modulated by the expression of the transcription factor KLF2.5 In the vasculature KLF2 is endothelial specific and its expression, which is modulated by different flow patterns, confers endothelial protection against inflammation, thrombosis and vasoconstriction.9 26 In fact, in atherosclerosis patients it has been demonstrated that vascular regions exposed to laminar shear stress highly express KLF2 and are thus resistant to atherosclerosis; whereas KLF2 expression is comparatively absent in atheroprone regions exposed to non-laminar shear stress.9
Our study demonstrates, for the first time, that the transcription factor KLF2 is highly expressed in the cirrhotic liver and, moreover, its expression is induced early during the progression of the disease. In particular, KLF2 gene and protein expression is upregulated in those animals with early and advanced cirrhosis (6 and 12 weeks of cirrhosis induction, respectively) compared with control animals.
Previous studies have demonstrated that in the vasculature KLF2 is specifically expressed in the endothelium; however, little is known about its expression in a given organ. The present study demonstrates that KLF2 expression within the liver is localised in the endothelium, and that its expression responds to flow stimulation. In fact, shear stress-derived KLF2 upregulation is significantly higher in hepatic endothelial cells from cirrhotic livers, suggesting that the hepatic endothelial phenotype in cirrhosis is partly primed to respond to this vasoprotective stimulus.
The main in-vivo biomechanical stimulus able to induce KLF2 expression is blood-derived shear stress, and interestingly it has been reported that shear stress upregulates the KLF2 target eNOS in the liver endothelium;15 27 therefore, we characterised the systemic and hepatic haemodynamics of all animals included in the present study. Animals with advanced cirrhosis, highly expressing KLF2, presented a marked increase in the quantity of blood flow entering the liver through the portal vein, thus favouring the expression of those endothelial genes upregulated by shear stress, including KLF2. However, the findings in animals treated with CCl4 for 6 weeks, showing no significant increment in PBF, suggest that other mechanisms, perhaps related to the intrahepatic architectural disturbances (including cell stretch), to metabolic changes or circulating factors (ie, bacterial products, growth factors, etc.) or hypoxic processes occurring during cirrhosis progression, could modify endothelium phenotype and upregulate KLF2 expression.9 28–30 The precise nature of these mechanisms needs to be characterised further.
Among other functions, healthy vascular endothelium maintains blood fluidity by producing different factors that promote fibrinolysis or inhibit blood coagulation. One of these is thrombomodulin, a factor involved in the generation of activated protein C through interactions with thrombin. KLF2 potently induces thrombomodulin expression by binding to its promoter being necessary for endothelial coagulant gene expression and function regulation.6 In the present study we demonstrate that increased hepatic KLF2 expression is accompanied by elevated hepatic thrombomodulin gene and protein expression, suggesting that KLF2 is active and induces the antithrombotic pathway in the cirrhotic liver. This may be of relevance delaying or attenuating vascular occlusion and subsequent parenchymal extinction lesions, which lead to morphological progression in cirrhosis.31 32 Our findings suggest that this process might be accelerated if the increase in the expression of KLF2 was not present.
As described above, increased IHR in the cirrhotic liver is partly due to a reduced bioavailability of vasodilators, mainly NO. Such an insufficient sinusoidal NO availability has been related to a reduced production by eNOS,13 22 together with an increase in its scavenging by elevated levels of superoxide radicals.14 Previous studies have shown that one of the more potent inducers of genes implicated in regulating vascular tone is KLF2. In fact, the expression of KLF2 stimulates eNOS expression and activity and also the expression of CNP.5 Our group and others have demonstrated that rats with advanced cirrhosis present reduced hepatic eNOS activity with no differences in total hepatic eNOS protein expression compared with controls.13 14 22 33 However, the present study adds new and potentially decisive data about eNOS expression regulation. This is demonstrated by our finding that, following KLF2 expression, eNOS mRNA is significantly induced both in rats with early and advanced cirrhosis compared with their matched controls, suggesting that in cirrhotic livers eNOS mRNA transcription could be deregulated. Similarly, hepatic mRNA expression of CNP is significantly induced during cirrhosis development, but no differences in its protein expression are observed. In addition, we observed that already at the stage of early cirrhosis, rats present a marked decrease in hepatic eNOS phosphorylation, indicating a reduction in its enzymatic activity. Interestingly, both data coincide with the haemodynamic observation that rats with early cirrhosis exhibit a significant increase in portal pressure, which could result from hepatic architectural disturbances present in these animals together with a reduced hepatic NO production by eNOS.
Statins, well-known inducers of endothelial KLF2 expression,34 35 represent one of the most promising drugs to ameliorate portal hypertension. In fact, it has been shown that statin administration to cirrhotic animals, partly by increasing NO production, reduces IHR and improves portal hypertension.16 36 Moreover, recent studies have validated the beneficial effects of statins on human portal hypertension;37 38 however, the underlying mechanisms involved are unknown. Herein we demonstrate that cirrhotic animals treated with simvastatin exhibit increased hepatic levels of KLF2 and its vasoprotective target genes, eNOS and thrombomodulin, compared with cirrhotic animals receiving vehicle. Our data suggest that statins' beneficial effects on hepatic haemodynamics in cirrhosis would be partly KLF2 mediated.
In summary, the present study demonstrates that the transcription factor KLF2 is induced early in the cirrhotic liver endothelium, inducing the expression of its vasoprotective target genes. These data suggest that the marked upregulation in KLF2 expression may represent an endothelial compensatory mechanism to improve the vascular disorders ongoing in the cirrhotic liver. Moreover, our data open the rationale to investigate whether this transcription factor is involved in the pathogenesis and resolution of liver diseases occurring with endothelial dysfunction.
Acknowledgments
The authors are grateful to Montse Monclús for excellent technical assistance.
References
Supplementary materials
Online only appendix
Footnotes
Funding JGS was supported by the Spanish Association for the Study of the Liver (AEEH) and the Catalan Digestology Society (SCD). The study was supported by grants from Instituto de Salud Carlos III (PI06-0623 and PI09-01261), Ministerio de Educación y Ciencia (SAF 07/61298) and NIH (HL-076686 and HL-090856). CIBERehd is funded by the Instituto de Salud Carlos III.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.