Article Text

Abstract
Objectives Emerging evidence from mouse models suggests that mutant Kras can drive the development of pancreatic ductal adenocarcinoma (PDA) precursors from acinar cells by enforcing ductal de-differentiation at the expense of acinar identity. Recently, human genome-wide association studies have identified NR5A2, a key regulator of acinar function, as a susceptibility locus for human PDA. We investigated the role of Nr5a2 in exocrine maintenance, regeneration and Kras driven neoplasia.
Design To investigate the function of Nr5a2 in the pancreas, we generated mice with conditional pancreatic Nr5a2 deletion (PdxCrelate; Nr5a2c/c). Using this model, we evaluated acinar differentiation, regeneration after caerulein pancreatitis and Kras driven pancreatic neoplasia in the setting of Nr5a2 deletion.
Results We show that Nr5a2 is not required for the development of the pancreatic acinar lineage but is important for maintenance of acinar identity. Nr5a2 deletion leads to destabilisation of the mature acinar differentiation state, acinar to ductal metaplasia and loss of regenerative capacity following acute caerulein pancreatitis. Loss of Nr5a2 also dramatically accelerates the development of oncogenic Kras driven acinar to ductal metaplasia and PDA precursor lesions.
Conclusions Nr5a2 is a key regulator of acinar plasticity. It is required for maintenance of acinar identity and re-establishing acinar fate during regeneration. Nr5a2 also constrains pancreatic neoplasia driven by oncogenic Kras, providing functional evidence supporting a potential role as a susceptibility gene for human PDA.
- PANCREATIC CANCER
- PANCREATITIS
- PANCREATIC DAMAGE
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Pancreatic ductal adenocarcinoma (PDA) precursor lesions (pancreatic intraepithelial neoplasia) can originate from pancreatic acinar cells that have undergone de-differentiation into duct-like cells.
-
Genome-wide association studies have identified the NR5A2 gene as a significant susceptibility locus for human pancreatic cancer.
-
Nr5a2 encodes a nuclear receptor and is a critical mediator of adult acinar function.
What are the new findings?
-
Nr5a2 is critical for maintenance of acinar cell differentiation and its loss leads to acinar cell duct-like de-differentiation.
-
Nr5a2 is required for pancreatic regeneration and re-establishing acinar cell integrity following caerulein pancreatitis.
-
Loss of acinar Nr5a2 cooperates with oncogenic Kras in preneoplastic transformation of the pancreas.
How might it impact on clinical practice in the foreseeable future?
-
Our findings provide functional support for NR5A2 as a PDA susceptibility locus and the notion that impaired acinar cell differentiation favours the development of preneoplastic lesions in the context of oncogenic signals.
-
In humans, inherited or acquired defects in NR5A2 may promote PDA initiation.
Introduction
Pancreatic ductal adenocarcinoma (PDA) and its precursor lesions pancreatic intraepithelial neoplasia (PanIN) morphologically resemble duct cells and express markers of ductal differentiation. However, evidence from animal models suggests that mutant Kras, one of the most frequently mutated genes in PDA, can direct the specification of PanINs from non-ductal compartments, including acinar cells.1 ,2 In these models, PanIN development is preceded by acinar to ductal metaplasia (ADM), a process in which acinar cells lose elements of terminal acinar cell differentiation and inappropriately activate ductal markers and pathways critical for embryonic pancreas development.3 ,4 Therefore, maintenance of acinar differentiation might be an important feature protecting the pancreas against neoplastic transformation. In line with this hypothesis, mouse models in which acinar differentiation is compromised by damage (eg, chronic or acute pancreatitis)3 ,5 or by loss of genes central to terminal acinar differentiation6 ,7 display dramatically accelerated development of Kras driven, acinar derived PanINs.
Evidence from mouse models comparing Cre-mediated recombination of oncogenic Kras in distinct, adult pancreatic compartments suggests that acinar cells, but not ductal or centroacinar cells, are the predominant cell of origin for Kras driven PanIN formation.8 Although it still remains to be elucidated if the acinar cell is also the cell of origin for human PanINs and PDA, recent genome-wide association studies (GWAS) suggest that defects in maintenance of acinar integrity may be involved in PDA initiation. These studies identified several PDA susceptibility loci in genes involved in embryonic pancreas development, and notably, maintenance of acinar cell identity.9 ,10 Included in this set of developmentally relevant genes was NR5A2, a nuclear receptor that cooperates with the pancreas transcription factor 1-L complex (PTF1-L) to maintain the secretory functions of acinar cells.11 The most significant single-nucleotide polymorphisms (SNPs) that were identified in this gene are non-coding and located either in the first two introns or in a region up to 90 kb upstream of NR5A2.9 ,10 It is presently unclear if susceptibility-associated Nr5a2 SNPs effect its function or expression levels, although an additional study has correlated one polymorphism with decreased survival.12
Since GWAS do not provide functional information underlying the role of susceptibility genes in carcinogenesis, we used a loss of function model to investigate the mechanistic involvement of Nr5a2 in pancreatic acinar cell plasticity and neoplastic transformation. Specifically, we investigated the effects of loss of Nr5a2 on acinar cell integrity, pancreatic regeneration and PanIN development in the context of oncogenic Kras. Conditional pancreas specific deletion of Nr5a2 in the mouse did not prevent the development of the endocrine, ductal or acinar compartments, but resulted in compromised terminal acinar differentiation. Furthermore, Nr5a2 was critical for acinar regeneration in the context of acute pancreatitis, and constrained the development of Kras driven ADM and PanIN development. These results highlight the importance of Nr5a2 for maintaining acinar identity in mature pancreas and re-establishing acinar integrity after injury, thereby providing critical functions during pancreatic regeneration and protection against neoplastic initiation.
Methods
Mouse lines
The following mice were mated to generate experimental mice: PdxCrelate (gift of Pedro Herrera, University of Geneva Medical School, Geneva, Switzerland), Ptf1aCre,13 Ptf1aCreER14 or ElastaseCreERT2 mice (gift of Doris Stoffers, University of Pennsylvania, Philadelphia, Pennsylvania, USA) with KrasG12D (gift of Dave Tuveson, Cancer Research, UK Cambridge Research Institute, Cambridge, UK), R26REYFP15 and Nr5a2flox (gift of Steven A Kliewer, University of Texas Southwestern Medical Center, Dallas, USA16) mice. Mice were maintained on a mixed background. Cre− mice and Cre+ mice without any additional genetic modification served as controls. The UCSF Institutional Animal Care and Use Committee (IACUC) approved all mouse experiments.
Immunofluorescence and immunohistochemical staining
After overnight fixation in Z-FIX (Anatech), pancreas tissue was embedded in paraffin. Antigen retrieval was performed on 5 µm sections using citra solution (biogenex). Primary antibodies were incubated overnight at 4° and secondary antibodies were incubated for 1 h at room temperature. The following primary antibodies were used: goat anti-CPA1 (R&D), rabbit anti-CK19 (Epitomics), mouse anti-insulin (Sigma), goat anti-Clusterin (Santa Cruz), rabbit anti-Nr5a2 (Sigma), chicken anti-green fluorescent protein (abcam), rat anti-CD45 (Biolegend) and rabbit anti-Sox9 (Millipore). Nuclear staining was performed using 4′,6-diamidino-2-phenylindole (DAPI) containing mounting medium (Vector labs). For immunohistochemistry (IHC), biotin-conjugated secondary antibodies were used. Further development was performed with the ABC kit (Vector labs) and DAB kit (Vector labs). Counterstaining was performed with haematoxylin. For Nr5a2 IHC stainings, Envision system (DAKO) was used for secondary antibody and development. H&E and Alcian blue stainings were performed according to standard protocols.
Caerulein treatment
For the study on Nr5a2 expression on Ptf1aCre; KrasG12D and control mice, pancreatitis was induced using the previously described ‘staggered protocol’ that consists of 6 hourly caerulein (Sigma) injections (50 μg/kg) on day -2 and day 0 in 6-week-old mice.3 ,17 To induce pancreatitis in the regeneration study of PdxCrelate; Nr5a2c/c and control mice, 6-week-old mice were injected with 8 hourly caerulein (American Peptide) injections (2 μg/injection) on two consecutive days.18 The final day of caerulein injection was considered day 0.
Tamoxifen treatment
For Ptf1aCreER mice, tamoxifen citrate (TEVA Pharmaceuticals, USA) was administered to 5–6-week-old mice by oral gavage. In total, each mouse received 30 mg tamoxifen citrate given by three individual gavages (each 10 mg) on alternating days. This regimen resulted in recombination of the vast majority of the acinar cell population. For ElastaseCreERT2 mice, tamoxifen (Sigma) was administered as described previously.3
RNA isolation and quantitative RT-PCR
RNA was isolated using the RNA-easy kit (Qiagen) and subsequent cDNA synthesis was performed using Superscript III First-Strand Synthesis Kit (Invitrogen). Quantitative real-time PCR (RT-PCR) was performed using Taqman Gene Expression Assays (Applied Biosystems) and expression levels were normalised to mouse Cyclophilin A (Genome Analysis Core, UCSF Helen Diller Family Comprehensive Cancer Center). All RT-PCR experiments were performed with n=3–6 individual biological samples per group.
Acinar cell culture
For acinar cell cultures, the protocols described previously by Means et al19 and Pinho et al20 were used with modifications. Briefly, a part of the mouse pancreas was cut into small pieces and further digested with collagenase P (Roche) solution (0.4 mg/ml). After two washing steps with Hank's Balanced Salt Solution (HBSS) containing 5% fetal bovine serum (FBS), tissue suspension was filtered through a 100 µm cell strainer. The flow through was pipetted on a HBSS+30% FBS solution and centrifuged. The pellet was resuspended in a 1 : 2 media:matrigel Becton, Dickinson (BD) solution and plated on a rat-tail collagen Type I (BD)-coated plate. The media for culturing was Roswell Park Memorial Institute media containing 10% FBS, 1× penicillin/streptomycin, 100 µg/ml soybean trypsin inhibitor (Sigma) and 1 µg/ml dexamethasone (Sigma).
Pancreatic duct cell culture
Pancreas of a 3-week-old PdxCrelate; KrasG12D; Nr5a2c/c mouse was dissected and minced followed by sequential digestion steps with collagenase D (Roche), trypsin (Invitrogen) and dispase (Invitrogen). The cell suspension was filtered through a 40 µm mesh and plated on a collagen-coated plate (BD). The duct cells were cultured in pancreatic duct cell culture media as described previously.21
PCR analysis for allele recombination
Nr5a2c recombination was analysed using the following primers: 5′-CATAAGGGCTCAGTGGCAC-3′ and 5′-CGCAGCATTCTTCGGCAG-3′. Allele specific PCR for KrasG12D was performed as previously described by T Jacks (http://web.mit.edu/jacks-lab/protocols/KrasCond_tablesTWO.html). The PCR analysis was performed on low passage (<5) PDCs.
Statistical analysis
p Values were determined using Student t test. All statistical analyses were performed with GraphPad Prism (V.4.0c and V.5.0d).
Results
Downregulation of Nr5a2 characterises acinar cells undergoing duct-like de-differentiation
To understand the role of Nr5a2 in acinar cell plasticity, we analysed its expression during acinar regeneration and Kras driven ADM in response to acute, caerulein-induced pancreatitis by IHC, immunofluorescence (IF) and RT-PCR (figure 1A and online supplementary figure S1A). In response to caerulein-induced damage, control acinar cells expressing wild-type Kras (KrasWT) transiently downregulate markers of terminal acinar differentiation, briefly assuming duct-like morphology with expression of duct markers and elements of embryonic pancreas development. This regeneration-associated de-differentiation is rapidly resolved, and acinar morphology, function and expression of markers of terminal differentiation are restored.18 ,22 ,23 Strong nuclear Nr5a2 expression could be detected in differentiated acinar cells of 6-week-old control mice (see online supplementary figure S1A; arrows). Similar to downregulation of other adult acinar markers,18 ,23 nuclear Nr5a2 expression was frequently decreased in transient duct-like structures in the exocrine compartment of control mice 2 days after caerulein treatment, corresponding with activation of the stress marker Clusterin3 (see online supplementary figure S1A; arrowheads). At day 7 after caerulein treatment, Nr5a2 nuclear localisation was re-established in regenerated acinar cells (see online supplementary figure S1A; arrows). RT-PCR for Nr5a2 over the pancreatitis time course of control mice mirrored the pattern revealed by IHC and IF. Similar to the expression results described by Molero et al,23 lower Nr5a2 levels were found at day 2 after pancreatitis induction, with expression increasing towards levels found in phosphate buffered saline (PBS) treated pancreata at day 7 (see online supplementary figure S1B). Therefore, Nr5a2 expression during acinar regeneration correlates with the recovery of terminal acinar differentiation.
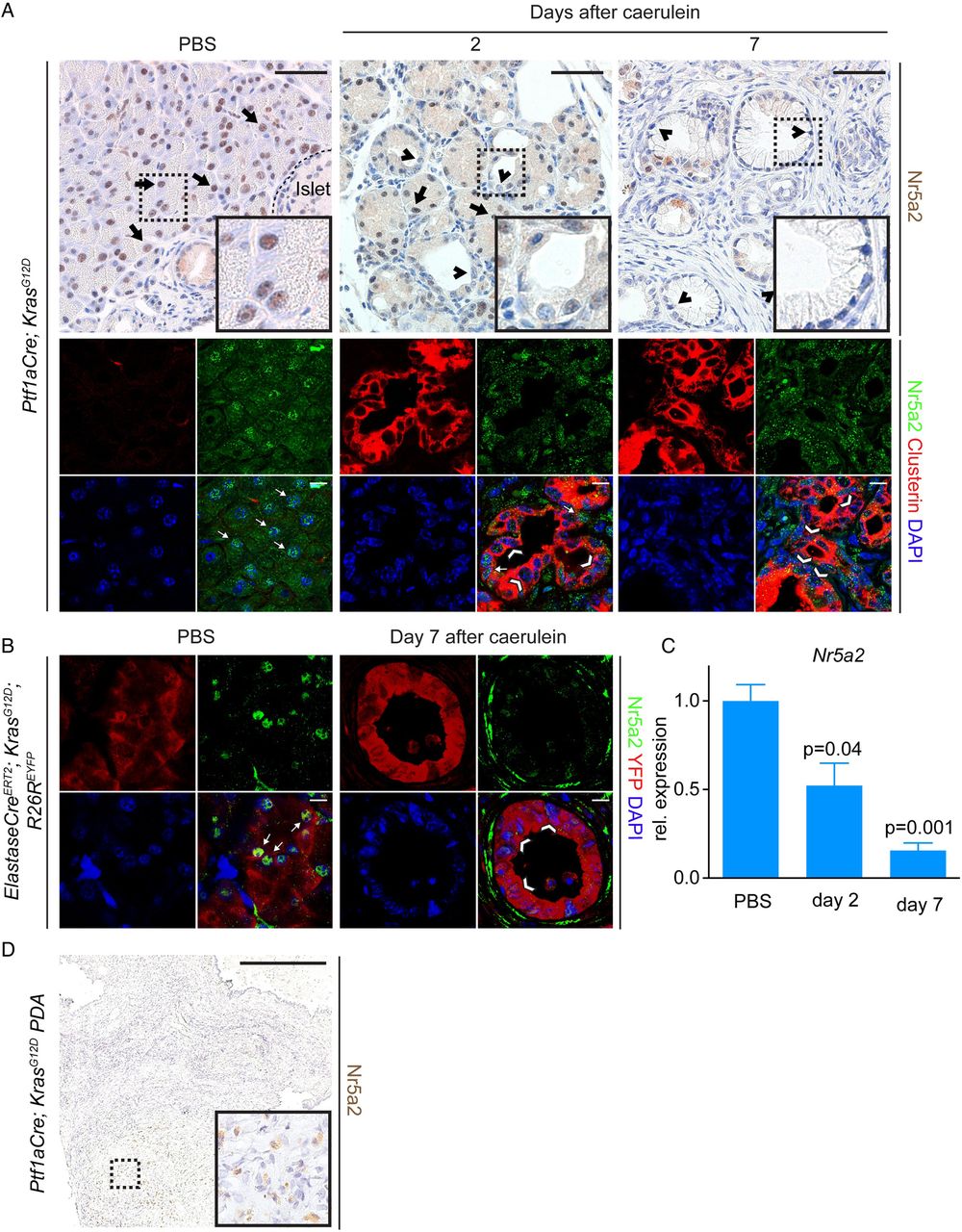
Nr5a2 is downregulated in acini expressing oncogenic Kras that undergo acinar to ductal metaplasia (ADM)/pancreatic intraepithelial neoplasia (PanIN) lesion formation and in pancreatic ductal adenocarcinoma (PDA). (A) Nr5a2 staining on pancreata at different time points (day 2 and 7) after pancreatitis induction or PBS injection of Ptf1aCre; KrasG12D mice; arrows mark Nr5a2 positive cells, arrowheads mark Nr5a2 negative cells. Upper panel: Immunohistochemistry staining for Nr5a2. Lower panel: Immunofluorescent co-staining of acinar tissue or ADM/PanIN lesions for CPA1, Clusterin and Nr5a2. Clusterin positive persistent ADM and early PanIN lesions exhibit strong reduction of nuclear Nr5a2. Note that the specific Nr5a2 staining is nuclear, whereas cytoplasmic staining is unspecific background. (B) Nr5a2 staining on pancreata 7 days after caerulein induction or PBS injection of ElastaseCreERT2; KrasG12D; R26REYFP mice. Depicted is an YFP positive PanIN lesion lacking nuclear Nr5a2, which occurred 7 days after caerulein induction; arrows mark Nr5a2 positive cells, arrowheads mark Nr5a2 negative cells. (C) RNA expression of Nr5a2 during indicated time points after pancreatitis induction in Ptf1aCre; KrasG12D mice. p Values are relative to PBS treated animals; values are shown as mean±SEM. (D) Nr5a2 staining of a PDA derived from a Ptf1aCre; KrasG12D mouse. (A) Upper panel: scale bar 50 µm. Lower panel: scale bar 10 µm. (B) Scale bar 10 µm. (D) Scale Bar 500 µm.
In contrast to KrasWT acinar cells, regeneration of acinar cells expressing mutant Kras (KrasG12D) is compromised, resulting in persistent ductal de-differentiation and the development of ADM and PanIN lesions following caerulein pancreatitis.3 ,5 ,24 ,25 To determine the dynamics of Nr5a2 expression in Kras driven ADM, we analysed its expression following caerulein pancreatitis in Ptf1aCre; KrasG12D mice. Ptf1aCre drives Cre expression during pancreatic development, resulting in recombination in nearly all acini and a subset of duct and endocrine cells.26 As previously shown,3 the exocrine pancreas of 6 weeks old Ptf1aCre; KrasG12D mice displays predominantly morphologically normal acinar cells with some areas of ADM and PanIN. Similar to control mice, morphologically normal acinar cells expressing oncogenic Kras also displayed nuclear Nr5a2 expression (figure 1A; arrows). Mirroring damage to KrasWT exocrine cells following caerulein, duct-like structures 2 days after treatment displayed decreased nuclear Nr5a2 expression (figure 1A, arrowheads). In contrast to regenerating KrasWT acinar cells, nuclear Nr5a2 remained absent in the persistent ductal metaplasia replacing the normal exocrine parenchyma at day 7 in Ptf1aCre; KrasG12D mice (figure 1A; arrowheads). RT-PCR analysis also revealed progressive loss of Nr5a2 expression during Kras driven ADM (figure 1C). Cell lineage tracing experiments were performed in ElastaseCreERT2; KrasG12D; R26REYFP to confirm that Nr5a2 levels decrease in adult acinar cells undergoing persistent Kras driven ductal reprogramming.3 Transgenic animals were treated with tamoxifen to induce Cre-mediated activation of Kras and expression of YFP. Nuclear Nr5a2 expression was observed in YFP-labelled acini in PBS control adult mice (figure 1B, arrows). In contrast, nuclear Nr5a2 expression was diminished in YFP positive ductal structures found 7 days after caerulein treatment (figure 1B; arrowheads).
To determine Nr5a2 expression in murine pancreatic cancer cells, PDA tissues derived from Ptf1aCre; KrasG12D mice were stained. All analysed individual cancer samples (n=5) showed weak or absent nuclear Nr5a2 (figure 1D). Together, these results indicate that changes in Nr5a2 expression correlate with acinar cell plasticity: transient reduction of nuclear Nr5a2 occurs during transient dedifferentiation of WT acini, whereas persistent downregulation is observed in acini that have undergone permanent, Kras driven ductal reprogramming.
Nr5a2 is not required for acinar lineage formation but maintains acinar differentiation
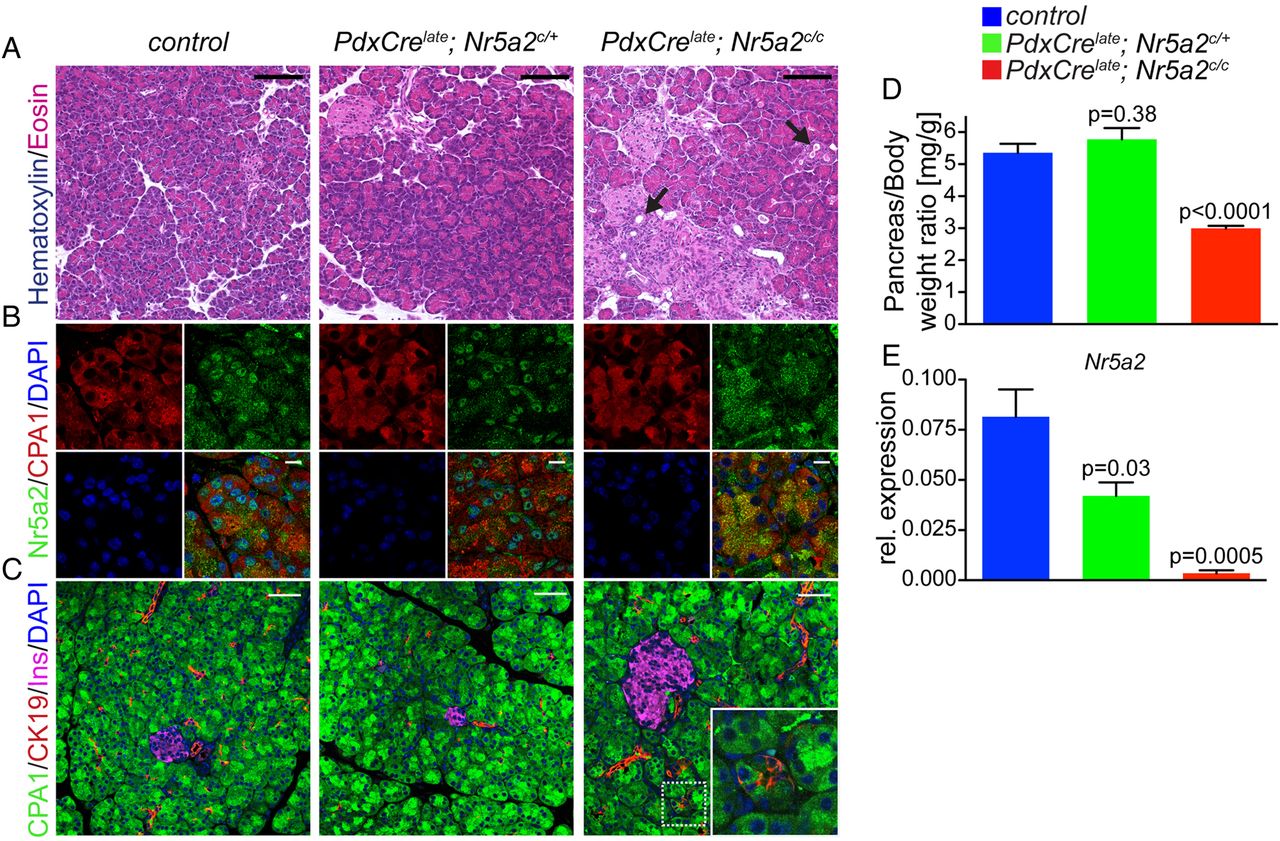
To further explore the functional role of Nr5a2 for acinar cell identity, we employed a genetic mouse model of Nr5a2 loss of function. We first aimed to investigate the requirements of Nr5a2 for acinar lineage development and maintenance of exocrine integrity. Therefore, we conditionally deleted Nr5a2 in mice expressing the Cre recombinase under the control of promoter elements of Pdx1, a transcription factor required for pancreas development (PdxCrelate; Nr5a2c/c).16 ,26 Our previous work has shown that the PdxCrelate line used in these experiments stimulates recombination in the embryonic pancreas at approximately E12.5. Cre activity targets the majority of acinar cells, a subset of endocrine cells and few duct cells.26 To ensure this strategy resulted in efficient recombination of Nr5a2 we performed IF in postnatal (p3) pancreas. In control mice, nuclear Nr5a2 staining was observed in acinar cells at p3 (see online supplementary figure S2A). In contrast, Nr5a2 was widely eliminated from acinar cells at p3 in PdxCrelate; Nr5a2c/c mice (see online supplementary figure S2A). All three pancreatic compartments appeared morphologically normal in PdxCrelate; Nr5a2c/c mice, and markers of each cell type (duct (CK19), endocrine (insulin) and acini (Cpa1) were distributed similarly to control mice (see online supplementary figure S2B,C). Therefore, PdxCrelate-mediated Nr5a2 deletion does not alter pancreatic lineage specification or development of acinar cells.
To investigate whether Nr5a2 plays a role in maintenance of the acinar compartment we analysed the effect of Nr5a2 deletion at 3 weeks of age. At this stage, PdxCrelate; Nr5a2c/c mice presented with significant reduction in pancreas size (figure 2D), a phenotype not observed in Nr5a2 heterozygous mutant mice. Nr5a2 deletion at 3 weeks was confirmed by RT-PCR and IF staining, displaying clear loss of nuclear Nr5a2 in acini (figure 2B,E). Morphologically, the exocrine and endocrine pancreatic lineages were grossly comparable among control, Nr5a2 heterozygous and homozygous deficient mice (figure 2A). However, biphenotypic cells staining positive for both acinar and duct markers were occasionally observed in PdxCrelate; Nr5a2c/c mice (figure 2C, inset), along with the appearance of metaplastic ductal lesions (figure 2A, arrow). Notably, neither cells displaying combined acinar and ductal features nor ADM was observed in PdxCrelate; Nr5a2c/+ mice. In addition, RT-PCR revealed significantly decreased expression of the acinar specific genes Hnf1a, Ptf1a, Mist1 and CPA1, as well as upregulation of the duct marker CK19 (figure 3A) in PdxCrelate; Nr5a2c/c mice, whereas expression of Gata6, a transcription factor important for maintenance of acinar differentiation,27 was not significantly different between the groups. Therefore, Nr5a2 appears to be required for maintaining terminal acinar differentiation and preventing de-differentiation towards a duct-like state.

Nr5a2 is essential for proper maintenance of acinar cell differentiation. (A) H&E staining and (B) immunofluorescence staining for Nr5a2 and CPA1 (marks acinar cells). (C) Immunofluorescence staining for acinar (CPA1), ductal (CK19) and β cells insulin (INS) of pancreata of the indicated genotypes at 3 weeks of age (arrow marks ductal metaplasia). (D) Pancreas weight to body weight ratio; p values are compared with control, values are shown as mean±SEM. (E) Real-time PCR (RT-PCR) for Nr5a2 relative to Cyclophilin A; p values represent comparison with control mice, values are shown as mean±SEM. All RT-PCR analyses in this figure were performed on 3-week-old animals. (A) Scale bar 100 µm, (B) scale bar 10 µm and (C) scale bar 50 µm.

Loss of Nr5a2 compromises acinar differentiation. (A) Real-time PCR for transcriptions factors and markers of acinar differentiation (Gata6, Hnf1a, Ptf1a, Mist1 and CPA1) and the duct gene Ck19; p values represent comparison with control mice; values are shown as mean±SEM. (B) Acinar culture of isolated acinar clusters; representative picture of acinar culture at day 2 and CPA1/CK19 co-staining; scale bar 25 µm. (C) Quantification of acinar clusters; p values are compared with acini from control mice for each time point, values are shown as mean±SEM. All analyses in this figure were performed on 3-week-old animals.
To directly test the propensity of Nr5a2-null acini to de-differentiate and acquire ductal characteristics, isolated acinar clusters were grown in matrigel in vitro. Under these conditions, control acini underwent ductal reprogramming within 3 days (figure 3B). In support of the notion that Nr5a2 mutant acinar cells are compromised in their differentiation state, acinar cells from PdxCrelate; Nr5a2c/c mice displayed accelerated acinar to ductal transdifferentiation, with nearly 100% of acinar clusters taking on a duct-like state within 2 days with frequent occurrence of complete CK19+/CPA1− negative ductal structures (figure 3B,C). Collectively, these results suggest a critical role for Nr5a2 in maintaining acinar differentiation and preventing duct-like de-differentiation.
Nr5a2 is required for efficient acinar regeneration after caerulein pancreatitis
Nr5a2 expression fluctuates with loss and restoration of acinar differentiation during caerulein pancreatitis (see online supplementary figure S1).23 Therefore, we asked whether Nr5a2 function was required for acinar regeneration following caerulein treatment. While caerulein treatment leads to the formation of transient, biphenotypic acinar/ductal metaplasia in both control and PdxCrelate; Nr5a2c/c mice (figure 4B), acinar re-differentiation was blocked in the absence of Nr5a2. Instead, PdxCrelate; Nr5a2c/c pancreata displayed persistent ductal metaplasia in the setting of pancreatic fibrosis combined with severe atrophy (figure 4A–C and see online supplementary figure S3). Thus, Nr5a2 function is important for maintenance of the acinar differentiation state and critical for acinar re-differentiation upon pancreatitis insult.

Loss of Nr5a2 blocks pancreas regeneration. (A) Representative H&E staining at the indicated time points after caerulein-induced acute pancreatitis of control and PdxCrelate; Nr5a2c/c mice. (B) Immunofluorescent co-staining for CPA1 and CK19 and (C) pancreas weight to body weight ratio (n=3 per group) during the time course of pancreatitis of the indicated genotypes; values are shown as mean±SEM. p Values are calculated comparing control and PdxCrelate; Nr5a2c/c mice of each time point. (A) Scale bar 100 µm and (B) scale bar 50 µm.
Loss of Nr5a2 cooperates with oncogenic Kras to drive ADM and PanIN development
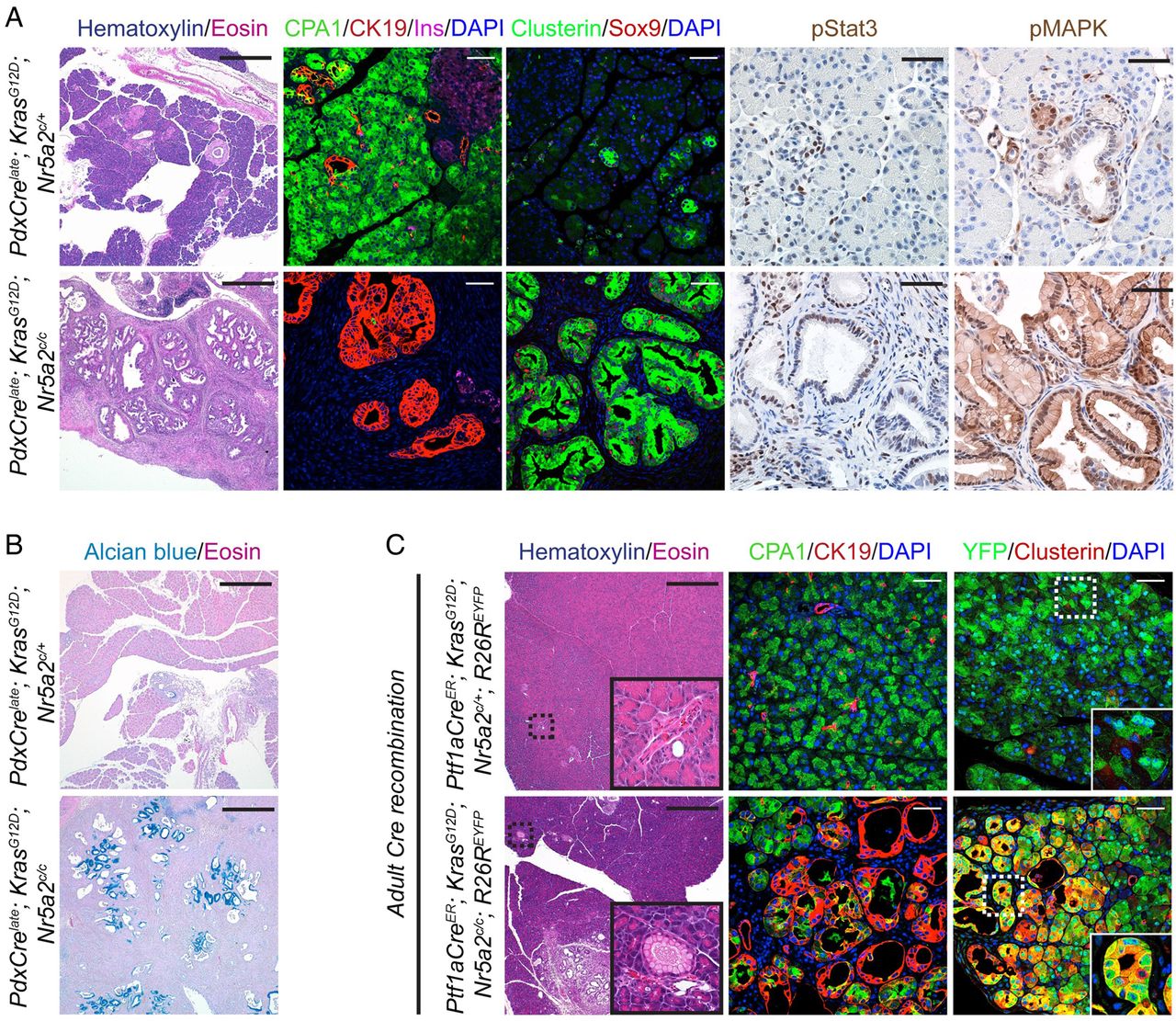
Compromised acinar differentiation has been shown in other genetic mouse models to cooperate with oncogenic Kras to drive ADM and PanIN development.7 To determine whether Nr5a2 dependent maintenance of acinar identity affects Kras driven ADM and PanIN formation we used the above described transgenic mice with combined pancreas specific loss of Nr5a2 and expression of oncogenic Kras (PdxCrelate; KrasG12D; Nr5a2c/c). Analysis of these compound transgenic animals revealed near complete replacement of the pancreatic parenchyma by metaplastic duct structures and mucinous PanIN lesions (Alcian blue positive) at 3 weeks of age when compared with age-matched PdxCrelate; KrasG12D; Nr5a2c/+ heterozygous mice (figure 5A,B). Preneoplastic lesions in mice lacking Nr5a2 in the context of oncogenic Kras possessed markers characteristic of Kras driven PanINs, including phospho-mitogen-activated protein kinase (MAPK) signalling,25 Sox9,8 pStat328 and the stress-associated protein Clusterin3 (figure 5A). This unconstrained Kras driven neoplastic transformation of the pancreas was accompanied by complete loss of the acinar compartment, resulting in severe exocrine insufficiency, pancreatic hyperplasia with marked fibrosis and death of the mice soon after weaning (see online supplementary figure S4, and data not shown). To ensure that the metaplastic duct structures (ADM and PanIN lesions) of the PdxCrelate; KrasG12D; Nr5a2c/c mice had recombined both Nr5a2 conditional alleles and were therefore Nr5a2 null, we established pancreatic duct cell cultures of 3-week-old whole pancreas. PCR analysis performed on these cells confirmed recombination within the metaplastic duct structures for the KrasG12D and both Nr5a2c alleles (see online supplementary figure S5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Loss of Nr5a2 accelerates oncogenic Kras driven neoplastic initiation. (A) H&E staining and immunofluorescence co-staining for CPA1/CK19/insulin/DAPI and for Clusterin/Sox9/DAPI, and immunohistochemical staining for pMAPK and pStat3 in 3-week-old mice of the indicated genotypes. (B) Alcian blue staining in 3-week-old mice of the indicated genotypes. (C) H&E staining and immunofluorescence co-staining for CPA1/CK19/DAPI and YFP/Clusterin/DAPI of the indicated genotypes 2 weeks after tamoxifen induction. (A) H&E staining: scale bar 500 µm; immunofluorescent/immunohistochemical staining: scale bar 50 µM, (B) scale bar 500 µm and (C) scale bar for H&E 500 µm and for immunofluorescence stainings 50 µm.
The dramatic acceleration of preneoplastic transformation in PdxCrelate; KrasG12D; Nr5a2c/c mice resulted in rapidly decaying body condition, likely due to severe exocrine insufficiency, which precluded the examination of later stages of disease. Therefore, we investigated if Nr5a2 heterozygosity can lead to faster PanIN development and progression. We compared PdxCrelate; KrasG12D; Nr5a2c/+ with PdxCrelate; KrasG12D mice at 12 weeks of age. At this time point, we detected a dramatic increase in ADM, PanIN incidence and PanIN grade in the Nr5a2 heterozygous compared with the Nr5a2 wild-type mice (see online supplementary figure S6).
To confirm that the acinar cells are the source for the observed ADM and PanIN lesions in PdxCre; KrasG12D; Nr5a2c/c mice, we made use of the Ptf1aCreER allele that allows tamoxifen inducible Cre recombination restricted to adult acinar cells.8 ,14 With this strategy we compared the occurrence of preneoplastic lesions in Ptf1aCreER; KrasG12D; Nr5a2c/+; R26REYFP and Ptf1aCreER; KrasG12D; Nr5a2c/c; R26REYFP mice. Two weeks after tamoxifen induction, almost no ADM and PanIN lesions could be observed in Ptf1aCreER; KrasG12D; Nr5a2c/+; R26REYFP mice. In contrast, we observed multiple ADM and PanIN lesions at this early time point, when oncogenic Kras was activated in the absence of Nr5a2 (figure 5C). Similar to the preneoplastic lesions observed in PdxCrelate; KrasG12D; Nr5a2c/c mice, these lesions were CK19 and Clusterin positive. Furthermore, lineage tracing of the R26REYFP allele showed overlap of the Clusterin positive preneoplastic lesions with YFP, confirming an acinar origin (figure 5C). Together, these results indicate that Nr5a2 inhibits Kras driven pancreatic neoplasia and further support a role for acinar differentiation as a critical determinant of neoplastic initiation in the pancreas.
We also used the Ptf1aCreER model to investigate potential non-cell autonomous effects of Nr5a2 deletion that could potentiate preneoplastic transformation. Inflammation is a key factor promoting Kras driven ADM/PanIN formation,29 and therefore we examined immune cell infiltration associated with developing exocrine lesions 2 weeks after tamoxifen. We observed a marked CD45 positive inflammatory infiltrate around newly formed ADM and PanIN lesions in Ptf1aCreER; KrasG12D; Nr5a2c/c; R26REYFP mice 2 weeks after induction (see online supplementary figure S7). In contrast, the areas around the few sporadic ADM lesions that occurred in Ptf1aCreER; KrasG12D; Nr5a2c/+; R26REYFP mice displayed a less pronounced inflammatory reaction (see online supplementary figure S7). These results indicate that Nr5a2 deletion could also support Kras driven transformation by promoting pro-neoplastic inflammation.
Discussion
The identification of NR5A2 in human GWAS as a susceptibility locus for PDA has elevated the gene as a candidate of interest in the formation and progression of this cancer.9 ,10 While GWAS can be useful in identifying new genes important for PDA development the technique does not provide evidence regarding functional relevance of the factors with regard to pancreas carcinogenesis.
Our initial results showed that Nr5a2 is downregulated in acinar cells undergoing duct-like de-differentiation, a prerequisite for acinar derived PDA initiation.8 This finding prompted us to investigate whether Nr5a2 plays a functional role in regulating acinar plasticity. We found that Nr5a2 is critical for maintaining acinar differentiation. As a consequence of Nr5a2 loss, we observed downregulation of elements of terminal acinar differentiation and increased capacity to undergo ADM in vitro. Of note, these defects are similar to other models in which Mist1, another gene important for the maintenance of acinar identity, is compromised.7 ,30 Nr5a2 has been shown to cooperate with the Ptf1-L complex11 to regulate expression of acinar specific genes.31 ,32 However, the precise function of Nr5a2 in maintaining acinar differentiation is unclear and future studies are needed to explore the hierarchical role of Nr5a2 in orchestrating acinar differentiation.
Nr5a2 was also required for the regeneration of de-differentiated acinar cells after caerulein-induced pancreatitis. These functions correlate well with the increased expression of Nr5a2 in acinar cells undergoing the re-differentiation phase of regeneration and support the notion that the nuclear receptor is important for re-establishing acinar fate after pancreatic damage. Although SNPs in NR5A2 have been associated with susceptibility to PDA,9 ,10 GWAS studies analysing whether such SNPs are associated with pancreatitis have not been performed. Since chronic pancreatitis is a significant pancreatic disease and a risk factor for PDA,33 ,34 it would be interesting to conduct future studies investigating whether SNPs in NR5A2 are linked to the absence of pancreatic regeneration observed in chronic pancreatitis.
The role of Nr5a2 in maintaining and restoring acinar differentiation may have implications for understanding Kras driven pancreatic transformation. Mouse models have shown that targeting mutant Kras to the pancreatic epithelium recapitulates the sequence of PanIN and PDA progression observed in human patients.35 Numerous studies have aimed to define the main cell type within the pancreas epithelium that serves as the progenitor cell for PDA.1 ,5 ,36 ,37 Interestingly, recent evidence from genetic models suggests that acinar cells are significantly more sensitive to Kras driven PanIN specification than centroacinar and duct cells.8 Therefore, understanding the molecular requirements for mutant Kras that force acinar cells into a duct-like state with formation of ADM and PanIN lesions is potentially important for preventing, detecting or treating PDA. ADM and PanIN formation is dramatically accelerated by insults that compromise acinar differentiation, including caerulein pancreatitis.3 ,5 ,24 Importantly, these insults reduce expression of genes that maintain acinar differentiation, including the transcription factor Mist1.6 ,7 Our data support the concept that loss of acinar differentiation via Nr5a2 elimination provides a permissive environment for Kras driven ADM and PanIN development.
It is currently unclear whether acinar cells play a role in the initiation of human PDA. While Kras mutations have not been detected in human acinar cells or isolated ADM, they have been identified in ADM associated with PanIN lesions.38 However, human acinar cells undergo de-differentiation and activation of a duct-like programme similar to what is found in cultured mouse acinar cells,39 suggesting they may also possess the capacity to undergo persistent ductal reprogramming in vivo. The recent identification of the key regulators of acinar differentiation NR5A2 and HNF1A23 as susceptibility loci for PDA development in GWAS9 may reflect a role for these genes in maintaining appropriate exocrine specification in the context of oncogenic signals. Additionally, it is conceivable that Nr5a2 impairments might cooperate in oncogenic Kras driven neoplastic transformation by further promoting inflammation. This is supported by the observation of marked inflammatory infiltrates surrounding Nr5a2 null ADM/PanIN lesions and by the study of Flandez et al40 that identified an enhanced inflammatory response in the context of Nr5a2 heterozygosity.
While it is currently unknown whether the SNPs identified in NR5A2 in PDA patients affect gene function, our study and the study of Flandez et al40 support the concept that they may result in decreased function or expression of the protein, leading to fragile acinar cell differentiation and an inflammatory environment that promotes neoplastic initiation by oncogenic Kras. An alternative explanation could be that these SNPs lead to a gain of function in Nr5a2, which could promote progression of established PDA. This scenario cannot be excluded since Nr5a2 was found in human PDA and specific knockdown of the gene in human PDA cell lines resulted in growth inhibition.41 Last, it is conceivable that Nr5a2 plays a dual role in PDA development: impaired protein function promotes PDA initiation by compromising acinar differentiation, whereas at later stages overexpression results in PDA growth advantage. Future studies exploring the effect of the individual SNPs on the protein function of Nr5a2 and the temporal role of the gene during PDA initiation and progression need to be performed to address these questions.
Acknowledgments
We thank Debbie Ngow for expert technical assistance and Sapna Puri as well as Ray MacDonald for discussions and comments on the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
Footnotes
-
Contributors GvF: study concept and design, acquisition and analysis of data, drafting of the manuscript. JPM IV: technical support, interpretation of data, critical revision of the manuscript. CVEW: generation of the Ptf1aCre and Ptf1aCreER mouse model and critical revision of the manuscript. MH: study supervision, including study design, analysis and interpretation of data, critical revision of the manuscript.
-
Funding This work was supported by a grant of the Deutsche Forschungsgemeinschaft (DFG-FI1719/1-1) and a Klein Family Foundation Fellowship to GVF and NIH-R01CA112537 to MH. Image acquisition was supported by the imaging core of the UCSF Diabetes and Endocrinology Research Center (DERC) NIH grant P30DK63720.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.