

Abstract
Although glial cells in the optic nerve head undergo a reactivation process in glaucoma, the role of glial cells during glaucomatous neurodegeneration of retinal ganglion cells is unknown. Using a coculture system in which retinal ganglion cells and glial cells are grown on different layers but share the same culture medium, we studied the influences of glial cells on survival of retinal ganglion cells after exposure to different stress conditions typified by simulated ischemia and elevated hydrostatic pressure. After the exposure to these stressors, we observed that glial cells secreted tumor necrosis factor-α (TNF-α) as well as other noxious agents such as nitric oxide into the coculture media and facilitated the apoptotic death of retinal ganglion cells as assessed by morphology, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling, and caspase activity. The glial origin of these noxious effects was confirmed by passive transfer experiments. Furthermore, retinal ganglion cell apoptosis was attenuated ∼66% by a neutralizing antibody against TNF-α and 50% by a selective inhibitor of inducible nitric oxide synthase (1400W). Because elevated intraocular pressure and ischemia are two prominent stress factors identified in the eyes of patients with glaucoma, these findings reveal a novel glia-initiated pathogenic mechanism for retinal ganglion cell death in glaucoma. In addition, these findings suggest that the inhibition of TNF-α that is released by reactivated glial cells may provide a novel therapeutic target for neuroprotection in the treatment of glaucomatous optic neuropathy.
During development and maintenance of the nervous system there exists a complex interdependency between neurons and glial cells. The glial cells maintain normal functioning of the nervous system both by controlling the extracellular environment and by supplying metabolites and growth factors. However, recent evidence challenges the view that glial cells are purely neuroprotective and rather suggests that they could participate in damaging neurons. For example, after focal cerebral ischemia or during the course of neurodegenerative diseases or trauma, reactive astrocytes as well as microglia within the CNS produce cytokines, reactive oxygen species, and nitric oxide (NO), which are implicated as mediators of tissue injury (Hewett et al., 1994; Dugan et al., 1995;Ridet et al., 1997; Vandenberghe et al., 1998; Viviani et al., 1998;Raivich et al., 1999).
The astrocytes located at the optic nerve head undergo a reactivation process in glaucoma (Hernandez and Pena, 1997). In glaucomatous optic neuropathy, apoptosis is implicated in the death of retinal ganglion cells (Quigley et al., 1995), but the precise pathogenic mechanisms leading to apoptotic cell death are unknown. Although the relationship of glial reactivation to neurodegeneration in glaucoma has not been established, increased production of some neurotoxic substances by optic nerve head astrocytes has been identified in glaucomatous eyes. For example, increased production of nitric oxide synthase (NOS;Neufeld et al., 1997) and tumor necrosis factor-α (TNF-α; Yan et al., 2000) has been described in the glaucomatous optic nerve head. In addition, expression of the inducible isoform of NOS (iNOS) and TNF-α by chemically reactivated retinal glial cells has been observed in rat models of hereditary retinal diseases (Cotinet et al., 1997; de Kozak et al., 1997; Goureau et al., 1999). Therefore, we hypothesize that retinal astroglial reactivation may lead to the increased production of neurotoxic substances and thereby participate in neuronal damage in glaucoma.
Although neuron–glia interactions have been examined in experimental models of degenerative diseases of the CNS, there is no direct evidence suggesting that glial cells are harmful to the survival of retinal ganglion cells. Using a coculture system, we studied the effects of glial cells on the survival of retinal ganglion cells after exposure to different stress conditions. This coculture system provides a good model to study neuron–glia interactions because it permits quantitative assessment of the effects of different stimuli on neuronal and glial cells separately. During these experiments we used elevated hydrostatic pressure and simulated ischemia as stress conditions, because elevated intraocular pressure and ischemia are common stress factors identified in glaucomatous eyes, which are thought to facilitate retinal ganglion cell apoptosis (Hart et al., 1979; Quigley et al., 1980; Hayreh, 1985).
Here we present novel evidence that elevated hydrostatic pressure as well as simulated ischemia can initiate the apoptotic cell death cascade in retinal ganglion cells, mainly because of the reactivity of glial cells in response to these stressors. Apoptosis-promoting substances, including TNF-α secreted by reactivated glial cells after exposure to stress, contribute directly to neuronal cytotoxicity.
MATERIALS AND METHODS
Retinal ganglion cell cultures. Primary cultures of retinal ganglion cells were derived from newborn rat retinas, using a protocol similar to that recently described (Tezel et al., 1999). All experiments were performed in accordance with the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by The Animal Studies Committee of Washington University. Sprague Dawley rats (5–7 d old) were anesthetized, and their eyes were enucleated. The eyes were rinsed with CO2-independent culture medium (Life Technologies, Grand Island, NY), and retinas were dissected mechanically under a microscope. To prepare retinal cell suspensions, we dissociated tissues in Eagle's MEM containing 20 U/ml papain, 1 mml-cysteine, 0.5 mm EDTA, and 0.005% DNase (Worthington, Lakewood, NJ) at 37°C for 40 min. Then the retinas were rinsed in an inhibitor solution containing Eagle's MEM, 0.2% ovomucoid (US Biological, Swampscott, MA), 0.04% DNase, and 0.1% bovine serum albumin (Sigma, St. Louis, MO). At the end of the treatment period the tissues were triturated through a 1 ml plastic pipette to yield a suspension of single cells. The retinal cells were spun at 400 × gfor 10 min, resuspended in Eagle's MEM containing 0.05% bovine serum albumin, and incubated in a tissue culture incubator until their immediate use for subsequent separation by immunomagnetic selection.
Immunomagnetic selection of the retinal ganglion cells was performed by using magnetic, 2.8 ± 0.2 μm diameter, polystyrene beads coated with biotinylated rat monoclonal antibody against mouse IgG1 (Dynal, Oslo, Norway) in a two-step process. In the first step, after a washing with PBS solution containing 0.1% bovine serum albumin, 1 × 107beads/ml were added to the monoclonal antibody against macrophage surface antigens (100 μg/ml; Sigma). After incubation at room temperature on a rotator for 30 min, the beads were washed with a specially designed magnet (Dynal). Coated beats were incubated with retinal cell suspension with gentle rotation for 15 min and then removed from the cell suspension to remove the bound macrophages. As a second step, fresh magnetic beads were added to monoclonal antibody (IgG1) specific to Thy-1.1 (Chemicon, Temecula, CA) to obtain a final concentration of 100 μg/ml. After incubation at room temperature for 30 min and washing, the coated beads were incubated with the macrophage-depleted retinal cell suspension for 15 min. Because the monoclonal antibody was attached to beads via streptavidin and a DNA linker, the attached cells were separated from the beads by incubation with DNase-releasing buffer (50 U/μl) at 37°C for 15 min. Then the cells were seeded on extracellular matrix-coated 24-well plates (Fisher Scientific, Pittsburgh, PA) at a density of 4 × 104 cells/well and were cocultured with glial cells. Cultures were incubated in a tissue culture incubator with a humidified atmosphere of 5% CO2/95% air at 37°C.
A retinal glial cell line was prepared with the retinal cells that were depleted for microglial and ganglion cells by following the magnetic selection process described above. After the loss of residual neuronal cells by two or three cycles of replating, these cultures contained essentially glial cells as previously described, which were identified as astrocytes and Müller glial cells, as presented in Results. The retinal glial cells were seeded on tissue culture inserts (Fisher Scientific) at a density of 3 × 104cells/well and placed in the wells in which the retinal ganglion cells were seeded. These inserts contain polyethylene terephthalate membrane with 0.4 μm pore size and allow for the transport of secreted molecules while preventing cell migration.
The serum-free culture medium was prepared by using B27-supplemented Neurobasal (Life Technologies) as previously described (Barres et al., 1988; Brewer et al., 1993). The medium also contained (in μg/ml) 100 bovine serum albumin, 5 insulin, 16 putrescine, and 100 transferrin; 1 mm pyruvate, 1 mm glutamine; (in ng/ml) 60 progesterone, 40 sodium selenite, 30 triiodo-thyronine, 50 BDNF, 20 CNTF, and 10 bFGF; 5 μm forskolin, 100 μminosine, and antibiotics (Jo et al., 1999). All supplements were purchased from Sigma.
Retrograde labeling of retinal ganglion cells. Under general anesthesia that used a mixture of 80 mg/kg ketamine (Fort Dodge Laboratories, Fort Dodge, IA) and 12 mg/kg xylazine (Butler, Columbus, OH) given intraperitoneally and with immobilization of the rats in a stereotaxic apparatus, bilateral microinjections of Fluoro-Gold (1.5 μl of a 5% solution of Fluoro-Gold in 0.9% sodium chloride; Fluorochrome, Englewood, CO) into the superior colliculi were performed according to the previously described methods (Selles-Navarro et al., 1996). One week after Fluoro-Gold application the retinas were dissected and dissociated. After a selection of retinal ganglion cells via the immunomagnetic method, selected and unselected cells were examined by flow cytometry after double immunolabeling of Fluoro-Gold and Thy-1.1.
Flow cytometry. Retinal cells were fixed with 2% paraformaldehyde solution for 20 min at room temperature. After centrifuge and resuspension of the cells, they were permeabilized in Triton X-100 (0.4% in PBS solution) for 30 min. Washed cells then were incubated with a mixture of rabbit antibody against Fluoro-Gold (Fluorochrome) and mouse antibody against Thy-1.1 at a 1:100 dilution for 30 min. After washing, the cells were incubated with a mixture of FITC- and Cy3-conjugated secondary antibodies (Sigma) for another 30 min. Then the cells were washed, resuspended at 106 cells/ml, and counted with a FACScan flow cytometer/CELLQuest software system (Becton Dickinson, San Jose, CA).
Study design. The retinal ganglion cells exhibiting contact of the neuritic processes and glial cells that had been grown to approximate confluence were incubated under stress conditions or normal conditions. For simulated ischemia the cells were exposed to reduced oxygen tensions in a medium lacking glucose. Hypoxia was maintained by placing the cultures in a dedicated culture incubator with a controlled flow of 95% N2/5% CO2. A closed pressurized chamber equipped with a manometer was used to expose the cells to elevated hydrostatic pressure. The pressure was elevated to 50 mmHg. The chamber was placed in a regular tissue culture incubator at 37°C. To examine the time course of cellular responses, we maintained the simulated ischemia or elevated pressure for 6, 12, 24, 48, or 72 hr. Control cells from an identical passage of cells were incubated simultaneously in a regular tissue culture incubator at 95% air/5% CO2 at 37°C. To examine glial sources of noxious insults on retinal ganglion cells, we collected conditioned medium from glial cells that were cultured alone after their incubation in the presence or absence of stress conditions for 72 hr. Retinal ganglion cells that were cultured alone then were incubated with the conditioned medium of glial cells for 24 hr.
In addition, to examine the role of TNF-α and NO on cell survival, we performed incubations under stress conditions in the presence or absence of specific inhibitors. A neutralizing antibody (AF510NA) was used to inhibit TNF-α activity (R&D Systems, Minneapolis, MN). The ability of this antibody to neutralize the bioactivity of recombinant rat TNF-α in the L-929 cell line in the presence of actinomycin D revealed that the neutralization dose50 was ∼0.3–0.9 μg/ml in the presence of 0.025 ng/ml of recombinant rat TNF-α. The neutralizing antibody of TNF-α activity was neuroprotective in in vitro or in vivoexperiments (Barone et al., 1997; Lavine et al., 1998; Downen et al., 1999). In addition, [N-(3-[aminomethyl]benzyl)acetamidine dihydrochloride] (1400W; Alexis, San Diego, CA), a selective inhibitor of iNOS, was used to inhibit inducible synthesis of NO (Garvey et al., 1997). We used 10 μg/ml of the neutralizing antibody of TNF-α and 2.5 μm of the iNOS inhibitor 1400W, because these were optimum conditions to inhibit TNF-α and iNOS, respectively, in our cocultures on the basis of concentration–response experiments (data not shown). At the end of the incubation period the cells were subjected immediately to the experiments described below, which were repeated at least three times for each condition.
The viability of the cells was determined with the Live/Dead Kit (Molecular Probes, Eugene, OR), which contains calcein-AM and ethidium homodimer (Haugland and Larison, 1994). Calcein-AM is a cell-permeable fluorogenic esterase substrate. The kit relies on the intracellular esterase activity within living cells to hydrolyze calcein-AM to a green fluorescent product, calcein. In dead cells ethidium can pass easily through the compromised plasma and nuclear membranes and attach to the DNA, yielding red fluorescence. Cells were counted in at least 10 random fields of each well at 200× magnification (∼150 ganglion cell per well) with a fluorescence microscope (Olympus, Tokyo, Japan). The viability of the cells was expressed as the average ratio of esterase (+) cells to the total number of cells multiplied by 100.
Morphological analysis of apoptosis. An in situcell death detection kit (Boehringer Mannheim, Mannheim, Germany) was used to identify the apoptotic cells by the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) technique (Gavrieli et al., 1992). Briefly, after fixation, permeabilization, and blocking steps the air-dried cells were incubated with a mixture of fluorescein-labeled nucleotides and terminal deoxynucleotidyl transferase for 1 hr. Terminal deoxynucleotidyl transferase catalyzes the polymerization of labeled nucleotides to free 3′-OH terminals of DNA fragments. Cells incubated with fluorescein-labeled nucleotide mixture without the presence of terminal deoxynucleotidyl transferase served as a negative control. Cells previously treated with DNase I (1 mg/ml) to induce breaks in the DNA strands served as a positive control. TUNEL-positive cells were counted in triplicate wells under a fluorescence microscope, and the percentage of apoptosis was calculated by using the total number of cells in these wells.
Western blotting. After the cells were washed with PBS solution, they were lysed in sample buffer (1% SDS, 100 mmdTT, 60 mm Tris, pH 6.8, and 0.001% bromophenol blue). Protein concentrations were determined via the bicinchoninic acid (BCA) method (Sigma). The samples were boiled for 5 min before they were subjected to electrophoresis.
Samples were separated by electrophoresis in 10–15% SDS polyacrylamide gels at 160 V for 1 hr and electrophoretically transferred to polyvinylidene fluoride membranes (Millipore, Marlboro, MA) via a semi-dry transfer system (Bio-Rad, Hercules, CA). After transfer the membranes were blocked in a buffer (50 mm Tris HCl, 154 mm NaCl, and 0.1% Tween-20, pH 7.5) containing 5% nonfat dry milk for 1 hr and then overnight in the same buffer containing a dilution of primary antibody and sodium azide. Primary antibodies were monoclonal antibodies to TNF-α (R&D Systems), isotypes of NOS (neuronal NOS and iNOS; Transduction Laboratories, Lexington, KY), caspase-8, or polyclonal antibody to caspase-3 (PharMingen, San Diego, CA); they were used at a dilution of 1:1000. After several washes and a second blocking for 20 min, the membranes were incubated with a dilution of secondary antibodies conjugated with horseradish peroxidase (Fisher Scientific) at 1:2000 for 1 hr. Immunoreactive bands were visualized by enhanced chemiluminescence with the use of commercial reagents (Amersham Life Science, Arlington Heights, IL).
Examination of caspase activity. Caspase-3-like activity was examined in situ after staining with Phiphilux-G6D2 (Alexis, San Diego, CA). Phiphilux-G6D2is a cell-permeable fluorogenic substrate that is cleaved to produce rhodamine molecules and that can be used to detect caspase-3-like activity in living cells (Finucane et al., 1999). For staining, the washed cells were incubated with a substrate solution of 10 μm for 20 min at 37°C. Rhodamine fluorescence was visualized under a fluorescence microscope.
In addition, caspase-3-like protease activity was measured in a fluorometric assay by measuring the extent of cleavage of the fluorometric peptide substrate as previously described (Cheng et al., 1998; Tezel and Wax, 1999). Briefly, cell lysates were incubated with Ac-Asp-Glu-Val-Asp-7-amino-4-trifluoro-methyl coumarin (Ac-DEVD-AMC) fluorometric substrate (50 μm). Positive controls included purified recombinant caspase-3 (0.1 μg; Upstate Biotechnology, Lake Placid, NY). Fluorescence was measured at an excitation wavelength of 360 nm and an emission wavelength of 460 nm in a fluorescent plate reader at different time points up to 180 min. The protease activity was expressed as picomoles of substrate per milligram of protein per minute as calculated by using the linear range of the assay.
Immunocytochemistry. Cells were washed in PBS solution and fixed with 4% paraformaldehyde solution for 30 min at room temperature. After washing, they were permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate solution on ice for 4 min. Then the cells were treated with 3% bovine serum albumin for 30 min to block the nonspecific binding sites. Triplicate wells were incubated with monoclonal antibodies against TNF-α (R&D Systems) or isotypes of NOS (neuronal NOS and iNOS; Transduction Laboratories) at 37°C for 2 hr. Next the samples were washed and incubated with the appropriate second antibodies conjugated with Cy3 (Sigma). After washing, they were examined with a fluorescence microscope.
For examination of the purity of cultures the cells were double-immunolabeled with specific cell markers. For double immunofluorescence labeling after the fixation, permeabilization, and blocking steps, the cultures were incubated with a mixture of two antibodies (one rabbit and one mouse antibody) against Thy-1.1, neurofilament protein, glial fibrillary acidic protein, or S-100 protein (Sigma) at 1:100 dilution for 30 min. After being washed, the cells were incubated with a mixture of Cy3- and FITC-conjugated secondary antibodies (Sigma) for another 30 min. Negative controls were performed by replacing the primary antibody with nonimmune serum or by incubating the cells with each primary antibody, followed by the inappropriate secondary antibody to determine that each secondary antibody was specific to the species it was made against. Then the cultures were examined with a fluorescence microscope.
Enzyme-linked immunosorbent assay (ELISA). We used a kit to measure TNF-α levels in conditioned medium by quantitative sandwich ELISA technique (R&D Systems). Conditioned medium was incubated in microwells coated with monoclonal antibody specific for rat TNF-α. After a wash, horseradish peroxidase-conjugated polyclonal antibody specific for rat TNF-α was added to the wells. After another wash, a substrate solution containing hydrogen peroxide and tetramethylbenzidine was added. The enzyme reaction was terminated by the addition of hydrochloric acid solution, and absorbance was measured at 450 nm. Using a standard curve prepared from seven dilutions of recombinant rat TNF-α, we calculated the concentrations of TNF-α in conditioned medium. The sensitivity was <5 pg/ml.
Colorimetric assay. To measure breakdown products of NO in conditioned medium, we used a colorimetric assay kit (R&D Systems). This assay determined NO on the basis of the enzymatic conversion of nitrate to nitrite by nitrate reductase. The reaction was followed by a colorimetric detection of nitrite as an azo dye product of the Griess reaction. As an additional step, lactate dehydrogenase and pyruvic acid were used before color formation to oxidize the excess of NADPH because NADPH, an essential cofactor for the function of NOS enzyme, interferes with the chemistry of Griess reagents. Because the relative levels of nitrate and nitrite can vary substantially, depending on the ambient conditions and redox state of the biological fluids, for the most accurate determination of total NO production both the nitrate and nitrite levels were measured. The absorbance was read at 540 nm, and the concentrations of breakdown products of NO were calculated by using a standard curve. The sensitivity of the nitrite assay was <0.22 μmol/l, and the sensitivity of the nitrate assay was <0.54 μmol/l.
RESULTS
Cell morphology and viability
Retinal ganglion cells were identified on the basis of retrograde labeling with Fluoro-Gold, morphology, and expression of cell markers. After retrograde labeling with Fluoro-Gold and the selection of retinal ganglion cells by an immunomagnetic separation method, the cells were immunolabeled by antibodies against Fluoro-Gold and Thy-1.1. Using flow cytometry, we observed that the immunolabeling by Fluoro-Gold and Thy-1.1 antibodies was colocalized in >90% of these cells; >95% of the cells were positive for Thy-1.1 (Fig.1A). Cells unselected by sorting were negative for both Fluoro-Gold and Thy-1.1 (Fig.1B).

Cultured retinal cells. After retrograde labeling by Fluoro-Gold and the selection of retinal ganglion cells by the use of an immunomagnetic separation method, the selected cells were immunolabeled with antibodies against Fluoro-Gold and Thy-1.1; they were examined with flow cytometry. A, Immunolabeling with Fluoro-Gold (FL1-H) and Thy-1.1 (FL3-H) antibodies was colocalized in >90% of these cells; >95% of these cells were positive for Thy-1.1.B, Unselected cells were negative for both Fluoro-Gold (FL1-H) and Thy-1.1 (FL3-H). Cultured retinal ganglion cells had round or oval cell bodies with a diameter of 10–20 μm, phase-bright appearance, and branched neuritis of uniform caliber and varying length. C, A retinal ganglion cell derived from newborn rat retina. D, Fluorescence microscope image of the retinal ganglion cell shown in C after labeling for neurofilament protein. E, Fluorescence microscope image of the retinal ganglion cell shown in C after labeling for Thy-1.1. F, Glial cells derived from newborn rat retina. G, Fluorescence microscope image of the retinal glial cells shown in F after labeling for glial fibrillary acidic protein. H, Fluorescence microscope image of the retinal glial cells shown in F after labeling for S-100. Scale bars: C–E, 20 μm;F–H, 60 μm.
Cultured retinal ganglion cells had round or oval cell bodies with a diameter of 10–20 μm, a phase-bright appearance, and branched neurites of uniform caliber and varying length (Fig. 1C), as previously identified (Barres et al., 1988). In addition, the purity of cultured retinal cells was examined by using immunolabeling for specific markers. The retinal ganglion cells were homogenously positive for Thy-1.1 and neurofilament protein, but negative for glial markers. Glial cells were homogenously labeled for glial fibrillary acidic protein, selectively labeled for S-100, but unlabeled for neuronal markers (Fig. 1).
At the beginning of the incubation of the cocultures under stress conditions, the percentage of living retinal ganglion cells and glial cells was 96.69 ± 1.6 and 97.84 ± 1.9%, respectively. The cell viability decreased to 69.69 ± 2.0 and 76.64 ± 1.9% in retinal ganglion cells after 72 hr of incubation in the presence of simulated ischemia or elevated hydrostatic pressure, respectively. However, the viability of glial cells was 96.24 ± 2.1% at the end of incubation period under either simulated ischemia or elevated hydrostatic pressure.
Induction of apoptosis in retinal ganglion cells in cocultures exposed to simulated ischemia or elevated hydrostatic pressure
Apoptosis was induced in retinal ganglion cells after the incubation of the cocultures in the presence of simulated ischemia or elevated hydrostatic pressure for as long as 72 hr. Specific morphological changes of apoptotic cell death included cell body shrinkage and compaction of the nucleus (Fig.2A–C). In addition, apoptotic cell death was examined via the TUNEL technique. The apoptotic retinal ganglion cells exhibited bright labeling of fragmented nuclear DNA by TUNEL (Fig. 2D–F). However, there was no evidence of apoptosis in glial cells in the cocultures that were incubated under stress conditions by either morphological examination or TUNEL technique (Fig.2G–L).

Morphological analysis of apoptotic cell death in cocultures of retinal ganglion cells and glial cells.A–C, Phase-contrast microscope images of retinal ganglion cells that were incubated under different conditions for 72 hr. A, Normal conditions. B, Simulated ischemia. C, Elevated hydrostatic pressure. Fluorescence microscope images of TUNEL in D–F correspond to retinal ganglion cells seen in A–C, respectively.G–I, Phase-contrast microscope images of glial cells that were incubated under different conditions for 72 hr.G, Normal conditions. H, Simulated ischemia. I, Elevated hydrostatic pressure. Fluorescence microscope images of TUNEL in J–L correspond to glial cells seen in G–I, respectively. After the incubation of cocultures under stress conditions, apoptosis was induced in retinal ganglion cells, although there was no evidence of apoptosis in glial cells.
Quantitative examination of apoptotic cell death after incubation under stress conditions revealed that the percentage of positive TUNEL was approximately three times greater in retinal ganglion cells cocultured with glial cells as compared with retinal ganglion cells cultured alone. After incubation under stress conditions for 72 hr, >20% of the retinal ganglion exhibited positive TUNEL; in the retinal ganglion cells that were cultured alone the rate of positive TUNEL was <7% (Mann–Whitney U test; p = 0.006 andp = 0.04 for simulated ischemia and elevated hydrostatic pressure, respectively; Fig.3A). In addition, after incubation under stress conditions apoptosis was induced in retinal ganglion cells in cocultures in a time-dependent manner (Fig.3A). Although the rate of positive TUNEL was 24.10 ± 6.0 and 19.90 ± 5.4% in retinal ganglion cells in cocultures that were exposed to simulated ischemia or elevated hydrostatic pressure for 72 hr, respectively, retinal ganglion cells in control cultures that were incubated under normal conditions exhibited apoptosis in <2% of the cell population (Mann–Whitney Utest, p = 0.017, p = 0.023, respectively). However, the percentage of positive TUNEL was virtually absent in glial cells that were incubated in the absence or presence of stress conditions (0.94 ± 0.6 and 1.12 ± 1.0%, respectively; p > 0.05 for both conditions).

A, Quantitative analysis of positive TUNEL in retinal ganglion cells that were incubated under simulated ischemia or elevated hydrostatic pressure. After incubation in the presence of simulated ischemia or elevated hydrostatic pressure for 72 hr, the rate of positive TUNEL was higher in retinal ganglion cells in cocultures compared with that in retinal ganglion cells that were cultured alone (RGCs; p = 0.006 and p = 0.04, respectively). In addition, the rate of positive TUNEL was higher in retinal ganglion cells in cocultures exposed to simulated ischemia or elevated hydrostatic pressure for 72 hr, respectively, compared with that in retinal ganglion cells in cocultures incubated under normal conditions (Mann–WhitneyU test; p = 0.017 andp = 0.023, respectively). B, Quantitative analysis of positive TUNEL in retinal ganglion cells after passive transfer experiments. Conditioned medium of glial cells that were cultured alone was collected after their incubation in the presence or absence of simulated ischemia or elevated hydrostatic pressure for 72 hr. Then retinal ganglion cells that were cultured alone were incubated with the glial-conditioned medium for 24 hr. The rate of positive TUNEL was higher in retinal ganglion cells that were incubated with the conditioned medium of stressed glial cells as compared with that in retinal ganglion cells that were incubated with the conditioned medium of control glial cells (p = 0.04 and p = 0.02 for simulated ischemia and elevated hydrostatic pressure, respectively).
In addition, we performed passive transfer experiments to examine the glial source of noxious insults on retinal ganglion cells. For this purpose, the conditioned medium of glial cells that were cultured alone was collected after their incubation in the presence or absence of simulated ischemia or elevated hydrostatic pressure for 72 hr. Retinal ganglion cells that were cultured alone then were incubated with the glial conditioned medium for 24 hr. The TUNEL was positive in ∼17% of retinal ganglion cells that were incubated with the conditioned medium of stressed glial cells, whereas <2% of retinal ganglion cells exhibited positive TUNEL in cultures that were incubated with the conditioned medium of glial cells incubated under normal conditions (Mann–Whitney U test; p = 0.04 andp = 0.02 for simulated ischemia and elevated hydrostatic pressure, respectively; Fig. 3B).
Caspase activation accompanying retinal ganglion cell apoptosis
To examine caspase activation, we used lysates of retinal cells in Western blotting. Western blot analysis demonstrated cleavage of caspase-8 and caspase-3 in retinal ganglion cells after exposure of the cocultures to simulated ischemia or elevated hydrostatic pressure. Western blots that used the lysates of retinal ganglion cells incubated under stress conditions revealed a 55 kDa immunoreactive band corresponding to caspase-8 and ∼30 and 20 kDa cleaved products. The presence of caspase-3 activation was assessed by the observation of 17 kDa subunit that was derived from the cleavage of the 32 kDa pro-enzyme caspase-3. No cleavage of caspase-8 or caspase-3 was detected by using the lysates of glial cells that were incubated under stress conditions (Fig. 4A,B).

Examination of caspase activity in cocultures incubated under simulated ischemia or elevated hydrostatic pressure. A, Western blot analysis of caspase-8 expression in cocultures. B, Western blot analysis of caspase-3 expression cocultures. Column 1, Control retinal ganglion cells; column 2, retinal ganglion cells incubated under simulated ischemia for 72 hr; column 3, retinal ganglion cells incubated under elevated hydrostatic pressure for 72 hr; column 4, control glial cells; column 5, glial cells incubated under simulated ischemia for 72 hr;column 6, glial cells incubated under elevated hydrostatic pressure for 72 hr. Western blots revealed that the 55 kDa immunoreactive band corresponding to caspase-8 cleaved to 30 and 20 kDa products in retinal ganglion cells that were incubated under stress conditions. In addition, 32 kDa pro-enzyme caspase-3 cleaved to a 17 kDa active subunit in retinal ganglion cells. No cleavage of caspase-8 or caspase-3 was detected with the use of the extracts of glial cells. Caspase activation also was examined, in situ, using Phiphilux-G6D2 in retinal ganglion cells that were incubated under different conditions for 72 hr. C, Normal conditions. D, Simulated ischemia.E, Elevated hydrostatic pressure. Fluorescence microscope images seen in F–H correspond to phase-contrast images of the retinal ganglion cells seen inC–E, respectively. Rhodamine fluorescence (red) indicates caspase-3-like activity in retinal ganglion cells that were incubated under stress conditions.I, The amount of DEVD-AMC cleaving activity with the use of fluorometric analysis was higher in retinal ganglion cells in cocultures that were incubated under simulated ischemia or elevated hydrostatic pressure for 72 hr as compared with cocultures that were incubated under normal conditions (Mann–Whitney U test;p = 0.006 and p = 0.04, respectively).
In addition, we examined caspase-3-like protease activity in cocultures. In situ examination that used the fluorogenic substrate Phiphilux-G6D2 demonstrated caspase-3-like activity in living retinal ganglion cells exposed to simulated ischemia or elevated hydrostatic pressure for 72 hr (Fig. 4C–H). We also performed fluorometric analysis that used lysates of retinal ganglion cells to measure the cleavage of Ac-DEVD-AMC, which reflects caspase-3-like activity. The amount of DEVD-AMC cleaving activity was ∼10 times higher in retinal ganglion cells in cocultures that were incubated under simulated ischemia or elevated hydrostatic pressure for 72 hr (38.67 ± 7.4 and 31.00 ± 6.2 pmol/mg protein/min, respectively) compared with control cultures that were incubated under normal conditions (3.50 ± 1.1 pmol/mg protein/min; Mann–WhitneyU test; p = 0.006 and p = 0.04, respectively; Fig. 4I).
Increased production of TNF-α and NO by glial cells in response to stressors
We examined the possibility that the production of TNF-α and NOS by glial cells in cocultures exposed to stress conditions was involved directly in facilitating retinal ganglion cell apoptosis. Western blot analysis that used cell lysates revealed that the expression of TNF-α and iNOS was undetectable in retinal ganglion cells incubated under either normal or stress conditions. However, the expression of TNF-α and iNOS increased in glial cells in cocultures exposed to simulated ischemia or elevated hydrostatic pressure (Fig.5A,B). Immunocytochemistry similarly demonstrated the increased expression of TNF-α and iNOS in glial cells in cocultures that were incubated under stress conditions (Fig. 5C–H).

Examination of TNF-α and iNOS expression in cocultures that were incubated under simulated ischemia or elevated hydrostatic pressure. Both Western blot analysis (A, B) and immunocytochemistry (C–H) revealed increased expression of TNF-α and iNOS in glial cells, but not in retinal ganglion cells, in cocultures that were incubated under stress conditions. A, Western blot analysis of TNF-α expression. B, Western blot analysis of iNOS expression.Column 1, Control retinal ganglion cells; column 2, retinal ganglion cells that were incubated under simulated ischemia for 72 hr; column 3, retinal ganglion cells that were incubated under elevated hydrostatic pressure for 72 hr;column 4, control glial cells; column 5, glial cells that were incubated under simulated ischemia for 72 hr;column 6, glial cells that were incubated under elevated hydrostatic pressure for 72 hr. C–E, TNF-α expression in glial cells that were incubated under different conditions for 72 hr. C, Normal conditions. D, Simulated ischemia. E, Elevated hydrostatic pressure.F–H, iNOS expression in glial cells that were incubated under different conditions for 72 hr. F, Normal conditions. G, Simulated ischemia. H, Elevated hydrostatic pressure.
We measured the levels of TNF-α and the end products of NO in conditioned medium of the cocultures. TNF-α levels in the conditioned medium measured by ELISA were approximately eight times higher in cocultures that were exposed to simulated ischemia or elevated hydrostatic pressure as compared with cocultures that were incubated under normal conditions (Mann–Whitney U test,p = 0.003; Fig.6A). As measured by a colorimetric assay, breakdown products of NO in conditioned medium increased approximately sevenfold in cocultures that were exposed to stress conditions compared with cocultures that were incubated under normal conditions (p = 0.003; Fig.6B).

Measurement of TNF-α and end products of NO in conditioned medium of cocultures that were incubated under stress conditions. A, Titers of TNF-α in conditioned medium as measured by ELISA. B, Titers of end products of NO in conditioned medium as measured by a colorimetric assay. Levels of both TNF-α and NO end products were higher in the conditioned medium of cocultures that were exposed to simulated ischemia or elevated hydrostatic pressure as compared with cocultures that were incubated under normal conditions (Mann–Whitney U test;p = 0.003 and p = 0.003, respectively).
We also performed experiments in which cocultures were incubated under stress conditions in the presence of specific inhibitors of TNF-α or iNOS. Our experiments revealed that inhibitors of TNF-α or iNOS were able to diminish apoptotic cell death in retinal ganglion cells induced by simulated ischemia or elevated hydrostatic pressure. Inhibition of the bioactivity of TNF-α by a specific neutralizing antibody (AF510NA; 10 μg/ml) resulted in a decreased rate of positive TUNEL from 24 to 8% (67%) in cocultures that were incubated under simulated ischemia and from 20 to 5% (75%) in cocultures that were incubated under elevated hydrostatic pressure (Mann–Whitney U test;p = 0.0002). Treatment of cocultures with 2.5 μm of the selective inhibitor of iNOS, 1400W, decreased the rate of positive TUNEL ∼50% in cocultures that were incubated under simulated ischemia and ∼35% in cocultures that were incubated under elevated hydrostatic pressure (p= 0.003; Fig. 7). Inhibition of apoptosis by a neutralizing antibody against TNF-α was more prominent than that by 1400W (p = 0.008).
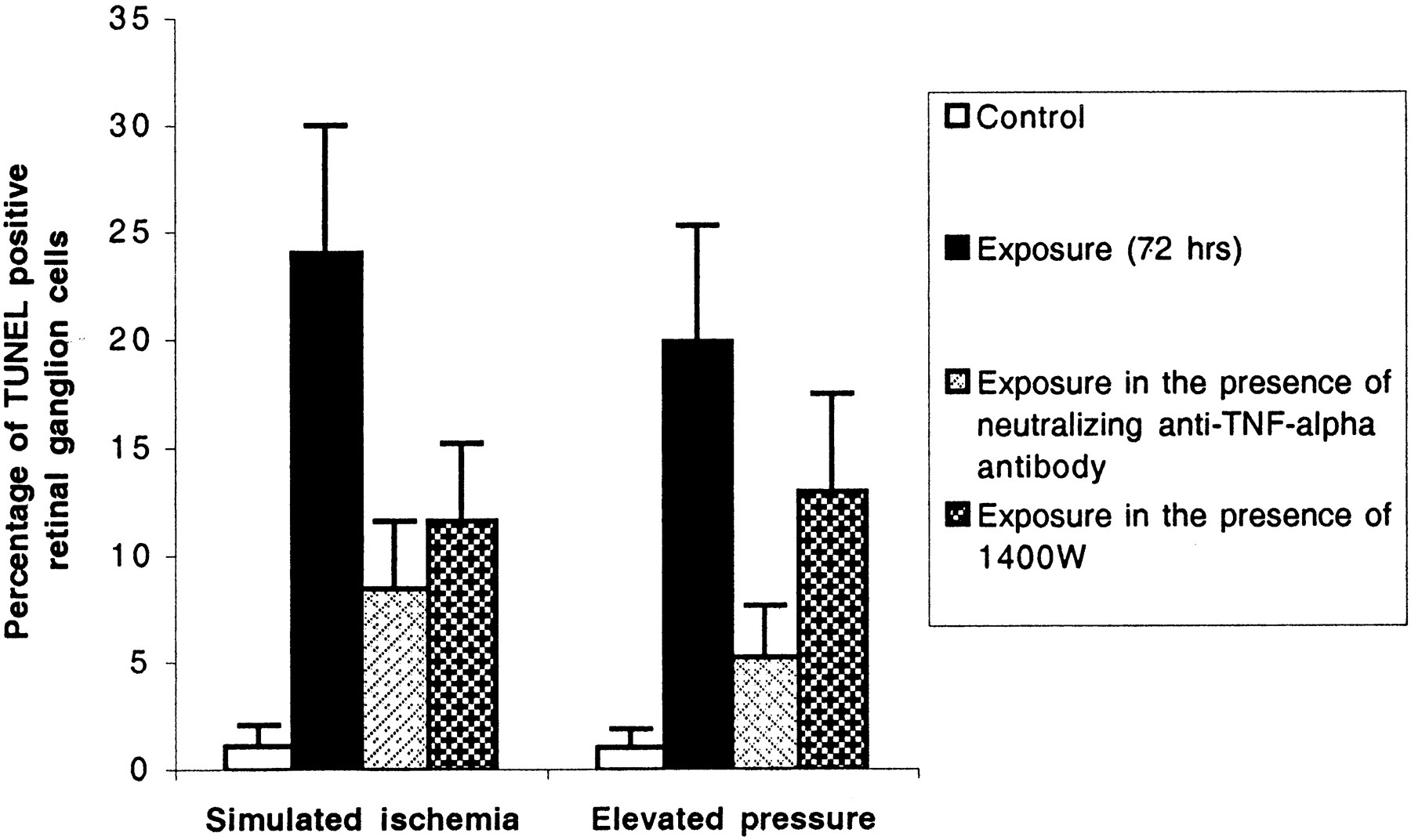
Inhibition of apoptosis in retinal ganglion cells in cocultures that were incubated under stress conditions in the presence of specific inhibitors of TNF-α or iNOS. Treatment of cocultures with a specific antibody neutralizing the activity of TNF-α (10 μg/ml) or with a selective inhibitor of iNOS, 1400W (2.5 μm), decreased the rate of positive TUNEL after incubation under stress conditions (Mann–Whitney Utest; p = 0.0002 and p = 0.003, respectively). Inhibition of apoptosis by a neutralizing antibody against TNF-α was more prominent than that by 1400W (p = 0.008).
DISCUSSION
Both elevated intraocular pressure and ischemia are common stress factors identified in glaucomatous eyes that are thought to affect retinal ganglion cell survival adversely (Hart et al., 1979; Quigley et al., 1980; Hayreh, 1985). However, the pathophysiological mechanisms by which elevated intraocular pressure leads to neuronal damage in glaucoma are unknown. The blockade of axoplasmic flow at the lamina cribrosa in the optic nerve head and thereby the blockage of neurotrophin transport to the retinal ganglion cells (Anderson and Hendrickson, 1974; Minckler et al., 1976; Quigley and Addicks, 1980;Pease et al., 2000) as well as ischemia secondary to elevated intraocular pressure (Hayreh, 1985; Flammer, 1994; Chung et al., 1999) have been suggested to be two mechanisms contributing to retinal ganglion cell death in glaucoma.
Although some evidence indicates that apoptotic cell death can be triggered by elevated hydrostatic pressure itself, as shown in human lymphoblasts (Takano et al., 1997), studies on the direct effect of elevated pressure on neuronal survival are limited [Agar A, Hill M, Coroneo MT (1999). Abstract. Invest Ophthalmol Vis Sci. 40:A265]. Our experiments provide evidence that environmental stress created by elevated hydrostatic pressure as well as simulated ischemia could affect neuronal survival. These findings are in accordance with previous observations that different cells exposed to biomechanical forces, including elevated hydrostatic pressure, exhibit functional as well as morphological changes. For example, hydrostatic pressure induces cytokine expression in a chondrocyte-like cell line, which includes TNF-α (Takahashi et al., 1998). In the eye, human lamina cribrosa astrocytes have been shown to react to pressure changes in their environment by modulating the production and secretion of extracellular matrix macromolecules (Hernandez and Pena, 1997). In addition, recent evidence suggests that cell lines derived from several intraocular tissues, including retinal cells and optic nerve head astrocytes, respond to acute and sustained levels of elevated hydrostatic pressure by changing their cell morphology as well as by increasing basal adenylyl cyclase activity (Wax et al., 2000).
In the current study, cell survival was examined in primary cocultures of retinal ganglion cells and glial cells that had been exposed to elevated hydrostatic pressure for a longer period (up to 72 hr), and it was demonstrated that the elevated hydrostatic pressure decreased neuronal survival. Increased production of apoptosis-promoting substances by retinal glial cells after exposure to elevated hydrostatic pressure or simulated ischemia accounts, in part, for the increased rate of cell death in cocultured retinal ganglion cells. Passive transfer experiments confirmed that the source of noxious insults on retinal ganglion cells was retinal glial cells that had been exposed to stress conditions. We therefore propose that alterations in the cellular functions of glial cells in the presence of environmental stress created during the course of glaucomatous optic neuropathyin vivo may lead to retinal ganglion cell death in these eyes. Reactivated glial cells in the outer retina have been implicated similarly in the ensuing death of photoreceptor cells (Cotinet et al., 1997; de Kozak et al., 1997; Goureau et al., 1999). Here, we focused on TNF-α and NO among several soluble factors secreted by stressed glial cells on the basis of findings obtained from glaucomatous eyes (Neufeld et al., 1997; Yan et al., 2000).
Tumor necrosis factor-α is known as a potent immunomediator and proinflammatory cytokine that is upregulated rapidly in the brain after injury (Liu et al., 1994; Barone et al., 1997). TNF-α is produced by reactivated macrophages, astrocytes, microglia, and retinal glial cells (Lieberman et al., 1989; Semenzato, 1990; Brenner et al., 1993; de Kozak et al., Meda et al., 1995; Cotinet et al., 1997). The dramatic increase in TNF-α production after ischemic and excitotoxic brain injury suggests an important role for this cytokine in modifying the neurodegenerative process. TNF-α also is known to be a potent activator of neurotoxic substances such as NO and excitotoxins (McGeer et al., 1993; Rothwell and Hopkins, 1995; Martin-Villalba et al., 1999). Its excessive synthesis after trauma has been correlated with poor outcome (Ertel et al., 1995), and its inhibition is accompanied by reduced brain damage (Shohami et al., 1996). In addition, a new concept in neurodegeneration exclaims that picogram concentrations of TNF-α, which is known to be noncytotoxic, induces cell death via the “silencing of survival signals” (Venters et al., 2000). Regarding optic nerve, TNF-α has been thought to account for axonal degeneration and glial changes that have been observed in the optic nerves of patients with AIDS (Lin et al., 1997). Furthermore, intravitreal injections of TNF-α into rabbit eyes produced axonal degeneration in their optic nerves (Madigan et al., 1996). Because our cultures were depleted of microglia, astrocytes and retinal Müller glial cells appeared to be the likely sources of TNF-α secreted into the conditioned medium. TNF-α released by chemically reactivated glial cells has been implicated similarly in the increased rate of apoptosis in cocultured neurons (Viviani et al., 1998; Downen et al., 1999).
Tumor necrosis factor-α is an inducer of apoptotic cell death via TNF-α receptor-1 occupancy in a caspase-mediated pathway, which includes the activation of caspase-8 (Hsu et al., 1995). Our observation of caspase-8 activation suggests the involvement of TNF-α as a mediator of the apoptosis of retinal ganglion cells. Furthermore, it has been demonstrated recently that the expression of TNF-α and its receptor are increased in glaucomatous optic nerve head. Although the TNF-α is expressed mostly in astroglial cells, the expression of TNF-α receptor-1 is more prominent in nerve bundles located in the anterior region of the glaucomatous optic nerve head (Yan et al., 2000). These observations support our current findings that retinal neuronal tissue is an important target for the effects of TNF-α that are produced by glial cells.
In addition to TNF-α, we found that increased production of NO in retinal glial cells that have been exposed to different stress conditions induced cell death in cocultured retinal ganglion cells. Previous studies suggested that glial expression of iNOS caused delayed neurotoxicity in mixed cultures of cortical neuronal and glial cells (Dawson et al., 1994). Similar to TNF-α, NO, which is formed froml-arginine by NOS, has been implicated in several neurodegenerative diseases. Although two isoforms, neuronal NOS and endothelial NOS, are expressed constitutively, iNOS is induced after infection and trauma (Bredt and Snyder, 1994). Induction of NOS in brain tissue results in neuronal cell death (Iadecola et al., 1995) in which astrocytes and microglia are major sources of the iNOS production (Liu et al., 1996). Inducible NOS produced by glial cells also is thought to cause retinal neuronal cell death in different retinal diseases and after optic nerve axotomy (Cotinet et al., 1997; de Kozak et al., 1997; Koeberle and Ball, 1999). In addition, intravitreal injection of the NO donor has been shown to cause retinal ganglion cell and photoreceptor loss, whereas reduction of NO levels by systemic inhibition of NOS reduces retinal ganglion cell loss in a rat model of retinal ischemia (Lam and Tso, 1996). Nitric oxide also has been suggested to play a role in the neurodegeneration process in glaucoma (Neufeld et al., 1997, 1999).
Our experiments that used inhibitors of TNF-α or iNOS revealed an inhibition of apoptotic cell death in retinal ganglion cells in cocultures that had been exposed to simulated ischemia or elevated hydrostatic pressure. Although iNOS inhibition provided partial protection against apoptotic cell death in cocultures, a more prominent inhibition of apoptosis was observed after the inhibition of TNF-α. These results indicate a crucial role for endogenous TNF-α in mediating neurotoxicity in cultured retinal ganglion cells. Because TNF-α induces NO secretion (Goureau et al., 1997; Shafer and Murphy, 1997; Heneka et al., 1998), inhibition of its activity indirectly could decrease the harmful effect created by NO as well. Similar to our observations, neutralizing anti-TNF-α antiserum, rather than a NOS inhibitor, inhibited neurotoxicity of cytokine-induced production of iNOS and TNF-α in neuron–astrocyte cultures that were derived from human fetal cerebrum (Downen et al., 1999). Furthermore, the inhibition of TNF-α has been shown to reduce iNOS expression and NOS activity (Perkins et al., 1998). In addition, in vivo observations support the idea that neutralization of systemic TNF-α ameliorates target organ damage in brain ischemia or in experimental autoimmune uveoretinitis (Dick et al., 1996; Sartani et al., 1996; Barone et al., 1997; Lavine et al., 1998). However, selective inhibition of iNOS does not prevent the organ injury/dysfunction that is caused by endotoxin (Wray et al., 1998). These findings emphasize the importance of further research to determine the potential neuroprotective role of TNF-α or iNOS inhibition in stressed retinal ganglion cells under in vivo conditions, as occurs in glaucoma, and to identify strategies that are feasible for patient treatment.
In conclusion, our findings provide evidence that the functional state of glial cells determined by environmental factors may be important for determining the ultimate role of glial cells as either neuroprotective or neurotoxic. The retinal glial cells exposed to stress conditions similar to that created in vivo during the process of glaucoma, such as elevated hydrostatic pressure or simulated ischemia, have a neurotoxic influence on retinal ganglion cells. Because of increased production of death-promoting substances, including TNF-α, alterations in the functional state of glial cells in response to these stressors lead to retinal ganglion cell death. These findings reveal a novel pathogenic mechanism for retinal ganglion cell death in glaucoma and provide a novel therapeutic target for neuroprotection in the treatment of glaucomatous optic neuropathy.
Footnotes
This study was supported in part by The Glaucoma Foundation (New York, NY; to G.T.), National Eye Institute Grant EY12314 (Bethesda, MD), Glaucoma Research Foundation (San Francisco, CA; to M.B.W.), and an unrestricted grant to Washington University School of Medicine, Department of Ophthalmology and Visual Sciences, from Research to Prevent Blindness (New York, NY).
Correspondence should be addressed to Dr. Martin B. Wax, Department of Ophthalmology and Visual Sciences, Washington University School of Medicine, Box 8096, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail: Wax{at}vision.wustl.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}