

Abstract
The cytokines that signal through the common receptor subunit gp130, including ciliary neurotrophic factor (CNTF), interleukin-6, leukemia inhibitory factor (LIF) and oncostatin M, have pleiotropic functions in CNS development. Given the restricted expression domain of the CNTF receptor α (CNTFR) in the developing forebrain germinal zone and adult forebrain periventricular area, we have examined the putative role of CNTFR/LIFR/gp130-mediated signaling in regulating forebrain neural stem cell fate in vivo and in vitro. Analysis ofLIFR-deficient mice revealed that a decreased level of LIFR expression results in a reduction in the number of adult neural stem cells. In adult LIFR heterozygote (+/−) mice, the number of neural stem cells and their progeny in the forebrain subependyma and TH-immunoreactive neurons in the olfactory bulb were significantly reduced. Intraventricular infusion of CNTF into the adult mouse forebrain, in the absence or presence of epidermal growth factor (EGF), enhanced self-renewal of neural stem cells in vivo. Analyses of EGF-responsive neural stem cells proliferating in vitro found that CNTF inhibits lineage restriction of neural stem cells to glial progenitors, which in turn results in enhanced expansion of stem cell number. These results suggest that CNTFR/LIFR/gp130-mediated signaling supports the maintenance of forebrain neural stem cells, likely by suppressing restriction to a glial progenitor cell fate.
- neural stem cells
- ciliary neurotrophic factor
- leukemia inhibitory factor receptor-deficient mice
- gp130
- stem cell maintenance
- astrocyte differentiation
Neural stem cells that can self-renew and give rise to various types of neurons and glia may play a major role in mammalian CNS development and continue to function throughout adulthood (for review, see Alvarez-Buylla and Temple, 1998;Gage, 2000; van der Kooy and Weiss, 2000). Epidermal growth factor (EGF)-responsive cells in the embryonic and postnatal forebrain germinal zone, later in the adult subependyma, behave as neural stem cells in vivo and in vitro (Reynolds and Weiss, 1992; Morshead et al., 1994) and can be propagated for extended periodsin vitro in the presence of EGF (Reynolds and Weiss, 1996). However, neural stem cells in the adult subependyma are relatively quiescent (Morshead et al., 1994). The average cell-cycle time is ∼15 d, and the number of neural stem cells does not appear to change throughout adult life, whereas proliferation of their progeny is decreased during aging (Tropepe et al., 1997; Morshead et al., 1998). Despite extensive analysis of factors that regulate proliferation and differentiation of neural stem cells and their progeny (for review, seeCameron et al., 1998; Lillien, 1998a,b), the mechanisms that maintain neural stem cells in an undifferentiated state are largely unknown. The principle exception is Notch signaling which, through lateral inhibition, may regulate the commitment of stem cells in CNS development (de la Pompa et al., 1997; Ohtsuka et al., 1999; Wakamatsu et al., 1999; Nakamura et al., 2000; Gaiano et al., 2000) (for review, see Artavanis-Tsakonas et al., 1999; Kageyama and Ohtsuka, 1999).
Cytokines related to IL-6, such as cardiotropin-1, ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), and oncostatin M (OSM), all transmit their signals into a cell through their respective receptor complex containing either homodimers of gp130 or heterodimers comprising gp130 and a partner: LIFR (for cardiotropin-1, CNTF, LIF, and OSM) or the OSM-specific receptor (OSMR) (for review, see Taga and Kishimoto, 1997; Heinrich et al., 1998). Numerous in vitroand in vivo studies have shown that these cytokines have pleiotropic actions on many different cell types (for review, seeHeinrich et al., 1998; Turnley and Bartlett, 2000). In vitrostudies of the developing CNS have shown that the activation of gp130 by these cytokines promotes differentiation and/or survival of astrocytes (Hughes et al., 1988; Johe et al., 1996; Bonni et al., 1997;Murphy et al., 1997; Gadient et al., 1998; Koblar et al., 1998; Rajan and McKay, 1998; Yanagisawa et al., 1999), oligodendrocytes (Mayer et al., 1994; Gard et al., 1995; Barres et al., 1996; Murphy et al., 1997;Gadient et al., 1998; Marmur et al., 1998), and specific types of neurons (Ip et al., 1991; Oppenheim et al., 1991; Martinou et al., 1992; Richards et al., 1996; Marz et al., 1997; Murphy et al., 1997;Galli et al., 1999). The compelling evidence that these cytokines are essential for maintenance of embryonic stem cells (Smith et al., 1988;Williams et al., 1988; Conover et al., 1993; Rose et al., 1994; Yoshida et al., 1994; Pennica et al., 1995) prompted us to consider whether these cytokines may be candidate molecules for regulating the maintenance of neural stem cells. Although analysis of null mutants ofCNTFR, LIFR, or gp130 has confirmed that these receptors are necessary for differentiation of astrocytes and survival of motor neurons in vivo (DeChiara et al., 1995; Li et al., 1995; Ware et al., 1995; Nakashima et al., 1999), no information regarding their putative roles in the maintenance of neural stem cells has been reported.
To test the hypothesis that gp130-mediated signaling plays a role in the maintenance of neural stem cells, we first used LIFRknock-out mice. We find that LIFR/gp130-mediated signaling is necessary for the maintenance of neural stem cells in vivo. Moreover, we analyzed the actions of CNTF (which activates LIFR/gp130 complex through CNTFR) on self-renewal and expansion of EGF-responsive neural stem cells in vivo and in vitro. We find that CNTF supports the self-renewal of EGF-responsive neural stem cells by suppressing their lineage restriction to glial progenitor cells.
MATERIALS AND METHODS
Animals and genotyping. LIFR+/− mice generated on the B6,129/J genetic background and C57BL6 STOCK (for expanding theLIFR+/− mice) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were then bred to allow for the generation of homozygote, heterozygote, and wild-type littermates (Koblar et al., 1998). The genotyping of mice carryingLIFR mutations was performed as described previously (Ware et al., 1995). CD-1 mice stocks were maintained in the University of Calgary Bioscience Animal Resources Center.
Neural stem cell culture and growth factors. Generation and differentiation of spheres from embryonic and adult forebrain were performed as described previously with minor modifications (Reynolds and Weiss, 1992; Reynolds et al., 1992). Briefly, striato-pallidum complexes were removed from mouse embryos at E14 and collected into PBS containing 0.6% glucose, penicillin (50 U/ml), and streptomycin (50 U/ml; both from Life Technologies, Gaithersburg, MD) and then transferred into the standard culture medium composed of DMEM–F-12 (1:1), glucose (0.6%), glutamine (2 mm), sodium bicarbonate (3 mm), and HEPES buffer (5 mm), insulin (25 μg/ml), transferrin (100 μg/ml), progesterone (20 nm), putrescine (60 μm), and selenium chloride (30 nm) (all from Sigma, St. Louis, MO, except glutamine from Life Technologies). For adult neural stem cell cultures, medial and lateral portions of the lateral ventricle subependyma from the adult brain were dissected from both hemispheres, pooled together, subsequently cut into 1 mm2 fragments, and transferred into the standard culture medium containing 1.33 mg/ml trypsin, 0.67 mg/ml hyaluronidase, and 0.2 mg/ml kynurenic acid (all from Sigma). After 30 min at 37°C, the tissue was transferred to the standard culture medium containing 0.7 mg/ml trypsin inhibitor (Roche Diagnostics, Laval, Quebec, Canada). Tissue pieces were mechanically dissociated with micropipettes. Cells were seeded at various densities into the standard culture medium, which also contained EGF (human recombinant; Peprotech, Rocky Hill, NJ) and/or CNTF (rat recombinant; kindly provided by Dr. Robert Dunn, Montreal General Hospital Research Institute, Peprotech or R & D, Minneapolis, MN) where indicated. Cells were cultured for 7 d in vitro (DIV) and formed floating cell clusters (spheres), then subsequently processed for various types of experiments described in the text. All the mice for culture experiments were killed by cervical dislocation.
Implantation of the osmotic pumps and growth factor infusion. Sixteen 8-week-old CD-1 mice (Charles-River, Laval, Quebec, Canada) were anesthetized with sodium pentobarbital (120 mg/kg, i.p.) and implanted with osmotic pumps (Alzet 1007D; Alza, Palo Alto, CA). The cannulas were located in the right lateral ventricle (anteroposterior +0.2 mm, lateral +0.8 mm to bregma, and dorsoventral −2.5 mm below dura with the skull leveled between lambda and bregma). Human recombinant EGF (33 μg/ml) and/or rat recombinant CNTF (33 μg/ml) were dissolved in 0.9% saline containing 1 mg/ml mouse serum albumin (Sigma). Each animal was infused for 6 d with either vehicle alone, EGF, CNTF, or EGF plus CNTF at a flow rate of 0.5 μl/hr, resulting in a delivery of 400 ng/d of each growth factor.
Bromodeoxyuridine labeling and detection. To identify the constitutively proliferating population in the adult mouse subependyma of the lateral ventricles, mice were injected with bromodeoxyuridine (BrdU) (Sigma) (120 mg/kg, i.p.; dissolved in 0.007% NaOH in phosphate buffer) every 2 hr for 10 hr and killed 0.5 hr after the last injection. Brains were processed for immunohistochemistry as described below. Rat monoclonal anti-BrdU (1:50; Sera-Lab, Sussex, UK) (primary antibody) and biotinylated-donkey anti-rat (1:200; Jackson ImmunoResearch, West Grove, PA) (secondary antibody) with streptavidin–Cy3 (1:2000; Jackson ImmunoResearch) were used for BrdU detection. To assess cell proliferation within spheres, 1 μm of BrdU was administrated to the cultures at 3 DIV. After 24 hr of incubation, spheres were mechanically dissociated and plated onto poly-l-ornithine-coated coverslips for 2 hr, then processed for immunocytochemistry using Amersham Cell Proliferation Assay kit (Amersham-Pharmacia, Oakville, Ontario, Canada) according to the manufacturer's instructions.
Antibodies and immunohistochemistry. The primary antibodies (final dilution and source) used in this study were as follows: rabbit polyclonal anti-bovine GFAP serum (1:400; Biomedical Technologies Inc., Stoughton, MA); rabbit polyclonal anti-human GFAP IgG (1:50; Sigma); mouse monoclonal anti-O4 IgM (1:20; Roche Diagnostics); mouse monoclonal anti-bovine S100β IgG (1:100; Sigma); rabbit polyclonal anti-tyrosine hydroxylase (1:100; Pel-Freez Biologicals, Rogers, AR); and mouse monoclonal anti-β-III tubulin IgG (1:100 or 1000; Sigma).
Adult mice were anesthetized and perfused transcardially with 4% paraformaldehyde and 0.16 m phosphate buffer, pH 6.9. Brains were post-fixed in the perfusing solution overnight at 4°C. Brains were cryoprotected for at least 24 hr in 15% sucrose in 0.1m PBS, pH 7.2. Serial 14 μm coronal cryosections (−20°C) of mouse forebrain were mounted directly onto gelatin-coated slides, then processed for immunohistochemistry. Sections were post-fixed with 100% acetone for 30 sec at room temperature, then washed three times (10 min each) with PBS. For the detection of BrdU labeling, sections were initially treated with 1 m HCl for 30 min at 65°C to denature cellular DNA before the immunohistochemistry. Sections were then incubated for 24 hr (4°C) in primary antibody diluted in washing solution containing 0.3% Triton X-100. After incubation in the primary antibody, sections were washed (as above) and incubated with the regular secondary antibodies conjugated to fluorescein or rhodamine for 2 hr or with the biotinylated secondary antibodies 1 hr at room temperature followed by incubation with streptavidin–Cy3 (1:2000; Jackson ImmunoResearch) or streptavidin-horseradish peroxidase (HRP) (1:5000; Chemicon Temecula, CA) for 1 hr at room temperature, in the presence of Hoechst 33258 nuclear stain (0.015 mg/ml stock solution diluted to 0.001 mg/ml; Roche Diagnostics). Sections were washed three times (5 min each), and where appropriate incubated in DAB containing HRP substrate solution (Vector Laboratories, Burlingame, CA) for 5 min followed by a rinse with water, then coverslipped with FluorSave (Calbiochem, La Jolla, CA) or Permount (Fisher Scientific, Pittsburgh, PA) and examined under a Zeiss axiophot microscope. Immunostaining for neurons, astrocytes, and oligodendrocytes in differentiated, sphere-derived cells (on coverslips) was performed as first described by Reynolds and Weiss (1996).
RESULTS
Reduced numbers and function of neural stem cells in the adultLIFR+/− forebrain
Neural stem cells can proliferate in vitro in the presence of EGF and/or FGF-2 to form floating cell clusters termed spheres. These neural stem cells can be maintained for extended periods of time through multiple passages (Reynolds and Weiss, 1996). Clonal analysis has demonstrated that these spheres can be passaged individually, and the resultant secondary spheres generate neurons, astrocytes, and oligodendrocytes. Thus, one can conclude that the majority (if not all) of the sphere-forming cells express the features of neural stem cells. We used this culture system to examine the function of LIFR/gp130 signaling on the maintenance of neural stem cells. Initially, we asked whether self-renewal or expansion of neural stem cells derived from developingLIFR−/− mice, assessed by sphere formation assay in vitro, would differ from those from wild-type littermates (wt). The number of primary, EGF-responsive sphere-forming cells was not reduced in the ganglionic eminence of LIFR−/− mice at embryonic day 14 (E14) (∼1% of the cells plated in bothwt and LIFR−/−; data not shown). However, when LIFR−/−neural stem cells were maintained in EGF-containing media in populations of spheres for multiple passages, the number of sphere-forming cells cultures decreased to ∼2% of total cells plated after five passages (Fig.1A), whereupon it declined precipitously, and sphere-forming was entirely lost after seven passages. However, the decrease (between first and second passage) in the sphere formation by wt neural stem cells reached a plateau (Fig. 1A) (4–5% of total cells plated) at the second passage and remained at this level for as many as 10 passages (extent of our analysis). Moreover, during the passage of the LIFR−/− cells at high density, by the sixth passage the spheres developed into cells possessing a flattened morphology, which gradually attached to the culture flask. Finally, after seven passages, theLIFR−/− cells appeared as proliferating fibroblastic-like cells forming monolayers (Fig.1B). When these fibroblastic-like cells were plated in differentiation conditions after 10 passages, most of them quickly died (within 3 d) without any sign of differentiation, whereaswt cells from passage 10 spheres were able to differentiate into neurons and glia (data not shown) (Reynolds and Weiss, 1996). These results suggest that LIFR is required for long-term maintenance of sphere-forming neural stem cells in vitro.

Limited self-renewal and expansion of EGF-responsive stem cells derived from embryonicLIFR−/− mice. Dissociated cells from E14 ganglionic eminences (GE) of wild-type (wt) andLIFR−/− (−/−) mice were plated at an initial density of 2 × 105cells/ml and cultured in EGF-containing growth medium. Resultant spheres were passaged at a density of 5 × 104cells/ml every 7 d. At each passage a portion of the spheres (wt vs −/−) was assayed for self-renewal and expansion by counting the number of secondary spheres per total cells formed in low-density cultures (plated at 1 × 103 cells/ml).A, Frequency of sphere-forming cells in each passage of the cultures. *p < 0.05 versus −/− cultures; two-tailed t test (n = 4).B, Morphological changes of the cells and spheres derived from wt andLIFR−/− (−/−) mice after seven and 10 passages. P7, Passage 7; P10, passage 10.
Given the results above and that EGF-responsive neural stem cells emerge late in development (E12–E14) (Tropepe et al., 1999), we hypothesized that function of LIFR/gp130 signaling on the maintenance of neural stem cells in vivo may only be apparent in the postnatal to the adult period. However, because homozygous mutants die immediately at birth (as described previously by Ware et al., 1995), we could only analyze heterozygotes for postnatal forebrain neural stem cell numbers and function. Initially we examined the number of EGF-responsive sphere-forming cells derived from the postnatal day 0 (P0) lateral ventricles of LIFR+/−and wt mice. We did not observe any difference between the number of spheres derived from wt andLIFR+/− mice (data not shown). Therefore, we decided to perform further sphere-formation assays and immunohistochemical analysis in adult forebrains fromLIFR-deficient mice. In adultLIFR+/− mice (7–12 months old) we found a significant reduction in the constitutively proliferating population of cells, which are the in vivo progeny of EGF-responsive forebrain neural stem cells (Morshead et al., 1994), in the subependyma of the lateral ventricles (Fig.2). Bromodeoxyuridine immunohistochemistry showed a 55% reduction in the number of cells in the constitutively proliferating population in the subependyma of heterozygotes (Fig. 2A,D,G). In addition, the volume of the periventricular area was significantly reduced (Fig.2B,C,E,F,Table 1), whereas there was no significant change in the pattern of S100β expression in the ependyma or GFAP expression in the subependyma. We then asked whether the numbers of EGF-responsive neural stem cells would be reduced in heterozygote forebrains. We found that the number of EGF-responsive sphere-forming cells in the periventricular area of the lateral ventricle was reduced by 37% (Fig. 2H) in 4- to 8-month-old LIFR+/− mice, compared with wt. We did not observe any change in the expansion and multipotency of spheres derived fromLIFR+/− mice (data not shown). These results clearly support the involvement of the LIFR in the maintenance of EGF-responsive neural stem cells and in turn the generation of the constitutively proliferating population in the adult forebrain.

Reduction in the number of EGF-responsive neural stem cells and subependyma cell proliferation in theLIFR+/− mouse forebrain.A–F, Immunofluorescence micrographs of coronal sections through the periventricular area of the adult forebrain lateral ventricle in a wt and aLIFR+/− mouse. The entire constitutively proliferating population was labeled by 10 hr injections of BrdU (red). GFAP-immunoreactive cells in subependyma and S100β-immunoreactive ependymal cells are visualized by FITC (green) fluorescence. Each section was counterstained by Hoechst 33258 (blue). Scale bars, 50 μm. G, Quantification of the number of proliferating (BrdU+) cells in the subependyma of wild-type andLIFR+/− mice (7- to 12-month-old females). The total number of BrdU+ cells in the subependyma between the rostral tip of the genu of the corpus callosum and the crossing of the anterior commissure from every 10th section per brain were counted.H, Reduced generation of spheres from the periventricular area of the lateral ventricles inLIFR+/− (4- to 7-month-old female) mice. Lateral and medial aspects of the periventricular area were dissected from both hemispheres and cultured in EGF containing growth media. *p < 0.05 and **p < 0.01 versus wt, two-tailed t test (n = 3).
Reduction in the volume of the periventricular layers of the lateral ventricle in LIFR+/− adult mice
A subpopulation of neuronal precursors produced by neural stem cells in the lateral ventricles migrates toward the olfactory bulb through the rostral migratory stream (Lois and Alvarez-Buylla, 1994). The ultimate fates of these precursors are as olfactory interneurons localized to the granule and periglomerular layers. Thus, one would hypothesize that the reduction of neural stem cells and their progeny in the periventricular area of the lateral ventricle would result in a reduction in the number of these interneurons. This assumes that the general integrity of the rostral migratory stream is not disrupted, and this is what we observed when comparing sagittal sections of LIFR+/− andwt adult mice (data not shown). To ascertain the effect of the reduction of neural stem cells (because of reduced expression of LIFR) on adult neurogenesis in the subependyma of the lateral ventricle, we compared the number of TH-immunoreactive interneurons in the periglomerular layers of olfactory bulbs betweenLIFR+/− and wt adult mice (7–12 months old). As shown in Figure3, the number of TH-immunoreactive cells was significantly reduced (33%) inLIFR+/− mice. Specifically, it appears as though the number of migrating TH-immunoreactive neurons in the external plexiform layer is reduced (Fig. 3A,B) along with a reduced density of total cells (wt: +/− = 1073 ± 9, 925 ± 18* cells/mm2; *p = 0.0016; n = 3). Total cell number was ascertained by Nissl staining in five 0.04 mm2 fields in the fifth coronal section from the anterior end of the cortex. This suggests that the reduction of TH-containing neurons rather than reduced expression of TH protein expression itself underlies the reduction in the number of TH-immunoreactive cells. Thus, the reduction of the adult forebrain neural stem cell number was indeed correlated with the reduction in the number of TH-immunoreactive interneurons of the olfactory bulb. We could not observe any gross morphological changes in the size or cytoarchitecture of the olfactory bulb inLIFR+/− mice (data not shown).

Reduction in the number of tyrosine hydroxylase-expressing interneurons in the olfactory bulbs ofLIFR+/− mice. TH immunohistochemistry of the coronal sections (30-μm-thick) of olfactory bulbs from wild-type (A) andLIFR+/− (B) mice (7–12 months old) are shown. D, Quantification of the TH-immunoreactive neurons in the periglomerular layer. *p < 0.01 versus wt, two-tailedt test (n = 3). All the TH-IR cells in the medial half of the olfactory bulb in five 30-μm-thick sections of every fifth section from the anterior end of the cortex illustrated in C were counted. epl, External plexiform layer; gl, glomerular layer;OB, olfactory bulb; ov, olfactory ventricle.
CNTF enhances self-renewal of EGF-responsive neural stem cellsin vivo
The maintenance of a neural stem cell could be supported by two distinct biological activities: cell survival and/or self-renewal. Because failure of either one would result in the extinction of neural stem cells, accompanied by the reduction of their differentiated progeny in the long-term, it is difficult to distinguish which activity is regulated by any one extrinsic factor. However, if such a factor supports survival of neural stem cells, a short exposure to the exogenous factor should not result in significant increases in stem cell number, because forebrain neural stem cells divide very slowly (average cell-cycle time is ∼15 d) and maintain their number throughout adulthood (Tropepe et al., 1997; Morshead et al., 1998). Conversely, if the factor supports self-renewal of neural stem cells, exposure to the exogenous factor should increase the number of stem cells in a manner that is dependent on the frequency of cell division. To begin addressing the mechanism by which LIFR/gp130-mediated signaling supports the maintenance of forebrain neural stem cells, we infused CNTF into the lateral ventricles of adult mice for 6 d, in the absence or presence of EGF. We chose CNTF to activate the gp130 signaling in subependymal cells, because the ligand and its specific receptor subunit CNTFR are expressed in the developing and adult CNS (Ip et al., 1993; Seniuk-Tatton et al., 1995). Particularly noteworthy is that CNTFR is predominantly expressed in a restricted manner in the adult forebrain periventricular area (Ip et al., 1993), where EGF-responsive neural stem cells reside (Morshead et al., 1994). Previous studies have shown that continuous infusion of EGF enhances proliferation and expansion of neural stem cells in vivo as well as in vitro (Craig et al., 1996; Kuhn et al., 1997). In the present study (Fig. 4) a 6 d infusion of CNTF into the lateral ventricles resulted in a slight increase (24%; p = 0.029 vs vehicle infusion;n = 3) in the number of subsequently derived EGF-responsive sphere-forming cells. When the in vivoproliferation of neural stem cells were enhanced by the infusion of EGF, co-infusion of CNTF resulted in a much greater increase (41%;p = 0.0033; n = 4) in the number of subsequently derived sphere-forming cells. These results suggest that CNTFR/LIFR/gp130-mediated signaling supports self-renewal of neural stem cells rather than their survival, given that the actions of CNTF were greater (41 vs 24%) when the frequency of neural stem cell proliferation was increased by concomitant exposure to EGF.

CNTF enhances the expansion of EGF-responsive neural stem cells in vivo. Sixteen 8-week-old CD-1 mice were infused with the following: vehicle alone, rat recombinant CNTF (33 μg/ml), human recombinant EGF (33 μg/ml), or EGF plus CNTF into the right lateral ventricle for 6 consecutive days, followed by sphere formation assay as described in Materials and Methods and legend to Figure 2. *p < 0.05 versus VEH and **p < 0.01 versus EGF, two-tailed ttest (n = 3 or 4).
CNTF enhances the expansion of EGF-responsive neural stem cellsin vitro
The enhancement of neural stem cell self-renewal may be a result of either increased proliferation or an inhibition of lineage commitment–restriction during proliferation. Because there are no unambiguous markers for neural stem cells, such analyses of the mechanisms of CNTF actions would be difficult to examine in vivo. To determine how CNTF supports the self-renewal of neural stem cells, we analyzed EGF-responsive stem cells in vitroin the presence of EGF plus CNTF or EGF alone. We used primary cultured spheres from E14 forebrain ganglionic eminences (striatal-pallidal primordia), grown in the EGF-containing growth media, as an enriched source of EGF-responsive neural stem cells for the following experiments, unless otherwise noted. The characteristics of E14 neural stem cells expanded in vitro are virtually identical to those derived from the adult forebrain, regarding self-renewal expansion and multipotency (Reynolds and Weiss, 1992, 1996). The ability to easily generate large numbers of enriched neural stem cells is why we chose the embryonic counterparts to ascertain the mechanism of action of CNTF (presumably not specific in vitro to adult neural stem cells) on the expansion of EGF-responsive neural stem cells. Also, because primary cultures initially contain small number of neural stem cells (<0.1%) and a large number of differentiated neurons, we used an enriched (relatively purified to 10–20% neural stem cells; Reynolds and Weiss, 1996) population selectively expandedin vitro in the presence of EGF to minimize indirect effects. The basic protocol is illustrated in Figure5.

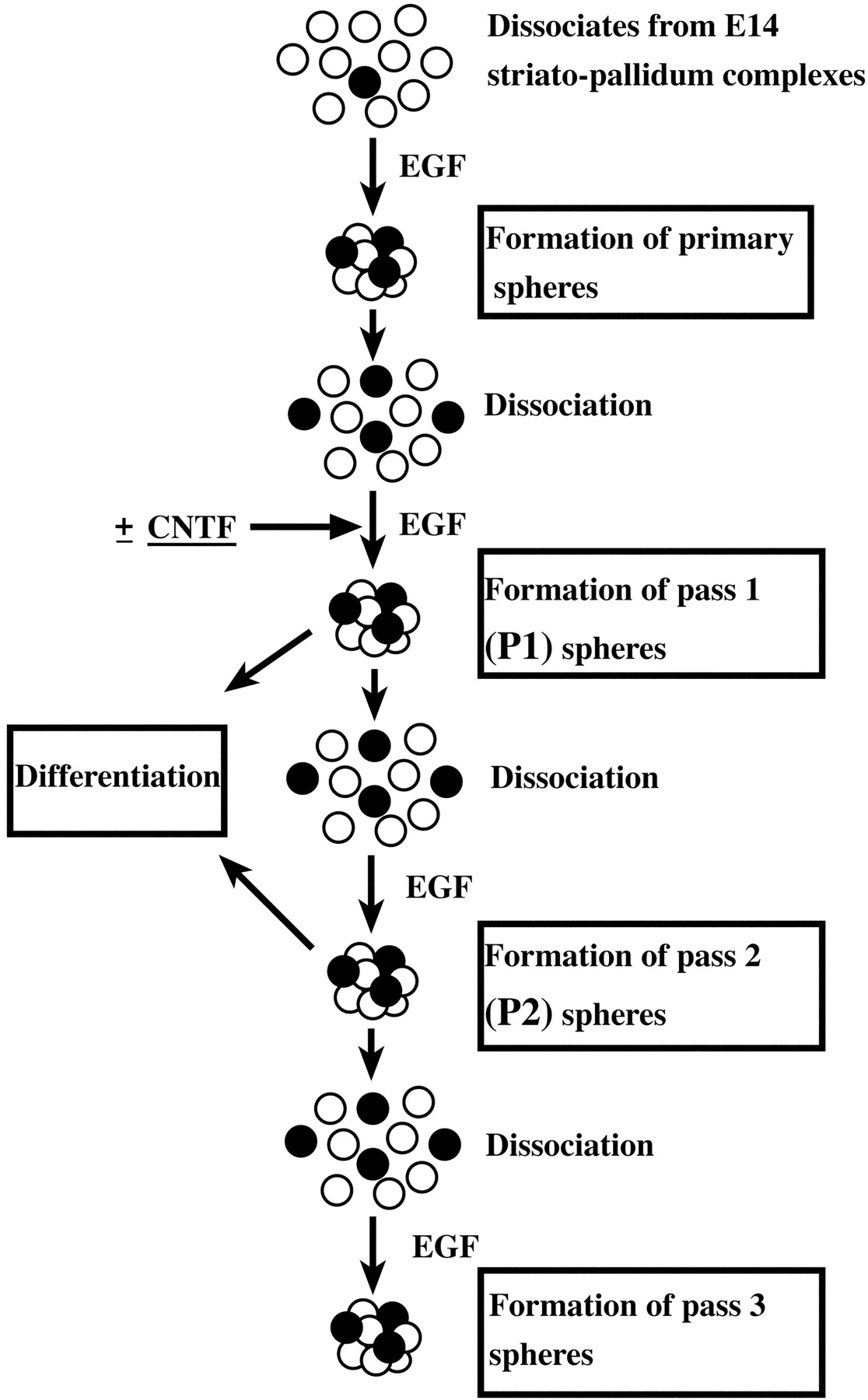
The basic experimental protocol for the in vitro assessment of neural stem cell activity in this study. A neural stem cell (black circle) can be expanded by the formation of a clonally derived cell cluster termed a sphere in EGF-containing growth medium. Neural stem cells are thus enriched by generation of primary spheres from dissociates of the E14 striato-pallidum complexes. These primary spheres containing a number of neural stem cells (∼20%; Reynolds and Weiss, 1996) are dissociated and cultured in populations (5 × 104cells/ml) for 7 d or clonally (150 cells/ml per 9.6 cm2) for 12–13 d, in EGF or EGF plus CNTF-containing medium, to obtain the next generation of spheres (P1). P1 spheres are then assessed for the following: (1) their expandability by dissociation and generation of secondary sphere (P2) formation and (2) multipotency by plating and allowing them to differentiate for 7 d in the absence of mitogens or cytokines. P2 spheres from clonally derived P1 spheres are similarly assessed for their expandability (to form P3 spheres) and multipotency.
Single-cell suspensions (derived from spheres generated in primary culture) were used as the source of neural stem cells and plated at a density of 5 × 104 cells/ml or at clonal density of 150 cells/ml). These cells generated P1 spheres (Fig. 5) during a further 7 d in vitro, in EGF-containing culture medium, in the absence or presence of CNTF (20 ng/ml). No P1 spheres formed in the presence of CNTF alone (data not shown). There was no difference in the absolute number of P1 spheres generated, at clonal density, in the presence of EGF compared with EGF plus CNTF (Fig. 6A). This confirms the contention that CNTF does not act as a neural stem cell survival factor. Furthermore, an examination of BrdU incorporation (Fig. 6B) or total cell number (Fig. 6C) within P1 spheres found no significant difference between those generated in EGF compared with those grown in EGF plus CNTF. These results suggest that the general proliferation and survival of cells within P1 spheres is not affected by the presence of CNTF.

CNTF enhances expansion of EGF-responsive neural stem cells in vitro. Primary EGF-derived spheres, which contain stem cells from E14 ganglionic eminences, were dissociated into a single cell suspension and plated onto 6-well plates or 25 cm2 flasks at a density of 150 or 5 × 104 cells/ml, respectively. These cells were grown and formed P1 spheres in the presence of either EGF (20 ng/ml) or EGF plus CNTF (20 ng/ml each) for 7 d in vitro.A, The effect of CNTF on the survival of EGF-responsive neural stem cells was examined. The absolute number of P1 EGF-generated spheres was compared with the absolute number of P1 EGF plus CNTF-generated spheres, at a density of 150 cells/ml per 9.6 cm2 (two-tailed Student's t test;n = 5 independent cultures). B, BrdU labeling was performed to examine the effect of CNTF on the proliferation of EGF-responsive neural stem cells. One micromolar BrdU was administrated into P1 cultures at 3 d in vitro. Twenty-four hours later, populations of spheres were mechanically dissociated and plated onto poly-l-ornithine-coated coverslips at a density of 1 × 105cell/cm2. The cells were processed for BrdU immunocytochemistry 2 hr after plating (two-tailed Student'st test; n = 4 independent cultures).C, Total cell numbers at 7 d in vitro. Population of 7 d in vitro P1 EGF-generated spheres and P1 EGF plus CNTF-generated spheres were trypsinized and mechanically dissociated to count the number of cells (two-tailed Student's t test; n = 4 independent cultures). D, During the growth of P1 spheres with EGF, concomitant exposure to CNTF was constitutive (full 7 d), transient (first 3 d), or delayed (last 4 d). Two hundred micrometer diameter P1 spheres were then exposed to 0.25% trypsin-EDTA solution for 5 min followed by quenching the enzymatic digestion by trypsin inhibitor (1 mg/ml; Sigma) containing media and then transferred individually into 96-well plates filled with EGF-containing growth media. Each P1 sphere was mechanically dissociated into a single cell suspension using micropipettes and cultured for another 7 d. Then resultant P2 spheres were counted, as their number assesses expansion of neural stem cells proliferated in each condition. **p < 0.01 versus control EGF cultures, Tukey's honestly significant difference (HSD) test (n = 120 from four independent cultures). Note that the media in all the cultures shown in the right panel were washed out and replaced at 3 d in vitro.
To ascertain whether CNTF could regulate the expansion of neural stem cells in vitro as we had observed in vivoand to determine if continuous exposure to the cytokine was necessary, we generated P1 spheres in the absence or presence of CNTF for various periods of time (constitutively, for the full 7 d; transient, for the first 3 d; or delayed, for the last 4 d). To quantify the possible expansion actions of CNTF, the P1 spheres from the different culture conditions were dissociated individually in 96 well plates and were allowed to form P2 spheres (Fig. 5) in culture medium containing EGF only (no CNTF) for 7 d. After 7 d in vitro, the numbers of P2 spheres per well were counted. As shown in Figure6D, exposure of neural stem cells to CNTF during the formation of P1 spheres resulted in a significant increase in the number of P2 spheres derived from a single P1 sphere. A 59% increase was observed with P1 spheres that had been generated with a constitutive exposure to CNTF, whereas either transient (first 3 d) or delayed (last 4 d) exposure were somewhat more effective (141 and 168% increases, respectively). However, when dissociates from P1 spheres were exposed to CNTF only for the first 24 hr (before the first cell division), we did not observe a significant increase in the number of P2 spheres (data not shown). These results suggest that CNTF actions on the expansion of EGF-responsive neural stem cells require cell division and/or cell–cell interaction. To confirm the results obtained by the analysis of individual spheres described above and to gain an appreciation of the actual numbers of sphere-forming cells, we performed the following experiments. Dissociated P1 spheres, grown in the presence of EGF alone or EGF plus CNTF, were plated at clonal density in the presence of EGF only. After 7 d we counted the number of P2 spheres generated, relative to the total number of cells plated. Similar to our results with single sphere analysis, we found that the P1 spheres grown in EGF plus CNTF produced 70% more P2 spheres (9.7 ± 1.8% of total cells) than those grown in EGF only (5.7 ± 1.8% of total cells; p = 0.0064;n = 3). Taken together, these results suggest that CNTF enhances self-renewal of EGF-responsive neural stem cells (production of secondary neural stem cells) without affecting cell proliferation or survival.
CNTF increases neural stem cell number by suppressing their lineage restriction to glial progenitors
In an effort to understand how CNTF enhances expansion of neural stem cells, we performed an examination of single P1 spheres grown at clonal density. Single-cell suspensions (Fig. 5) were plated at clonal density (<15 cells/cm2) and grown into P1 spheres as before in EGF-containing culture medium in the absence or presence of CNTF. P1 sphere formation under these clonal density experimental conditions required 12–14 d. Virtually all P1 spheres grown at this density should be clonally derived (Tropepe et al., 1999). The media in all culture conditions were replaced with EGF-containing culture medium (no CNTF) at 6 d in vitro, so that growing P1 spheres were exposed to CNTF only transiently. The resultant P1 spheres were individually dissociated into EGF alone to form P2 spheres to examine self-renewal and expansion. Also, individual P1 spheres were allowed to differentiate intact for 7 d to examine their multipotent phenotype. As shown in the left-hand column of Table 2, when P1 spheres that had been generated in the presence of EGF plus CNTF were dissociated and replated in EGF alone, a significantly greater number of P2 spheres (22 ± 2 P2 spheres/P1 sphere) were produced when compared with P1 spheres grown in EGF alone and subcultured into P2 spheres in EGF alone (13 ± 1 P2 spheres/P1 sphere). This is similar to what was observed in the high-density cultures (Fig.6D), suggesting that CNTF acts directly on neural stem cells and/or through cell–cell interactions within individual spheres. The difference in the average number of secondary spheres from a single P1 sphere, grown in EGF alone, between high-density cultures (Fig. 6D) and clonal cultures (Table 2) may be attributable to differences in the plating density and/or culture periods. On the other hand, the percentage of P1 spheres that were capable of expansion (produce more than one P2 sphere) when dissociated and plated in EGF alone was not different when comparing P1 EGF spheres (85.9 ± 4.6%) and P1 EGF plus CNTF (97.9 ± 1.0%) spheres. Moreover, when intact P1 spheres were examined for the presence of neurons, astrocytes, and oligodendrocytes (N plus A plus O) again, no difference was observed in the percentage of P1 EGF spheres (78.2 ± 4.1%) and P1 EGF plus CNTF spheres (85.3 ± 1.5%) that contained all three CNS cell types. Thus, P1 spheres generated in EGF plus CNTF produced a greater number of secondary neural stem cells (P2 spheres) than those generated in EGF alone, whereas their ability to produce the three principal differentiated cell types remained unaffected.
Clonal analysis suggests that CNTF suppresses a lineage restriction of EGF-responsive neural stem cells to glial progenitors
Two possible mechanisms (at least) could explain the increased number of secondary stem cells (increased P2 spheres/single P1 sphere) caused by the presence of CNTF. Either CNTF enhances the numbers of symmetric cell divisions of neural stem cells within a growing P1 sphere or CNTF prevents the lineage restriction of newly generated neural stem cells (within a P1 sphere) to a more differentiated (less expandable, less multipotent) phenotype. In both scenarios, one would see a greater number of P2 spheres (secondary neural stem cells) generated from a single P1 sphere. However, if this increase was simply attributable to an increased number of symmetric cell divisions, then one would predict that the phenotype of P2 spheres (expansion and multipotency) should not be different, whether derived from P1 EGF spheres or P1 EGF plus CNTF spheres. On the other hand, should CNTF prevent the lineage restriction of newly generated multipotent cells to a more differentiated phenotype, P2 spheres derived from P1 EGF plus CNTF spheres should be more expandable and multipotent than P2 spheres derived from P1 EGF spheres. We tested this by examining the expansion and multipotency of P2 spheres, derived (in EGF alone) from single P1 EGF spheres or single P1 EGF plus CNTF spheres (right-hand column of Table 2). P2 spheres derived from EGF plus CNTF spheres could expand (71.3 ± 6.0%) to a greater extent than P2 spheres derived from EGF spheres (47.9 ± 4.8%; p < 0.05). Also, P2 spheres derived from EGF plus CNTF spheres were multipotent (87.4 ± 2.7%) to a greater extent than P2 spheres derived from EGF spheres (62.5 ± 6.7%; p < 0.05). At the same time, P2 spheres derived from EGF plus CNTF spheres were astrocyte- and oligodendrocyte-producing (A plus O) only (7.2 ± 2.2%) to a lesser extent than P2 spheres derived from EGF spheres (30.0 ± 4.2%; p < 0.05).
Taken together, these results show that when subcultured as individual spheres (P1 → P2) (Fig. 5), a proportion of EGF-responsive neural stem cells becomes restricted to glial progenitors. However, if exposed to CNTF during the formation of P1 spheres, the lineage restriction of a significant number of the EGF-responsive neural stem cells is suppressed, resulting in enhanced self-renewal and expansion in vitro.
CNTF enhances astrocyte differentiation but not commitment
It has been proposed that CNTFR/LIFR/gp130 signaling instructs stem cells to differentiate into astrocytes and inhibits neurogenesis (Johe et al., 1996; Bonni et al., 1997; Koblar et al., 1998). In contrast, we observed a CNTF-induced increase in the number of P2 sphere-forming neural stem cells (approximately twofold) during the proliferation of P1 neural stem cells (as shown above). The majority of cells in spheres are themselves not sphere-forming neural stem cells (Reynolds and Weiss, 1996), suggesting that the fate of most cells is more restricted. To understand the exact effect of CNTF on lineage commitment and differentiation of astrocytes during neural stem cell proliferation, we assessed the generation of astrocytes by EGF-responsive neural stem cells exposed to CNTF for different periods of time. First, we examined astrocyte production within growing spheres over time. Single cell suspensions (from primary EGF-derived spheres) were cultured in population in EGF-containing culture media in the absence or presence of CNTF for 3, 5, or 7 d in vitro. The resultant spheres were dissociated and then differentiated on poly-l-ornithine coated coverslips for 7 d to obtain full differentiation, followed by immunocytochemistry and cell counting. Surprisingly, we found that administration of CNTF during the proliferation of neural stem cells did not change astrocyte production in resultant spheres (Fig.7A). The number of astrocytes, characterized by GFAP expression and astrocytic morphology, was consistent at ∼60% of total cell number in both conditions throughout the culture period. We then examined the effect of CNTF on astrocyte differentiation. This was an effort to reconcile our findings with those previously reported (Johe et al., 1996; Bonni et al., 1997;Bartlett et al., 1998). Normally, GFAP-expressing cells are first detected 3 d after plating of spheres in differentiation conditions (data not shown). When cells were exposed to CNTF constitutively for 7 d during the growth of spheres, then dissociated and allowed to differentiate, we observed a significantly greater number of GFAP-expressing astrocytes just 2 hr after plating (Fig. 7B). On the other hand, there was no significant increase in GFAP-expressing cells when growing spheres were exposed to CNTF transiently (for the first 3 d), despite both these conditions giving the same number of secondary neural stem cells. These results suggest that CNTF enhances differentiation of precursors already committed to the astroglial lineage. On the other hand, our data suggest that CNTF does not promote uncommitted precursors, generated during the proliferation of EGF-responsive forebrain neural stem cells, to the astroglial lineage.

Effect of CNTF on astrocyte generation within spheres. A, Populations of spheres grown in the presence of either EGF or EGF plus CNTF (as described in Fig. 6) were allowed to differentiate at 3, 5, or 7 d in vitro. After mechanical dissociation into a single cell suspension, cells were plated onto poly-l-ornithine-coated coverslips in the EGF and CNTF-free media (see Materials and Methods) at a density of 1 × 105 cells/cm2 and cultured a further 7 d. Fully differentiated cells were fixed and processed for GFAP immunocytochemistry to assess astrocyte generation by neural stem cells. B, CNTF was administrated constitutively, first 3 d (transient) or last 4 d (delayed) during growth of spheres (as described in Fig. 6D) and then dissociated and plated as described in A. Two hours after, plated cells were fixed and processed for GFAP immunocytochemistry. **p < 0.01 versus control EGF cultures, Tukey's HSD test (n = 4).
DISCUSSION
CNTFR/LIFR/gp130 mediated signaling supports self-renewal of neural stem cells
The results of previous studies suggest that forebrain neural stem cells initially expand their numbers during early development, these numbers are reduced during the early postnatal period, and then maintained without any further reduction into adulthood (Tropepe et al., 1997, 1999; Morshead et al., 1998; Martens et al., 2000). Because forebrain neural stem cells appear to be localized principally to the periventricular area, the microenvironment in this area could be important in maintaining stem cell number. In this study, we found limited long-term self-renewal and expansion of EGF-responsive stem cells derived from E14LIFR−/− mice (Fig. 1) and a reduction in the number of neural stem cells and their progeny (Figs.2, 3) in the forebrain of adult mice with a 50% reduction in LIFR expression (LIFR+/− mouse). This suggests that there is a requirement for LIFR/gp130-mediated signaling in the maintenance of forebrain EGF-responsive neural stem cells. The first question that arises is how this signaling supports the maintenance of these neural stem cells. The maintenance of a neural stem cell could be supported by two distinct biological activities: cell survival and/or self-renewal. To address this issue, we focused on an examination of CNTF actions, because of its receptor (CNTFR) being expressed in a restricted manner in the developing and adult perventricular area (Ip et al., 1993), where EGF-responsive neural stem cells reside (Morshead et al., 1994). Our in vivo andin vitro studies suggest that CNTF enhances self-renewal of stem cells but not their survival. First, a 6 d infusion of CNTF into the lateral ventricle of the adult mouse forebrain resulted in a slight increase (24%) in the number of sphere-forming cells (Fig. 4). The fact that this action is significantly augmented (up to 41%) when the proliferation of neural stem cells was stimulated by the infusion of EGF suggests that CNTF does not support survival but rather self-renewal of neural stem cells. If CNTF was merely acting as a survival factor, one would expect that the relative increase in neural stem cell number should be similar, with or without the actions of EGF. Second, our in vitro analyses also showed no evidence suggesting CNTF is a survival factor to neural stem cells. The fact that the absolute number of P1 spheres generated by EGF plus CNTF (Fig.6A) is not different than that generated by EGF alone (as opposed to the differences in the number of P2 neural stem cells within the generated P1 spheres) (Fig. 6D, Table 2) most directly argues against an action of CNTF on survival. Thus, the reduction in the number of neural stem cells inLIFR+/− mice is likely to be caused by diminished self-renewal. It is still possible that environmental changes caused by the reduction in LIFR expression might have affected neural stem cell survival. In fact, the number of cell layers in the periventricular area was significantly reduced, which might have in turn modified the cytoarchitectural and chemical environment. Furthermore, there is a possibility that the neural stem cells might have simply reduced their responsiveness to mitogens such as EGF and FGF-2, which should have resulted in reduced sphere formation in vitro. However, the fact that CNTF does not affect cell proliferation but rather the number of secondary neural stem cells within spheres grown in the presence of EGF in vitrosuggests that this is not likely to be the case. Thus, taking bothin vitro and in vivo observations into account, CNTFR/LIFR/gp130-mediated signaling appears to support self-renewal of EGF-responsive neural stem cells rather than their survival.
Maintenance of the neural stem cell state
The next issue arising from our results is how CNTFR/LIFR/gp130-mediated signaling supports self-renewal of neural stem cells. It has been proposed that EGFR-mediated signaling biases late cortical and retinal progenitor cells toward glial lineages, in particular astrocytes, depending on the receptor and ligand concentration (Burrows et al., 1997), although in that study it was not clear how the EGFR signal influenced fate choice. When examined at clonal density (Table 2), EGF-responsive neural stem cells exhibited a gradual, time-dependent lineage restriction to glial progenitors, a process accompanied by a limitation of their expandability. It seems as though CNTFR/LIFR/gp130-mediated signaling antagonizes this process, resulting in the enhanced self-renewal of neural stem cells. In turn, this suggests that the EGFR signal may instruct EGF-responsive neural stem cells toward the glial lineage. On the other hand, it has been shown that the postnatal brain of theEGFR−/− mouse exhibited reduced proliferation and delayed differentiation of astrocytes yet was cytoarchitecturally normal at birth (Kornblum et al., 1998; Sibilia et al., 1998). Also, an in vitro study using EGFR-specific tyrosine kinase inhibitor suggested that the increased bias toward glial differentiation during development does not depend on EGFR signaling (Zhu et al., 2000). Taken together, it may be that the EGFR signal merely increases the frequency of deterministic fate restriction of EGF-responsive neural stem cells to the glial lineage by an increased number of cell divisions. If this is the case, CNTFR/LIFR/gp130-mediated signaling could support self-renewal of EGF-responsive neural stem cells by suppressing the lineage restriction to glial progenitor cells. In other words, this signal is required for the maintenance of the neural stem cell state.
Several studies have suggested that LIFR/gp130-mediated signaling instructs neural stem cells to the astrocytic lineage (Johe et al., 1996; Bonni et al., 1997; Bartlett et al., 1998), possibly as a result of a reduction in the number of differentiated neurons produced. However, none of those studies showed exactly which type of cells differentiated into astrocytes and how reduced neurogenesis appeared (e.g., fate change or suppression of differentiation) in response to gp130 signaling. Here, we found that CNTF enhances differentiation of astrocyte precursors (Fig. 7) but not the commitment of neural stem cells to an astrocytic fate. A recent report (Molne et al., 2000) that shows that early cortical precursors [which only include neural stem cells and neuroblasts but few glial precursors (Davis and Temple, 1994)] do not undergo LIF-mediated astrocytic differentiation, supports our observations. An alternative explanation is that some of the previous observations may be attributable to LIFR/gp130 signal enhancing GFAP gene and protein expression in neural stem cells and/or multipotent progenitors. Indeed, we have observed enhanced GFAP expression within spheres growing in the presence of EGF plus CNTF (data not shown). This may not be surprising given that GFAP is expressed in neural stem cells of the adult subependyma (Doetsch et al., 1999). In fact, GFAP gene expression is directly regulated by STAT3 (Bonni et al., 1997; Nakashima et al., 1999), a downstream intracellular mediator of the LIFR/gp130 signal.
Analysis of neural stem cell number and their proliferative cell-producing activities in vivo, inLIFR-deficient mice, indicates that the requirement of this signaling system for the maintenance of neural stem cells is relevant in the postnatal to adult period rather than during embryogenesis. There are a couple of possible explanations for this observation. First, some other signaling may compensate for the function of LIFR/gp130 signaling and may principally contribute to the self-renewal of more primitive neural stem cells that generate mostly neurons (Qian et al. 2000). In this case, it is likely not a different gp130 complex-mediated signaling, because there is no significant change in the number of neural stem cells in gp130-deficient mice at E14 (Ohtani et al., 2000). In fact, such an alternative maintenance pathway has been proposed for embryonic stem cells that can be maintained in the absence of LIF and the LIFR (Dani et al., 1998). Second, EGF-responsive neural stem cell to glial progenitor lineage restriction model, as proposed above, could be applicable in explaining this stage dependent effect of LIFR/gp130 signaling. The peak period of gliogenesis, perhaps largely contributed to by EGF-responsive neural stem cells exclusively localized in the subependyma (Martens et al., 2000), takes place primarily in the early postnatal period (Altman, 1966). During this period, the subependyma predominantly contains glial-restricted progenitors (Levison and Goldman, 1993; Levison et al., 1993; Young and Levison, 1996). If the lineage restriction of neural stem cells to glial progenitors occurs exclusively during this period and if LIFR/gp130 signaling suppresses that lineage restriction specifically, the significant effect of reduced expression of LIFR should not appear before the peak period of gliogenesis. We have summarized our hypothesis regarding when and how LIFR/gp130 signaling regulates the maintenance of neural stem cells in Figure8. To ascertain precisely when and how the actions of the LIFR/gp130 signal on self-renewal of neural stem cells begins, conditional timed disruption (complete) of LIFR or gp130 needs to be done. This is particularly so, given that (1) the difference in the number of neural stem cells in adultLIFR+/− brain is only 37% of wild-type littermates and (2) the number of neural stem cells robustly decrease during postnatal maturation of the forebrain (from ∼200,000 per forebrain at P0, when no difference is seen with LIFR disruption, to <1000 in the mature adult). These types of conditional, timed disruptions would also reveal detailed mechanisms, which could explain the stage-dependent responsiveness of neural stem cells to LIFR/gp130-mediated signaling and its correlation to the transition from neurogenesis to gliogenesis during CNS development.

Possible mechanism of action of LIFR/gp130-mediated signaling on the maintenance of neural stem cells during forebrain development. Primitive neural stem cells (Sp) may principally generate neurons (N) during early embryonic development and then begin the generation of glia (G) after their maturation (Qian et al., 2000) in late embryogenesis. Some unknown factors (?) likely support the self-renewal of primitive neural stem cells. The peak period of the gliogenesis may be in the early postnatal period (Altman, 1966) after the expansion of the mature multipotent neural stem cells (Sm). The contribution of LIFR/gp130 signaling to self-renewal and maintenance likely appears after maturation of neural stem cells. Specifically, the LIFR/gp130 signal is proposed to inhibit the lineage restriction of mature multipotent neural stem cells to the glial lineage during the early postnatal period to adulthood.
Function of adult forebrain neural stem cells
In postnatal rodents, neuronal precursors generated by neural stem cells of the lateral ventricle migrate into the olfactory bulb and differentiate into GABAergic and dopaminergic interneurons in the granular and periglomerular layers (Lois and Alvarez-Buylla, 1994;Morshead et al., 1994; Betarbet et al., 1996). Therefore, the function of these newly generated interneurons may in turn reflect, in part, the function of the adult neural stem cells. Recently, by analyzingNCAM-deficient mice that have a defect in the rostral migration of neuronal precursors generated in the subependyma at early postnatal days and adulthood (Tomasiewicz et al., 1993; Cremer et al., 1994; Chazal et al., 2000), it was found that the newly generated interneurons located in the granule cell layer may be involved in odor discrimination (Gheusi et al., 2000). However, it is still unclear whether the TH-positive interneurons located in the periglomerular layer are involved in olfactory discrimination, because it has not been determined whether their numbers are reduced inNCAM-deficient mice. In the present study, we observed a significant reduction in the number of TH-positive interneurons in the olfactory bulb of LIFR-deficient heterozygotes. It is reasonable to conclude that the reduction of neural stem cell number and the constitutively proliferative population contributed to the reduction of TH-positive neurons in the olfactory bulb. We cannot rule out, however, that the reduction of TH-positive neurons could be, in part, a result of the migration or survival of the neurons or their precursors being altered in LIFR+/−mice. In any case, analysis of the function of TH-positive interneurons in periglomerular layer may further provide clues regarding the role or roles of adult forebrain neural stem cells. Although it is known that these periglomerular neurons receive innervation from olfactory receptor cells and regulate the activities of mitral and tufted cells (Shepherd, 1994), their actual function in olfactory behavior remains to be determined.
Footnotes
This work was supported by the Canadian Institutes of Health Research. T.S. was supported by a fellowship from the Neuroscience Network of the Canadian Network of Centers of Excellence. S.W. is an Alberta Heritage Foundation for Medical Research Scientist. We thank Drs. Hideyuki Okano, Keiko Nakao, and Derek van der Kooy, and the Weiss and van der Kooy laboratories, for critical reading of an earlier version of this manuscript. Special thanks to Andrew Chojnacki for suggestions and assistance with the figures. We also thank Joy Goldberg and Dorothea Livingstone for excellent technical assistance.
Correspondence should be addressed to Dr. Weiss at the above address. E-mail: weiss{at}ucalgary.ca.
T. Shimazaki's present address: Department of Physiology, Keio University School of Medicine, 35 Shinanomachi, Shinjyuku-ku, Tokyo 160-8582, Japan.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}