

Abstract
α-Synuclein physiologically chaperones SNARE-complex assembly at the synapse but pathologically misfolds into neurotoxic aggregates that are characteristic for neurodegenerative disorders, such as Parkinson's disease, and that may spread from one neuron to the next throughout the brain during Parkinson's disease pathogenesis. In normal nerve terminals, α-synuclein is present in an equilibrium between a cytosolic form that is natively unfolded and monomeric and a membrane-bound form that is composed of an α-helical multimeric species that chaperones SNARE-complex assembly. Although the neurotoxicity of α-synuclein is well established, the relationship between the native conformations of α-synuclein and its pathological aggregation remain incompletely understood; most importantly, it is unclear whether α-synuclein aggregation originates from its monomeric cytosolic or oligomeric membrane-bound form. Here, we address this question by introducing into α-synuclein point mutations that block membrane binding and by then assessing the effect of blocking membrane binding on α-synuclein aggregation and neurotoxicity. We show that membrane binding inhibits α-synuclein aggregation; conversely, blocking membrane binding enhances α-synuclein aggregation. Stereotactic viral expression of wild-type and mutant α-synuclein in the substantia nigra of mice demonstrated that blocking α-synuclein membrane binding significantly enhanced its neurotoxicity in vivo. Our data delineate a folding pathway for α-synuclein that ranges from a physiological multimeric, α-helical, and membrane-bound species that acts as a SNARE-complex chaperone over a monomeric, natively unfolded form to an amyloid-like aggregate that is neurotoxic in vivo.
Introduction
α-Synuclein is an abundant neuronal protein that is highly enriched in presynaptic terminals and functions as a SNARE-complex chaperone (George et al., 1995; Iwai et al., 1995; Burré et al., 2010). Pathologically, α-synuclein is a major component of Lewy bodies in neurodegenerative disorders, including Parkinson's disease (PD; Spillantini et al., 1997; Wakabayashi et al., 1997; Arawaka et al., 1998; Gai et al., 1998). Moreover, point mutations (Polymeropoulos et al., 1997; Krüger et al., 1998; Zarranz et al., 2004; Kiely et al., 2013; Proukakis et al., 2013; Pasanen et al., 2014) and duplications and triplications of the α-synuclein gene (Singleton et al., 2003; Ibáñez et al., 2004) cause early-onset PD. Wild-type α-synuclein forms amyloid-like fibrils during prolonged incubation in solution; aggregation is enhanced by disease-related mutations (Conway et al., 1998; El-Agnaf et al., 1998; Narhi et al., 1999; Conway et al., 2000; Greenbaum et al., 2005; Fredenburg et al., 2007; Uversky, 2007; Yonetani et al., 2009). α-Synuclein aggregates exhibit various structures, ranging from soluble oligomeric forms in ring-like, string-like, or spherical shapes (protofibrils) to insoluble fibrils (Rochet et al., 2000; Ding et al., 2002; Lashuel et al., 2002). These fibrils are believed to form the basis of Lewy bodies, although it is still controversial if smaller protofibrils or larger amyloid fibrils are the toxic species of α-synuclein causing neuronal cell death (Breydo et al., 2012; Lashuel et al., 2013). Additionally, α-synuclein pathology may spread throughout the brain in a prion-like manner by propagation of neurotoxic α-synuclein aggregates from one neuron to the other (Desplats et al., 2009; Volpicelli-Daley et al., 2011; Luk et al., 2012; Rey et al., 2013).
In presynaptic terminals, α-synuclein cycles between a soluble and a membrane-bound state on synaptic vesicles (Maroteaux et al., 1988; Iwai et al., 1995; Kahle et al., 2000). Cytosolic α-synuclein is natively unfolded and monomeric, whereas membrane-bound α-synuclein assumes an α-helical conformation that mediates its association with membranes and its SNARE-complex chaperoning activity (Weinreb et al., 1996; Kim, 1997; Chandra et al., 2003; Jao et al., 2004; Ulmer and Bax, 2005; Ulmer et al., 2005; Burré et al., 2010; Lokappa and Ulmer, 2011; Fauvet et al., 2012; Burré et al., 2013). Whether aggregation of α-synuclein initiates from its lipid-bound α-helical or from its unstructured state remains controversial. Membranes have been reported to both accelerate (Narayanan and Scarlata, 2001; Cole et al., 2002; Lee et al., 2002) and inhibit (Narayanan and Scarlata, 2001; Zhu and Fink, 2003; Jo et al., 2004) α-synuclein fibril formation. Understanding how and where α-synuclein aggregates form is crucial for insight into its role in neurodegeneration. However, the relationship of membrane binding of α-synuclein to its neurotoxic activity remains uncharacterized. Here, we show that membrane binding of α-synuclein protects α-synuclein from aggregation and inhibits its neurotoxicity. Our data define a molecular folding pathway for α-synuclein whereby α-synuclein physiologically functions as a membrane-bound α-helical multimer but can pathologically assemble into neurotoxic β-stranded aggregates when it dissociates from the membranes in an unstructured monomeric form.
Materials and Methods
Recombinant protein expression and purification.
Full-length human α-synuclein cDNA was inserted into modified pGEX-KG vectors (GE Healthcare), containing an N-terminal TEV protease recognition site. α-Synuclein contained an extra N-terminal glycine after cleavage with TEV protease in addition to the normal N terminus of α-synuclein. Mutant α-synuclein constructs were generated by site-specific mutagenesis, according to the protocol of the manufacturer (Stratagene). All proteins were expressed as GST fusion proteins in bacteria (BL21 strain), essentially as described previously (Burré et al., 2010). Bacteria were grown to optical density 0.6 (measured at 600 nm), and protein expression was induced with 0.05 mm isopropyl β-d-thiogalactoside for 6 h at room temperature. Bacteria were harvested by centrifugation for 20 min at 4000 rpm, and pellets were resuspended in solubilization buffer [PBS, 0.5 mg/ml lysozyme, 1 mm PMSF, DNase, and an EDTA-free protease inhibitor mixture (Roche)]. Cells were broken by sonication, and insoluble material was removed by centrifugation for 30 min at 7000 average centrifugal force (gav) and 4°C. Proteins were affinity-purified using glutathione Sepharose bead (GE Healthcare) incubation overnight at 4°C, followed by TEV protease (Invitrogen) cleavage overnight at room temperature. His-tagged TEV protease was removed by incubation with Ni-NTA (Qiagen) overnight at 4°C. Protein concentration was assessed using the BCA method according to the protocol of the manufacturer (Thermo Fisher Scientific).
Liposome preparation, liposome binding assay, and liposome aggregation assay.
Liposomes were prepared as described previously (Burré et al., 2010). For lipid binding assays, 1 mg of brain phosphatidylcholine (PC; Avanti Polar Lipids) or 0.7 mg of PC and 0.3 mg of brain phosphatidylserine (PS; Avanti Polar Lipids) in chloroform were dried in a glass vial under a nitrogen stream. For aggregation studies, 84 mg of brain PC and 36 mg of brain PS were used. Residual chloroform was removed by lyophilization for 2 h. Small unilamellar vesicles were formed by sonicating 1 mg/ml (for lipid binding studies) or 120 mg/ml (for aggregation studies) of lipids in PBS on ice (Barenholz et al., 1977). For lipid binding studies, 5 μg protein of α-synuclein were incubated with 100 μg of liposomes for 2 h at room temperature. Samples were then either subjected to a liposome flotation assay (Burré et al., 2010) or crosslinking experiments. For aggregation assays, 3 μg of α-synuclein was incubated with or without 60 μg of liposomes in 1 m NaCl in PBS with protease inhibitors at 37°C and 300 rpm for indicated time periods. A total of 5 μl of sample was mixed with 125 μl of 100 μm K114 (Santa Cruz Biotechnology) in 100 mm glycine-NaOH, pH 8.45, and K114 fluorescence was measured using a Mithras LB 940 plate reader (Berthold Technologies; excitation, 390 nm and emission, 535 nm). Images of α-synuclein aggregates were acquired on a DFC400 Leica microscope.
Size exclusion chromatography.
Purified recombinant α-synuclein (90 μl of 6 mg/ml starting concentration) was separated using a Superdex 200 10/300 GL column on an AKTA FPLC system (GE Healthcare), equilibrated with PBS. To estimate molecular masses, molecular mass standards thyroglobulin, apoferritin, β-amylase, alcohol dehydrogenase, albumin, and carbonic anhydrase (all from Sigma) were separated under the same conditions. Relative mobility of α-synuclein was determined using linear regression of molecular mass versus migration distance (log molecular weight vs Rf value, measured as running distance of protein/total running distance). Blue dextran was used to measure the void volume.
Chemical crosslinking.
Crosslinking experiments were performed using glutaraldehyde (TCI America) as described previously (Burré et al., 2014). Briefly, 30 μl of recombinant myc-tagged α-synuclein (2 μg on liposomes) were incubated for 5 min with 10 μl of glutaraldehyde. Reaction was stopped by addition of 10 μl of 1 m Tris, pH 8.8. Samples were boiled before separating via SDS-PAGE.
Protein expression levels and in situ aggregation.
HEK293T cells or N2a neuroblastoma cells (both from ATCC) were transfected with cDNA encoding human wild-type or mutant α-synuclein using calcium phosphate as described previously (Burré et al., 2012). For measuring protein expression levels, 48 h after transfection, cells were washed twice in PBS and solubilized in 2× Laemmli's sample buffer. Proteins were separated using SDS-PAGE and immunoblotted for α-synuclein and β-actin. For immunocytochemistry, cells were washed twice in PBS containing 1 mm MgCl2, followed by fixation with 4% paraformaldehyde (PFA) in PBS for 20 min at room temperature, washing in PBS, and solubilization in 0.1% Triton X-100 in PBS for 5 min at room temperature. After washing cells with PBS, cells were blocked in 5% BSA in PBS for 20 min at room temperature and were incubated with anti α-synuclein antibodies over night at 4°C. The next day, cells were washed with PBS, blocked again, and incubated with Alexa Fluor-488-labeled secondary antibody and DAPI for 1 h at room temperature in the dark. Cells were washed in PBS and mounted using Vectashield (Vector Laboratories). Imaging was performed on a DFC400 Leica microscope for HEK293T cells and on an Eclipse 80i Nikon microscope for N2a neuroblastoma cells.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed essentially as described previously (Mosmann, 1983), with minor modifications. HEK293T or N2a neuroblastoma cells were transfected with cDNA encoding human wild-type or mutant α-synuclein in a 12-well format using calcium phosphate as described previously (Burré et al., 2012). To measure metabolic activity, 48 h after transfection, medium was replaced with 500 μl of fresh medium and 50 μl of 5 mg/ml MTT (VWR) in PBS, and cells were incubated for 1 h at 37°C. Medium was removed, and reduction of MTT to purple formazan was visualized after solubilization with 200 μl of 40 mm HCl in isopropanol for 2 min at room temperature by measuring absorption at 560 nm in a spectrophotometer (Synergy H1 Hybrid Reader; BioTek). Metabolic activity was determined by subtracting absorption at 620 nm (reference wavelength). Each condition was done in duplicate, and each well was measured in duplicate.
Expression of α-synuclein in substantia nigra.
Lentiviral vector L302 containing an IRES-driven GFP reporter (containing myc-tagged α-synuclein mutants or empty vector), vesicular stomatitis virus glycoprotein, the protein Rev, and Rev-responsive element were co-transfected in 1:1:1:1 molar ratio into HEK293T cells (ATCC) using calcium phosphate. Medium containing the viral particles was collected 48 h later and centrifuged for 10 min at 2000 rpm to remove cellular debris. Viral particles were concentrated by centrifugation for 90 min at 50,000 × gav, and viral pellet was resuspended in neuronal medium containing 4 mg/ml polybrene (Sigma-Aldrich) at 1% of the precentrifugation volume, snap frozen in liquid nitrogen, and kept at −80°C. Male CD1 mice (P40–P45; Charles River Laboratories) were anesthetized by intraperitoneal injection of 125–150 mg/kg Avertin (tribromoethanol; Sigma-Aldrich). One microliter of viral solution was delivered through a glass pipette at a flow rate of 0.15 μl/min unilaterally (left hemisphere) at the following coordinates: anteroposterior, 2.6–3.3 mm posterior to bregma (determined based on lambda-to-bregma distance of each mouse); lateral, 1 mm from midline suture; ventral, 4.2 mm below brain surface. After 10 d of recovery, analysis of mouse behavior was commenced.
Behavioral studies.
The beam-walking test was used to record deficits in balance and limb control (accuracy and strength in limb placement). The animal's ability to navigate across a beam to return to its home cage was tested using a wooden dowel (cylindrical beam 60 cm in length, 1 cm in diameter). The beam was fixed steadily on both ends 40 cm above the ground leading to a small cage filled with bedding from the animal's home cage. Mice were placed onto one side of the beam and were left to cross the beam to reach the cage. Mice that escaped into the cage were picked up and placed on the opposite side again for a total of three trials with 1 min intertrial intervals. The number of foot slips for each mouse per trial was scored. Force-plate actometry, a sensitive and quantitative method (Fowler et al., 2001), was used to document changes in locomotor activity induced by injection of lentivirus expressing α-synuclein mutants in substantia nigra. From the force-plate traces/coordinate records, total distance traveled, continuous distance traveled, low-mobility bouts, and spatial confinement per 6 min session were determined.
Immunohistochemistry.
Anesthetized mice were perfused with ice-cold 4% PFA in PBS, followed by removal of the brain and overnight fixation in 4% PFA in PBS (room temperature). Fixed brains were cryopreserved in 30% sucrose in PBS for 2 d and frozen in Tissue-Tek Optimal Cutting Temperature embedding medium (Sakura Finetechnical). Coronal brain sections (20 μm) were cut at −20°C (CM3050S cryostat; Leica), picked up on slides, and heat adhered at 37°C for 30 min. For immunostaining, slides were incubated in blocking solution (3% BSA and 0.1% Triton X-100 in PBS) for 1 h, followed by overnight incubation with primary antibodies (4°C). Slides were washed three times in PBS (5 min each) and incubated in blocking buffer containing Alexa Fluor 488-coupled, 546-coupled, or 633-coupled secondary antibodies (Invitrogen) for 3 h at room temperature. After six washes in PBS, slides were mounted with Vectashield hard-set mounting medium with DAPI (Vector Laboratories), followed by fluorescence microscopy. All quantitations of immunofluorescence images were done with the image processing and analysis software NIH ImageJ. Total tyrosine hydroxylase (TH)-positive neurons, NeuN-positive puncta, and DAPI puncta were counted in each section containing GFP fluorescence. NeuN puncta were normalized to total DAPI puncta in the same brain section. Image acquisition and thresholding parameters were kept constant across each experiment.
Gel electrophoresis and protein quantitation.
Protein samples were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Blots were blocked in Tris-buffered saline containing 0.1% Tween 20 (Sigma) and 3% fat-free milk for 2 h at room temperature. The blocked membrane was incubated in blocking buffer containing primary antibody for 1 h, followed by five washes. Membranes were incubated in blocking buffer containing horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000; MP Biomedicals) for 2 h at room temperature. HRP immunoblots were developed using enhanced chemiluminescence (GE Healthcare). Quantitative immunoblotting experiments were performed with either fluorescent secondary antibodies (1:20,000; LI-COR) for 1 h at room temperature or iodinated secondary antibodies (1:1000; PerkinElmer Life and Analytical Sciences) overnight at room temperature as described previously (Rosahl et al., 1995). Fluorescent immunoblots were scanned using an Odyssey CLx (LI-COR). 125I blots were exposed to PhosphorImager screens (GE Healthcare) overnight and scanned using a Typhoon scanner (GE Healthcare), followed by quantification with ImageQuant software (GE Healthcare).
Primary antibodies.
Monoclonal antibodies used were α-synuclein (610786; BD Biosciences Transduction Laboratories), β-actin (A1978; Sigma), myc (clone 6E10; Developmental Studies Hybridoma Bank), and NeuN (MAB377; Millipore). Polyclonal antibodies used were α-synuclein (U1127) and TH (AB112; Abcam).
Statistical analyses.
Curves obtained from in vitro aggregation assays were analyzed by two-way repeated-measures ANOVA, using GraphPad Prism (GraphPad Software). All other data shown are means ± SEMs and were analyzed by Mann–Whitney U test to compare the data groups.
Results
Design of lipid-binding deficient mutants of α-synuclein
The finding that α-synuclein multimerizes on lipid membranes (Burré et al., 2014) raises the question whether such α-helical multimers directly transition into β-strand-containing neurotoxic aggregates or whether it is the unstructured soluble monomeric α-synuclein species that gives rise to such aggregates. To address this central question, we generated α-synuclein mutants that were designed to lack lipid binding based on previous studies (Burré et al., 2012). We generated two different double point mutations (A11P/V70P and T44P/A89P), targeting at the same time both lipid-binding α-helices in α-synuclein and a quadruple point mutation combining the two double point mutations (A11P/T44P/V70P/A89P; Fig. 1A,B). We found that all of these mutations obliterated phospholipid binding (Fig. 1C,D). In agreement with previous results showing that membrane binding is required for α-synuclein to form multimers in the presence of liposomes (Burré et al., 2014), all three lipid-binding deficient mutants did not form multimers in the presence of liposomes, confirming the design strategy (Fig. 1E).

Design of lipid-binding deficient mutants of α-synuclein. A, α-Synuclein domain structure (red, lipid-binding domain; blue, protein-interaction domain), with lipid-binding deficient mutations marked in red and 11-mer sequences highlighted with black boxes. B, SDS-PAGE analysis of purified recombinant mutant α-synuclein (5 μg/lane), stained with Coomassie Brilliant Blue. C, D, Lipid binding of wild-type and mutant α-synuclein. C, Recombinant α-synuclein was incubated with negatively charged liposomes (composition: 30% phosphatidylserine (PS) and 70% phosphatidylcholine (PC)) and subjected to a flotation assay. Eight fractions were collected from top to bottom of the flotation gradient, and equal volumes of each fraction were separated by SDS-PAGE and immunoblotted for α-synuclein. D, The top two fractions were defined as lipid bound and quantitated as percentage of total α-synuclein (means ± SEMs; **p < 0.001 by Mann–Whitney U test; n = 3). E, Analysis of the effect of α-synuclein mutations on membrane-binding induced α-synuclein multimerization. Recombinant α-synuclein was incubated with negatively charged liposomes (composition: 30% PS, 70% PC) and exposed to increasing concentrations of the chemical crosslinker glutaraldehyde (concentration: 0–0.5%). Equal volumes of crosslinked proteins were analyzed by immunoblotting. Arrowheads indicate α-synuclein multimers.
Binding to liposomes protects α-synuclein from aggregation in vitro
We then asked whether liposomes increase or decrease aggregation of wild-type α-synuclein and how liposomes affect aggregation of mutant α-synuclein that does not bind to membranes. We used an in vitro assay in which we incubated wild-type and mutant recombinant α-synuclein at 37°C over a period of 35 d, in either solution or presence of charged liposomes and monitored aggregation by microscopy. We found that, in the absence of liposomes, wild-type α-synuclein and all three types of mutant α-synuclein formed visible aggregates, which appeared to be smaller and formed more slowly for mutant α-synucleins than for wild-type α-synuclein (Fig. 2A). Strikingly, addition of liposomes completely blocked aggregation of wild-type α-synuclein but had no effect on the aggregation of the lipid-binding deficient α-synuclein mutants (Fig. 2B). These experiments suggest that α-synuclein aggregates form from its monomeric soluble state.

Aggregation of α-synuclein in vitro. A, B, Recombinant wild-type α-synuclein or mutant α-synuclein unable to bind to liposomes were incubated in buffer (left panels) or in the presence of charged liposomes (composition: 30% PS, 70% PC; right panels) at 37°C and 300 rpm. At the indicated time points, aggregation of α-synuclein was assessed by light microscopy.
Lewy bodies include amyloid-type aggregates of α-synuclein (Spillantini et al., 1998). Therefore, we quantified the degree of amyloid formation of α-synuclein using the fluorescent dye K114. We found formation of amyloid fibrils for wild-type α-synuclein only in the buffer condition (Fig. 3A,B), confirming the absence of visible aggregates in the previous experiment (Fig. 2B). Mutants of α-synuclein that are unable to bind to liposomes formed K114-detectable amyloid fibrils in the buffer condition at a later time point than wild-type α-synuclein and did not exhibit the same size of the fluorescence signal (Fig. 3A). However, in the presence of liposomes, no time-dependent increase of K114 fluorescence was observed for either wild-type or mutant α-synuclein, possibly because the liposomes may sequester the hydrophobic K114 fluorescence dye (Fig. 3B). This is suggested by the observation that the relative fluorescence of K114 in the presence of liposomes was ∼10-fold higher than in the buffer condition, which may mask aggregation of α-synuclein on liposomes, and makes it impossible to use K114 for monitoring amyloid formation in the presence of membranes.

In vitro aggregation of α-synuclein (αSyn). A, B, Amyloid formation of α-synuclein. Recombinant α-synuclein was incubated as described for Figure 2. At the indicated time points, wild-type and mutant α-synuclein were analyzed for amyloid fibril formation in buffer (A) or in the presence of charged liposomes (B), using the dye K114 (means ± SEMs; *p < 0.05, **p < 0.01, ***p < 0.001 by Mann–Whitney U test; ##p < 0.01, ###p < 0.001 by two-way ANOVA; n = 7; n.s., not significant). C, D, Analysis of loss of monomer of α-synuclein. At the indicated time points, the same volumes of wild-type and mutant α-synuclein aggregating in buffer or in the presence of charged liposomes were analyzed by immunoblotting, and loss of monomer was quantitated (means ± SEMs; **p < 0.01 by Mann–Whitney U test; ###p < 0.001 by two-way ANOVA; n = 7; n.s., not significant). E, Analysis of wild-type and mutant α-synuclein by gel filtration. At the indicated time points, 90 μl of aggregating recombinant wild-type and mutant α-synuclein were analyzed by gel filtration. Visible aggregates were removed before loading via centrifugation to avoid clogging of the gel filtration column.
To circumvent this problem, we quantified the degree of aggregation under these in vitro conditions in parallel by measuring the loss of monomeric α-synuclein species as a function of incubation time using SDS-PAGE. This assay is based on the observation that α-synuclein aggregates are SDS resistant and do not enter an SDS gel. These quantifications confirmed the dramatic inhibition of wild-type α-synuclein aggregation by liposomes and clearly demonstrated that mutant α-synuclein aggregated completely independent of whether or not liposomes were present, consistent with their lack of liposome binding (Fig. 3C,D). Similar to wild-type α-synuclein, aggregates of mutant α-synuclein could not be resolved by SDS-PAGE (Fig. 3C,D) or by gel filtration (Fig. 3E), likely because of formation of very large aggregates of a broad range of sizes, which were removed before gel filtration or did not enter the gel matrix in SDS-PAGE. Viewed together, these experiments suggest that, in the absence of membranes, the lipid-binding deficient α-synuclein mutants are no more prone to aggregation than wild-type α-synuclein but that the presence of membranes blocks aggregation of wild-type but not lipid-binding deficient mutant α-synuclein.
α-Synuclein mutants deficient in lipid binding reveal increased aggregation in situ and increased cellular toxicity
To test the question whether lipid-binding deficient mutants of α-synuclein form aggregates in a cellular environment, we transfected HEK293T cells with cDNAs expressing wild-type α-synuclein or mutant α-synucleins that are unable to bind to lipid membranes. Despite the presence of membranes, wild-type α-synuclein aggregates when overexpressed in transfected HEK293T cells, probably because there is an excess of α-synuclein. However, we found that lipid-binding deficient α-synuclein mutants aggregated approximately twofold more strongly than wild-type α-synuclein in transfected HEK293T cells (Fig. 4A,B). The expression levels of full-length wild-type and mutant α-synucleins were similar in the transfected HEK293T cells, and no proteolytic α-synuclein species were observed, suggesting that the α-synuclein mutants did not aggregate more readily because they were expressed at higher levels or because they were proteolytically processed (Fig. 4C,D).

Aggregation and toxicity of α-synuclein in HEK293T cells. A, B, Aggregation of wild-type and mutant α-synuclein in HEK293T cells transfected with equal amounts of expression vectors encoding wild-type and mutant α-synuclein. A, Two days after transfection, cells were fixed and immunostained with antibodies against the myc-epitope. B, The number of immunopositive aggregates per field was quantitated and compared with wild-type levels (means ± SEMs; ***p < 0.001 by Mann–Whitney U test; n = 3 independent cultures). C, D, Expression of wild-type and mutant α-synuclein (α-Syn) in HEK293T cells. Two days after transfection, expression levels were analyzed by immunoblotting with antibodies against the myc-epitope (C), normalized to β-actin levels, and quantitated as percentage of wild-type levels (D; means ± SEMs; n = 4 independent cultures). E, Metabolic activity of HEK293T cells transfected with wild-type and mutant α-synuclein. Two days after transfection, cells were subjected to an MTT assay. Data were normalized to wild-type α-synuclein levels (means ± SEMs; *p < 0.05 by Mann–Whitney U test; n = 6 independent cultures).
We additionally measured the metabolic activity of transfected HEK293T cells to assess whether α-synuclein mutants impair cellular function and thereby health. We found that all lipid-binding deficient variants of α-synuclein reduced metabolic activity (Fig. 4E). In parallel, we repeated the same experiments in transfected N2a neuroblastoma cells. N2a cells transfected with cDNAs expressing wild-type α-synuclein or mutant α-synucleins that are unable to bind to lipid membranes revealed multiple aggregates (Fig. 5A). Yet we were unable to quantitate α-synuclein aggregates because we could not visually isolate individual aggregates. Similar to transfected HEK293T cells, the expression levels of full-length wild-type and mutant α-synucleins were similar (Fig. 5B,C), and mutant α-synucleins reduced metabolic activity in N2a neuroblastoma cells (Fig. 5D). Note that, as observed previously (Burré et al., 2012), mutants containing the A11P substitution revealed slightly reduced SDS-PAGE mobility for unclear reasons.

Aggregation and toxicity of α-synuclein in N2a neuroblastoma cells. A, Expression of wild-type and mutant α-synuclein in N2a neuroblastoma cells. Two days after transfection, cells were fixed and immunostained with antibodies against the myc epitope. Nuclei were visualized using DAPI. B, C, Expression of wild-type and mutant α-synuclein (α-Syn) in N2a neuroblastoma cells. Two days after transfection, expression levels were analyzed by immunoblotting with antibodies against the myc epitope (B), normalized to β-actin levels, and quantitated as percentage of wild-type levels (C; means ± SEMs; n = 6 independent cultures). D, Metabolic activity of N2a neuroblastoma cells transfected with wild-type and mutant α-synuclein. Two days after transfection, cells were subjected to an MTT assay. Data were normalized to wild-type α-synuclein levels (means ± SEMs; *p < 0.05, **p < 0.01 by Mann–Whitney U test; n = 6 independent cultures).
Viewed together, these data suggest that α-synuclein aggregates form exclusively from the soluble monomeric unstructured conformation of α-synuclein and suggest that blocking lipid binding of α-synuclein increases aggregation in a cellular environment. Moreover, these data provide a possible explanation for the pathogenecity of increased α-synuclein expression because increased levels of total α-synuclein would be expected to increase the levels of soluble α-synuclein as a starting point for α-synuclein aggregation.
Mice expressing lipid-binding deficient α-synuclein mutants exhibit impaired motor function
Is in vitro aggregation of α-synuclein related to its in vivo neurotoxicity? The lipid-binding mutants provide a unique opportunity to test this question because these mutants are devoid of physiological activity yet aggregate. We stereotactically injected lentiviruses expressing wild-type or mutant α-synuclein unilaterally into substantia nigra of wild-type mice (Fig. 6A). We then analyzed the mice starting at 10 d after the injections for motor deficits using force-plate actometry and the beam-walking assay and killed the mice at 45 d after the injections for immunohistochemical studies.

Lentiviral expression of wild-type and mutant α-synuclein in dopaminergic substantia nigra neurons. A, Schematic overview of the stereotactic injection experiments. Wild-type mice (40–45 d old) were stereotactically and unilaterally injected into the substantia nigra (left). Mice were monitored every 5 d from 10 to 45 d after injection, when mice were killed for histochemical analysis (right). B, C, Beam-walk assay. B, Motor defects were assayed using the beam-walk task in which foot slips on a beam walk were measured. Three rounds of beam walk were analyzed per session. Averaged foot slips were recorded. C, Quantitation of average foot slips at 45 d after injections. Data are means ± SEMs (*,#p < 0.05 by Mann–Whitney U test; n = 5 mice).
Compared with mice expressing only GFP (control), mice expressing wild-type α-synuclein revealed an increasing number of foot slips in the beam-walk assay as a function of time after the injections. Importantly, mice expressing mutant α-synuclein unable to bind to lipids exhibited an aggravated phenotype (Fig. 6B,C). Similarly, expression of wild-type α-synuclein produced an overall decrease in motor performance, as demonstrated by an increase in spatial confinement, reduced total and continuous distance traveled, and showed an increase in low mobility bouts (Fig. 7). Mice expressing lipid-binding deficient mutants of α-synuclein exhibited impairments in these motor performance assays that were significantly more severe than the impairments observed in mice expressing wild-type α-synuclein.

Motor impairments of mice injected with lentiviral particles expressing α-synuclein variants. A, Representative traces of force-plate analyses. Mice were injected and analyzed as described for Figure 5. B–I, Analysis of spatial confinement, total and continuous distance traveled, and low mobility bouts, as calculated from the force-plate data obtained with multiple identically injected mice. Every 5 d, mice were subjected to behavioral analyses (B, D, F, H), and data were plotted as the difference at 45 d after injection (C, E, G, I). Data are means ± SEMs (*,#p < 0.05, **,##p < 0.01 by Mann–Whitney U test; n = 5 mice).
Lack of lipid binding of α-synuclein increases neurotoxicity
Next, we analyzed whether the impairments in motor function observed after expression of wild-type and mutant α-synuclein are attributable to a loss of neurons in the injected areas. We first analyzed dopaminergic cell loss by quantitating TH-positive neurons (Fig. 8A). GFP produced by the virus via an internal ribosomal entry site was used to identify the injection site. Compared with control injections, brains expressing wild-type α-synuclein exhibited a reduction in the number of dopaminergic neurons in the injected area, in a similar time frame and to a similar extent as demonstrated previously (Fig. 8B; Kirik et al., 2002; Klein et al., 2002; Lo Bianco et al., 2002; Kirik et al., 2003; Lauwers et al., 2003; Yamada et al., 2004; St Martin et al., 2007; Azeredo da Silveira et al., 2009; McFarland et al., 2009; Koprich et al., 2010; Winner et al., 2011; Burré et al., 2012; Decressac et al., 2012; Gombash et al., 2013; Oliveras-Salvá et al., 2013). Again, the lipid-binding mutation aggravated this phenotype. We also assessed loss of NeuN-positive neurons by measuring the ratio of NeuN-positive to DAPI-positive cells, which monitors the survival of non-TH-positive neurons. Again, we observed the same relationship between the expression of wild-type or mutant α-synuclein and neuronal cell loss (Fig. 8C,D). Viewed together, these data show that mutant α-synuclein unable to bind phospholipids is more neurotoxic than wild-type α-synuclein, suggesting that neurotoxicity induced by α-synuclein derives from its soluble form.

Neuron loss in mice expressing α-synuclein (αSyn) mutants in the substantia nigra. A, C, Control lentiviruses (control) or lentiviruses expressing wild-type or mutant α-synucleins were stereotactically and unilaterally injected into the substantia nigra of 40- to 45-d-old mice. Forty-five days after injection, injected areas were immunostained for either tyrosine hydroxylase (TH; left) or NeuN (right) and DAPI (blue). IRES-driven GFP marks the injection site. B, D, The density of dopaminergic neurons was quantitated by immunostaining for TH (B), and the density of NeuN-positive (non-dopaminergic) neurons was quantitated by immunostaining for NeuN (D). Data are means ± SEMs (*,#p < 0.05 by Mann–Whitney U test; n = 5 mice).
Discussion
Extensive studies over the past decade have revealed that α-synuclein, when overexpressed, is neurotoxic, that neurotoxicity correlates with α-synuclein aggregation, that α-synuclein mutations observed in familial PD cause increased aggregation, and that α-synuclein neurotoxicity and aggregation may spread from one neuron to the next (Spillantini et al., 1997; Wakabayashi et al., 1997; Singleton et al., 2003; Ibáñez et al., 2004; Desplats et al., 2009; Volpicelli-Daley et al., 2011; Burré et al., 2012; Rey et al., 2013). At the same time, α-synuclein was shown to physiologically bind to membranes and to enhance SNARE-complex assembly (Iwai et al., 1995; Jo et al., 2000; Chandra et al., 2003; Burré et al., 2010). These two sets of data raised two overall questions. First, what folding pathway mediates α-synuclein aggregation and neurotoxicity—does α-synuclein aggregate by misfolding of its multimeric membrane-bound form or its monomeric cytoplasmic form? Second, what is the relationship between physiological functions of α-synuclein and its pathological effects—are they interdependent or unrelated?
In the present study, we have made two principal sets of observations that address these basic questions. First, we described two different double point mutations of α-synuclein that block phospholipid binding. These mutations were different from all of the many previously described mutations that do not block phospholipid binding and show that blocking lipid-binding by α-synuclein enhances its aggregation in a cellular environment, whereas addition of liposomes to wild-type α-synuclein inhibits aggregation (Figs. 1⇑⇑⇑–5). It is important to note that our data do not suggest that these mutations increase the rate of aggregation of α-synuclein as such. They do not have the same effect as PD mutations in this sense; instead, these mutations only increase aggregation in a cellular environment presumably because the block of lipid binding by these mutations increases the amount of soluble monomeric α-synuclein. Thus, our results indicate that α-synuclein aggregation is increased when α-synuclein is in its soluble unstructured monomeric form and is blocked when α-synuclein is in its membrane-bound α-helical multimeric form. Long-range intramolecular interactions between the N termini and C termini of α-synuclein may be crucial for the process of fibril formation (Ulrih et al., 2008). Possibly, binding of the 95 N-terminal helical residues of α-synuclein to lipid membranes and binding of the 45 C-terminal residues of α-synuclein to synaptobrevin-2 on the synaptic vesicle membrane (Burré et al., 2010) prevents contacts between the N and C termini of α-synuclein and thereby impedes aggregation.
Second, we show that blocking phospholipid-binding by α-synuclein enhances its neurotoxicity (Figs. 6⇑–8). These results are consistent with the notion that neurotoxicity is attributable to α-synuclein aggregation, although these results do not reveal what type of α-synuclein aggregate is neurotoxic. Based on these results, it seems to us that the neurotoxicity of α-synuclein is produced by a gain-of-toxic function effect of α-synuclein when it misfolds into aggregates and that its physiological and pathogenic activities are unrelated to each other. This conclusion supports the finding that α-synuclein mutations that cause PD accelerate α-synuclein aggregation (Conway et al., 2000; Pandey et al., 2006; Uversky, 2007).
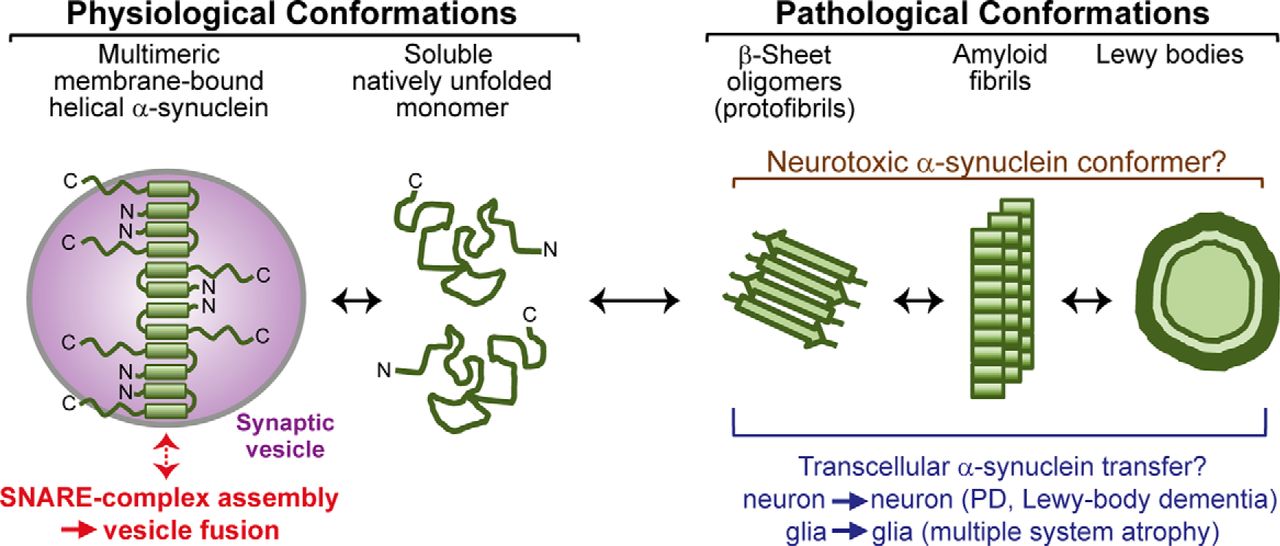
Viewed together, our data point to a folding pathway whereby α-synuclein physiologically is primarily phospholipid bound in presynaptic terminals, in which it normally functions to chaperone SNARE-complex assembly (Fig. 9). A small pool of α-synuclein is probably always soluble and monomeric in vivo. However, overexpression of α-synuclein dramatically increases the relative amount of soluble α-synuclein because the synaptic vesicle binding sites become saturated. α-Synuclein protein levels are increased with aging in the substantia nigra and correlate with decreased TH immunostaining (Chu and Kordower, 2007). Furthermore, alleles within a Rep1 polymorphic region 10 kB upstream of the a-synuclein gene promoter that confers risk for sporadic PD (Maraganore et al., 2006) have been associated with increased expression of α-synuclein mRNA in human and mouse neurons (Chiba-Falek et al., 2003; Cronin et al., 2009) and in the temporal neocortex and substantia nigra in humans (Linnertz et al., 2009). Together with the link of gene duplications and triplications to early onset PD (Singleton et al., 2003; Ibáñez et al., 2004), these findings suggest that increased concentrations of α-synuclein mRNA and protein constitute a triggering factor for PD pathogenesis in the brains of patients with PD. Soluble α-synuclein spontaneously forms aggregates at a low rate; this rate is increased with an increase in the α-synuclein concentration but more importantly with the presence of a nucleating amount of preformed aggregates, such as could be transferred from another neuron during the proposed cell-to-cell spread (Desplats et al., 2009; Volpicelli-Daley et al., 2011; Luk et al., 2012; Rey et al., 2013). Therapeutically, these considerations suggest that α-synuclein toxicity could be combated not only by inhibiting cell-to-cell spread or aggregation but also by chaperoning of soluble α-synuclein, decreasing α-synuclein synthesis, increasing α-synuclein degradation, or increasing α-synuclein phospholipid binding, which are avenues that could be pursued productively.

Schematic of α-synuclein conformations associated with its physiological function and pathological activities. Soluble α-synuclein is natively unstructured and monomeric. After binding to highly curved membranes such as synaptic vesicles, α-synuclein undergoes a conformational change and folds into a broken amphiphathic α-helix, which is associated with multimerization and mediates its SNARE-complex chaperoning function. Under pathological conditions, soluble α-synuclein forms β-sheet-like oligomers (protofibrils), which convert into amyloid fibrils and eventually deposit into Lewy bodies. Protofibrils and fibrils may propagate from neuron to neuron in PD and Lewy body dementia and from glia to glia in multiple system atrophy.
Footnotes
This work was supported by National Institutes of Health Grant AG010770 and NS077906 (T.C.S.). We thank Christian Burré (CircumFlex Computer Systems) for help with data analysis.
The authors declare no competing financial interests.
- Correspondence should be addressed to either of the following: Jacqueline Burré, Appel Institute for Alzheimer's Disease Research, Brain and Mind Research Institute, Weill Cornell Medical College, 413 East 69th Street, New York, NY 10021, jab2058{at}med.cornell.edu; or Thomas C. Südhof, Lorry I. Lokey Stem Cell Research Building, G1021, 265 Campus Drive, Stanford, CA 94305. tcs1{at}stanford.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}