

Abstract
Persistent firing of entorhinal cortex (EC) pyramidal neurons is a key component of working and spatial memory. We report here that a pro-brain-derived neurotrophic factor (proBDNF)-dependent p75NTR signaling pathway plays a major role in excitability and persistent activity of pyramidal neurons in layer V of the EC. Using electrophysiological recordings, we show that proBDNF suppresses persistent firing in entorhinal slices from wild-type mice but not from p75NTR-null mice. Conversely, function-blocking proBDNF antibodies enhance excitability of pyramidal neurons and facilitate their persistent firing, and acute exposure to function-blocking p75NTR antibodies results in enhanced firing activity of pyramidal neurons. Genetic deletion of p75NTR specifically in neurons or during adulthood also induces enhanced excitability and persistent activity, indicating that the proBDNF-p75NTR signaling cascade functions within adult neurons to inhibit pyramidal activity. Phosphatidylinositol 4,5-bisphosphate (PIP2)-sensitive transient receptor potential canonical channels play a critical role in mediating persistent firing in the EC and we hypothesized that proBDNF-dependent p75NTR activation regulates PIP2 levels. Accordingly, proBDNF decreases cholinergic calcium responses in cortical neurons and affects carbachol-induced depletion of PIP2. Further, we show that the modulation of persistent firing by proBDNF relies on a p75NTR-Rac1-PI4K pathway. The hypothesis that proBDNF and p75NTR maintain network homeostasis in the adult CNS was tested in vivo and we report that p75NTR-null mice show improvements in working memory but also display an increased propensity for severe seizures. We propose that the proBDNF-p75NTR axis controls pyramidal neuron excitability and persistent activity to balance EC performance with the risk of runaway activity.
SIGNIFICANCE STATEMENT Persistent firing of entorhinal cortex (EC) pyramidal neurons is required for working memory. We report here that pro-brain-derived neurotrophic factor (proBDNF) activates p75NTR to induce a Rac1-dependent and phosphatidylinositol 4,5-bisphosphate-dependent signaling cascade that suppresses persistent activity. Conversely, using loss-of-function approaches, we find that endogenous proBDNF or p75NTR activation strongly decreases pyramidal neuron excitability and persistent firing, suggesting that a physiological role of this proBDNF-p75NTR cascade may be to regulate working memory in vivo. Consistent with this, mice rendered null for p75NTR during adulthood show improvements in working memory but also display an increased propensity for severe seizures. We propose that by attenuating EC network performance, the proBDNF-p75NTR signaling cascade reduces the probability of epileptogenesis.
Introduction
The entorhinal cortex (EC) plays an important role in working and spatial memory (Young et al., 1997; Hahn et al., 2012). During working-memory tasks, principal neurons in the EC of rodents and primates display persistent firing (PF) and this sustained activity is believed to represent a mnemonic property with intrinsic and network components (Suzuki et al., 1997; Young et al., 1997). Despite the importance of PF in sustaining memory traces, the cellular and molecular mechanisms that establish, maintain, and extinguish working memory in the EC remain largely unknown. The EC is the major gateway between the hippocampus and the neocortex, with output from the hippocampal formation targeting mainly layers V and VI of the EC. These EC neurons in turn extend projections to widespread areas of cortex and to subcortical regions. The EC also receives cholinergic inputs, mainly from the medial septum and from the diagonal band of Broca (Lucas-Meunier et al., 2003) and these have a crucial impact on memory acquisition and retention. A large component of the cholinergic input is mediated by postsynaptic muscarinic receptor-induced changes in the intrinsic firing properties of cortical neurons (Egorov et al., 2002; Reboreda et al., 2007; Zhang et al., 2011). We previously showed that these changes reflect enhanced activity of transient receptor potential canonical (TRPC) channels (Zhang et al., 2011) induced by activation of muscarinic or glutamatergic mGluR5 receptors expressed in layer V EC pyramidal neurons and here we explore mechanisms that regulate persistent activity in the EC.
The neurotrophins are a family of trophic factors that in mammals is composed of nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4). Neurotrophins are synthesized as precursors or proforms and can be cleaved intracellularly or secreted as proneurotrophins (J. Yang et al., 2009). The mature processed neurotrophins bind to the p75 neurotrophin receptor (p75NTR) but also exhibit selective binding to the TrkA, TrkB, and TrkC tyrosine kinase receptors. In contrast, the proneurotrophins have low affinity for the Trk receptors and instead bind to receptor complexes that are composed of p75NTR and VPS10 family members (Lee et al., 2001; Chao, 2003). In this study, we investigated the role of p75NTR and its ligand proBDNF in neuronal excitability and PF in the EC. We report here that proBDNF functions as a neuromodulator that activates p75NTR to decrease excitability of layer V EC pyramidal neurons and inhibit PF. We also observe that layer V EC neurons of p75NTR-null mice display a large increase in excitability and generate persistent activity more readily than wild-type controls. We show that the proBDNF-p75NTR cascade functions postsynaptically to increase phosphatidylinositol 4,5-bisphosphate (PIP2) levels through a Rac1-dependent pathway. We also demonstrate that these changes do not reflect a developmental circuit defect but instead reflect an acute physiological effect of p75NTR. Finally, we report that p75NTR-null mice show improvements in short-term memory and display an increased propensity for severe epileptic seizures. Together, these data indicate that the proBDNF-p75NTR axis negatively regulates excitability and mnemonic activity in the EC.
Materials and Methods
Chemicals and solutions.
All drugs were purchased from Sigma-Aldrich, except 6-methyl-2-(phenylethynyl) pyridine, (S)-3,5-dihydroxyphenylglycine hydrate, and NSC23766 from Tocris Bioscience, mBDNF and proBDNF (wild-type mouse, #B240) from Alomone Labs, p75NTR antibody (AB-N01) from Advanced Targeting Systems, and proBDNF antibody (5H8) from Santa Cruz Biotechnology. All drug stocks were freshly diluted to the desired concentration. Final concentration of DMSO never exceeded 0.01%. Extracellular solution in current-clamp experiments contained (in mm) the following: 124 NaCl, 3 KCl, 26 NaHCO3, 1.8 MgSO4, 1.25 NaH2PO4, 10 glucose, 1.6 CaCl2; pH was maintained at 7.4 by constant bubbling with carbogen (95% O2, 5% CO2). Intracellular solution was composed of the following (in mm): 120 K-gluconate, 10 HEPES, 0.2 EGTA, 20 KCl, 2 MgCl2, 7 diTrisP-creatine, 4 Na2ATP, and 0.3 NaGTP (pH adjusted to 7.3 with KOH). Synaptic transmission was blocked using kynurenic acid (2 mm) and picrotoxin (100 μm).
Animals.
All experimental procedures were approved by the McGill University Animal Care Committee and were in compliance with the guidelines of the Canadian Council on Animal Care. Male C57BL/6 mice (6–8 weeks old) were obtained from Charles River Canada. p75NTR−/− (Lee et al., 1992) and p75NTR fl/fl mice have been previously described (Bogenmann et al., 2011). Tα1-cre mice (Coppola et al., 2004) were provided by Dr. Lino Tessarollo (National Cancer Institute at Frederick, Maryland) and CMV-ER-cre mice (Metzger and Chambon, 2001) were provided by Dr. Josephine Nalbantoglu (Montreal Neurological Institute, Montreal, Quebec, Canada). Animals were housed under standard conditions with a 12 h light/dark cycle and had ad libitum access to water and food.
Brain slice preparation.
Acute brain slices were obtained from 6–8-week-old male C57BL/6 mice (Charles River Canada) and prepared according to a protocol previously described (Tahvildari and Alonso, 2005). Briefly, mice were anesthetized with ketamine/xylamine mixture (60 mg/kg) and perfused with ice-cold choline chloride-based artificial CSF (ACSF) containing the following (in mm): 110 choline-Cl, 1.25 NaH2PO4, 25 NaHCO3, 7 MgCl2, 0.5 CaCl2, 2.5 KCl, 7 glucose, 3 pyruvic acid, and 1.3 ascorbic acid; pH 7.4, bubbled with carbogen (O2 95%, CO2 5%). Semicoronal horizontal slices (300 μm) containing the EC from the retrohippocampal region were obtained using a VT1000 vibratome (Leica) in the same choline chloride-based solution. The slices were allowed to settle down for 1 h in ACSF containing the following (in mm): 124 NaCl, 3 KCl, 26 NaHCO3, 1.8 MgSO4, 1.25 NaH2PO4, 10 glucose,1.6 CaCl2.
Recording procedures.
Brain slices were placed in a recording chamber mounted on the stage of an upright microscope Axioskop (Zeiss) equipped with a 63× water-immersion objective and differential contrast optics. A near-infrared charged-coupled device (CCD) camera (Sony XC-75) was used to visualize the neurons. Brain slices were stabilized using a U-shaped stainless steel anchor with Lycra threads at 1.5 mm spacing (Warner Instruments). Layer V entorhinal neurons selected for recording were located close to the lamina dissecans. Brain slices were perfused by gravity at a speed of 1–2 ml/min. The temperature of perfusion solution was maintained at 32−34°C using a TC-324B temperature controller (Warner Instruments). Patch pipettes (5–9 MΩ) were pulled on a Brown Flaming puller (P-97, Sutter Instruments) using borosilicate glass electrode (Sutter Instruments). Tight seals (>5 GΩ) were obtained by applying constant negative pressure. Electrical signals were amplified using an Axopatch 200B amplifier (Molecular Devices), low-pass filtered at 10 kHz, digitized at 50 kHz via a Digitadata 1322A interface (Molecular Devices), and stored on a computer using pClamp9.2 software (Molecular Devices) for off-line analysis. In this study, all neurons displayed a resting membrane potential ranging from −55 to −75 mV. Neurons with a resting membrane potential more positive than −55 mV were discarded. In current-clamp recordings, the holding current of ∼0 pA was slightly adjusted to obtain a membrane potential of −60 mV. Series resistance (<20 MΩ) was not compensated. Input resistance was assessed by injecting negative current pulses (−100 pA, 1 s) at −60 mV. A depolarizing current pulse (100 pA; duration, 1 s) was applied to induce repetitive spiking in whole-cell configuration. Expression of PF was defined by firing frequency and plateau potential amplitude.
Primary cell culture.
Cortical tissue was dissected in cold HBSS from embryonic day (E) 16 mice embryos of either sex, trypsinized (0.25% trypsin) for 15 min, triturated, and seeded on precoated plates with poly-d-lysine (50 μg/ml in borate buffer, 0.1 m, pH 8.5; Sigma-Aldrich) in 35 mm plates with coverslips at 2 × 105 per plate. Cultures were maintained in Neurobasal (Invitrogen) supplemented with 1% v/v l-glutamine (Wisent), 2% v/v B27 (Invitrogen), and 1% v/v penicillin/streptomycin (Wisent) for 10 d. Between day 3 and 5, 20 μm 5-fluoro-2′-deoxyuridine (Sigma-Aldrich) was added.
Live imaging in cortical neurons expressing GFP-PLCΔ-PH.
Neurons were dissociated from cerebral cortices isolated from E15 embryos of C57BL/6j mice of either sex. Briefly, brains of E15 embryonic mice were placed in cold Ca2+-free and Mg2+-free Hank's solution containing 33 mm glucose, 10 mm HEPES, and 1% penicillin/streptomycin. For each culture, 8–12 E15 cortices were mechanically minced by repetitive aspiration though a sterile and fire-polished Pasteur pipette. Then cells were placed in DMEM high-glucose (Hyclone) plus 10% FBS for nucleofection using the Amaxa neuron nucleofector kit in accordance with the manufacturer's protocol (Lonza). Nucleofection was performed with 2 μg of the plasmid GFP-PLCΔ-PH. Then, neurons were plated on glass coverslips precoated with poly-d-lysine and GFP fluorescence was observed 2–4 d after nucleofection under an inverted TE2000-U microscope (Nikon) equipped with a 40× oil-immersion objective [CFI super(S) fluor, Nikon]. Rhodamine-labeled wheat-germ agglutinin (5 μg/ml; Vector Laboratories) was added to the dishes 20 min before recordings and thereafter rhodamine fluorescence was used to ensure that the focus plane of the imaging remained constant during the recording session. Images from GFP-positive cells were captured at baseline, and then at 10 and 20 min after carbachol (CCh) addition. Images containing GFP-positive cells were converted into binary masks using ImageJ and the outer limits of each cell were defined as a two-pixel-wide plasma membrane zone. Data presented represent the total pixel count, normalized to the initial GFP fluorescence, within the plasma membrane zone. All experiments were performed at room temperature with drugs perfused by gravity at a flow rate of 1–2 ml/min.
Ca2+ imaging experiments.
Changes in the intracellular concentration of Ca2+ were recorded using ratiometric Fura-2-based single-cell fluorescence microscopy in primary culture of cortical neurons at room temperature. After removal of the culture medium, cells were washed twice with a solution containing the following (in mm): 140 NaCl, 5KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH 7.4 (NaOH). Cells then were incubated for 20 min at 37°C with this solution supplemented with Fura-2 AM in the dark. After loading, cells were washed twice with a dye-free saline and kept at 37°C for 30 min in the dark before being placed on the stage of an inverted TE2000-U microscope (Nikon) equipped with a 40× oil-immersion objective [CFI super(S) fluor, Nikon]. Fura-2 was excited at 340 and 380 nm every second and emission at 510 nm was detected with a high-resolution cooled CCD camera (Cool Snap-HQ, Roper Scientific/Photometrics). Changes in intracellular calcium levels were determined by the ratio of fluorescence at 340 and 380 nm (340:380 ratio) calculated using Metafluor 7.0 software (Molecular Devices). We normalized the Fura-2 response using the mean of the 340:380 ratio during the 2 min baseline.
Western blot.
Mouse cortical tissue was homogenized in RIPA buffer (150 mm NaCl, 1% NP40, 0.5% deoxycholate, 0.1% SDS, 50 mm Tris, pH 8), cleared by low-speed centrifugation, normalized for protein content, and then boiled in a standard Laemmli Tris-glycine-SDS sample buffer. Immunoblotting was performed as previously described (Unsain et al., 2013) using anti-actin (catalog #0869100), purchased from MP Biomedicals, and p75NTR antibodies (Majdan et al., 1997).
Immunohistochemistry.
Medial EC sections of 25 μm were prepared using a microtome (Leica). Floating sections were washed in PBS, treated with 0.05% citraconic anhydride, pH 7.2 (5 min at 94°C), then washed with PBS-gelatin (0.2%)-Triton (0.25%). They were incubated overnight at room temperature with a goat anti-choline acetyltransferase (ChAT, 1:400; Millipore). ChAT expression was detected by immunofluorescence with Cy3-conjugated secondary antibodies (1:400; Jackson ImmunoResearch) and observed in confocal microscopy (Zeiss LSM 510; emission intensity, 543 nm). Digital images acquired under the same conditions were analyzed with ImageJ. The number of ChAT-positive fibers was quantified and expressed as the number of surface area occupied in the delineated areas of interest. Quantification was performed blindly from the tissue preparation to the imaging stage (n = 4 mice/group, 2–3 slices/mouse, 2–4 images/slice).
Object recognition test.
p75NTRfl/fl:CMVER-cre mice aged 12–18 weeks, injected with vehicle or tamoxifen, were habituated to the testing room for 20 min. The test consisted of two trials. In trial 1, the mice were exposed to two identical objects (small bottles; height, ∼2.5 cm; width; 1 cm) in an arena (length, 43 cm; width, 21 cm) for 10 min. The mice were then removed from the arena for 1 h. In trial 2, mice were placed in the same arena, with one of the objects being familiar (i.e., the same bottle as during trial 1), while the second object was novel (three dice glued in the form of a pyramid; height, 2.5 cm; width, 1 cm). Both trials were video recorded and then analyzed with the TopScan 2.0 tracking program (CleverSys). Exploration was defined as the cumulative duration within a trial in which the animal's nose was within 3.5-cm-diameter circle of an object. A novelty-preference ratio (NPR) was then calculated: NPR = [N/(N + F)] × 100, where N is the exploration time of the novel object, and F is the exploration time of the familiar object. Data are expressed as means ± SEs.
Evaluation of seizure threshold by multiple pentylenetetrazole injections.
All experiments were designed to minimize the number of animals used and their suffering. Seizure threshold was measured in adult p75NTRfl/fl-CMV-ER-cre mice littermates that were treated with oil or tamoxifen for 2 weeks. Experiments were performed ≥5 weeks after the last oil or tamoxifen injection. Seizures were induced by multiple intraperitoneal injections of subthreshold doses of pentylenetetrazole (PTZ; Sigma-Aldrich) every 20 min (Lüttjohann et al., 2009). The procedure was performed by a trained investigator who was blinded to the animal's treatment. The first injection consisted of 25 mg/kg of body weight, and the subsequent ones of 12.5 mg/kg. PTZ was dissolved in saline solution (0.9% NaCl) at 12.5 mg/ml. During the entire procedure, mice behavior was observed in Plexiglas boxes to register latency, duration, and incidence of seizure-related behaviors. Seizure threshold was determined as the number of PTZ injections needed to reach generalized clonic–tonic convulsive seizures. Seizures were evaluated according to a modified version of the Racine stage 1–5 scale, with 5 being the most severe (Racine, 1972). Stage 1 consists of ear and facial twitching. Stage 2 consists of myoclonic body jerks. In stage 3, animals show low-intensity tonic–clonic seizures marked by unilateral forelimb myoclonus. Stage 4 also includes bilateral forelimb myoclonus and rearing while stage 5 is marked by the appearance of generalized, clonic–tonic convulsive seizures (i.e., bilateral forelimb and hindlimb myoclonus and transient loss of postural control and sometimes jumping). PTZ injections were stopped when animals reached stage 5 and, because of the rapid turnover of the drug, animals recovered normal behavior by 1 h after the last PTZ injection. The validity of relying on behavioral monitoring to assess seizure activity in the brain has been demonstrated previously by studies correlating electrophysiological measures with observed seizure behaviors (Tremblay et al., 1984).
Pilocarpine-induced seizures and seizure assessment.
These procedures were performed in adult C57BL/6 wild-type or p75NTR−/− littermates (10 animals per genotype). The p75NTR-exonIII −/− mice have been previously described (Lee et al., 1992). Seizures were induced by a single dose of pilocarpine hydrochloride (295 mg/ kg, i.p.; Sigma-Aldrich). To avoid peripheral cholinergic effects, scopolamine methyl-bromide (1 mg/kg, i.p.; ICN Biomedical) was administered 25 min before the application of pilocarpine. Following the initial pilocarpine injection, the animals were closely monitored to assess seizure activity. Seizures were evaluated according to a modified version of the Racine scale (see above) using stages 1–6, with 6 being the most severe (Racine, 1972). Stage 6 describes animals that died after a severe stage 5 generalized clonic–tonic seizure. Status epilepticus was defined as continuous limbic seizure activity (uninterrupted events from stages 3 to 5) which was terminated after 60 min by diazepam (10 mg/kg, i.p.; Hoffmann-La Roche). This dose of diazepam stopped motor convulsions in all mice, as confirmed by close observation of the mice over the 30 min recovery period. Latency time is defined as the time between pilocarpine injection and the first event corresponding to stages 3, 4, 5, or 6. A fraction of the pilocarpine-injected animals (∼20%) did not develop limbic seizures, and hence status epilepticus (i.e., will not exhibit seizures characteristic from stages 3–6 during the observation period).
Data analysis.
Electrophysiological data were analyzed using Clampfit 9.2.1.8 (Axon Instruments). Values are expressed as mean ± SEM. Firing frequency was defined as the average spiking frequency within 20 s after the depolarizing current pulse (duration, 1 s; 100 pA). Plateau potential amplitude was defined as the difference between the mean membrane potential (in millivolts) measured at baseline before the pulse and the mean membrane potential measured during the steady-state phase of PF. When the response was a short sustained PF (<20 s), the firing frequency was still determined as the average spiking frequency within 20 s after the depolarizing pulse and the amplitude of the plateau as the difference (in millivolts) between the baseline mean membrane potential before the pulse and the mean membrane potential during the 20 s period after the pulse. Statistical analysis was made with Sigma Stat (version 3.5, Systat Software). Differences were considered statistically significant when p < 0.05.
Results
We have previously shown that CCh-induced PF in the EC relies directly on activation of TRPC4/5 channels, with CCh-induced phospholipase C activation driving a decrease in PIP2 levels and thereby relieving TRPC4/5 channel inhibition (Zhang et al., 2011). However, other mechanisms that regulate PF in the EC are largely unknown. BDNF and proBDNF have been reported to play prominent roles in the CNS as modulators of neuronal excitability and memory process. We therefore asked whether these neurotrophins have an effect on PF. PF can be activated in acute EC brain slices by exposure to CCh and this effect is strongly dose dependent, with 5 μm CCh never eliciting PF whereas 10 μm CCh invariably activating it (Fig. 1A,B). PF elicited using 10 μm CCh was completely blocked by the addition 2 ng/ml of proBDNF (Fig. 1C), whereas 2 ng/ml of mature BDNF does not affect PF in this setting (Fig. 2A,B). The blockade of PF produced by proBDNF also did not occur in p75NTR−/− mice (Fig. 1D–F), suggesting that an endogenous proBDNF-p75NTR signaling cascade regulates PF. Under physiological conditions, proBDNF and mBDNF are coreleased (J. Yang et al., 2009). To test whether mBDNF affects proBDNF activity on persistent activity, we coapplied proBDNF and BDNF at a ratio of 1:10 under 10 μm of CCh. Figure 2C,D shows that mBDNF does not prevent the inhibitory effect of proBDNF on persistent firing, indicating that under physiological conditions proBDNF can downregulate neuronal activity.
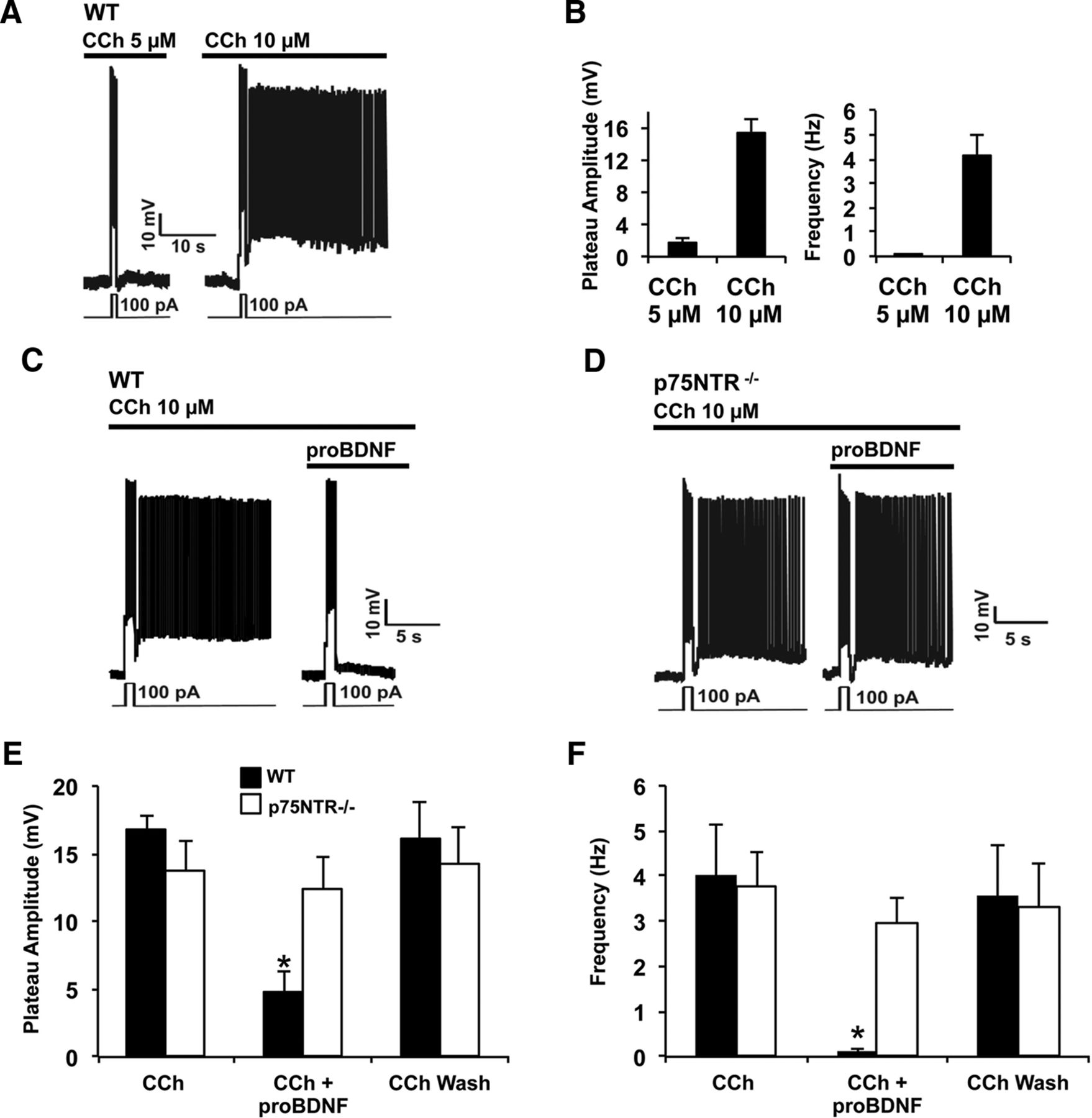
ProBDNF inhibits PF in entorhinal pyramidal neurons. Current-clamp recording from pyramidal neurons in layer V EC. A, Dose-dependent induction of PF by CCh in control condition: CCh at 10 μm (10 min), but not 5 μm (10 min), induces PF. B, Quantification of the plateau amplitude and the frequency of A (CCh: 5 μm, n = 16; 10 μm, n = 8). C, D, 10 min application of proBDNF at 2 ng/ml blocks PF in wild-type mice (C, WT, n = 5) but not in p75NTR−/− mice (D, n = 5). E, F, Quantification of the plateau amplitude (E) and the frequency (F) of C and D.

mBDNF does not block the effect of proBDNF on persistent firing. A, BDNF (10 min, 2 ng/ml) does not inhibit PF. B, Quantification of the plateau amplitude and the frequency of A (n = 4). C, PF is blocked in slices exposed to a combination of proBDNF [at 2 ng/ml (38 pm)] and BDNF [at 10.3 ng/ml (381 pm); n = 5]. D, Quantification of plateau amplitude and firing frequency from C. *p < 0.05, Student's t test.
The notion that proBDNF is normally secreted and functions as an extracellular ligand remains controversial. To address this, we identified function-blocking proBDNF antibodies (See Materials and Methods, data not shown) and used these to block the effect of secreted endogenous proBDNF. We predicted that the removal of endogenous proBDNF would increase the propensity for PF in the EC. Consistent with expectation, Figure 3A,B shows that 5 μm CCh elicits PF in slices preincubated with the proBDNF antibody, whereas PF was not elicited in slices incubated with heat-denatured proBDNF antibody (Fig. 3A).

Endogenous proBDNF regulates persistent firing and cell excitability. A, Slices treated with proBDNF antibodies showed PF under 5 μm CCh (n = 6/condition). B, Quantification of the plateau potential amplitude and firing frequency of the PF shown in A; −, mean heat-inactivated antibody; +, mean nondenatured antibody. C, Pyramidal cell response following pulses of current. A 10 min treatment with proBDNF (2 ng/ml) decreases the neuronal excitability (n = 12). D, Incubation with proBDNF antibodies increases excitability (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001, Student's t test.
We then compared the effects of proBDNF function-blocking antibodies and proBDNF addition on EC pyramidal neuron excitability, using a standard input–output curve protocol. Figure 3C shows that proBDNF sharply decreases baseline excitability of neurons, whereas proBDNF antibodies significantly increase neuronal excitability (Fig. 3D). Thus, endogenous proBDNF clearly affects the cell excitability (near rheobase) but does not alter the neuron's capacity for firing in response to strong depolarization stimuli in the absence of CCh. Together, these results indicate that endogenous proBDNF normally acts to suppress pyramidal neuron excitability and persistent firing within EC circuits.
To establish whether p75NTR plays a role in the regulation of EC pyramidal neuron activity, we assessed the threshold for PF induction in EC slices derived from p75NTR−/− animals. Figure 4A shows that subthreshold concentrations of CCh (5 μm) did not produce PF in slices derived from wild-type animals. Yet this level of CCh consistently induced PF in slices derived from p75NTR−/− littermates (Fig. 4D,E). To determine whether this change in PF reflects a selective contribution of neuronal p75NTR, we produced p75NTRfl/fl mice expressing cre from the neuron-specific Tα1-tubulin promoter to selectively delete p75NTR from neurons only. p75NTR levels are significantly reduced in EC lysates derived from these animals and the small amount of p75NTR remaining presumably reflects low levels of p75NTR expressed by non-neuronal cells in the EC (Fig. 4B). Figure 4D,E shows that 5 μm CCh evoked PF in neurons from p75NTRfl/fl:Tα1-cre mice. In contrast, we never observed sustained persistent activity in neurons from p75NTRfl/fl littermates that lacked the cre transgene.

Pyramidal neurons from p75NTR-null mice are highly sensitive to induction of PF by the muscarinic agonist CCh. A, Current-clamp recording of pyramidal neurons from brain slices of wild-type (WT, n = 16) and p75NTR-null mice (p75NTR−/−, n = 7). CCh (5 μm, 10 min) induced persistent firing in slices from p75NTR−/− mice but not from wild-type mice. B, Current-clamp recording of pyramidal neurons from brain slices of p75NTR fl/fl:Tα1-cre mice. cre− (n = 10): p75NTR is expressed in neuronal cells. cre+ (n = 8): p75NTR is suppressed in neuronal cells. CCh (5 μm, 10 min) induced persistent firing in neurons from mice lacking the p75NTR in neuronal cells but not in neurons from mice expressing the p75NTR. C, Current-clamp recording of pyramidal neurons from brain slices of p75NTRfl/fl:CMVER-cre mice injected with oil (n = 10) or with tamoxifen (n = 7). CCh (5 μm, 10 min) never induced persistent activity in slices from oil-injected mice but induced persistent firing in slices from tamoxifen-injected mice. A–C, The expression of p75NTR was measured by Western blot in cortex from mice of each genotype. D, E, Quantification of the amplitude (D) and frequency (E) of the persistent activity in the different models of mice. *p < 0.05, one-way ANOVA followed by a Bonferroni's test. F, Input–output curves, the number of action potentials in 1 s (AP/s) in response to stimulus from 0–100 pA have been counted in pyramidal neurons of the layer V of the EC from wild-type (WT) mice (n = 14), p75NTR−/− mice (n = 27), p75NTRfl/fl:CMVER-cre oil-injected mice (n = 12), and p75NTRfl/fl:CMVER-cre tamoxifen-injected mice (n = 11). *p < 0.05, Mann–Whitney test. G, ChAT immunostaining in the medial EC of p75NTRfl/fl:CMVER-cre oil-injected and p75NTRfl/fl:CMVER-cre tamoxifen-injected mice (scale bar, 50 μm). Right, Quantification of cholinergic fiber density in the medial EC (n = 3 mice/group).
Mice lacking neuronal p75NTR during development may have defects in central circuitry (Boskovic et al., 2014). To determine whether selectively deleting p75NTR in adult animals increases the propensity of EC pyramidal cells to exhibit PF, we generated p75NTRfl/fl:ER-cre mice to allow tamoxifen-dependent p75NTR gene excision. Adult animals that were administered tamoxifen showed a sharp drop in p75NTR expression within the cortex (Fig. 4C) and, importantly, subthreshold CCh (5 μm) readily evoked PF in EC pyramidal neurons within slices of tamoxifen-treated, but not vehicle-treated, littermates (Fig. 4C–E). We next asked whether p75NTR regulates EC neuron excitability. The input–output curves in Fig. 4F show that EC neurons rendered null for p75NTR, either by germ-line deletion or by tamoxifen-induced deletion during adulthood, are more readily excitable than their control counterparts. Previous studies have suggested that p75NTR deletion may increase cholinergic innervation in the CNS and we therefore assessed cholinergic fiber innervation of the medial EC in p75NTRfl:fl:CMVER-cre adult mice treated with tamoxifen or with vehicle. We did not observe any difference between the two groups (Fig. 4G) and conclude that the changes in excitability and firing pattern that result from p75NTR deletion in adults do not reflect an increase in cholinergic innervation of the medial EC. Rather, together, our findings indicate that a proBDNF-p75NTR signaling cascade acutely regulates the excitability of EC pyramidal neurons. To address this directly, we used antibodies directed against the p75NTR extracellular domain to determine whether acute p75NTR blockade can regulate PF. Figure 5A shows that subthreshold CCh (5 μm) readily evoked PF in slices exposed to the p75NTR antibody, whereas, after heat denaturation, the same antibody had no effect (Fig. 5A–C). Figure 5D shows that wild-type EC neurons acutely exposed to antibodies directed against the p75NTR extracellular domain are also more readily excitable than EC neurons treated with heat-inactivated p75NTR antibody.

Acute blockade of p75NTR facilitates persistent firing in EC pyramidal neurons. A, Current-clamp recording of pyramidal neurons from the layer V of the EC in brain slices incubated with vehicle (n = 16), with a heat-inactivated p75NTR antibody (1:500; n = 6), or with a functional p75NTR antibody (1:500) over 90 min (n = 5). CCh (5 μm, 10 min) induced persistent firing only in slices treated with the noninactivated antibody. B, C, Quantification of the amplitude (B) and of the frequency (C) of persistent activity following different treatments. *p < 0.05, one-way ANOVA followed by a Bonferroni's test. D, Input–output curves in pyramidal neurons from wild-type slices treated with a heat-inactivated p75NTR antibody (n = 11) or with a functional p75NTR antibody (n = 5). *p < 0.05, Mann–Whitney test.
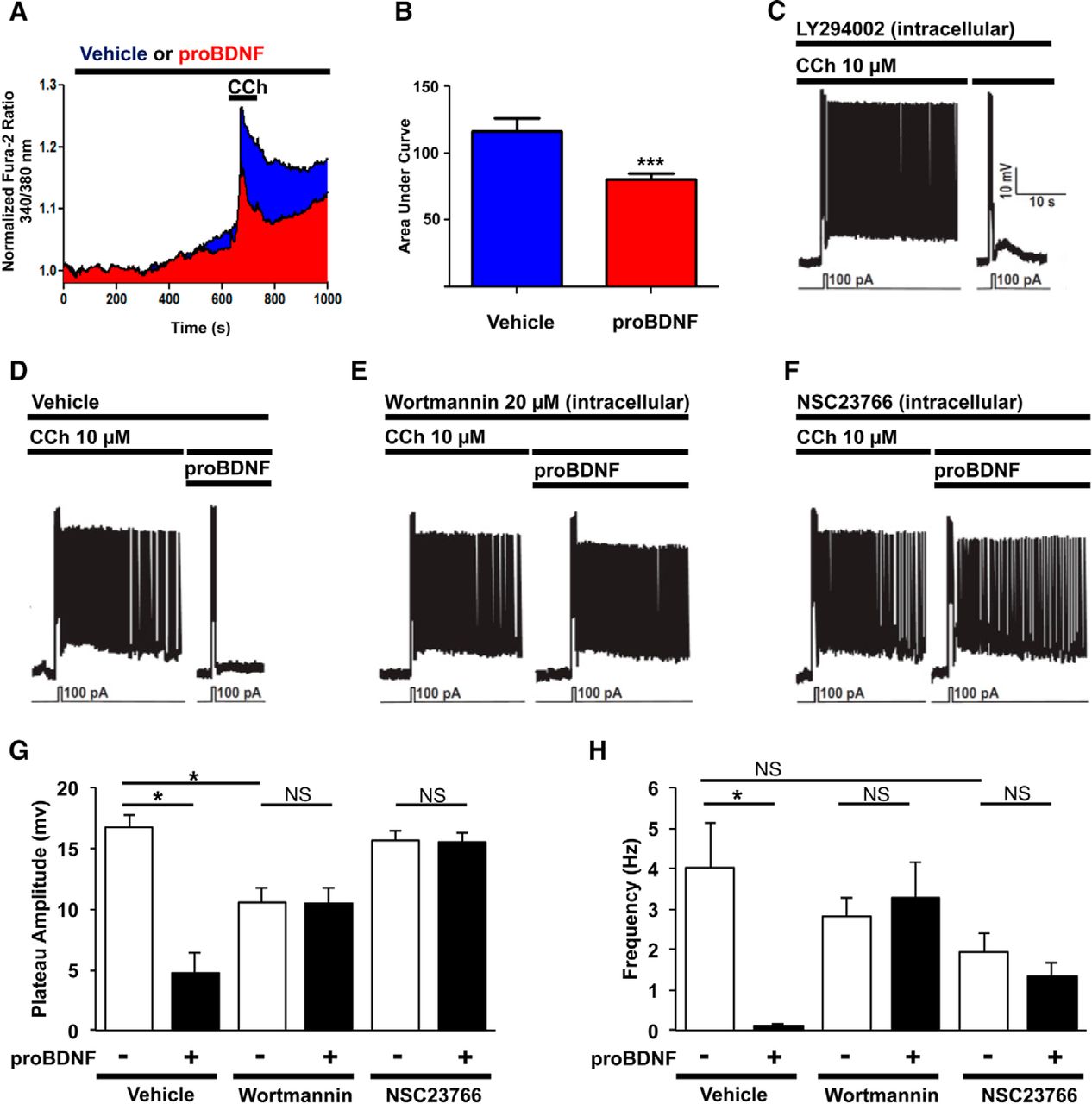
We have previously shown that calcium influx via TRPC4/5 channels is necessary for persistent activity in pyramidal neurons of the layer V EC (Zhang et al., 2011) and therefore predicted that proBDNF would decrease Ca2+ influx elicited by CCh. Using Fura2-based Ca2+ imaging in primary culture of mouse cortical neurons, we established that these dissociated cells display Ca2+ influx at higher concentrations of CCh (100 μm) than those required in slices. Nonetheless, CCh-induced Ca2+ entry is significantly decreased in neurons exposed to 2 ng/ml proBDNF (Fig. 6A,B).

ProBDNF inhibits cholinergic calcium responses and PF via a Rac1-PIP2 pathway. A, Normalized Fura-2 responses of cortical neurons treated with vehicle (n = 55) or proBDNF 2 ng/ml (n = 68) 10 min and then subject to CCh (100 μm) for 1 min. B, Area under curve of A. ***p = 0.0006, t test. C, Current-clamp recording from pyramidal neuron in wild-type mice; 30 min with LY294002 20 μm within the patch pipette avoid the cell to fire again. D, proBDNF blocks PF (Fig. 1E). E, Wortmannin (20 μm, 15 min) within the patch-pipette solution blocks the effect of the proBDNF. F, The Rac1 inhibitor NSC23766 (50 μm, 15 min) in the patch-pipette solution blocks the effect of the proBDNF. G, Quantification of the plateau amplitude of D–F. H, Quantification of the frequency of D–F. *p < 0.05, one-way ANOVA followed by a Bonferroni's test.
TRPC4/5 channel opening is strongly suppressed by PIP2 (Trebak et al., 2009). To confirm that PIP2 levels affect PF in the EC, we demonstrated that intracellular delivery of LY294002, a potent PI3K inhibitor that increases PIP2 levels by blocking its conversion to phosphatidylinositol 3,4,5-bisphosphate (PIP3), reduces the generation of PF (Fig. 6C,G,H). This result led us to hypothesize that activation of p75NTR by proBDNF blocks PF by elevating levels of PIP2, which in turn inhibits transient receptor potential canonical (TRPC) channels (Fig. 6D). To determine whether p75NTR alters the expression of TRPC channels, protein levels of TRPC4 and TRPC5 subunits were assessed by Western blot in wild-type and p75NTR−/− mice. No differences were observed between these two groups (data not shown).
PIP2 is produced from phosphatidylinositol (PI) in a two-step process, in which PI is converted into to phosphatidylinositol 4-phosphate (PIP) by PI4K, and thenconverted into PIP2 by phosphatidylinositol 4-phosphate 5-kinase (PIP5K). Selective PIP5K inhibitors are not available but high concentrations of wortmannin (20 μm) can be used to block phosphatidylinositol 4-kinase (PI4K). When supplied intracellularly by patch pipette to pyramidal neurons, 20 μm wortmannin completely blocked proBDNF-induced suppression of PF (Fig. 6E,G,H), whereas lower concentrations that are typically used to block phosphatidylinositol 3-kinase (PI3K; 100 nm) had no effect (data not shown).
Previous studies have established that Rac1 lies downstream of p75NTR and others have shown that activated Rac1 can regulate PIP2 generation (Carpenter et al., 1999; Hinchliffe, 2000; Osborne et al., 2001; Harrington et al., 2002; Weernink et al., 2004; Coulson et al., 2008). We therefore hypothesized that activation of p75NTR by proBDNF activates Rac1 activity, which then acts to induce PI4K production. Consistent with this, we found that intracellular delivery of NSC23766, a specific Rac1 inhibitor, strongly suppressed the effect of proBDNF on PF (Fig. 6F–H).
To test the hypothesis that proBDNF can increase PIP2 levels in neurons, we examined primary cultures of cortical neurons transfected with a plasmid encoding the PIP2 sensor GFP-PLCΔ-PH (Stauffer et al., 1998). To mimic the conditions used in our electrophysiological recordings of persistent activity in slices, we first determined the effect of CCh on membrane levels of GFP-PLCΔ-PH after 10 or 20 min exposure. The application of the muscarinic agonist CCh sharply depleted membrane levels of PIP2 in cortical neurons by 10 min, with a further statistically significant drop occurring between 10 and 20 min exposure (Fig. 7A,B). We then asked whether proBDNF could reduce the CCh-mediated reduction in plasma membrane GFP-PLCΔ-PH levels. To mimic the conditions used in our electrophysiological recordings, we asked whether proBDNF, added after 10 min CCh exposure, prevents the subsequent drop in plasma membrane PIP2 levels. Figure 7C,D shows that proBDNF exposure does indeed block the effects of CCh. This effect of proBDNF is prevented when cells were pre-exposed (for 2 h) to a p75NTR function-blocking antibody (Fig. 7E,F) or preincubated (15 min) with the Rac1 inhibitor NSC23766 (7G,H). Thus, our findings indicate that proBDNF inhibits PF by activating a Rac1-PI4K signaling cascade that lies downstream of p75NTR, elevating the levels of PIP2 and inhibiting the entry of extracellular calcium.

proBDNF increases PIP2 at the plasma membrane of cortical neurons via a p75NTR-Rac1 pathway. The surface level of PIP2 was monitored in live primary cultures of cortical neurons transfected with the GFP-PLCΔ-PH reporter and incubated with rhodamine-labeled wheat-germ agglutinin. A, Representative images of control condition where neurons were treated with CCh (100 μm) for 20 min. Pictures were taken before application of CCh (top), after 10 min application of CCh (middle), and after 20 min application of CCh (bottom). B, Quantification of A (n = 6) after normalization to the GFP fluorescence at the membrane before drug application (see also Material and Methods). *p < 0.05, paired t test. White asterisks represent areas of membrane where the GFP fluorescence disappeared after application of CCh. C, Same experiment as in A but after initial 10 min of CCh, proBDNF (2 ng/ml) was coapplied with CCh for subsequent 10 min. D, Quantification of C (n = 7). E, Same experiment as in C but dishes were pretreated 2 h with a p75NTR function-blocking antibody (1:500). F, Quantification of E (n = 8). **p < 0.01 paired t test. G, Same experiment than in C but the Rac1 inhibitor NSC23766 (50 μm) was bath-applied throughout the experiment. H, Quantification of G (n = 5). Scale bar, 10 μm.
The effect of p75NTR on PF suggests it may affect working memory in adult mice. To test this, we compared the performance of p75NTRfl/fl:CMVER-cre mice injected with tamoxifen or with vehicle in a novel-object recognition task (NOR; Fig. 8A,B). Tamoxifen-treated mice performed significantly better in the NOR test than p75NTRfl/fl:CMVER-cre mice treated with the control vehicle (Fig. 8C), consistent with the hypothesis that endogenous p75NTR signaling has a negative impact on short-term memory.

Deletion of p75NTR improves working memory but increases sensitivity to pilocarpine-induced and PTZ-induced epileptic seizures. A, p75NTRfl/fl:CMVER-cre mice were used for a novel-object recognition test. B, Mice were tested for object preference between the two objects used in this experiment. C, Mice injected with tamoxifen (n = 12 for the 3-min-delay group; n = 20 for the 60-min-delay group) spend more time around the novel object than mice injected with corn oil (n = 11 for the 3-min-delay group; n = 18 for the 60-were entered min-delay group). *p < 0.05, Mann–Whitney test. D, Quantification of the time latency to observe seizures in wild-type (p75NTR+/+, n = 6) and p75NTR−/− (n = 6) in minutes after a single-dose injection of pilocarpine. Only mice with seizures were entered into accounts. *p < 0.05, Mann–Whitney test. E, Response of wild-type (p75NTR+/+, n = 9) and p75NTR−/− (n = 11) mice to pilocarpine in percentages. Sx-y correspond to occurrences of stages x and y of the Racine scale. For each group the exact number of animals undergoing a specific stage is noted as X/total number of animals. F, p75NTRfl/fl:CMVER-cre mice injected with vehicle (n = 5) or with tamoxifen (n = 5) were subjected to a dose of 12.5 mg/kg of PTZ every 20 min over 3 h. The number of injections of PTZ necessary to reach stage 5 of the Racine scale was monitored.
Hyperactivity in the EC can promote uncontrolled discharges and epilepsy (Bear et al., 1996; Fountain et al., 1998). Given the modulatory effect of p75NTR on PF and cell excitability, we speculated that p75NTR deletion may increase propensity for seizures. We tested this by administering pilocarpine to p75NTR−/− mice and wild-type littermates, and monitored animals for seizure induction. We found that initial seizures were induced more rapidly in p75NTR−/− mice and that generalized clonic–tonic seizures occurred in the p75NTR−/− mice but not in wild-type littermates (Fig. 8D,E), suggesting that p75NTR may normally suppress hyperexcitability. To establish whether suppressing p75NTR in adult mice increases seizure sensitivity, we established the minimal dose of PTZ required to induce seizures in adult p75NTRfl/fl:CMVER-cre mice injected with tamoxifen or vehicle. Figure 8F shows that tamoxifen-injected p75NTRfl/fl:CMVER-cre mice were considerably more sensitive to PTZ-induced seizures compared with control animals injected with vehicle.
Discussion
This study demonstrates that proBDNF and p75NTR are important regulators of persistent activity in the adult mammalian brain slices. p75NTR−/− slices express persistent activity when exposed to subthreshold levels of the muscarinic agonist CCh and persistent firing can be inhibited in wild-type slices exposed to low levels of proBDNF. By selectively deleting a floxed p75NTR allele of p75NTR, we demonstrated that the effect of p75NTR on persistent activity is due to its neuronal expression in adult animals. Together with data showing that exposure to p75NTR antibodies, which block ligand binding, mimicked the effect of p75NTR deletion, we conclude that an endogenous proBDNF-p75NTR cascade operates tonically within the EC to control persistent activity.
Previous studies have shown that proBDNF and BDNF often have opposite effects on synaptic transmission and stability (Chao, 2003). Indeed, the conversion of proBDNF to BDNF that can occur extracellularly has emerged as an important regulatory event because it determines whether p75NTR or Trk receptors are activated (F. Yang et al., 2009). To confirm that the effects of exogenous proBDNF reported here reflects an activity of the uncleaved precursor, rather than mature BDNF, we directly compared the effects of BDNF versus proBDNF on persistent activity. proBDNF had a clear and obvious effect, blocking persistent firing, whereas identical concentrations of BDNF had no effect. To confirm a modulatory role for endogenous proBDNF on persistent activity of pyramidal neurons, we exposed slices to a proBDNF function-blocking antibody and demonstrated that this allows persistent activity to be generated by subthreshold levels of CCh. We conclude that endogenous proBDNF, working through p75NTR, functions tonically to regulate persistent firing in the EC.
Persistent activity in the EC requires TRPC channel activity (Zhang et al., 2011). We have previously reported that PIP2 functions as a potent suppressor of TRPC4/5 in the EC and proposed that a phospholipase C-dependent reduction in PIP2 level was a prerequisite for the emergence of persistent firing (Ayala et al., 2009; Zhang et al., 2011). Consistent with these previous results, we show here that LY294002, which blocks the conversion of PIP2 to PIP3, completely blocks CCh-induced persistent firing. This led us to hypothesize that proBDNF blocks persistent activity by activating signaling cascades that result in the postsynaptic accumulation of PIP2 and we provide evidence indicating that PI4K and Rac1 play a crucial role in the effect of proBDNF on persistent activity. Since PIP5K is activated by Rac1, the scenario that emerges is that PI is converted to PIP via PI4K and that proBDNF, working through p75NTR, activates PIP5K via a Rac1-dependent pathway. By enhancing the phosphorylation of PIP, this cascade leads to the accumulation of PIP2, which then inhibits TRPC4/5 and blocks persistent activity.
An alternative mechanism for the generation of PIP2 downstream of p75NTR could involve the conversion of PIP3 to PIP2, a reaction that is mediated by the phosphatase and tensin homolog (PTEN). A recent study has reported that proNGF rapidly increases levels of PTEN via a p75NTR/sortilin pathway (Song et al., 2010). This mechanism does not seem to overlap with that reported here as we did not observe proBDNF-induced changes in PTEN phosphorylation or protein levels (data not shown).
Two previous studies have shown that p75NTR−/− mice display improved performance in tests of spatial memory and those authors proposed that p75NTR acts as a negative regulator of hippocampal function (Greferath et al., 2000; Barrett et al., 2010). Cholinergic neurons express high levels of p75NTR and germ-line deletion of p75NTR has been reported to increase cholinergic innervation of the hippocampus (Yeo et al., 1997). However, a more recent study from the Coulson group has shown that deletion of p75NTR alters cholinergic innervation of barrel cortex but not hippocampus (Boskovic et al., 2014). We examined cholinergic innervation of the EC and found that innervation levels were not changed when p75NTR was deleted from adult mice. Therefore, we conclude that the physiological and behavioral changes observed reflect alterations intrinsic to the EC network.
Aberrant hyperactivity of pyramidal neurons in the EC is commonly associated with disorders such as epilepsy (Bear et al., 1996; Fountain et al., 1998). Because we observed an increase in seizure severity in p75NTR−/− mice and an increase in the susceptibility to generalized clonic–tonic seizures, we postulate that p75NTR normally functions to dampen cell excitability in the EC. In the model of pilocarpine-induced seizures used here, wild-type animals showing seizures entered status epilepticus, a condition of continuous seizure activity. In support of a role of p75NTR in the regulation of cell excitability, we found that p75NTR−/− mice experiencing seizures rapidly manifested severe clonic–tonic seizures that were invariably lethal. Notably, we and others have previously shown that p75NTR is highly expressed in the EC and hippocampus after seizure and that p75NTR−/− mice display reduced seizure-induced neuronal apoptosis (Roux et al., 1999; Troy et al., 2002), consistent with the well known role of p75NTR as a proapoptotic receptor. Together, these findings suggest that the proBDNF-p75NTR signaling pathway in the EC normally functions to restrain runaway activity.
To conclude, our data reveal that activation of p75NTR by proBDNF activates a signaling cascade that reduces pyramidal neuron excitability and inhibits persistent activity. Our findings suggest that this functions to balance EC performance in working memory with the risk of runaway activity that can result in epileptic seizure. These studies complement and extend other recent work that has suggested that p75NTR can regulate the firing pattern of Purkinje cells (Tian et al., 2014) and sympathetic neurons (Luther and Birren, 2009), with the emerging picture being that proneurotrophin-p75NTR signaling cascades play important physiological roles as neuromodulatory mechanisms that regulate neuronal firing in the peripheral nervous system and the CNS.
Footnotes
This work was supported by Canadian Institutes of Health Research (37850 to P.A.B.; MOP-86527 and Savoy Foundation for Epilepsy to P.S.). We are grateful to B. Sharif, A. Deleon, X.K. Tong, and E. Hamel for technical support and shared expertise.
The authors declare no competing financial interests.
- Correspondence should be addressed to either of the following: P. Séguéla at the above address. philippe.seguela{at}mcgill.ca; or P.A. Barker at the above address. phil.barker{at}mcgill.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}