

Abstract
A natural consequence of aging is a loss of dopamine function and associated deficits in working memory in both human and nonhuman primates. Specifically, deficiency of D1 receptor signaling has been implicated in age-related cognitive decline. Here, we report that an intermittent, sensitizing regimen of the D1 dopamine agonist ABT-431 dramatically enhances working memory performance in aged rhesus monkeys, while either producing impairment or having little effect on performance in young adult monkeys. Importantly, cognitive enhancement in the aged monkeys was still evident for >1 year after cessation of D1 treatment. Because intermittent exposure to low doses of amphetamine and other stimulants has been shown to enhance responsiveness to subsequent stimulant exposure, our findings suggest that sensitization of D1 signaling may provide a novel neurobiological mechanism for improving a core cognitive process in conditions in which dopamine function has deteriorated, such as in normal aging and Parkinson's disease.
Introduction
A pronounced loss of dopamine function occurs along with the natural aging process and is associated with a decline in short- and long-term memory capacity as well as a diminished quality of life for millions of individuals (Volkow et al., 1998; Bachman and Farde, 2001; Nieoullon, 2002). Dopamine dysfunction is integrally involved in the working memory impairments prominent in Parkinson's disease (Rinne et al., 2000; Cools et al., 2002), schizophrenia (Goldberg et al., 1991; Abi-Dargham et al., 2002), Huntington's chorea (Beckman et al., 1997), and attention deficit disorder (Levy and Farrow, 2001; Nieoullon, 2002), and may even constitute a prodromal sign of Alzheimer's disease (Welsh et al., 1991). Age-related decline in dopamine and working memory are also observed in nonhuman primates and other species (Arnsten et al., 1994; Enborg et al., 1998; El-Ghundi et al., 1999; Siddiqi et al., 1999). A variety of pharmacological strategies used to ameliorate these deficits have met with limited success.
Age-dependent decline in dopamine function encompasses decreases in the density of D1 and D2 dopamine receptor protein and mRNA, among a variety of other markers of deficiency (Goldman-Rakic and Brown, 1981; de Keyser et al., 1990; Rinne et al., 1990; Bannon and Whitty, 1997; Wang et al., 1998; Ma et al., 1999; Naoi and Maruyama, 1999; Harada et al., 2002; Henby and Trojanowski, 2003). The decline in D1 receptors is significant, because stimulation of this receptor improves working memory in humans (Davidson et al., 1990; Muller et al., 1998) and dopamine-deficient monkeys (Arnsten et al., 1994; Schneider et al., 1994; Cai and Arnsten, 1997). We recently demonstrated that intermittent D1 stimulation ameliorates working memory impairments in young adult monkeys in whom D1 receptors were experimentally downregulated by chronic haloperidol (Lidow and Goldman-Rakic, 1994; Castner et al., 2000). Remarkably, the cognitive improvement induced by this intermittent schedule persisted for >1 year after cessation of treatment. A restricted range for D1 receptor stimulation has also been found for the expression of spatially tuned delay activity or memory fields, in prefrontal neurons—the physiological underpinning of spatial working memory (Sawaguchi and Goldman-Rakic, 1991; Williams and Goldman-Rakic, 1995). These studies suggest that either too much or too little D1 stimulation can lead to impairments in spatial working memory (Lidow et al., 1998).
Whereas sensitization is readily produced in rats, monkeys, and humans by psychomotor stimulants such as amphetamine (Robinson and Becker, 1986; Kalivas and Stewart, 1991; Sax and Strakowski, 1998; Castner and Goldman-Rakic, 1999), there is also evidence that it can be induced by intermittent D1 agonist exposure in rats and monkeys (Pierce et al., 1996; Castner et al., 2000). Moreover, D1 receptor stimulation has also been implicated in cocaine craving (Giardina and Williams, 2001), and is required for the induction of amphetamine-induced behavioral and neurochemical sensitization (Stewart and Vezina, 1989; Bjijou et al., 1996; Vezina, 1996). Thus, the mechanisms by which intermittent D1 stimulation restored cognitive function in a chronic haloperidol-induced D1-downregulated state might be akin to those involved in the process of sensitization to psychomotor stimulants. Therefore, we examine here whether this process might be exploited for the treatment of endogenous dopamine deficiency in age-related cognitive decline.
Materials and Methods
Subjects. Cognitive performance was measured throughout the study in four aged (20-30+ years of age) and three young (7-10 years of age) female rhesus monkeys (Macaca mulatta). Males were not included in the study because of the limited availability of aged males. Monkeys were housed individually and maintained in accordance with Yale Animal Use and Care Committee and federal guidelines for the care of nonhuman primates. All of the monkeys received either a full bowl or a maintenance number of biscuits sufficient to maintain their normal body weight, as well as fruit and peanuts each day. On the basis of the medical histories available for the animals included in the present study, none of the subjects was directly related (i.e., siblings, parents, etc.). It should be noted that three of the four aged monkeys received acute low-dose drug treatments several years before the present study at various times. However, because the animals were all trained to a stable level of baseline performance before repeated D1 treatment, any previous drug experience in these monkeys should not have influenced the present results.
Spatial delayed-response task. Monkeys were trained to perform the delayed-response task, a test of spatial working memory capacity, in a Wisconsin General Testing Apparatus situated in a sound-attenuated room. In the initial training for this task, the monkey is presented with two spatially displaced wells, and the monkey watches while the experimenter baits one well; the well baited varies in a semirandom order on a trial-by-trial basis. After the well is baited, the wells are then covered with identical plaques, and an opaque screen is lowered between the experimenter and the monkey for a randomized delay (see below). During the delay, the monkey must hold in mind the spatial location of the baited well to respond appropriately and be rewarded. Task difficulty/memory load is increased by either increasing the length of delay or increasing the number of spatially displaced wells (i.e., the spatial resolution). This task involves the use of five delays that are pseudorandomly varied across trials within a given test session. Because there are 20 trials per test session, each of the five delays was tested on four trials. For a given number of wells (e.g., two-well testing board), delay lengths were defined as 0N, 1N, 2N, 3N, and 4N, where N can be increased from 1 to 10 sec. Thus, in the initial training, N = 1 sec, and the five delays used across trials correspond to 0, 1, 2, 3, and 4 sec. Once an animal performed at 90% at N = 1, N was increased to 2 until 90% correct performance was achieved, and so forth, until N = 10. If a monkey performed at an average of 90% correct across three consecutive test sessions at N = 10, then the task was made more difficult by adding an additional well, and N was lowered to 1 sec and then increased as described above. This process continued until the animals achieved a stable level of performance as defined by ∼70 ± 2.0% correct across a minimum of 20 consecutive test sessions (see Table 1). Throughout the 3+ years of this study, animals were tested 3-5 d per week as standard in this type of longitudinal study.
Individual delayed-response performance across testing conditions
D1 agonist treatment. Once baseline performance was stable at ∼70 ± 2.0% correct across a minimum of 20 consecutive test sessions, monkeys received intermittent treatment with the selective D1 agonist (-)-trans 9,10-acetoxy-2-propyl-4,5,5a,6,7,11-b-hexahydro-3-thia-5-azacyclopent- 1-ena[c]phenanthrene hydrochloride (ABT-431; provided by Hoechst Marion Roussel, now Aventis Pharmaceuticals), which is the diacetyl prodrug of the active metabolite A-86929 to which it is hydrolyzed in vivo (Giardina and Williams, 2001). Before injection, ABT-431 was dissolved in nitrogen-percolated, double-distilled, ice-cold water under dark conditions with 0.2% ascorbic acid. Individual aliquots of 1 ml were stored in light-protected containers at -70°C. During the end of the baseline testing period, the D1 agonist period, and the early part of the post-D1 agonist period, animals received an intramuscular injection of either drug or vehicle 30 min before testing. This was to ensure that the investigators were blinded to the treatment condition at all of the times, and that the monkeys were acclimated to intramuscular injections before receiving D1 agonist treatment. No changes in gross motor behavior or alertness were observed in response to either saline or drug injections. Furthermore, no adverse consequences of repeated D1 or saline treatment were observed in either the aged or young monkeys in the present study. The full D1 agonist ABT-431 (0.00001 or 0.0001 mg/kg) was given weight by volume; volume of vehicle in milliliters = body weight in kilograms/10. The doses and dosing regimen used in the present study were based on our previous work with ABT-431, in which the same doses and dosing regimen reversed chronic haloperidol-induced working memory deficits (Castner et al., 2000). For the D1 agonist period, all of the monkeys received treatment with the D1 agonist for 5 consecutive days followed by a 2-3 week washout, during which time they received the vehicle alone, and this regimen (i.e., 5 d of D1 agonist treatment followed by a 2-3 week washout is defined as a block) was repeated four additional times. Monkeys received five consecutive blocks of treatment. Thus, the D1 agonist period corresponded to ∼4 months of testing. Note: Monkey TYR became temporarily ill during the D1 agonist period and therefore did not receive the final two blocks of treatment but was still tested in the post-D1 agonist period (see Table 1).
Data analysis. Factorial ANOVA with Scheffe post hoc comparisons was performed on the percentage (mean ± SEM) of correct scores for each monkey for each testing period. Regression analysis (Pearson's) on the individual scores was used first to determine the stability of performance during the baseline period for the aged and young groups separately. This was then repeated across all five blocks of the D1 agonist period and for the post-D1 agonist period for the young and aged groups separately. The values for the correlation coefficient r referred to in Figure 1 were calculated from the average scores of the monkeys in each group, whereas the r values in the text refer to the total data for all of the individuals. Two-way ANOVA was used to examine group (aged vs young) × condition (D1 agonist blocks 1-5 in chronological order) effects on the percentage of test sessions for which delayed-response performance was significantly above baseline. Three-way ANOVA was also used to examine delay (0N, 1N, 2N, 3N, and 4N) × group (aged vs young) × testing condition (baseline; D1 agonist period; post-D1 agonist period) effects on the mean number of errors in the task.

Delayed-response performance across baseline, D1 agonist, and post-D1 agonist testing periods in aged and young monkeys. The filled triangles represent the average performance of four aged (a-c) and three young (d-f) monkeys on the spatial working memory task for individual test sessions across baseline, D1 agonist (all five blocks), and post-D1 agonist testing periods. Both aged (a) and young (d) monkeys maintained ∼70% ± 2% correct baseline performance for a minimum of 20 consecutive test sessions before treatment with the D1 agonist. Whereas the performance of the aged monkeys (b) significantly improved during repeated D1 agonist treatment, the performance of the young monkeys (e) remained unchanged or became impaired. During the post-D1 agonist testing period, the improved performance of the aged monkeys was sustained (c), and the young monkeys' performance returned to baseline (f). The r values in the graphs are based on the mean performance for all four aged or all three young monkeys on given test sessions within each testing period.
Results
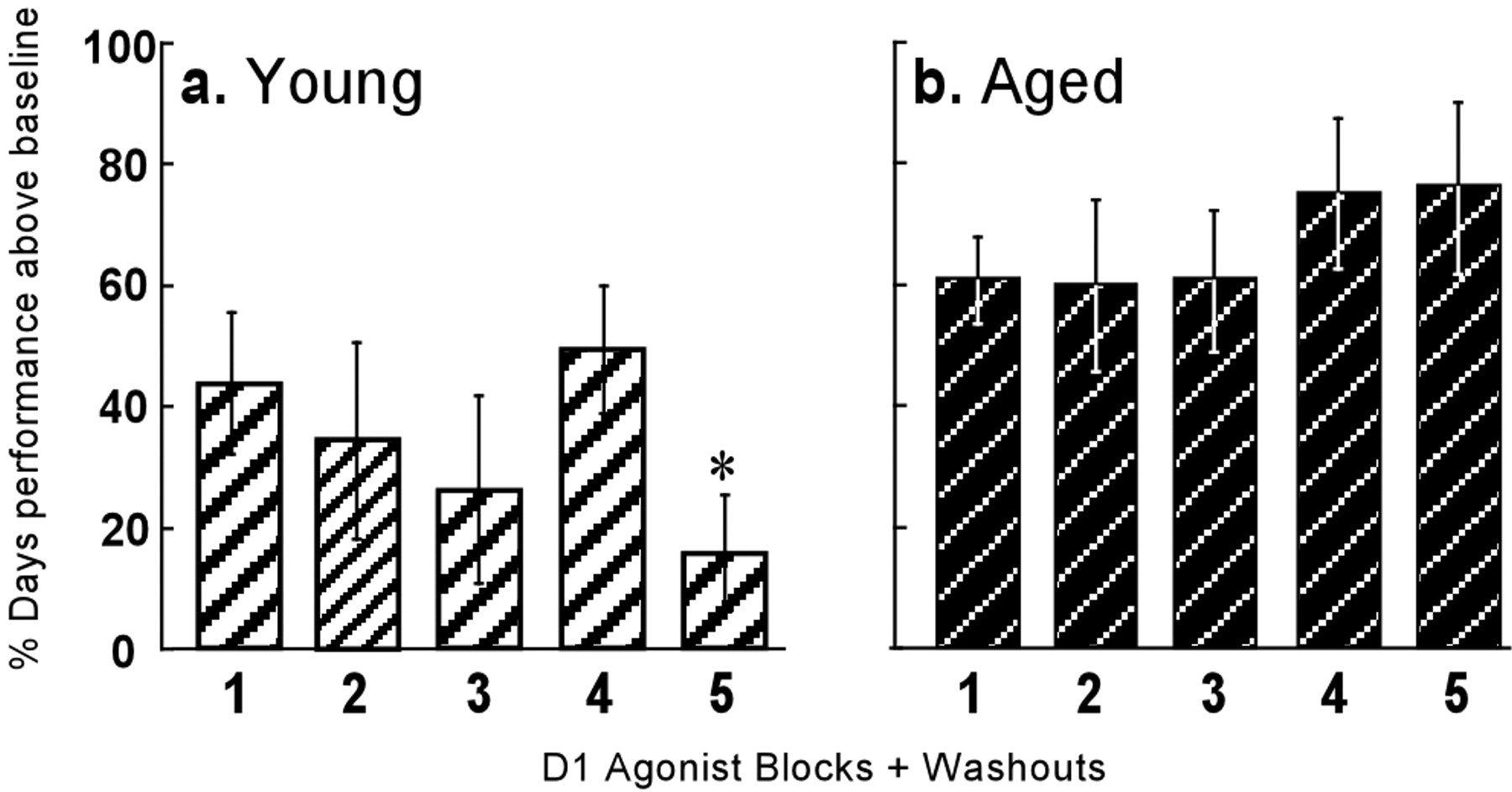
The aged and young monkeys all met a criterion of stable baseline performance (∼70 ± 2% correct) on delayed response for a minimum of 20 consecutive test sessions before D1 treatment (Fig. 1a) (aged, r = 0.02; F(1,109) = 1.84; p = 0.18) (d) (young, r = 0.00002; F(1,90) = 0.002; p = 0.97) (note that these r values pertain to the individual scores for monkeys in both groups; also see Table 1 for individual mean percent correct ± SEM). Despite the fact that the aged monkeys tended to make more errors than young monkeys at the longest delays (see Fig. 3a), the two groups reached a comparable level of baseline performance (Fig. 1, Table 1). Significant differences in delayed-response performance of young and aged monkeys were observed, however, during the D1 agonist period. In aged monkeys, D1 agonist treatment markedly improved cognitive performance (Fig. 1b) (r = 0.25; F(1,202) = 66.12; p = 4.216 × 10-14) (Table 1). Across the five blocks of D1 treatment, the aged monkeys' performance rose by 20.22 ± 3.82% compared with their mean baseline performance of 67.85 ± 0.87% (Table 1). In contrast, the performance of young monkeys either showed no change (N = 1) or was significantly impaired (N = 2) in response to the same D1 agonist regimen (Fig. 1e) (r = -0.007; F(1,129) = 0.08; p = 0.78) (Table 1). Remarkably, with each subsequent block of D1 treatment, the aged monkeys showed increasingly extended periods of sustained cognitive improvement that extended into the washout periods between D1 agonist administrations. By the fifth D1 block, the aged monkeys exhibited a significantly greater percentage of test sessions in which their performance was above their own baseline performance compared with the young monkeys (F(1,4) = 15.48; p = 0.0007) (Fig. 2). Indeed, after the fifth and final D1 agonist block, the performance of the aged monkeys remained improved (up to an average of 83.61 ± 0.68, or 23.22 ± 1.63% improvement relative to their baseline performance) for periods of >1 year (Fig. 1c) (r = 0.002; F(1,329) = 1.77; p = 0.18; no significant change in performance over time). In direct contrast, the post-D1 agonist performance of young monkeys was not significantly different from their baseline performance (mean ± SEM, 71.47 ± 4.71% correct during the post-D1 agonist testing period vs 72.65 ± 4.41% during the baseline testing period). During the post-D1 agonist period, the cognitive improvement shown by the two previously D1-impaired young monkeys (Fig. 1f) could be attributable to the recovery from agonist-induced cognitive deficits.

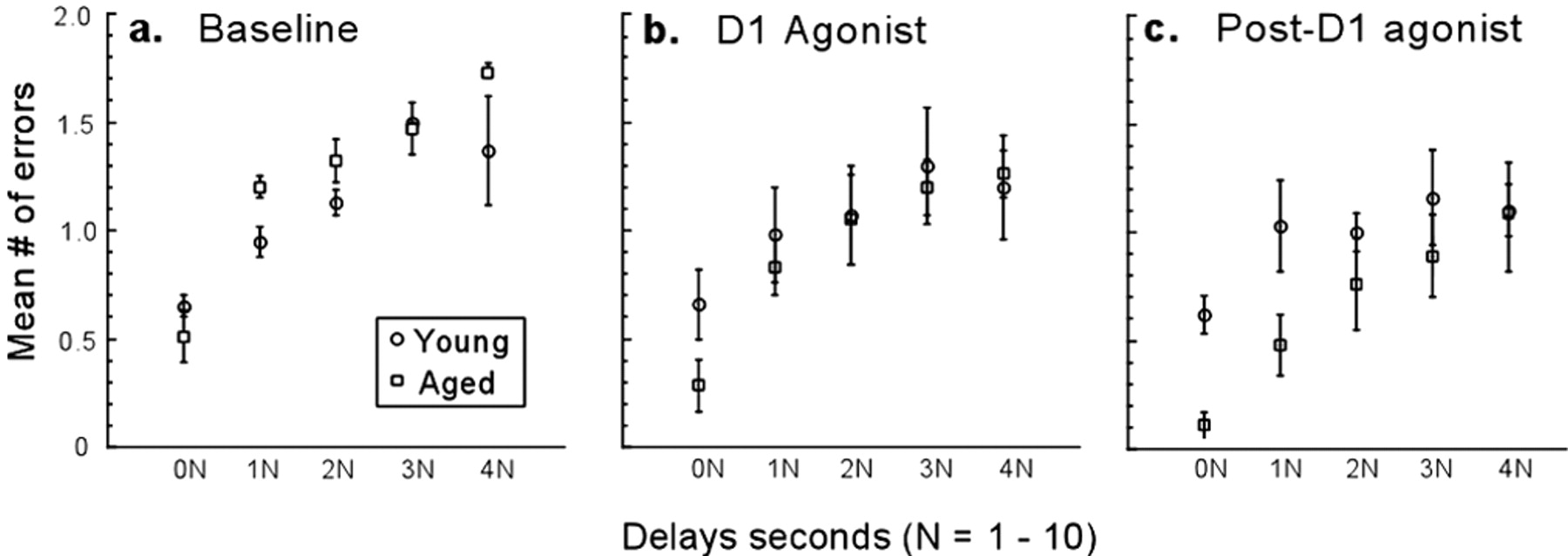
The mean number of errors for aged and young monkeys across delays during baseline, D1 agonist, and post-D1 agonist testing periods. Shown are the mean number of errors ± SEM (error bars) for aged (open squares) and young (open circles) monkeys across the five different delays (N ranging from 1 to 10 sec) during baseline (a), D1 agonist (b), and post-D1 agonist (c) testing periods. There was a significant effect of delay across all of the conditions for both aged and young monkeys, that is, both groups of monkeys made significantly more errors at longer delays. As a group, aged monkeys made significantly fewer errors than did young monkeys at the lowest delays and compared with their own baseline performance during the D1 agonist (b) and post-D1 agonist testing periods (c).

The percentage of days that delayed-response performance was significantly above baseline during the D1 agonist period. For young monkeys (a) and aged monkeys (b), the percentage (mean ± SEM) of test sessions in which performance was significantly above baseline for each of the five treatment blocks is shown. On the x-axis, the five D1 agonist blocks (each block corresponded to 5 d of D1 treatment and a 2-3 week washout period) are chronologically labeled 1-5. The average performance of the aged monkeys was significantly improved by the fifth D1 agonist block, whereas, as a group, the young monkeys actually showed a decline in performance, as evidenced by the fact that they performed above baseline on only <20% of test sessions for this block. *The performance of the young and aged monkeys differed significantly from one another during the fifth treatment block at an α-level of 0.05, as measured by two-way ANOVA with Scheffe post hoc comparisons. Error bars denote SEM.
There was a significant effect of delay across all of the conditions, that is, monkeys in both groups made more errors at longer delays (baseline, F(4,25) = 23.23, p < 0.0001; D1 agonist, F(4,25) = 8.63, p = 0.0002; post-D1 agonist, F(4,25) = 4.74, p = 0.006) (Fig. 3). However, no delay-dependent effects of treatment were observed. Delay-dependent effects of age were evident during the D1 agonist and post-D1 agonist testing periods. As a group, aged monkeys made significantly fewer errors than did young monkeys at the lowest delays during the D1 agonist period (F(1,1) = 12.84; p = 0.001) (Fig. 3b) as well as during the post-D1 agonist period (F(1,1) = 7.46; p = 0.01) (c). None of the monkeys showed any change in appetite, motivation, or general activity in response to D1 agonist treatment.
Discussion
The present findings provide evidence that a short-term, intermittent regimen of D1 stimulation improves spatial working memory performance in aged monkeys in whom the dopamine system is known to be compromised, but has minimal effects in young adult monkeys with presumed normal dopamine function. This result is consistent with our previous finding that short-term D1 stimulation ameliorates working memory deficits in a chronic haloperidol-induced D1-downregulated state (Castner et al., 2000) and with previous findings on the cognitive-enhancing effects of acute D1 agonist treatment in aged monkeys and other cases of dopamine deficiency, including schizophrenia (Davidson et al., 1990; Arnsten et al., 1994; Schneider et al., 1994; Cai and Arnsten, 1997; Muller et al., 1998; Castner et al., 2000). The present study did not assess the importance of the timing of intermittency per se, but there is reason to hypothesize that this is an important element in the treatment regimen.
The progressive and long-lasting cognitive enhancement evoked by repeated, intermittent treatment with a D1 agonist in aged monkeys is reminiscent of the development of behavioral sensitization (Robinson and Becker, 1986; Kalivas and Stewart, 1991; Sax and Strakowski, 1998; Castner and Goldman-Rakic, 1999). However, as demonstrated by the lack of an observable long-term effect of D1 treatment in young monkeys, it appears that sensitization to a selective D1 agonist may require a dopamine-deficient state such as that which occurs during the natural aging process or during chronic neuroleptic treatment. In fact, previous studies have shown that repeated D1 agonist exposure induces behavioral sensitization in 6-hydroxydopamine-lesioned animals, in which it has been postulated that a functional uncoupling of D1 and D2 receptors underlies this effect (Criswell et al., 1989, 1990). Whether such uncoupling occurs in response to age-related dopamine decline remains to be determined. In addition to age-related cognitive decline, this therapeutic D1 regimen has potential relevance for the treatment of cognitive deficits in other dopamine dysfunctional states such as schizophrenia, Parkinson's disease, and attention deficit disorder.
The sustained enhancement in the cognitive function of aged monkeys suggests that intermittent treatment with a D1 agonist may induce fundamental alterations in the functional circuitry that underlies working memory. These long-term changes may involve permanent alterations in the D1 signal transduction pathway. In aging, there is a significant reduction in striatum and frontal cortex in the ability of dopamine to stimulate cAMP (Puri and Volicer, 1977; Amenta et al., 1990). Given that cAMP production is elevated after repeated administration of psychomotor stimulants, such as amphetamine and cocaine, it is conceivable that enhancement of cAMP may contribute to the enduring cognitive enhancement observed in the aged monkeys in the present study. Furthermore, there is evidence that inositol 1,4,5-triphosphate and calcium signaling decline as a function of aging in the rodent brain (Sandhu et al., 2001), which raises the question as to whether D1 facilitation of this signaling via calcyon, a D1-interacting protein (Lezcano et al., 2000), is also downregulated in aging. Thus, one could hypothesize that the enduring cognitive improvement in the aged monkeys might be, at least in part, attributable to a D1-induced upregulation of signal transduction in this pathway. Morphological changes could also be involved. For example, there is evidence that repeated treatment with indirect dopamine agonists, such as amphetamine, increases the density of dendritic spines on pyramidal neurons in prefrontal cortex (Robinson and Kolb, 1999). Because D1 receptors are preferentially localized to spines, it is possible that the improvement in spatial working memory shown by aged monkeys after repeated, intermittent D1 agonist treatment could actually reflect an upregulation of D1 receptors. Moreover, repeated stimulation of the D1 receptor can also lead to an enhancement of D2 receptor function, and D2 agonists have also been shown to improve spatial working memory in aged monkeys (Arnsten et al., 1995; Pollack and Yates, 1999). Hence, the potential contribution of enhanced signaling via D2 receptors to the present findings cannot be ruled out, although ABT-431, which is the prodrug of A-86929, has itself a low affinity at the D2 receptor (Shiosaki et al., 1996). It remains to be seen, however, to what extent the effects attributable to the repeated systemic administration of the D1 agonist used in the present study reflect actions through the D5 versus the D1 signaling pathway. Thus, for future studies, it will be important to determine the optimal regimen of treatment and the cellular mechanisms by which repeated brief periods of D1 agonist stimulation lead to an enduring improvement in working memory in aging. These findings are highly significant to the enhancement of cognitive function in an ever-increasing elderly population and may provide the basis for drug development targeted toward this purpose.
Footnotes
Funding was provided in part by a grant from the National Institutes of Mental Health (MH44866). We thank Peter Vosler and Heather A. Findlay for their expert technical assistance, and Hoechst Marion Rousell (now Aventis Pharmaceuticals) for their kind gift of ABT-431.
Correspondence should be addressed to Dr. Stacy A. Castner, 322 South Green Street, Suite 202, Chicago, IL 60607. E-mail: scastner{at}miicro.com or scastner{at}uic.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/241446-05$15.00/0
↵† Deceased, July 31, 2003.





{kind=link}
{kind=link}
{kind=link}