

Abstract
The gene Dyrk1a is the mammalian ortholog of Drosophila minibrain. Dyrk1a localizes in the Down syndrome (DS) critical region of chromosome 21q22.2 and is a major candidate for the behavioral and neuronal abnormalities associated with DS. PFC malfunctions are a common denominator in several neuropsychiatric diseases, including DS, but the contribution of DYRK1A in PFC dysfunctions, in particular the synaptic basis for impairments of executive functions reported in DS patients, remains obscure. We quantified synaptic plasticity, biochemical synaptic markers, and dendritic morphology of deep layer pyramidal PFC neurons in adult mBACtgDyrk1a transgenic mice that overexpress Dyrk1a under the control of its own regulatory sequences. We found that overexpression of Dyrk1a largely increased the number of spines on oblique dendrites of pyramidal neurons, as evidenced by augmented spine density, higher PSD95 protein levels, and larger miniature EPSCs. The dendritic alterations were associated with anomalous NMDAR-mediated long-term potentiation and accompanied by a marked reduction in the pCaMKII/CaMKII ratio in mBACtgDyrk1a mice. Retrograde endocannabinoid-mediated long-term depression (eCB-LTD) was ablated in mBACtgDyrk1a mice. Administration of green tea extracts containing epigallocatechin 3-gallate, a potent DYRK1A inhibitor, to adult mBACtgDyrk1a mice normalized long-term potentiation and spine anomalies but not eCB-LTD. However, inhibition of the eCB deactivating enzyme monoacylglycerol lipase normalized eCB-LTD in mBACtgDyrk1a mice. These data shed light on previously undisclosed participation of DYRK1A in adult PFC dendritic structures and synaptic plasticity. Furthermore, they suggest its involvement in DS-related endophenotypes and identify new potential therapeutic strategies.
Introduction
Down syndrome (DS), the most common form of intellectual disability, is a chromosomal disorder resulting from the presence of all or part of an extra chromosome 21. The gene Dyrk1a is localized in the DS critical region of chromosome 21q22.2 and is overexpressed in DS (Delabar et al., 1993). As a member of the dual-specificity tyrosine phosphorylation-regulated kinase family, DYRK1A phosphorylates numerous proteins, including transcription factors, controls cell proliferation, participates in brain development (Becker, 2011; Guedj et al., 2012), and regulates >200 genes (Lepagnol-Bestel et al., 2009) and multiple signaling pathways (Guedj et al., 2012). Because of its pleiotropic functions in brain development, Dyrk1a is a strong candidate gene for the learning and neuronal abnormalities associated with DS (Becker, 2011; Guedj et al., 2012; Altafaj et al., 2013).
The prefrontal cortex (PFC) is essential to working memory, reasoning, action planning, and emotionally guided behaviors (Goldman-Rakic, 1990; Seamans et al., 1995). PFC malfunctions are a common denominator in several neuropsychiatric diseases, including DS (Goto et al., 2010). Neuropsychological evaluation of DS subjects revealed impairments of executive functions (Lanfranchi et al., 2010). The exact contribution of DYRK1A in PFC dysfunctions and the synaptic basis for the working memory defects reported in DS patients remain obscure (Pennington et al., 2003; Lanfranchi et al., 2010).
In the present study, we evaluated synaptic plasticity, biochemical synaptic markers, and dendritic morphology of pyramidal PFC neurons in adult mBACtgDyrk1a mice that overexpress Dyrk1a under the control of its own regulatory sequences (Guedj et al., 2012). We uncover morphofunctional abnormalities in the PFC of mBACtgDyrk1a mice that may underlie part of the multiple cognitive and behavioral deficits described in DS patients.
Materials and Methods
Animals.
All animal experiments were performed according to the criteria of the European Communities Council Directive (86/609/EEC).
To generate the Dyrk1a-overexpressing mouse and the murine bacterial artificial chromosome 189N3 (BAC-189N3) strain, the HM-1 embryonic stem (ES) cells was electroporated with the BAC-189N3 retrofitted with a G418 resistance gene. ES clone was selected for overexpression of Dyrk1a close to 1.5 ratio and injected into blastocysts (Guedj et al., 2012). All mice were weaned at 21 d. After weaning, they were caged socially in same-sex groups. Male Tg189N3 (mBACtgDyrk1a) mice and wild-type littermates were used at 4–6 months of age.
Green tea treatment.
Male (3–4 months old) wild-type and mBACtgDyrk1a mice were fed with water or with green tea extract (GTE)-containing water. GTE was administered orally, in the form of a solution containing 0.25% green tea decaffeinated extract (LifeExtension For Longer Life) and 0.25% glucose. GTE contained ∼45% epigallocatechin 3-gallate (EGCG). The GTE solution was freely available in home cage feeding bottles and was renewed every 24 h. Depending on daily intake, the dose ranged between 120 and 200 mg/kg. Administration of the diet started 4–6 weeks before experimentation.
Biochemistry.
Immunobloting was performed following standard protocols using either Western blots or slot blots (according to antibody specificity). PFC proteins were prepared in modified NP40 lysis buffer and subjected to SDS electrophoresis on acrylamide gels under reducing conditions before being transferred to Hybond-C Extra membrane (GE Healthcare Europe); alternatively, a slot blot apparatus (SHM-48; SCIE-Plas) was used to increase the number of samples with highly specific antibodies. After transfer, membranes were blocked in 10% nonfat dry milk in Tris-buffered saline-Tween (TBS-T; 1.5 mm Tris, 5 mm NaCl, 0.1% Tween 20) and probed overnight at 4°C with primary anti-DYRK1A (1/250; Abnova), PSD95 (1/1000; ThermoFischer Scientific), anti-CaMKII and anti pCaMKII (1/5000 and 1/1000; Abcam) antibodies, followed by HRP-labeled anti–species-specific antibodies. The membranes were rinsed in TBS-T, and the immunocomplexes were detected using Luminol Reagent (Santa Cruz Biotechnology). β-Actin (1/4000, Sigma-Aldrich) was the internal control for Western blots, and total protein stained with red Ponceau was used as standard for slot blots. Immunoblots were imaged by a LAS-3000 imaging system (Fuji Photo Film) and analyzed by densitometry (UnScan It, Silk Scientific).
PFC slice preparation.
PFC slices were prepared as described previously (Lafourcade et al., 2011). Mice were anesthetized with isoflurane and decapitated. The brain was sliced (300 μm) in the coronal plane (Integraslice, Campden Instruments) and maintained in physiological saline (4°C). Slices were stored for 30 min at 32–35°C in artificial CSF (ACSF) containing 126 mm NaCl, 2.5 mm KCl, 2.4 mm MgCl2, 1.2 mm CaCl2, 18 mm NaHCO3, 1.2 mm NaH2PO4, and 11 mm glucose, equilibrated with 95% O2/5% CO2. Slices were stored at 22 ± 2°C until recording.
Electrophysiology.
Whole-cell patch-clamp and field EPSP (fEPSP) were recorded from layer V/VI pyramidal cells in coronal slices of mouse prelimbic PFC (Lafourcade et al., 2011). For recording, slices were superfused (2 ml/min) with ACSF at 32–35°C. Picrotoxin (100 μm) was added to block GABAA receptors. To evoke synaptic currents, 150–200 μs stimuli were delivered at 0.1 Hz through a glass electrode placed in layer II/III. The glutamatergic nature of the fEPSP was confirmed at the end of the experiments using the glutamate receptor antagonist DNQX (20 μm), which blocked the synaptic component without altering the nonsynaptic component (data not shown). Long-term potentiation (LTP) was induced using a theta-burst stimulation (TBS) protocol consisting of five trains of burst with four pulses at 100 Hz, at 200 ms interval, repeated four times at intervals of 10 s (Hirsch and Crepel, 1990). LTD was induced by 10 min stimulation at 10 Hz (Lafourcade et al., 2011). For whole-cell patch-clamp, pyramidal neurons were visualized using an infrared microscope (BX-50, Olympus). Experiments were performed with electrodes containing 128 mm cesium methane-sulfonate (CH3O3SCs, voltage clamp) or potassium gluconate (KGlu, current clamp and voltage clamp), 20 mm NaCl, 1 mm MgCl2, 1 mm EGTA, 0.3 mm CaCl2, 2 mm Na2+-ATP, 0.3 mm Na+-GTP, 10 mm glucose buffered with 10 mm HEPES, pH 7.3, osmolarity 290 mOsm. Electrode resistance was 4–6 MOhm. If access resistance (no compensation, <25 MOhm) changed by >20%, the experiment was rejected. To perform current–voltage (I–V) curves and to test neuronal pyramidal neuron excitability, a series of hyperpolarizing and depolarizing current steps were applied immediately after breaking in the cell. For EPSP-spike coupling (ES-coupling), membrane potential was held at −65 mV.
Voltage clamp experiments.
Miniature EPSCs (mEPSCs) were recorded at −70 mV (KGlu-based solution), and tetrodotoxin (1 μm) was added in the perfusion bath. Evoked EPSCs were recorded at −70 mV (KGlu-based solution). To determine the ratio of AMPAR-mediated/NMDAR-mediated EPSC, neurons were first recorded at −70 mV and then at 40 mV (CH3O3SCs-based solution) (Kasanetz and Manzoni, 2009).
The progressive block of NMDAR-EPSCs by MK-801 (40 μm) allowed estimating the release probability of glutamate (Chavis and Westbrook, 2001). NMDAR-EPSCs were recorded at 40 mV in the presence of 20 μm DNQX, and synaptic inputs were activated every 10 s. After a 10 min baseline, MK-801 was added and synaptic stimulation stopped for 15 min. Stimulation was then resumed until total blockade of NMDAR-EPSCs. The progressive block of NMDAR-EPSCs by MK-801 was fitted using a double exponential: y = a*exp(−t/τfast) + b*exp(−t/τslow) where y is the normalized response amplitude, a and b are constants, t is the corresponding stimulus number, and τfast andτslow are the time constants (Chavis and Westbrook, 2001).
Data acquisition and analysis.
Because of the strong phenotypes, the experimenters were not blind to the genotypes, but no data were excluded before statistical analysis (GraphPad Software). Data were recorded on a MultiClamp700B, filtered at 2 kHz, digitized (20 kHz, DigiData 1440A), collected using Clampex 10.2, and analyzed using Clampfit 10.2 (all from Molecular Devices). Both area and amplitude of fEPSPs and EPSCs were analyzed. The magnitude of LTP was calculated 25–30 min after TBS as percentage of baseline responses.
To perform I–V curves and to test the excitability of MSN, a series of hyperpolarizing and depolarizing current steps were applied immediately after breaking in the cell. Membrane resistance was estimated from the I–V curve around resting membrane potential as previously described (Kasanetz and Manzoni, 2009).
For ES-coupling analysis, 10 traces were recorded for each stimulus. The value of ES-coupling obtained for each animal was calculated by averaging the spiking probability corresponding to each class of EPSP slope. EPSP slopes were measured during the first 2 ms, sorted in 0.5 mV/ms bins, and the firing probability was determined for each bin.
mEPSCs were filtered at 2 kHz and digitized at 20 kHz. EPSC amplitude and interinterval time were analyzed with Axograph X using a double exponential template: f(t) = exp(−t/rise) + exp(−t/decay), rise = 0.5 ms and decay = 3 ms. The threshold of amplitude detection was set at 5 pA (Kasanetz and Manzoni, 2009).
AMPAR/NMDAR ratio.
The AMPAR component was measured from EPSC at −70 mV. The NMDAR component was determined 40 ms after the start of dual component EPSC at 40 mV (Kasanetz and Manzoni, 2009).
Drugs.
Drugs were added at the final concentration to the ACSF. Picrotoxin was from Sigma. DNQX, MK801, JZL184, and d-APV were from the National Institute of Mental Health's Chemical Synthesis and Drug Supply Program. Ifenprodil, CP55940, and tetrodotoxin were from Tocris Bioscience.
Dendritic spine reconstruction and quantification.
Whole-cell recorded neurons were loaded with neurobiotin through patch pipettes. Slices were fixed overnight in 4% PFA and revealed with Texas Red-conjugated avidin for subsequent confocal imaging. Only pyramidal neurons within layer V/VI of the PFC showing proper filling of the distal dendritic tree were included in the analysis (Iafrati et al., 2013). Stack images were acquired using an Olympus Fluoview FV500 confocal microscope. Laser power and photomultiplier gain were adjusted to obtain few pixels with maximum intensity on dendrite shaft, and the offset range was tuned to cut off background noise. Tridimensional deconvolution of each stack was performed with AutoQuantX2 to compensate the spherical aberration and to correct the z-smear for reliable spine morphology. The tridimensional reconstruction and blind semiautomated analysis were performed with Imaris (Bitplane) (Iafrati et al., 2013) with the following detection settings for dendrites: dendrite starting: 1.333 μm/dendrite seed point: 0.333 μm/threshold: 1; and for spines: seed point: 0.333 μm /maximum length: 2.666 μm/threshold: 0.5.
Statistical analysis.
The value n corresponds to the number of cells for morphological experiments and to the number of individual cells or animals in the electrophysiology experiments (maximum 3 cells in a single mice). All values are given as mean ± SEM. Statistical analysis was performed with Prism 6.0 (GraphPad Software). Two sample comparisons were made with the Mann–Whitney t test. Kolmogorov–Smirnov tests were used for comparing the cumulative distributions of amplitude and interevent intervals of mEPSCs. Statistical significance was set at p < 0.05.
Results
As expected from the expression of an additional copy of the gene (Guedj et al., 2012), DYRK1A brain protein levels were increased in mBACtgDyrk1a mice compared with wild-type congeners (100.00 ± 7.49%, n = 8 wild-type vs 157.40 ± 5.28%, n = 12 mBACtgDyrk1a; p = 0.0002, Mann–Whitney U test).
Intrinsic properties of PFC pyramidal neurons are normal in mBACtgDyrk1a mice
Independently of their genotypes, all recorded pyramidal neurons showed similar membrane response profiles in response to a series of somatic current steps (Fig. 1A) and a strong inward rectification, shown as a shift from linearity in the I-V plots (Fig. 1B). The resting membrane potentials (Fig. 1C), the minimal current necessary to evoke action potential firing or rheobases (Fig. 1D), and the number of action potentials in response to somatic current steps (Fig. 1E) were similar in wild-type and mBACtgDyrk1a mice (Kasanetz and Manzoni, 2009). Thus, Dyrk1a overexpression did not impact on the intrinsic properties of PFC neurons.

Intrinsic properties of layers V/VI PFC pyramidal neurons in wild-type and mBACtgDyrk1a littermates. A, Typical membrane responses to somatic current steps of PFC pyramidal neurons from wild-type or mBACtgDyrk1a mice. B, Summary of all the current–voltage (I–V) curves recorded in the two genotypes showing no difference between wild-type (n = 24, black symbols) and mBACtgDyrk1a mice (n = 16, white symbols; p > 0.05, Mann–Whitney U test). C, The resting membrane potential (−71.08 ± 0.71 mV, n = 24 cells from wild-type mice, black symbols; −69.69 ± 1.14 mV, n = 16 cells from mBACtgDyrk1a mice, white symbols). D, The rheobase (133.33 ± 11.13 pA, n = 24 cells from wild-type mice, black symbols; 112.50 ± 9.68 pA, n = 16 cells from mBACtgDyrk1a mice, white symbols) was similar in both genotypes (p > 0.05, Mann–Whitney U test). Horizontal lines indicate the mean value. Error bars indicate SEM. n.s., Not significant. E, Summary of current-firing curves indicating that the number of evoked action potentials in response to somatic current steps was similar in pyramidal neurons from mBACtgDyrk1a (n = 24, black symbols) and wild-type mice (n = 16, white symbols; p > 0.05, Mann–Whitney U test).
Spine density is higher in PFC pyramidal neuron of mBACtgDyrk1a mice
An alteration of dendritic arborization and/or spine anomaly is common in neuropsychiatric diseases and constitutes a core feature of intellectual disability (Penzes et al., 2011). The effect of Dyrk1a gene over dosage on PFC pyramidal neurons spines has never been evaluated. PFC pyramidal neurons were filled with neurobiotin during the electrophysiological recordings to allow the confocal imaging and 3D reconstruction of dendritic morphology (Iafrati et al., 2013). Quantitative spine analysis was purposely performed on oblique dendrites because they form the principal target of excitatory synapses impinging on layer V/VI PFC pyramidal neurons.
Expression of an additional copy of the Dyrk1a gene almost doubled spine density in pyramidal PFC neurons (Fig. 2A,B). The average spine length was similar in both genotypes (data not shown; 1.38 ± 0.03 μm, n = 241 spines from 5 wild-type vs 1.38 ± 0.02 μm, n = 609 spines from 6 mBACtgDyrk1a; not significant, Mann–Whitney U test). We quantified the levels of PSD95, an essential postsynaptic scaffolding protein highly expressed in excitatory spines that allows the clustering and anchoring of numerous essential synaptic proteins (Feng and Zhang, 2009). The PSD95 protein levels were augmented in PFC extracts from mBACtgDyrk1a mice (Fig. 2C). To probe the functional maturity of spines, we compared fast AMPAR-mediated miniature EPSCs (mEPSC) in the two genotypes. Cumulative distribution plots showed that mEPSC amplitudes were larger in mBACDyrk1a mice compared with wild-type littermates (Fig. 2D). However, the mEPSC frequency was similar in both groups (Fig. 2E). Waveform analysis of averaged mEPSCs confirmed the increased amplitude in transgenic mice (28.01 ± 2.88 WT vs 41.23 ± 2.84), whereas other parameters remain unchanged (frequency: 1.05 Hz ± 0.11 vs 0.92 Hz ± 0.15; rise time, 10–90%: 0.68 ± 0.10 ms vs 0.84 ± 0.07 ms; decay time, 100–50%: 4.11 ± 0.26 ms vs 3.83 ± 0.27 ms; time to peak: 1.31 ± 0.07 ms vs 1.28 ± 0.10 ms; for wild-type, n = 10 and mBACtgDyrk1a, n = 8, respectively).

Increased spine density in PFC pyramidal neurons of mBACtgDyrk1a mice. A, Representative unprocessed images of spines and dendritic shafts from pyramidal PFC neurons of wild-type (left) and mBACtgDyrk1a mice (right). Scale bars, 2 μm. B, The density of spines on oblique dendrites of layer V/VI PFC pyramidal neurons increased in mBACtgDyrk1a mice compared with wild-type littermates (0.76 ± 0.07, n = 8 cells from 5 wild-type vs 1.41 ± 0.10, n = 10 cells from 6 mBACtgDyrk1a): ***p = 0.003 (Mann–Whitney U test). Error bars indicate SEM. C, Quantification of slot blot show that PSD95 level was increased in mBACtgDyrk1a mice (100.00 ± 6.53, n = 11 wild-type, black bar, vs 122.82 ± 5.44, n = 10 mBACtgDyrk1a, white bar): *p = 0.0378 (Mann–Whitney U test). D, Representative recordings of mEPSCs taken from both genotypes. Cumulative mEPSC amplitude distributions showed a significant increase in amplitude in mBACtgDyrk1a mice (white symbols, n = 8) compared with wild-type littermates (black symbols, n = 10): *p < 0.05 (Kolmogorov–Smirnov test). E, Cumulative mEPSCs interevent interval distributions showed no difference in frequency in mBACtgDyrk1a mice (white symbols, n = 8) compared with wild-type littermates (black symbols, n = 10): p > 0.05 (Kolmogorov–Smirnov test). Horizontal lines represent the mean value. Error bars indicate SEM.
Synaptic transmission and plasticity malfunctions often correlate with dendritic or spine alterations (Penzes et al., 2011; Iafrati et al., 2013), and we next searched for functional correlates in mBACtgDyrk1a mice.
LTP is ablated in mBACtgDyrk1a mice
Activity NMDAR-dependent LTP is probably the most widely expressed and extensively studied form of synaptic plasticity. TBS induces an LTP of extracellular fEPSP at pyramidal synapses that was completely prevented by the NMDAR antagonist d-AP5 (50 μm, data not shown, 87.95 ± 7.19% of basal transmission, n = 6; p > 0.05, Mann–Whitney U test), confirming its dependency on NMDAR (Iafrati et al., 2013).
We found that LTP was totally ablated in mBACtgDyrk1a mice (Fig. 3A). Although post-tetanus potentiation was observed in both genotypes (compare the first 3 min after TBS, Fig. 3A), the fEPSP rapidly returned to baseline levels in mBACtgDyrk1a mice but remained stably enhanced for the duration of the recording in wild-type littermates. These data reveal that the ability to maintain LTP is impaired in mBACtgDyrk1a mice and are in line with previous data showing the ablation of LTP at a specific subset of striatal synapses in the Td65Dn mouse model of DS (Di Filippo et al., 2010). The relative contribution of AMPAR and NMDAR to synaptic currents is an indicator of synaptic plasticity in particular in those depending on the incorporation or removal of AMPAR in the postsynaptic density (e.g., LTP or long-term depression, respectively) (Kasanetz and Manzoni, 2009). We measured the ratio between AMPAR- and NMDAR-evoked EPSCs and found this index indistinguishable between the two mice groups, suggesting that expression of an additional copy of Dyrk1a did not alter the overall gain of PFC excitatory synapses (Fig. 3B).

LTP is abolished in the PFC of mBACtgDyrk1a mice. A, LTP was abolished in mBACtgDyrk1a mice. Representative traces averaged from fEPSP response before (1) and 30 min after plasticity induction (2) in mice from either genotype. TBS of layers II/III induced an LTP of evoked fEPSP recorded in layer V/VI pyramidal neurons in wild-type mice (n = 12, black symbols; 98.60 ± 0.58, baseline vs 121.76 ± 3.21, 25–30 min; p < 0.0001, Mann–Whitney U test) but not in mBACtgDyrk1a mice (n = 8, white symbols; 98.02 ± 0.84, baseline vs 93.99 ± 3.98, 25–30 min; p > 0.05, Mann–Whitney U test). Average time courses of mean fEPSP were normalized to baseline. Error bars indicate SEM. B, Representative recordings of AMPA and NMDA EPSCs taken from both genotypes. Summary data showing that the AMPAR-mediated EPSC/NMDAR-mediated EPSC ratio was similar in PFC pyramidal neurons of wild-type (black symbols) and mBACtgDyrk1a mice (white symbols) (4.17 ± 0.65, n = 11 wild-type vs 3.53 ± 0.39, n = 10 mBACtgDyrk1a; p > 0.05, Mann–Whitney U test). Horizontal lines indicate the mean value. C, The fraction of the NMDAR-mediated EPSC sensitive to bath application of ifenprodil (3 μm, 15 min), a GluN2B-containing NMDAR inhibitor, was similar in PFC pyramidal neurons from wild-type (black symbols) or mBACtgDyrk1a mice (white symbols) (1.77 ± 0.18, n = 7 wild-type vs 2.33 ± 0.37, n = 7 mBACtgDyrk1a; p > 0.05, Mann–Whitney U test). Horizontal lines indicate the mean value. Error bars indicate SEM. n.s., Not significant. D, The pCaMKII/CaMKII ratio was decreased in the PFC of mBACtgDyrk1a mice (white symbols) compared with wild-type littermates (black symbols) (100.00 ± 12.66, n = 11 wild-type vs 54.78 ± 6.36, n = 11 mBACtgDyrk1a): *p = 0.0127 (Mann–Whitney U test). The CaMKII levels were identical in wild-type and mBACtgDyrk1a mice (data not shown; 100.00 ± 5.26, n = 11 wild-type, black bar, vs 113.84 ± 4.74, n = 12 mBACtgDyrk1a, white bar): p > 0.05 (Mann–Whitney U test). The pCaMKII levels were decreased in mBACtgDyrk1a mice (data not shown; 100.00 ± 11.15, n = 11 wild-type, black bar vs 62.54 ± 6.27, n = 12 mBACtgDyrk1a, white bar): *p = 0.0106 (Mann–Whitney U test). The data are normalized to the baseline level of wild-type mice. Horizontal lines indicate the mean value. Error bars indicate SEM.
The GluN2B subunit of the NMDAR has been implicated in PFC LTP and PFC-related learning (Iafrati et al., 2013). To evaluate its relative contribution to the NMDAR-EPSC in our two genotypes, we measured the fraction of the total NMDAR-EPSC that is sensitive to ifenprodil, a selective antagonist of GluN2B-containing NMDAR. This fraction was similar in wild-type and mBACtgDyrk1a littermates (Fig. 3C), excluding the possibility that GluN2B-containing NMDARs underlie the lack of LTP in mBACtgDyrk1a mice.
Finally, we measured the protein levels of CaMKII, a major synaptic protein that autophosphorylation regulates LTP (T286) (Lisman et al., 2012; Sanhueza and Lisman, 2013). In the PFC, CaMKII activation increases AMPAR synaptic currents (Gu et al., 2006) and induces an LTP of pyramidal neurons' intrinsic excitability (Chen et al., 2007). There was a marked reduction in the pCaMKII/CaMKII ratio in mBACtgDyrk1a mice (Fig. 3D), principally attributable to a decrease in pCaMKII levels (data not shown, 100.00 ± 11.15, n = 11 wild-type vs 62.54 ± 6.27, n = 12 mBACtgDyrk1a; p = 0.0106, Mann–Whitney U test) in the face of normal CaMKII levels (data not shown 100.00 ± 5.26, n = 11 wild-type vs 113.84 ± 4.74, n = 12 mBACtgDyrk1a; p > 0.05, Mann–Whitney U test). Thus, altered levels of this key memory molecule paralleled the inability to trigger LTP.
Dyrk1a overexpression ablates endocannabinoid (eCB)-dependent LTD
The eCB system is a major actor of synaptic plasticity that may participate to the etiology of PFC-dependent mood disorders (Hill et al., 2009). In response to neuronal activity, the two principal eCBs, anandamide and 2-arachidonoylglycerol (2-AG), are released by postsynapses and retrogradely activate a presynaptic CB1receptor (CB1R) (Robbe et al., 2002b; Puente et al., 2011).
In PFC slices prepared from wild-type mice, tetanic stimulation induced a robust eCB-mediated LTD of excitatory synapses onto layer V/VI pyramidal PFC neurons (Fig. 4A) (Lafourcade et al., 2007). This form of synaptic plasticity was totally ablated in mBACtgDyrk1a mice (Fig. 4A). To determine whether the Dyrk1a overexpression acted on the transduction pathways downstream of presynaptic CB1R, we took advantage of another presynaptic type of LTD mediated by the same presynaptic transduction pathways but triggered by mGluR2/3 receptors (Robbe et al., 2002a). The mGluR2/3-LTD activated by the agonist LY379268 (100 nm) was normal in mBACtgDyrk1a mice (Fig. 4B), strongly suggesting that Dyrk1a overexpression had no effect on the signaling cascades downstream of mGluR2/3 or CB1R.
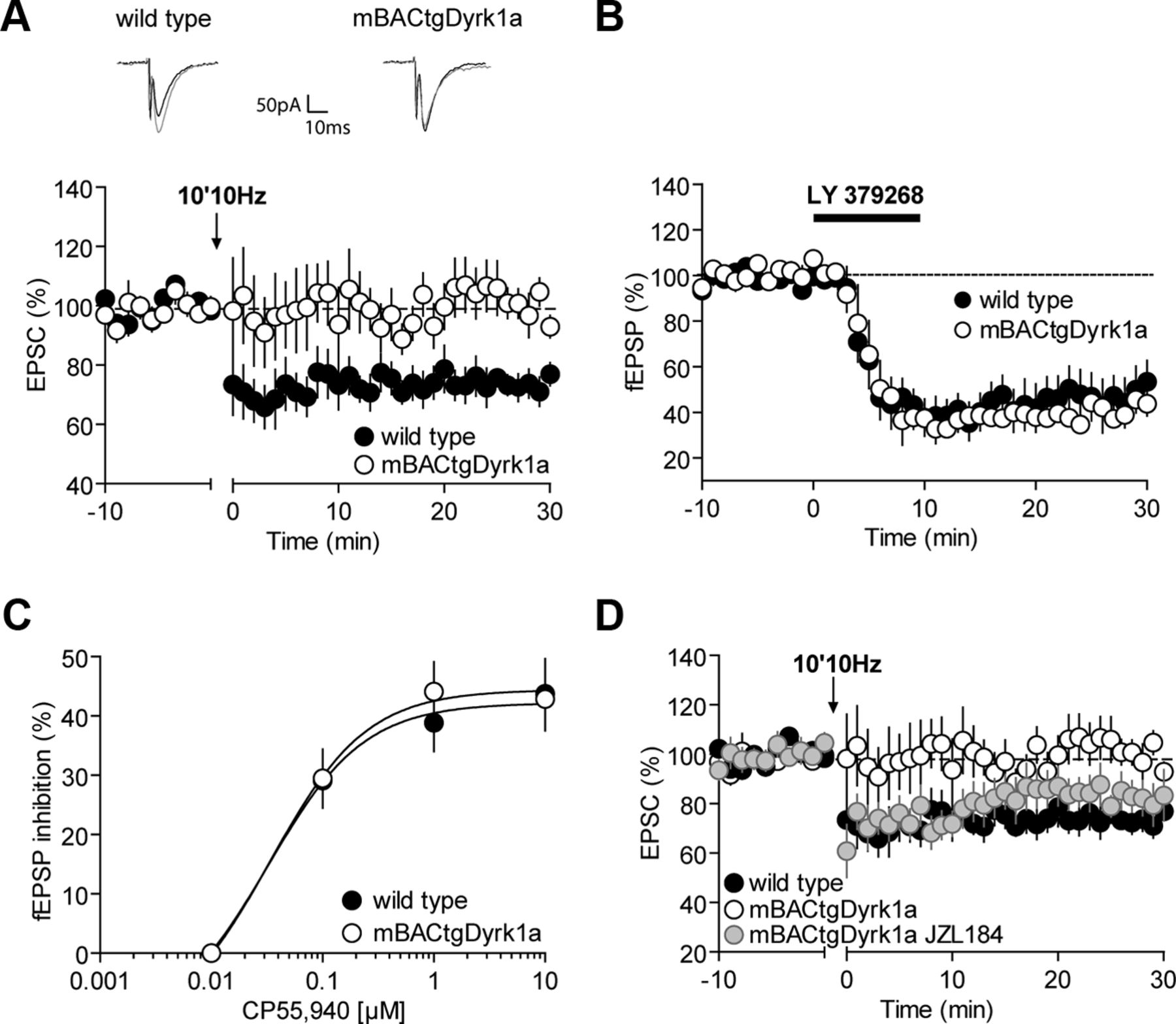
eCB-LTD is abolished in mBACtgDyrk1a mice. A, Representative traces averaged from EPSC response before (gray) and 30 min after plasticity induction (black) in mice from either genotype. Low frequency (10 min at 10 Hz) of layers II/III induced an LTD of evoked EPSCs recorded in layer V/VI pyramidal neurons in wild-type mice (black symbols) (100.90 ± 0.86, n = 8, wild-type baseline vs 73.72 ± 2.90, n = 7, wild-type 25–30; p = 0.003, Mann–Whitney U test) but not in mBACtgDyrk1a mice (99.03 ± 1.40, n = 8, mBACtgDyrk1a baseline vs 102.02 ± 4.50, n = 7, mBACtgDyrk1a 25–30; p > 0.05, Mann–Whitney U test). Average time courses of mean EPSCs were normalized to baseline. Error bars indicate SEM. B, Ten minute application of the mGluR2/3 agonist, LY379268 (100 nm), induced similar LTD of evoked fEPSPs recorded in layers V/VI pyramidal neurons in wild-type mice (black symbols 97.09 ± 0.98, n = 6, wild-type baseline vs 53.50 ± 9.07 n = 6, wild-type 25–30; p = 0.0022, Mann–Whitney U test) and in mBACtgDyrk1a mice (white symbols 96.53 ± 0.94, n = 4, mBACtgDyrk1a baseline vs 48.60 ± 5.02, n = 4, mBACtgDyrk1a 25–30; p = 0.0286, Mann–Whitney U test). Average time course of mean fEPSPs was normalized to baseline. Error bars indicate SEM. C, Dose–response curve for the cannabimimetic CP55940 in wild-type (black symbols) and mBACtgDyrk1a littermates (white symbols 0.1 mm: 29.09 ± 4.59, n = 9 vs 29.48 ± 4.90, n = 7; 1 mm: 38.87 ± 4.85, n = 10 vs 44.12 ± 5.00, n = 8; 10 mm: 43.72 ± 5.93, n = 8 vs 42.87 ± 5.32, n = 8; p > 0.05, Mann–Whitney U test). fEPSP amplitudes were measured 30 min after application of CP55940. Each point is expressed as the percentage of inhibition of its basal value. Error bars indicate SEM. D, Bath incubation with the selective MAG lipase inhibitor, JZL184 (1 μm), restored eCB-LTD in mBACtgDyrk1a mice (gray symbols 100.93 ± 1.71, n = 10, mBACtgDyrk1a JZL184 baseline vs 82.04 ± 6.54; n = 8, mBACtgDyrk1a JZL184 25–30; p = 0.043, Mann–Whitney U test). Average time courses of mean EPSCs were normalized to baseline. Error bars indicate SEM.
Dyrk1a overexpression may directly impair CB1 Rs functionality. To estimate CB1R presynaptic functions, we built dose–response curves for the cannabinoid agonist CP55940. In both genotypes, CP55940 inhibited synaptic transmission similarly to what we previously reported in wild-type mice (Lafourcade et al., 2007) (Fig. 4C).
Inhibition of intracellular 2-AG hydrolysis by the 2-AG-deactivating enzyme monoacylglycerol lipase with a selective inhibitor JZL184, potentiates eCB-mediated LTD in the PFC of wild-type mice and (Marrs et al., 2010) and normalizes eCB-LTD in the fmr1−/− mice model of fragile X (Jung et al., 2012). Brain slices were incubated with JZL184 (1 μm) for 45–90 min before LTD induction. This treatment was sufficient to restore synaptic induced eCB-LTD in slices prepared from mBACtgDyrk1a (Fig. 4D).
Blocking another key 2-AG-hydrolyzing enzyme called ABHD6 with its selective inhibitor WWL70 can normalize impaired eCB-LTD in fmr1−/− mice (Jung et al., 2012). However, acute blockade of ABHD6 did not reestablish eCB-LTD PFC slices from mBACtgDyrk1a mice (data not shown; 100.94 ± 2.29, n = 6, mBACtgDyrk1a WWL70, 10 μm baseline vs 97.95 ± 13.41 n = 4, mBACtgDyrk1a WWL70, 10 μm 25–30; p > 0.05, Mann–Whitney U test).
Synaptic properties of PFC pyramidal neurons in mBACtgDyrk1a mice
Finally, we evaluated basic synaptic properties in mBACtgDyrk1a PFC synapses. Input/output relationships were similar in wild-type and mBACtgDyrk1a mice, showing that the excitability of PFC synapses was unaltered (Fig. 5A) and that the lack of LTP and LTD is not the result of a reduction of the number of synapse recruited during the induction of synaptic plasticity in mBACtgDyrk1a.

Basal synaptic properties of layers V/VI PFC pyramidal neurons are intact in mBACtgDyrk1a littermates. A, There was no change in synaptic excitability in PFC of mBACtgDyrk1a mice. Input/output curves of evoked EPSCs in layer V/VI pyramidal neurons in wild-type (n = 9, black symbols) and in mBACtgDyrk1a mice (n = 7, white symbols) for increasing stimulation intensities. Average mean sEPSCs are represented. Mann–Whitney U test. B, EPSP-spike coupling was similar in wild-type (black symbols) and in mBACtgDyrk1a mice (white symbols) (Mann–Whitney U test). C, Representative traces averaged from NMDAR EPSC response before (gray) and 40 min after bath application of MK801 (black) in mice from either genotype. The probability of glutamate release was estimated from the rate of inhibition of the amplitude of consequent evoked NMDAR-mediated currents by the presence of MK-801 (40 μm, see Materials and Methods). The release probability at synapses impinging onto layer V/VI pyramidal neurons is identical in wild-type mice (black symbols) (100.10 ± 0.45, n = 12, wild-type baseline vs 7.33 ± 0.85, n = 10, wild-type 35–40; p < 0.005, Mann–Whitney U test) and in mBACtgDyrk1a mice (white symbols) (99.86 ± 0.25, n = 10, mBACtgDyrk1a baseline vs 5.46 ± 1.35, n = 6, mBACtgDyrk1a 35–40; p < 0.005, Mann–Whitney U test). Average time courses of mean NMDA EPSC were normalized to baseline. Error bars indicate SEM. The probability of glutamate release estimated from the rate of exponential two-phase decay (τ fast and τ slow) were τ slow: 47.08 ± 9.91, n = 6 cells from 5 wild-type versus 73.52 ± 16.59, n = 7 cells from 5 mBACtgDyrk1a; τ fast: 13.89 ± 3.28, n = 9 cells from 7 wild-type versus 11.47 ± 2.02, n = 9 cells from 5 mBACtgDyrk1a. D, Top, Representative recording of successive stimuli induced EPSCs in wild-type and mBACtgDyrk1a mice. Summarized results showing similar in paired-pulse ratio after Dyrk1a overexpression. Time interval between stimulations varied from 25 to 400 ms (wild-type, white bar, vs mBACtgDyrk1a, black bar, 25 ms: 1.96 ± 0.16, n = 9 vs 2.07 ± 0.11, n = 8; 50 ms: 1.73 ± 0.09, n = 12 vs 1.69 ± 0.09, n = 8; 100 ms: 1.46 ± 0.08, n = 10 vs 1.31 ± 0.10, n = 7; 200 ms: 1.22 ± 0.06, n = 9 vs 1.19 ± 0.06, n = 6; 300 ms: 1.15 ± 0.07, n = 8 vs 1.07 ± 0.04, n = 7; 400 ms: 1.16 ± 0.09 n = 4 vs 1.16 ± 0.06, n = 3; p > 0.05, Mann–Whitney U test). EPSC sizes were normalized to the amplitude of the first response. Histograms represent the mean value. Error bars indicate SEM.
Excitatory synaptic inputs are integrated into action potential and the EPSP-spike coupling (or E-S coupling) varies in complement to synaptic plasticity (Daoudal et al., 2002). We found that the E-S coupling was similar in wild-type and in mBACtgDyrk1a mice (Fig. 5B). We conclude that the lack of LTP in mBACtgDyrk1a mice has no or little repercussion to excitatory synaptic integration in PFC neurons.
To test whether the synaptic plasticity impairments are the result of impaired probability of glutamate release, we quantified the progressive inhibition of NMDAR-mediated EPSCs by an open-channel blocker of NMDAR (see Materials and Methods) (Chavis and Westbrook, 2001). The release probability at excitatory synapses impinging onto layer V/VI pyramidal neurons was identical in wild-type and in mBACtgDyrk1a mice (Fig. 5C). Accordingly, the paired pulse ratio a form of short-term synaptic plasticity that depends on release probability was identical at all intervals tested in both genotypes (Fig. 5D).
Together, these data indicate that the lack of LTP and LTD in mBACtgDyrk1a mice cannot readily be explained by modifications of the synaptic properties, release probability, excitation-spike coupling, or the NMDAR subunit composition of PFC pyramidal synapses in mBACtgDyrk1a mice.
GTEs normalizes LTP and pCaMKII/CaMKII ratio in adult mBACtgDyrk1a mice
DYRK1A kinase activity is inhibited in vitro by a natural polyphenol abundantly found in green tea, EGCG. Naturally occurring green tea polyphenols rescue behavioral and morphological functions associated with Dyrk1a overexpression: lifelong treatment with green tea infusion restored behavioral and morphological deficits in a polygenic model overexpressing Dyrk1a and four other HSA21 genes (YACtg152F7) (Guedj et al., 2009). More recently, EGCG was also shown to correct cognitive deficits in mouse DS models and humans (De La Torre et al., 2013).
To test whether green tea could restore synaptic plasticity in adult mBACtgDyrk1a mice, our 4-month-old mBACtgDyrk1a mice were given ad libitum access to water containing decaffeinated GTE (0.8 mg/ml EGCG) for 4–6 weeks.
Remarkably, this simple treatment restored synaptic LTP in mBACtgDyrk1a: in marked contrast with untreated littermates, TBS-LTP was normal in GTE-treated mBACtgDyrk1a mice (Fig. 6A). In support of the idea of a link between pCaMKII levels and the ability to successfully induce LTP, the rehabilitation of LTP was paralleled by the augmentation of pCaMKII levels (Fig. 6B) and the normalization of the pCaMKII/CaMKII ratio in the PFC of mBACtgDyrk1a mice treated with GTE (54.78 ± 6.36, n = 12 mBACtgDyrk1a vs 112.47 ± 11.34, n = 7 mBACtgDyrk1a GTE; p = 0.0008, Mann–Whitney U test). Although the GTE treatment had no significant effects on CaMKII levels, it elevated pCaMKII levels in wild-type littermates, reinforcing the link between DYRK1A and pCaMKII protein levels (Fig. 6B). In contrast, GTE treatment did not rescue eCB-LTD in the PFC of mBACtgDyrk1a mice (100.10 ± 0.91, n = 11, mBACtgDyrk1a GTE baseline vs 92.10 ± 11.25 n = 7, mBACtgDyrk1a GTE 25–30; p > 0.05, Mann–Whitney U test).

Drinking GTE restored normal LTP and spine density in adult mBACtgDyrk1a mice. Four- to 5-month-old mBACtgDyrk1a mice were treated for 1 month with GTE (drinking water). A, TBS of layers II/III induced an LTP of evoked fEPSP recorded in layer V/VI pyramidal neurons in wild-type mice (n = 12, black symbols; 98.60 ± 0.58, baseline vs 121.76 ± 3.21, 25–30 min; p < 0.0001, Mann–Whitney U test) but not in mBACtgDyrk1a mice (n = 8, white symbols; 98.02 ± 0.84, baseline vs 93.99 ± 3.98, 25–30 min; p > 0.05, Mann–Whitney U test). The In vivo GTE treatment restored normal LTP in mBACtgDyrk1a mice (n = 10, light green symbols; 99.80 ± 0.30, baseline vs 125.51 ± 2.09, 25–30 min; p < 0.0001, Mann–Whitney U test) but had no effect on wild-type mice (n = 8, dark green symbols; 99.06 ± 0.32 baseline vs 120.27 ± 4.62, 25–30 min; p = 0.003, Mann–Whitney U test). Average time courses of mean fEPSP were normalized to baseline. Top, GTE rescued LTP in mBACtgDyrk1a mice. Representative traces averaged from fEPSP response before (1) and 30 min after plasticity induction (2) in mice from either genotype. Error bars indicate SEM. B, GTE normalized pCaMKII levels. Slot blot of pCaMKII andCaMKII. Top bands represent the protein; bottom band represents the whole amount of protein marked with Ponceau red. The pCaMKII level was decreased in mBACtgDyrk1a mice (100.00 ± 11.15, n = 11 wild-type, black bar vs 62.54 ± 6.27, n = 12 mBACtgDyrk1a, white bar): *p = 0.0106 (Mann–Whitney U test). The GTE treatment largely increased pCaMKII level in mBACtgDyrk1a mice (62.54 ± 6.27, n = 12 mBACtgDyrk1a vs 119.48 ± 12.50, n = 7 mBACtgDyrk1a GTE, light green bar; **p = 0.0027, Mann–Whitney U test) and also in wild-type mice (100.00 ± 11.15, n = 11 wild-type vs 141.95 ± 9.50, n = 10 wild-type GTE, dark green bar; *p = 0.0124, Mann–Whitney U test). The CaMKII level was the same in wild-type and mBACtgDyrk1a mice (100.00 ± 5.26, n = 11 wild-type, black bar, vs 113.84 ± 4.74, n = 12 mBACtgDyrk1a, white bar; p > 0.05, Mann–Whitney U test). The GTE treatment had no effect on CaMKII level either in wild-type mice (100.00 ± 5.26, n = 11 wild-type vs 93.79 ± 7.34, n = 10 wild-type GTE, dark green bar; p > 0.05, Mann–Whitney U test) or in mBACtgDyrk1a mice (113.84 ± 4.74, n = 12 mBACtgDyrk1a vs 104.58 ± 10.90, n = 7 mBACtgDyrk1a GTE, light green bar; p > 0.05, Mann–Whitney U test). C, The GTE treatment restored normal spine density of mBACtgDyrk1a mice. Representative unprocessed images of spines and dendritic shafts from pyramidal PFC neurons of GTE-treated wild-type (left) and mBACtgDyrk1a mice (right). Scale bars, 2 μm. D, Summary histograms of the effects of GTE on spine density. The GTE treatment restored normal spine density of mBACtgDyrk1a mice (1.41 ± 0.10, n = 10 cells from 6 mBACtgDyrk1a, white bar, vs 0.96 ± 0.07, n = 14 cells from 8 mBACtgDyrk1a GTE, light green bar): **p = 0.002 (Mann–Whitney U test). The treatment has no effect on the spine density of wild-type mice (0.76 ± 0.07, n = 8 cells from 5 wild-type, black bar, vs 0.94 ± 0.07, n = 10 cells from 10 wild-type GTE, dark green bar): ***p > 0.05 (Mann–Whitney U test). Error bars indicate SEM. E, GTE normalized PSD95 levels. Slot blot of PSD95. Top bands represent the PSD95 protein; bottom band represents the whole amount of protein marked with Ponceau red. The PSD95 level was increased in mBACtgDyrk1a mice (100.00 ± 6.53, n = 11 wild-type, black bar, vs 122.82 ± 5.44, n = 10 mBACtgDyrk1a, white bar): *p = 0.0378 (Mann–Whitney U test). The GTE treatment brought PSD95 level in mBACtgDyrk1a mice back to normal levels (122.82 ± 5.44, n = 10 mBACtgDyrk1a vs 98.42 ± 7.84, n = 7 mBACtgDyrk1a GTE, light green bar): *p = 0.014 (Mann–Whitney U test). On the contrary, GTE had no effects on PSD95 in wild-type mice (100.00 ± 6.53, n = 11 wild-type vs 83.57 ± 5.84, n = 10 wild-type GTE, dark green bar): p > 0.05 (Mann–Whitney U test). The presented data are normalized to the baseline protein level of wild-type mice fed with water. Error bars indicate SEM.
Supporting our hypothesis that GTE can correct multiple deficits linked to Dyrk1a overexpression, we found that the GTE rehabilitation of LTP and pCaMKII/CaMKII levels was paralleled by concomitant normalization of both the spine density of PFC pyramidal neurons (Fig. 6C) and PSD-95 levels (Fig. 6D).
Our data demonstrate that a short treatment with GTE at the adult stage can rehabilitate both LTP and spine density in the PFC of transgenic mice expressing an extra copy of Dyrk1a.
Discussion
A primary goal of this study was to provide new neurophysiological basis to cognitive deficits associated with DS by directly tackling their morphological and physiological substratum. We took advantage of a newly generated mouse model of partial DS (Guedj et al., 2012) that expresses a third copy of Dyrk1a, a main candidate gene for the intellectual deficits of DS (Becker, 2011; Guedj et al., 2012). We discovered profound alterations of PFC synaptic plasticity and dendritic architecture in adult mBACtgDyrk1a. We also suggest new restorative routes to correct spine deficits and synaptic functions in adult mBACtgDyrk1a mice.
DYRK1A is important to cortical development and neuronal morphology (Guedj et al., 2012; Martinez de Lagran et al., 2012). The present observation of a marked increase in spine density is reminiscent of that reported in the fmr1−/− mouse model of Fragile X, another human genetic disease associated with severe intellectual impairment (Bagni and Greenough, 2005; Penzes et al., 2011). Subjects with DS present hippocampal dysfunctions (Pennington et al., 2003) and reduced LTP correlates with lower hippocampal-dependent spatial learning and alteration of dendritic organization in the Ts65dn mice model of DS (Fernandez et al., 2007; Belichenko et al., 2009; Guedj et al., 2009, 2012; Garner and Wetmore, 2012). By showing that similar anomalies take place at pyramidal PFC synapses, our data are consistent with and extend previous reports of hippocampus-related memory deficits and alteration of hippocampal synaptic plasticity in DS animal models (Guedj et al., 2009, 2012). We found a marked increase in the spine density at oblique dendrites of PFC pyramidal neurons of mBACtgDyrk1a. Our results may at first glance appear to contradict recent observations of reduced dendritic arborization and spine density measured in a different mice model of Dyrk1a overexpression (Martinez de Lagran et al., 2012). However, we propose that this apparent discrepancy is the result of differences in the cortical areas structures considered (here the PFC vs the motor cortex), the layers where the analysis were made (here deep layers vs superficial layers), the dendrites of interest (here oblique vs basal), or the type of promoter driving Dyrk1a expression (here endogenous vs exogenous). It is also important to note that, in the motor cortex, both Dyrk1a overexpression and happloinsufficiency reduced basal dendritic spine density (Benavides-Piccione et al., 2005; Martinez de Lagran et al., 2012), showing the remarkable sensitivity of this cortical area to DYRK1A activity.
In all cases, our structural data are correlated to biochemical and functional evidence. In agreement with a local increase in synaptic spine densities and in line with the idea that PSD95 levels and AMPAR synaptic responses covary (Stein et al., 2003), we found that postsynaptic PSD95 levels and mEPSC amplitudes were significantly increased in PFC extracts from mBACtgDyrk1a mice. However, the lack of change in mEPSC frequency indicates that many of the new dendritic spines observed in mBACtgDyrk1a mice do not establish functional synapses.
Together, all available data converge to show that adequate Dyrk1a gene dosage is necessary to establish and maintain proper spine arborization and that there is a tight relationship between the number of copies of Dyrk1a and spine abnormalities (present data; Benavides-Piccione et al., 2005; Martinez de Lagran et al., 2012).
The spine anomalies were paralleled by impairments in two major forms of synaptic plasticity. First, NMDAR-LTP is absent in the PFC of mBACtgDyrk1a mice.
Because there was a large increase of both spine density and mEPSC amplitude, the inability to induce LTP is theoretically compatible with a ceiling effect that would occlude LTP. However, we measured similar AMPAR/NMDAR ratio in both genotypes, arguing against the idea of a preexisting increase in the gain of mBACtgDyrk1a synapses. In search for a mechanistic understanding of the modified LTP, we found marked alterations in the expression levels and/or phosphorylation of two essential postsynaptic proteins involved in LTP and spine plasticity. First, as already discussed, we found that PFC PSD95 levels were significantly elevated in mBACtgDyrk1a mice. This is likely to participate in the lack of LTP because overexpression of PSD95 completely occludes LTP in pyramidal cells of the hippocampus (Stein et al., 2003). Second, the pCaMKII/CaMKII ratio was reduced in mBACtgDyrk1a PFC. Normally, CaMKII activity increases the conductance of AMPARs and their synaptic binding to PSD95 (Sanhueza and Lisman, 2013). When autophosphorylated at T286, CaMKIIα subunits are in an “autonomous' state” and LTP induction is prevented (Lledo et al., 1995). DYRK1A has the potential to control multiple presynaptic and postsynaptic targets directly and/or at the gene regulation level, and determining which precise targets underlie the present effects remains beyond the scope of the present study. Our results showing that the strong imbalance of the pCaMKII/CaMKII ratio is principally attributable to a decrease in pCaMKII (T286) levels are compatible with a scenario where DYRK1A overactivity augments the quantity of autonomous CaMKIIα subunits, saturates LTP, and inhibits further LTP induction (Stein et al., 2003; Lisman et al., 2012). Together, these results suggest that common mechanisms link DYRK1A activity to spine stabilization: PSD95, CaMKIIα, AMPAR, and LTP.
In line with previous recent work showing the potential of EGCG contained in GTE on biochemical, morphological (Guedj et al., 2012), and behavioral deficits in mouse DS models (De La Torre et al., 2013), we observed that EGCG-rich GTE restores normal spine density, LTP, and associated biochemical markers when administered in adult Dyrk1a-overexpressing mice. These data favor the idea that the origin of the DYRK1A-related PFC deficits is not merely developmental and that continuous excessive DYRK1A activity is detrimental to optimal PFC functioning. A primary goal of preclinical research is to propose innovative therapeutic approaches; thus, our present study provides new neurophysiological basis to the current interest in the therapeutic and prophylactic use of green tea to alleviate some of the cognitive deficits associated with DS by directly tackling their morphological and physiological substratum.
Facts have rapidly accumulated that the eCB system may be an important target in neuropsychiatric diseases, notably intellectual disability and autism (Jung et al., 2012; Busquets-Garcia et al., 2013). Here, we provide clear evidence that eCB-mediated LTD is abolished in the PFC of DYRK1A overexpressing mice. This finding is reminiscent of our recent demonstration that eCB-LTD is absent in fmr1−/− mice (Jung et al., 2012). Our experiments showed normal CB1R efficacy and its downstream signaling, as well as normal excitability and synaptic properties in the mBACtgDyrk1a mice. Additional research beyond the scope of this study will be necessary to clarify how DYRK1A activity controls retrograde 2-AG LTD at PFC synapses. In particular, one should test the link between enhanced DYRK1A phosphorylation activity and proteins involved in eCB synthesis/degradation. In this context, in support of our previous observation that pharmacological blockade of 2-AG degradation normalizes LTD in fmr1−/− mice (Jung et al., 2012), we found that blocking 2-AG hydrolysis using the monoacylglycerol lipase inhibitor JZL184 restored LTD in mBACtgDyrk1a mice. In contrast, inhibition of the postsynaptic 2-AG-hydrolyzing enzyme ABHD6 did not restore LTD (Jung et al., 2012). Together, these new data indicate that modulation of the eCB system may have beneficial effects in DS.
In conclusion, our present work shows that correct Dyrk1a gene dosage is necessary for structural and functional organization of PFC synapses and reveals new synaptic functions for DYRK1A kinase which could be target for therapeutic strategy in DS.
Footnotes
This work was supported by Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, ANR “DsTher” (J.D., and O.J.M.), Fondation Jérôme Lejeune (O.J.M., A.T., and J.D.), and Fondation pour la Recherche Médicale (A.T. and O.J.M.). We thank Dr. H. Martin for proofing our manuscript and the National Institute of Mental Health's Chemical Synthesis and Drug Supply Program (Rockville, Maryland) for providing DNQX, MK801, JZL184, and d-APV.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Olivier Manzoni, Institut National de la Santé et de la Recherche Médicale U901, INMED, Marseille, 13009, France. olivier.manzoni{at}inserm.fr





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}