

Abstract
Within the dorsal lateral geniculate nucleus (dLGN) of the thalamus, retinal ganglion cell (RGC) projections excite thalamocortical (TC) cells that in turn relay visual information to the cortex. Local interneurons in the dLGN regulate the output of TC cells by releasing GABA from their axonal boutons and specialized dendritic spines. Here we examine the functional role of these highly specialized interneurons and how they inhibit TC cells in mouse brain slices. It was widely thought that activation of metabotropic glutamate receptor type 5 (mGluR5) on interneuron spines leads to local GABA release restricted to sites receiving active RGC inputs. We reexamined experiments that supported this view, and found that in the presence of TTX, mGluR5 agonists evoked GABA release that could instead be explained by interneuron depolarization and widespread intracellular calcium increases. We also examined GABA release evoked by RGC activation and found that high-frequency stimulation induces a long-lasting subthreshold afterdepolarization, persistent firing, or prolonged plateau potentials in interneurons and evokes sustained GABA release. mGluR5 antagonists virtually eliminated sustained spiking and the resulting widespread calcium-signals, and reduced inhibition by >50%. The remaining inhibition appeared to be mediated by a fraction of interneurons in which plateau potentials produced large and widespread calcium increases. Local calcium signals required for local GABA release were not observed. These findings indicate that, contrary to the previous view, RGC activation does not simply evoke localized GABA release by activating mGluR5, rather, synaptic activation of mGluR5 acts primarily by depolarizing interneurons and evoking widespread dendritic GABA release.
Introduction
The dorsal lateral geniculate nucleus (dLGN) conveys visual information from the retina to the cortex: retinal ganglion cells (RGCs) activate thalamocortical (TC) cells that in turn excite cortical neurons (Sherman and Guillery, 1996). RGCs also activate interneurons within the dLGN that inhibit TC cells, thereby sharpening their receptive fields (Norton and Godwin, 1992), and controlling their firing (Usrey et al., 1999; Blitz and Regehr, 2005). These local interneurons have long been of interest because, in additional to conventional axodendritic synapses, they inhibit TC cells via specialized dendrodendritic synapses that involve the convergence of an RGC axon, an interneuron dendritic spine, and a relay cell dendrite, collectively known as a triad (Rafols and Valverde, 1973; Ohara et al., 1983; Hamos et al., 1985). RGC inputs to the interneuron spine are in close proximity to GABA release machinery and to metabotropic glutamate receptors type 5 (mGluR5s) expressed in interneurons (Godwin et al., 1996). This, along with the observation that mGluR5 agonists evoke GABA release from interneuron dendrites in the absence of spiking (Cox et al., 1998; Cox and Sherman, 2000; Govindaiah and Cox, 2004; Errington et al., 2011), suggested that RGC axons trigger local GABA release by activating mGluR5 on interneuron spines and locally elevating calcium (Netzeband et al., 1997; Tempia et al., 1998). Experiments with caged glutamate support the potential mechanism of local release in the absence of firing (Crandall and Cox, 2012). Local inhibition would enable interneuron spines to be functional units of feedforward inhibition, providing precise control of TC cell activity at the location of the excitatory RGC input.
However, mGluRs are often coupled to TRP channels (Crépel et al., 1994; Congar et al., 1997; Abramowitz and Birnbaumer, 2009), suggesting that mGluR activation could depolarize dLGN interneurons. Moreover LGN interneuron dendrites support sodium and calcium action potentials (Acuna-Goycolea et al., 2008), and sustained plateau potentials (Antal et al., 2010). This suggests that RGC could activate mGluRs and depolarization of dLGN interneurons, and trigger active responses that evoke widespread GABA release. Most studies of mGluR signaling have not tested this possibility because they examined the effects of bath-applied mGluR agonists on inhibition onto TC cells in the presence of TTX (Cox et al., 1998; Cox and Sherman, 2000; Errington et al., 2011).
Here we test the prevailing view that mGluR5 activation leads exclusively to GABA release localized to sites receiving active RGC inputs. In contrast to previous studies, we find that mGluR activation depolarizes dLGN interneurons and produces widespread calcium signals. We also find that high-frequency RGC stimulation evokes a long-lasting subthreshold afterdepolarization, persistent firing, or prolonged plateau potentials in interneurons, and evokes sustained GABA release. mGluR5 antagonists eliminated sustained spiking and the associated calcium increases, and greatly reduced disynaptic inhibition. Local calcium signals within the spines of LGN interneurons were not observed. Although these findings do not preclude the possibility that local release could occur under some conditions, they establish that synaptic activation of mGluR5 depolarizes interneurons and evokes widespread dendritic GABA release. Thus, contrary to the previous view, local GABA release is not the predominant means of releasing GABA.
Materials and Methods
Animals and slice preparation.
Mice (22–32 d old, either sex, C57BL/J6) were deeply anesthetized with Isoflurane or Halothane, their brains were quickly removed and placed in ice-cold oxygenated (95% oxygen/5%CO2) dissection solution containing (in mm): 130 K-Gluconate, 15 KCl, 0.05 EGTA, 20 HEPES, 25 glucose. The pH was set to 7.4 using NaOH, and with 2.5 μm 3-((R)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid [(R)-CPP] added (Dugué et al., 2005; Kanichay and Silver, 2008; Hull and Regehr, 2012). A block of brain tissue containing the optic tract (OT) and the dLGN was gently removed and transferred to a vibratome where 250 μm pseudosagittal slices were cut. Corticothalamic and thalamic reticular nucleus (nRT) feedback was eliminated by severing the connections of these regions to the dLGN (Blitz and Regehr, 2005). The slices were then moved to a chamber filled with oxygenated artificial CSF (ACSF) that contained (in mm): 124 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 25 glucose. After 30 min of recovery at 32°C, brain slices containing close to a millimeter of the OT and the dLGN were placed in the recording chamber mounted on an Olympus BX51WI microscope equipped with DIC and fluorescence capabilities. The temperature in the recording chamber was kept between 34 and 35°C using an inline heater controller (Warner Instruments). The tissue was continuously superfused with oxygenated ACSF at ∼5 ml/min flow rate.
Electrophysiological recordings and data analysis.
The inhibitory interneurons within the dLGN were identified by selective GFP expression in C57BL/J6 GAD67-GFP transgenic animals (Tamamaki et al., 2003), whereas for most TC cell recordings, wild-type C57BL/J6 animals were used. Green fluorescent protein-expressing cells within the dLGN were identified using appropriate fluorescence filters.
Whole-cell voltage and current-clamp recordings were performed by using 3–5 MΩ borosilicate glass pipettes pulled with a Sutter horizontal puller (Sutter Instruments). For current-clamp recordings, we filled the pipettes with an intracellular solution that contained the following (in mm): 150 KMeSO4, 1 MgCl2, 10 HEPES, 0.5 EGTA, 2 Mg-ATP, 0.4 Na2-GTP, and 10 phosphocreatine di(tris), pH 7.3 with KOH. For Ca-imaging recordings, we used an internal solution that contained (in mm): 150 K-gluconate, 1 MgCl2, 10 HEPES, 8 NaCl, 2 Mg-ATP, 0.4 Na3GTP, and 10 phosphocreatine di(tris), pH 7.3 with KOH. Voltage-clamp recordings from TC cells were made with an intracellular solution that contained (in mm): 150 Cs-gluconate, 1 MgCl2, 10 HEPES, 0.5 EGTA, 2 Mg-ATP, 0.4 Na3GTP, and 10 Na2-phosphocreatine, pH 7.3 with CsOH. Access resistance was continuously monitored throughout the experiments and only those cells with stable access resistance (changes <10%) were used for analysis.
The recordings were performed with a Multiclamp 700B amplifier (Molecular Devices) and were controlled with custom software written in IGOR (generously provided by Matthew Xu-Friedman University of Buffalo, Buffalo, NY).
Extracellular RGC axon stimulation was performed with stainless steel Teflon-coated bipolar electrodes (tip separation ∼0.1–0.4 mm). Stimulation electrodes were placed in the OT at least 1 mm away from the ventral border of the dLGN (300 μs, 0–1 mA pulses, WPI A395 Linear Stimulus Isolator). In most experiments synaptically evoked responses were recorded at intervals of 60 s. Stimulus artifacts were removed, and stimuli were noted with an asterisk (see Figs. 2⇓⇓⇓⇓–7).
To quantify the long-lasting afterdepolarizations in dLGN interneurons, the integral of the membrane potential change was calculated over 30 s following stimulation. Detection and quantification of IPSCs was performed using custom software written in IGOR Pro in which, first, the current trace from the voltage-clamped TC cell was filtered at 500 Hz. Then, the first derivative of the current traces was used to identify the rapid rise times of the IPSCs. A thresholding procedure was then performed for detection of events. Events were then binned in time groups of 500 ms and plotted (see Figs. 2, 4).
Two-photon imaging.
Two-photon calcium imaging experiments were performed as described previously (Brenowitz and Regehr, 2007; Acuna-Goycolea et al., 2008) using a custom two-photon laser-scanning microscope with a 60× objective (0.9 numerical aperture) (Olympus Optical) and a Ti:sapphire laser (Mira; Coherent, Inc.). The recording pipette for these cells contained the red dye Alexa Fluor 594 (20–40 μm) for visualizing the interneurons' dendrites and spines, and fluo-5F (100 μm; KD of 585 nm at 35°C) for measuring Ca fluorescence. A single excitation wavelength of nearly 790 nm was effective for simultaneous excitation of both green and red fluorophores. Fluorescent signals were converted to [Ca] by using values of Rmin and Rmax measured in the same imaging system (Brenowitz and Regehr, 2007). The Rmax values used were measured in neurons in response to high-intensity stimulation and they were 25% larger than those measured in a cuvette with internal solution in the presence of 3 mm Ca2+; Rmin values were measured in a cuvette with internal solutions containing 3 mm EGTA.
The composite low-magnification images presented (see Figs. 1, 6, 7) are compressed 2-photon z-stacks, with colors inverted, and contrast enhanced for clarity. These images are shown to illustrate the gross morphology of the neuron.
Statistical significance was determined using the Student's t test, or (see Fig. 3), the Kolmogorov–Smirnov test. Data are presented as mean ± SEM.
Results
Responses of dLGN neurons to mGluR activation
The role of mGluR5 activation in feedforward inhibition onto TC cells was studied using pseudosagittal slices of the mouse dLGN. These slices contain TC cells, LGN interneurons, and the optic tract, which contains RGC axons (Fig. 1A). Whole-cell and on-cell recordings were made from visually identified TC cells that were identified using DIC optics, and LGN interneurons that were identified with GFP fluorescence in GAD67 GFP transgenic mice (Tamamaki et al., 2003).

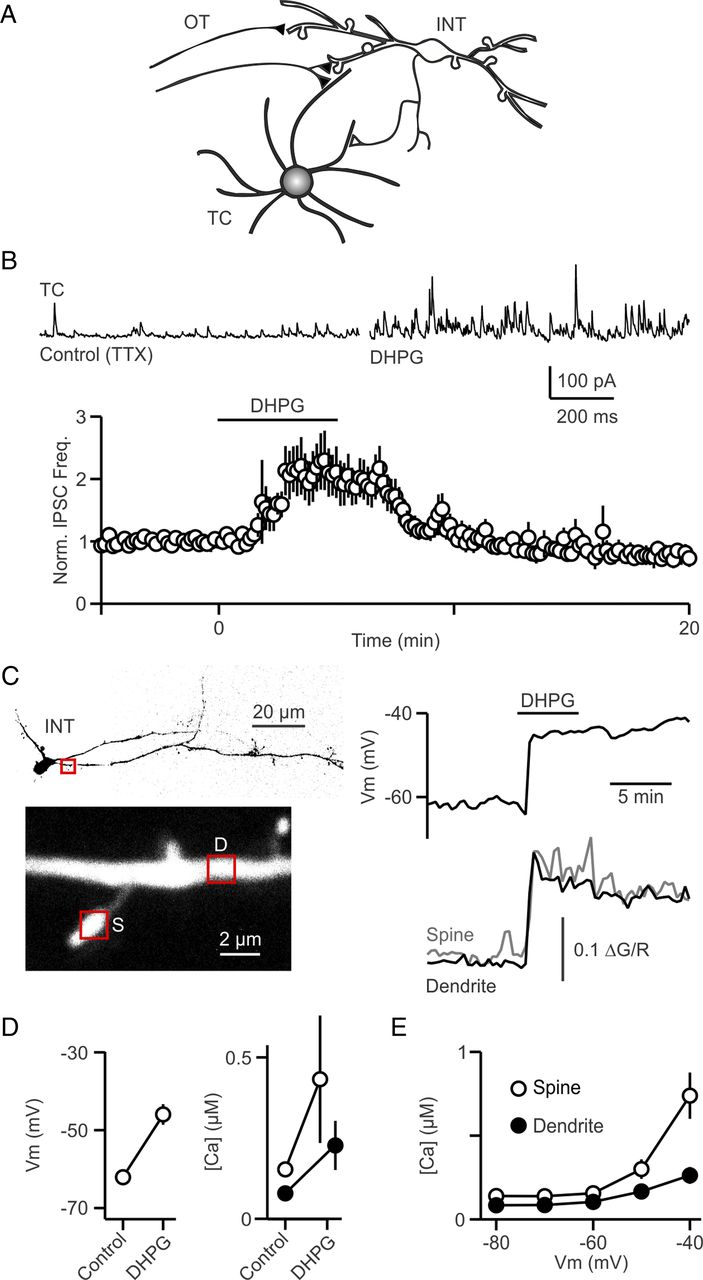
mGluR activation depolarizes interneurons and evokes persistent inhibition in TC cells. A, A schematic shows RGC axons that project to the dLGN via the OT, where they excite interneurons and TC cells. dLGN interneurons inhibit TC cells via both axonal and dendrodendritic synapses. B, Top, IPSCs were recorded in a TC cell (at 0 mV) in control conditions and in the presence of the group I mGluR agonist DHPG (5 μm). Bottom, A summary plot of normalized IPSC frequency after bath application of DHPG (n = 7 cells). C, A representative interneuron experiment in which DHPG (5 μm) was bath applied and the membrane potential was measured in current clamp (right) and intracellular calcium from a spine and a dendrite (lower left) was measured (lower right). D, Summary of the effects of DHPG (5–100 μm) on interneuron membrane potential (n = 14) (left) and resting Ca (right) for dendrites (n = 14) and spines (n = 7). E, A plot of resting calcium as a function of membrane potential in dendrites (n = 19) and spines (n = 14). TTX was present in all experiments in B–D.
To explore the role of mGluR activation in thalamic inhibition, we first repeated the key experiment that had provided support for mGluR triggering local release of GABA in the absence of action potential firing (Cox et al., 1998). We performed voltage-clamp recordings in the presence of TTX (500 nm); the group I mGluR agonist (S)-3,5-dihydroxyphenylglycine (DHPG) elevated the frequency of spontaneous IPSCs (sIPSCs) in TC neurons (Fig. 1B, top). DHPG (5 μm) more than doubled sIPSC frequency (Fig. 1B, bottom; 203 ± 30% of baseline in DHPG, n = 7; p < 0.03). Based on the observation that the reversal potential of these events was at the chloride reversal, and their sensitivity to GABAA receptor antagonists (Cox et al., 1998; Cox and Sherman, 2000; Govindaiah and Cox, 2004; Errington et al., 2011) these events were IPSCs mediated by GABAA receptors. These are similar to previous findings, which had interpreted the inhibition as arising from increased GABA release from the dendrites of dLGN interneurons. The rationale is that, of the potential sources of inhibition onto TC neurons, mGluR5 are located only on the spines and dendrites of dLGN interneurons, and not on the axon terminals of either dLGN interneurons or nRT interneurons (Godwin et al., 1996). It was also thought that in the presence of TTX, membrane potential changes that might arise in the dendrites following mGluR activation would not lead to sufficient axonal depolarization to trigger GABA release (Bloomfield and Sherman, 1989; Cox et al., 1998).
An alternative hypothesis is that activation of mGluR5 depolarizes interneurons sufficiently to open voltage-gated calcium channels that can evoke global transmitter release (Acuna-Goycolea et al., 2008). We tested this possibility by determining whether mGluR activation can depolarize interneurons even in the presence of sodium channel blockers. Whole-cell current-clamp recordings were obtained from interneurons in the presence of TTX (500 nm) and the effect of mGluR activation by DHPG was assessed. DHPG (5–100 μm) depolarized all neurons tested, as shown for a representative experiment (Fig. 1C, upper right; 5 μm DHPG) and for a summary of 14 neurons (Fig. 1D, left; from −62.1 ± 0.9 mV to −45.9 ± 2.6 mV; p < 0.001). We also measured calcium levels, and found that dendritic and spine calcium levels were elevated in these experiments (Fig. 1D, right; from 80 ± 10 nm to 230 ± 80 nm in 14 dendrites, p < 0.03); and 150 ± 10 nm to 430 ± 200 nm in 7 spines; p = 0.20). We assessed whether depolarization was sufficient to account for the observed calcium increases by measuring dendritic and spine calcium increases produced by somatic depolarization (Fig. 1E). Depolarizing the soma to −50 mV and −40 mV increased calcium levels in spines and dendrites (Fig. 1E; to 160 ± 40 nm and 260 ± 50 nm in 19 dendrites; and 300 ± 60 nm and 740 ± 140 nm in 14 spines). These experiments show for the first time that mGluR agonists actually depolarize dLGN interneurons, and that depolarization is sufficient to produce widespread voltage-dependent calcium increases. This novel finding suggests that the elevation of sIPSC frequency in TC neurons in response to DHPG, may not reflect the local activation of mGluR5, but could be a result of interneuron depolarization that results in widespread activation of voltage-gated calcium channels.
Although we have shown that mGluR activation depolarizes interneurons, this is an artificial situation in which all group I mGluRs are activated in a prolonged manner by the bath application of an agonist. It remains unclear whether RGC synapses can activate mGluRs sufficiently to depolarize interneurons and trigger GABA release. Based on the properties of mGluR signaling described at other synapses (Batchelor et al., 1994; Tempia et al., 1998; Karakossian and Otis, 2004), we focused our initial studies of synaptically activated mGluR signaling in the dLGN on high-frequency bursts of high-intensity stimulation that activated many RGC axons. Based on previous studies, it is likely that some visual stimuli evoke transient high-frequency firing in many RGCs (Kuffler et al., 1957; Enroth-Cugell et al., 1966; Barlow and Levick, 1969; Cleland et al., 1973; Mastronarde, 1989; Meister and Berry, 1999). Also, groups of RGCs in close proximity to each other fire synchronously in response to visual stimulus (Mastronarde, 1983; Meister et al., 1995; Berry et al., 1997). In fact, geniculate neurons typically receive input from several retinal afferents, all with >50% overlapping receptive fields (Usrey et al., 1999). We therefore examined the effect of stimulating many RGCs with a high-frequency burst (20 stimuli at 200 Hz). We then measured the resulting increase in sIPSC frequency in voltage-clamped TC neurons, and determining the pharmacological sensitivity of sIPSCs (Fig. 2). The sIPSC frequency was elevated for tens of seconds after stimulation (Fig. 2A). In control conditions RGC stimulation increased sIPSC frequency by 41.9 ± 3.4 Hz (n = 48; p < 0.0001). We found that the increase in sIPSC frequency evoked by stimulating RGC axons (200 Hz × 20 stimuli, 1 mA) every 2 min was stable (the increase in sIPSC frequency was 98 ± 7% of initial values 30–36 min later, n = 4). We then tested the involvement of mGluR5 using the selective antagonist MPEP (50 μm; (Salt et al., 1999), and found that it reduced the persistent inhibition to 43 ± 16% of control levels (n = 4, p < 0.05 in these wash-in experiments; Fig. 2B,C). This indicates that high-frequency synaptic activation of RGCs activates mGluR5 receptors on interneurons sufficiently to promote GABA release. The additional application of NBQX (10 μm) and (R)-CPP (5 μm) reduced the persistent inhibition to 8 ± 7% of control levels (p = 0.001). The initial application of NBQX and (R)-CPP to block ionotropic glutamate receptors also strongly attenuated the sustained inhibition of TC neurons (Fig. 2D,E), and reduced the persistent inhibition to 41 ± 11% (n = 4, p < 0.05 in these wash-in experiments). These data indicate that metabotropic and ionotropic glutamate receptors work together to promote sustained GABA release. In Figure 2, C and E, data were normalized to respective baseline values of ΔIPSC frequency in control just before drug application.

mGluRs play a role in the persistent inhibition onto TC cells produced by high-frequency activation of RGCs. A, High-frequency RGC stimulation (20 stimuli at 200 Hz indicated by *) triggers persistent inhibition in TC cells. In these experiments the inhibition is disynaptic and reflects RGCs stimulating LGN interneurons to trigger GABA release. B–E, Experiments were conducted to determine the contribution of mGluRs and iGluRs in triggering persistent inhibition. In some experiments mGluR5 antagonists were applied followed by a coapplication of mGluR5 antagonists and iGluR antagonists (B, C), and in other experiments iGluR antagonists were applied first (D, E). Representative experiments with traces and analyzed IPSC frequencies (B, D) and summaries of multiple experiments (C, E) are shown (n = 4 for each).
The sustained inhibition evoked in TC neurons indicates that high-frequency RGC stimulation promotes persistent GABA release from LGN interneurons, but how does brief synaptic activation lead to sustained GABA release? We determined whether synaptic activation leads to sustained interneuron spiking by recording interneuron activity. We used on-cell recordings to determine the response of many interneurons because this approach is noninvasive and allows rapid sampling of many interneurons. We found, in direct opposition to the previous literature (Cox et al., 1998; Cox and Sherman, 2000; Govindaiah and Cox, 2004), that RGC activation with a high-frequency burst evokes sustained firing in interneurons, as in Figure 3A. Similar spiking was also evoked in whole cell recordings from LGN interneurons (see Figs. 5⇓–7).

mGluRs help to mediate persistent firing of interneurons produced by RGC activation. The firing of LGN interneurons was monitored with on-cell recordings. A, High-frequency RGC stimulation (20 stimuli at 200 Hz) evokes persistent firing in interneurons. B, C, As shown in representative experiments, an mGluR5 antagonist eliminated the persistent firing (B) and blocking iGluRs strongly attenuated persistent firing (C). D, E, Similar experiments were performed on multiple cells in control (n = 35) and to assess the effects of an mGluR5 antagonist (n = 27) or iGluR antagonists (n = 25) on firing and the results are summarized in a histogram showing the average number of spikes as a function of time (D), and by plotting the cumulative fraction of cells in which RGC stimulation evoked a given number of action potentials (E).
In control conditions, the time course of the increase in interneuron spiking (Fig. 3D) was qualitatively similar to the time course of the increase of IPSC frequency onto TC neurons, suggesting that persistent interneuron firing contributes to the sustained inhibition. However, there were small quantitative differences, with the half-decay time for spiking (Fig. 3D) of 10.5 s compared with 12.5 s for IPSCs frequency (Fig. 4A). It is also possible that a buildup of dendritic calcium could produce asynchronous GABA release that outlives spiking, as is observed conventional synapses following sustained periods of presynaptic activity (Zucker and Regehr, 2002). In addition prolonged plateau potentials in interneurons could trigger GABA release that outlives elevations in interneuron spiking (Antal et al., 2010; see below).

Input parameters control persistent inhibition onto TC cells and persistent firing in LGN interneurons. A, B, RGC axons were activated with high-intensity stimulation (20 stimuli, at frequencies of 10–200 Hz, at time 0), and the resulting IPSC frequency recorded in TC cells (A) and frequency of LGN interneuron firing (B) were quantified in control conditions and in the presence of blockers of mGluR5 and/or ionotropic receptors. C, The normalized peak IPSC frequency is plotted as a function of the peak number of action potentials measured in LGN interneurons. In A, for control conditions (n = 48 for 200 Hz, n = 25 for the rest), for MPEP (n = 12 for 200 Hz, n = 8 for the rest), for NBQX/CPP (n = 11 for 200 Hz, n = 5 for the rest) and for MPEP + NBQX/CPP (n = 17 for 200 Hz, n = 7 for the rest), and in B, for control conditions (n = 15), for MPEP (n = 11), and for NBQX/CPP (n = 13).
Persistent firing was eliminated by an mGluR5 antagonist (Fig. 3B,D), and was greatly attenuated by blocking ionotropic glutamate receptors (Fig. 3C,D). In control conditions, persistent firing was evoked in 57% of the cells (20 of 35), and 35% of those cells fired >50 spikes (7 of 20) (Fig. 3E). During the stimulus train, control cells fired 6.4 ± 0.7 action potentials (APs). When mGluR5 was inhibited 1 of 27 cells persistently fired [Control vs MPEP, Kolmogorov–Smirnov (KS) test p < 0.001), and during the stimulus train, they fired 2.1 ± 0.4 APs (p < 0.0001 compared with control). When ionotropic glutamate receptors were blocked, 5 of 25 cells fired persistently (Control vs NBQX/CPP, KS test p < 0.03; MPEP vs NBQX/CPP, KS test p = 0.85), and during the stimulus train, they fired 0.6 ± 0.2 APs (p < 0.0001 compared with control; p < 0.002 compared with MPEP). These findings indicate that RGC stimulation and synaptic activation of mGluR5 can promote sustained spiking in interneurons. In some cases, even when ionotropic glutamate receptors are blocked, activation of mGluR5 is sufficient to promote sustained spiking in interneurons.
Thus, in contrast to the previous view (Cox et al., 1998; Cox and Sherman, 2000; Govindaiah and Cox, 2004), we find that physiological trains of stimuli activate mGluRs on interneurons leading to a prolonged depolarization and action potential firing. This in turn generates prolonged inhibition of the post synaptic TC cell.
The stimulus parameters governing persistent inhibition onto TC cells
We showed high-frequency RGC input generates persistent inhibition onto TC cells. However, the ultrastructure of the RGC à dLGN interneuron synapse is unusual in that the presynaptic RGC bouton and many release sites along with an interneuron spine and TC cell dendrite, are ensheathed by glia (Hamos et al., 1985; Sillito, 1992; Godwin et al., 1996). This has the potential to promote glutamate pooling, thereby influencing the manner in which RGC activation drives mGluR signaling and regulates dLGN circuitry (Rafols and Valverde, 1973; Ohara et al., 1983; Hamos et al., 1985; Godwin et al., 1996). Glutamate pooling is expected to be highly frequency dependent. This suggests that the glutamate signal will also be frequency dependent, and because ionotropic and metabotropic receptors have different properties, it raises the possibility that metabotropic and ionotropic glutamate receptors could be differentially activated in a frequency-dependent manner.
We therefore tested the hypothesis that metabotropic and ionotropic glutamate receptors are specialized to convey inhibitory responses determined by the frequency of the stimuli. We examined the frequency dependence of GABA release by stimulating RGCs at 10 Hz to 200 Hz (200 Hz, n = 48; n = 25 for other frequencies) and monitoring the resulting inhibition in voltage-clamped TC neurons. While the magnitude of persistent inhibition increased with increases in stimulus frequency, engagement of the interneuron by mGluRs was seen at frequencies as low as 10 Hz (Fig. 4A). Half-maximal inhibition occurred between 20 and 50 Hz and the magnitude of inhibition began to saturate at 200 Hz. The blockade of mGluR5 greatly reduced the inhibition evoked at all stimulus frequencies (200 Hz, n = 12, p < 0.0001; 100–10 Hz, n = 8, p < 0.0001; compared with 200 Hz control). The blockade of ionotropic glutamate receptors also attenuated the inhibition, but to a lesser extent (200 Hz, n = 11, p < 0.04; 20 Hz. n = 5, p < 0.001; 10 Hz, n = 5, p < 0.0001). The coapplication of ionotropic and metabotropic receptor antagonists almost completely eliminated the persistent inhibition (200 Hz, n = 17; n = 7 for other frequencies; p < 0.0001 compared with 200 Hz control). The frequency dependence of the inhibition mediated by mGluR5 alone (in the presence of NBQX and CPP) or ionotropic receptors alone (in the presence of MPEP), was not markedly different. The blockade of NMDARs, AMPARs and mGluR5 with NBQX, CPP and MPEP eliminated the increase in inhibition evoked by 50 Hz stimulation and lower frequencies, but a small residual component remained at 100 and 200 Hz. It is not known whether this small component of inhibition remaining in blockers of metabotropic and ionotropic glutamate receptors reflects incomplete block of glutamate receptors, or whether it is mediated by another receptor. These findings indicate that there is not a marked difference in the frequency dependence of the contributions of ionotropic and metabotropic receptors with regard to conveying inhibition to TC neurons.
In on-cell recordings in control conditions, activation of RGCs with 20 stimuli at different frequencies (n = 15) evoked spiking in LGN interneurons that had a frequency dependence similar to the inhibition recorded in TC neurons (Fig. 4B). MPEP eliminated sustained firing (n = 11), and blockade of ionotropic receptors greatly attenuated firing at all frequencies (n = 13).
A comparison of the LGN interneuron spiking (Fig. 4B, right) and disynaptic inhibition onto TC neurons (Fig. 4A, right) raises some interesting issues. Although there is an approximately linear relationship in control conditions, in the presence of an mGluR5 antagonist there is still considerable inhibition present even though there is greatly reduced persistent spiking in interneurons (Fig. 4C, replotting Fig. 4A,B on the same axis, error bars removed for clarity); under these conditions, GABA release is not triggered by action potentials and it must be triggered in some other way. There are two leading possibilities: (1) GABA release could occur locally with minimal sustained depolarization, or (2) GABA release could be widespread and reflect a large sustained depolarization that does not trigger action potentials.
As a first step to discriminating between these possibilities, we monitored the responses of LGN interneurons that generated sustained activity using whole-cell current clamp (Fig. 5). We recorded from a total of 19 neurons and found that in 13 of these neurons high-intensity RGC activation (200 Hz × 20 stimuli, 1 mA intensity) triggered an initial series of spikes (Fig. 5A, inset) and after a delay the cell depolarized and fired a second more prolonged series of action potentials, as in Figure 5A. As the stimulus intensity was reduced the sustained response was also reduced and a further reduction in stimulus intensity revealed a small slow afterdepolarization. Sustained spiking was also eliminated by hyperpolarizing cells (13 of 13 neurons), and a large afterdepolarization remained, as in Figure 5B.

mGluR5 mediates prolonged responses of LGN interneurons that are triggered by RGC activation. A, B, Activation of RGCs synapses (20 stimuli at 200 Hz) triggered sustained spiking in some LGN interneurons that was suppressed by reducing the number of RGC activated (A), or by hyperpolarizing the interneuron (B). C, D, For other interneurons, RGC activation evoked a prolonged plateau potential that was eliminated by reducing the number of RGCs activated (C), or by hyperpolarizing the cell (D). A–D show the initial responses on an expanded scale. E, F, The contributions of ionotropic and metabotropic glutamate receptors to the after depolarization observed in hyperpolarized LGN interneurons were assessed by blocking mGluR5 (n = 4) or ionotropic glutamate receptors (n = 4). Action potentials are truncated in E, F.
In 6 of 19 neurons, high-intensity OT stimulation also triggered an initial series of spikes, but this was followed by a plateau potential that did not trigger action potentials, as in Figure 5C. Even moderate stimulus intensities could evoke plateau potentials, but weak stimulus could not. Hyperpolarization also suppressed the plateau potentials, as in Figure 5D. These findings suggest that brief high-frequency RGC activity produces a long-lasting depolarization that in some cases is subthreshold, in some cases triggers a series of sodium action potentials, and in other cases triggers a long-lasting plateau potential. This also begins to reconcile the issue we find in Figure 4; perhaps the long-lasting plateau potential is contributing to GABA release in the absence of spiking.
We investigated the contributions of mGluR5 and ionotropic glutamate receptors to the prolonged depolarization that underlies the sustained output modes by hyperpolarizing cells, as in Figure 5B (lower trace). Blocking either mGluR5 or AMPAR/NMDARs strongly reduced the magnitude of the afterdepolarization (compared with control: MPEP reduction to 42.3 ± 5%, p < 0.001; NBQX/CPP reduction to 23.9 ± 8%, p < 0.003), and coapplication of the antagonists eliminated (reduced to 2.4 ± 3%, p < 0.0001) the afterdepolarizations (Fig. 5E,F top, examples shown with action potentials truncated; Fig. 5E,F bottom, summary plots, n = 4 for each). These findings indicate that mGluR5 makes an important contribution to afterdepolarizations evoked by RGC activation, which likely triggers the plateau potential and sustained firing modes.
Calcium increases in dLGN interneurons evoked by high-frequency activation
There are three primary types of responses evoked by RGC stimulation of 20 stimuli at 200 Hz. The first is a subthreshold afterdepolarization that lasts for tens of seconds. The second is persistent firing that is evoked if the afterdepolarization is sufficiently large. The third is a prolonged plateau potential that lasts longer than subthreshold afterdepolarizations, does not trigger sodium spikes, and appears to involve regenerative currents. We measured dendritic and spine calcium signals evoked by RGC stimulation to provide insight into overall efficacy and spatial properties of GABA release evoked by these different firing modes.
We first examined the different types of responses evoked in control conditions (Fig. 6). The most common type of response was persistent firing (Fig. 6A), which was accompanied by a global and sustained calcium increase in spines (150 ± 20 nm, half decay time, t1/2 = 11.6 s, n = 38; p < 0.0001) and dendrites (98 ± 20 nm, t1/2 = 5.6 s, n = 16; p < 0.001) for 1 s, 4 s after stimulation. This is consistent with the observation that sodium spikes produce global calcium transients throughout the dendrites and axons of dLGN interneurons (Acuna-Goycolea et al., 2008). When persistent spiking was prevented by reducing the stimulus intensity (Fig. 6A, right) prolonged calcium increases were not evoked in either in spines (9 ± 4 nm, n = 49) or dendrites (6 ± 5 nm, n = 18). This was also the case for cells that were hyperpolarized sufficiently to prevent persistent firing (as in Fig. 5B,D), and was also not accompanied by a prolonged calcium increase in either the spines or dendrites (Fig. 6C, lower left; pooled data of hyperpolarized cells and cells with lower intensity stimulation).

RGC activation that triggers persistent firing and plateau potentials evokes widespread calcium increases in dLGN interneurons. A, B, Representative experiments showing voltage and Ca transients in cells where strong RGC stimulation (200 Hz, 20 stimuli) produces persistent firing (A) or plateau potentials (B). Low-magnification and high-magnification images (A, top; B, top) are shown for the representative cells, and the voltage for the cells as well as the Ca measurements in the indicated regions are shown below. Additionally, lower-intensity stimulation that was insufficient to generate persistent firing or plateau potentials is shown (A, right; B, right). Note in A and B that sustained activity generates large Ca transients in dendrites and spines, and decreased stimulation does not generate prolonged Ca responses. C, Mean sustained Ca transients in spines (gray traces) and dendrites (black traces) are plotted in cells that respond to RGC activation with persistent firing (n = 38 spines, n = 16 dendrites) or by generating long-lasting calcium plateaus (n = 13 spines, n = 4 dendrites). Additionally, note the lack of a sustained Ca transient in these cells when hyperpolarized (n = 46 spines, n = 16 dendrites) or when stimulated with decreased intensity (n = 39 spines, n = 14 dendrites) to reveal subthreshold afterdepolarizations.
In these experiments we cannot know whether the synapse onto a given spine is activated. However, we never saw a long-lasting calcium signal in the spines of cells with subthreshold afterdepolarizations, and it is likely that some of the spines had activated RGC inputs. This suggests that afterdepolarizations do not evoke prolonged calcium signals in spines and are unlikely to evoke local GABA release. In contrast, prolonged plateau potentials (as in Fig. 5C,D) were accompanied by extremely large calcium signals throughout the dendrites and spines (Fig. 6B). In all cases in which we measured calcium signals evoked by such plateaus in control conditions (n = 3 cells), we saw large calcium signals in spines (390 ± 80 nm, t1/2 = 10.6 s, n = 13; p < 0.001) and dendrites (310 ± 130 nm, t1/2 = 13.7 s, n = 4; p = 0.097) (Fig. 6C, right). The average calcium signals during plateau potentials were larger than those associated with persistent firing (for spines: p < 0.001; for dendrites: p < 0.01), and both the plateau potential-derived Ca (for spines: p < 0.0001; for dendrites: p < 0.0001) and the persistent firing-derived Ca (for spines: p < 0.0001; for dendrites: p < 0.0001) were greater than the Ca elevation seen due to subthreshold afterdepolarizations. These results suggest that plateau potentials are accompanied by widespread calcium signals and that they are well suited to evoking long-lasting, global GABA release—even in the absence of sustained action potential firing. Last, local sustained Ca signals—required for local GABA release, in response to high-frequency stimulation, were not observed.
We went on to determine the effect of blocking glutamate receptors on the calcium signals evoked in LGN interneurons (Fig. 7). In these experiments, after evoking responses in control conditions (in 9 of 10 cells), a glutamate receptor antagonist was washed in as the calcium indicator filled the neuron. Glutamate receptor antagonists reduced resting Ca levels from 118 ± 8 nm (n = 138 regions, 9 cells) in control conditions, to 89 ± 13 nm (n = 65 regions; p < 0.05 compared with control) when mGluR5 was inhibited, and 29 ± 4 nm (n = 36 regions; p < 0.001 compared with control) when ionotropic receptors were blocked. This reduction in baseline Ca levels in glutamate receptor antagonists likely contributes to the reduction in spontaneous IPSC rate seen in TC cells when these antagonists are applied (Figs. 2, 4).

Glutamate receptors do not alter Ca influx during sustained responses. Representative experiments in mGluR and iGluR antagonists, showing voltage and Ca transients in cells where strong RGC stimulation produced sustained firing in control. A cell fires persistent action potentials in control, and in MPEP, the same stimulation at the same membrane potential only produces a modest afterdepolarization that lacks a prolonged Ca transient (A). Plateau potentials that were generated by RGC activation were presumably unaffected by iGluR (n = 1) or mGluR antagonists (n = 2). Note the long-lasting Ca transients seen in dendrites and spines in the presence of MPEP (B) and NBQX/CPP (D). Persistent firing that was unaffected by iGluR (n = 2) antagonists also evoked long-lasting Ca transients in dendrites and spines (C).
We examined the calcium signaling evoked by RGC activation (20 stimuli, 200 Hz) in a rather small number of cells to complement the larger datasets in which the current-clamp responses were characterized (Fig. 5) and calcium signaling produced by different firing modes was characterized (Fig. 6). In cells in which persistent firing was evoked, we found that MPEP completely abolished the persistent firing (Fig. 7A, n = 4 cells). In cells in which RGC stimulation evoked an afterdepolarization in the presence of MPEP, a transient calcium signal was observed in dendrites (34 ± 10 nm, t1/2 = 5.9 s, n = 14) and spines (35 ± 7 nm, t1/2 = 4.9 s, n = 41). The amplitude of Ca entry was slightly higher than control afterdepolarizations for spines (p < 0.002) and dendrites (p < 0.02). Long-lasting plateaus evoked in control conditions were not inhibited by mGluR5 application (n = 2 cells), and we observed long-lasting Ca signals in spines (300 ± 80 nm, t1/2 = 5.8 s, n = 8) and dendrites (330 ± 250 nm, t1/2 = 5.4 s, n = 2) similar to that found in control (Fig. 7B). Similarly, when ionotropic glutamate receptors were blocked (Fig. 7C,D), long-lasting plateaus were not affected (n = 1) and large calcium signals were elevated in spines (680 ± 50 nm, t1/2 = 13.2 s, n = 4) and dendrites (370 nm, t1/2 = 17.3 s, n = 1). In the cases (n = 2 of 3 cells) when ionotropic glutamate receptors were unable to block persistent firing in response to strong synaptic activation, we also saw elevated calcium in spines (64 ± 10 nm, t1/2 = 21 s, n = 8) and dendrites (51 ± 10 nm, t1/2 = 17.4 s, n = 3). Subthreshold afterdepolarizations in these blockers (n = 4 cells) only generated minute Ca signals in spines (1 ± 1 nm, n = 15) and dendrites (2 ± 2 nm, n = 5). The amplitude of Ca entry was similar to control afterdepolarizations for spines (p = 0.28) and dendrites (p = 0.68). These findings suggest that the effects of glutamate receptor antagonists on calcium signals are consistent with the effects they have on the firing modes of the cells. Long-lasting plateaus and the large calcium signals they produce can be evoked when either mGluR5 is blocked or ionotropic glutamate receptors are blocked, and when strong inputs generate persistent firing even in the presence of these blockers, prolonged Ca increases are still found in spines and dendrites.
Discussion
Our main finding is that mGluR5 activation leads primarily to widespread GABA release from the entire dendritic arbor of LGN interneurons, which is in sharp contrast to the prevailing view (Cox et al., 1998; Cox and Sherman, 2000; Shepherd, 2004; Govindaiah and Cox, 2004, 2006; Sherman, 2004; Sherman and Guillery, 2006; Errington et al., 2011; Crandall and Cox, 2012) in which GABA release is localized to synaptically activated dendritic spines. The widespread nature of GABA release is a natural consequence of depolarization mediated by mGluR5.
A new interpretation for the effect of mGluR activation in dLGN interneurons
Several lines of evidence supported the prevailing view of mGluR5 signaling that emphasized local GABA release. First, previous studies found that mGluR agonists increase spontaneous inhibition onto TC cells, the extent of inhibition is insensitive to TTX, and when examining interneurons, they did not observe a depolarization in mGluR agonists (Cox et al., 1998; Cox and Sherman, 2000; Govindaiah and Cox, 2004; Errington et al., 2011). Second, it was found that RGC activation evoked very few action potentials and no long-lasting afterdepolarizations were observed in interneurons (Govindaiah and Cox, 2004), which reinforced the view that mGluR5-mediated inhibition is not dependent on depolarization and must therefore be localized. Third, it was recently concluded that focal glutamate uncaging could evoke local GABA release onto TC cell dendrites without evoking spikes in interneurons (Crandall and Cox, 2012).
Our results lead to a very different view. By directly recording from dLGN interneurons, we have shown that activating mGluR5 with an agonist sufficiently depolarizes interneurons to open voltage-gated calcium channels and evoke widespread Ca increases in the dendrites of dLGN interneurons (Fig. 1). We also found that synaptic activation of RGCs could evoke sustained firing and plateau potentials (Fig. 5), and that both of these firing modes were associated with widespread calcium signaling well suited to producing widespread GABA release (Fig. 6). We found that inhibiting mGluR5 eliminated sustained firing of interneurons (Fig. 5), reduced sustained calcium signals in interneurons (Fig. 7), and strongly reduced sustained inhibition of TC neurons (Fig. 2). The remaining inhibition seems to be mediated by the fraction of neurons that produce sustained plateau potentials in response to stimulation, even in mGluR5 antagonists. Our use of a pseudo-sagittal slice orientation in which the optic tract is preserved allows stimulation of more RGC inputs than is generally possible with the slice orientation used in other studies (Govindaiah and Cox, 2004; Errington et al., 2011).
Our findings do not rule out the possibility that under some circumstances local release can occur. However, our data show that for more physiological mGluR activation, global release dominates and interneurons release GABA from both axons and spines. In cases where persistent firing is blocked by mGluR or ionotropic GluR (iGluR) antagonists, persistent inhibition can still be generated by the more robust plateau potentials generated in some interneurons. Indeed, we find no evidence for focal signaling of any sort under our experimental conditions.
Regulation of inhibition by metabotropic and ionotropic receptors
It appears that both metabotropic and ionotropic glutamate receptors are activated by ambient glutamate levels in the slice: antagonists of either mGluR5 or ionotropic glutamate receptors decrease resting calcium levels in LGN interneurons (Fig. 7 results), reduce the frequency of spontaneous IPSCs measured in TC neurons (Figs. 2, 4), and reduce the amplitude of evoked IPSCs (Fig. 2). These findings are consistent with the high-affinity of mGluR5 and NMDA receptors (Pin and Duvoisin, 1995; Zorumski et al., 1996; Scanziani et al., 1997). These findings indicate that these receptors are well suited to respond to even modest levels of glutamate, and it is likely that elevations of glutamate levels by ongoing RGC activity will be highly effective at regulating the output of LGN interneurons.
Multiple firing modes allow dLGN interneurons to dynamically inhibit TC cells
In contrast to brain regions such as the hippocampus, in which many types of inhibitory interneurons provide distinctive inhibition onto the principle cells, within the dLGN a single type of inhibitory interneuron is present in the dLGN (Sherman and Guillery, 1996; Jones, 2007) that regulates the visual information from the retina that reaches the cortex (Sillito and Kemp, 1983; Norton et al., 1989; Norton and Godwin, 1992; Usrey et al., 1999; Blitz and Regehr, 2005). Previous studies have shown that there is axonal and dendritic release, and that sodium action potentials invade dendrites and axons (Acuna-Goycolea et al., 2008). When interneurons are hyperpolarized, calcium spikes can be generated that can trigger long-lasting plateaus (Antal et al., 2010). In this investigation we expand our understanding of how these interneurons can regulate the flow of information from the retina to the cortex. We describe an output mode in which persistent firing is triggered in interneurons. We show that this persistent firing mode enhances tonic inhibition for tens of seconds following high-frequency RGC stimulation. We also find that high-frequency activation generates long-lasting afterdepolarizations in interneurons that are activated by moderate strength inputs, and when interneurons are hyperpolarized. Thus, mGuR5 activation contributes to afterdepolarizations and persistent firing that allow heightened periods of RGC activity to enhance inhibition for long periods of time.
The mechanisms underlying the different response modes of LGN interneurons are likely similar to those involved in sustained firing of entorhinal cortex pyramidal cells (Egorov et al., 2002) and olfactory bulb Blanes cells (Pressler and Strowbridge, 2006). In the entorhinal cortex and the olfactory bulb a nonselective cation current (known as ICAN and thought to be mediated by TRP channels) generates a sustained depolarization (Egorov et al., 2002; Pressler and Strowbridge, 2006; Zhang et al., 2011). In dLGN interneurons, it is likely that mGluR5 activates TRP channels directly, and NMDARs contribute by generating Ca influx, thereby activating TRP channels and producing a subthreshold afterdepolarization. When the afterdepolarization is sufficiently large it can evoke persistent firing. Finally, if the postsynaptic cell is in an appropriate state, an afterdepolarization is produced that is so large that the LGN interneuron is depolarized to such an extent that the cell is unable to fire action potentials. The depolarization produces sufficient calcium entry to activate TRP channels, and thereby generate a prolonged plateau potential that outlives the subthreshold afterdepolarization. As previously shown, activation of muscarinic M2 receptors on LGN interneurons gates plateau potential firing (Antal et al., 2010). Prolonged plateaus are only generated in a few interneurons (even though all interneurons are likely capable of generating prolonged plateau potentials), because M2-receptor activation is sufficient for only a small fraction of neurons in the quiescent brain slice.
Previous studies have shown that certain kinds of visual input can trigger periods of high-frequency RGC activity (Kuffler et al., 1957; Enroth-Cugell et al., 1966; Barlow and Levick, 1969; Cleland et al., 1973; Mastronarde, 1989; Meister and Berry, 1999). We predict that these high-frequency spike trains would activate interneuron iGluRs and mGluR5s. iGluR activation would generate fast spiking and Ca entry, especially through NMDARs, which would activate TRP channels. mGluR5 activation would also activate TRP channels, and this would depolarize the neuron further, which activates L-type Ca channels. When sufficient RGC axons onto an interneuron are activated the interneuron enters a persistent firing state or a prolonged plateau state, and generates sustained inhibition onto TC cells, either mediated by L-type Ca channels (Antal et al., 2010; Errington et al., 2011) or through a VDCC activated by L-type Ca channels, thereby setting the level of tonic inhibition in the circuit. Even if insufficient inputs are activated to trigger persistent firing, an afterdepolarization would be produced that would enhance fast disynaptic inhibition for tens of seconds. By controlling both tonic inhibition, and fast disynaptic inhibition in this manner, mGluR5 activation could dynamically regulate receptive field size and determine the timing of TC cell output to visual cortex.
Footnotes
We thank members of the Regehr laboratory for their comments on this manuscript, and Kevin Takasaki for assistance with Matlab software. We thank D. Blitz for input on early stages of this project.
- Correspondence should be addressed to Wade G. Regehr, Goldenson 308, Department of Neurobiology, Harvard Medical School, 220 Longwood Avenue, Boston, MA 02115. wade_regehr{at}hms.harvard.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}