Article Text

Abstract
Although the role of transforming growth factor β (TGFβ) in initiating fibrosis is well established, the role that TGFβ plays in maintaining fibrosis is unclear. The gene encoding connective tissue growth factor (ccn2; ctgf), which promotes fibrosis, is not normally expressed in dermal fibroblasts unless TGFβ is present. However, in dermal fibroblasts cultured from lesional areas of scleroderma, ccn2 (ctgf) is expressed constitutively. The contribution of several elements in the ccn2 (ctgf) promoter to basal and TGFβ induced ccn2 (ctgf) expression in normal and scleroderma fibroblasts has been investigated. A functional SMAD binding site in the ccn2 (ctgf) promoter that is necessary for the TGFβ mediated induction of this gene has been identified. The previously termed TGFβ responsive enhancer (TGFβRE) in the ccn2 (ctgf) promoter has been found to be necessary for basal promoter activity in normal fibroblasts. The SMAD element is not necessary for the high ccn2 (ctgf) promoter activity seen in scleroderma fibroblasts. However, mutation of the previously termed TGFβRE reduces ccn2 (ctgf) promoter activity in scleroderma fibroblasts to that seen in normal fibroblasts. Thus, the maintenance of the scleroderma phenotype, as assessed by a high degree of ccn2 (ctgf) promoter activity, appears to be relatively independent of SMAD action and seems to reflect increased basal promoter activity.
- connective tissue growth factor
- scleroderma
- transforming growth factor β
Statistics from Altmetric.com
Scleroderma (systemic sclerosis) is characterised by the progressive scarring of skin and certain internal organs.1 There is currently no effective treatment for scleroderma, and the cause of this disorder is unclear, but probably involves an autoimmune component.2 Because of the ability of transforming growth factor β (TGFβ) to promote fibroblast proliferation and matrix synthesis, attention has been focused on its potential role in initiating and maintaining the fibrotic phenotype (for review, see Blobe and colleagues3), including scleroderma.4 In acute drug or surgery induced animal models of fibrosis the neutralisation of TGFβ reduces the deposition of collagen and tissue fibrosis (for review, see Blobe and colleagues3).
In scleroderma, the importance of TGFβ in the maintenance of fibrosis is less clear—data supporting a role for TGFβ are circumstantial and somewhat contradictory, mainly depending on studies that have examined the histological distribution of TGFβ mRNA and protein. Mononuclear cells taken from bronchial lavage fluids of patients with scleroderma have raised TGFβ values.5 However, in skin lesions, TGFβ mRNA is localised to the leading edge of the scleroderma lesion—that is, the region of enhanced inflammation involved in the initiation of the fibrotic response.6, 8 Furthermore, fibroblasts taken from scleroderma lesions show increased amounts of collagen relative to their normal counterparts, yet show little difference in their ability to produce TGFβ or in their ability to bind TGFβ, and neither do they show enhanced sensitivity to TGFβ treatment.9, 10 In addition, differential display analysis between normal and scleroderma fibroblasts failed to identify genes known to be involved in the TGFβ signalling pathway, such as TGFβ receptors.11 Thus, although some data support the role of TGFβ in the onset of the fibrotic phenotype in scleroderma, its precise role in initiating or maintaining the scleroderma phenotype is unclear.
ccn2 (ctcf) is constitutively expressed in scleroderma fibroblasts
We have focused on identifying those genes playing a role in scleroderma. Using differential display analysis, we showed that connective tissue growth factor (ccn2; ctgf) was upregulated in dermal fibroblasts cultured from patients with scleroderma.11 We estimated that scleroderma fibroblasts in culture synthesise ccn2 (ctgf) mRNA at approximately 10–20 times the concentration seen in fibroblasts cultured from normal skin.11 ccn2 (ctgf) mRNA was also greatly increased in fibroblasts derived from unaffected skin from these patients.11 CCN2 (CTGF), encoded by a member of the CCN gene family,12, 13 is a heparin binding, 38 kDa, cysteine rich peptide that induces proliferation, collagen synthesis, and chemotaxis in mesenchymal cells,13–18 and has been shown to potentiate sustained fibrosis when injected together with TGFβ in an acute animal model.19 ccn2 (ctgf) mRNA and protein are expressed constitutively in numerous fibrotic disorders, both in skin and in internal organs, such as atherosclerosis and pulmonary and renal fibrosis.12, 20–24 In contrast to TGFβ, the expression of ccn2 (ctgf) correlates well with the severity of fibrosis and collagen synthesis in scleroderma.25, 26 Hence, in adult tissues constitutive ccn2 (ctgf) expression is perhaps a more clinically relevant molecular marker of fibrosis and is thought to be a mediator of the fibrotic phenotype.12–24
A SMAD binding element in the ccn2 (ctgf) promoter is necessary for TGFβ mediated induction but not for the increased expression seen in scleroderma fibroblasts
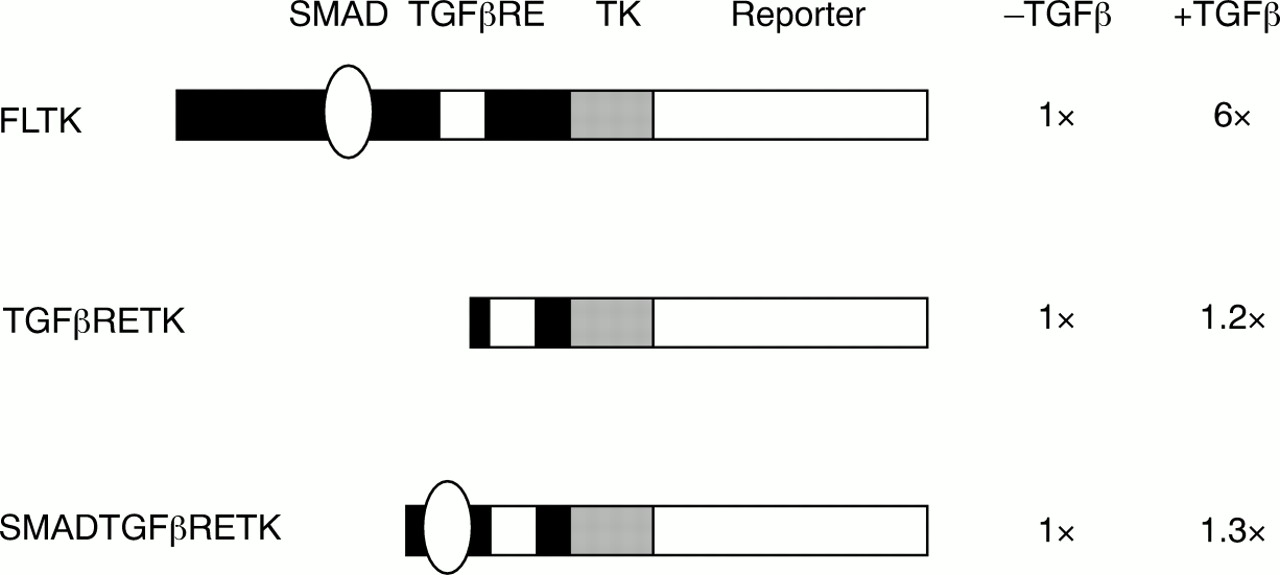
In contrast to the situation in fibrotic disorders, ccn2 (ctgf) is not expressed in fibroblasts unless cells are treated with TGFβ.14, 15, 20, 27 Induction by TGFβ is cell-type specific, because it occurs in connective tissue cells, but not in epithelial cells or lymphocytes.14, 16 The regulation of ccn2 (ctgf) expression by TGFβ appears to be controlled primarily at the level of gene transcription.20, 27 Grotendorst's laboratory identified an element in the ccn2 (ctgf) promoter necessary for the response to TGFβ—the TGFβ responsive enhancer (TGFβRE).27 However, this sequence by itself is insufficient to confer TGFβ responsiveness to a heterologous promoter (fig 1).
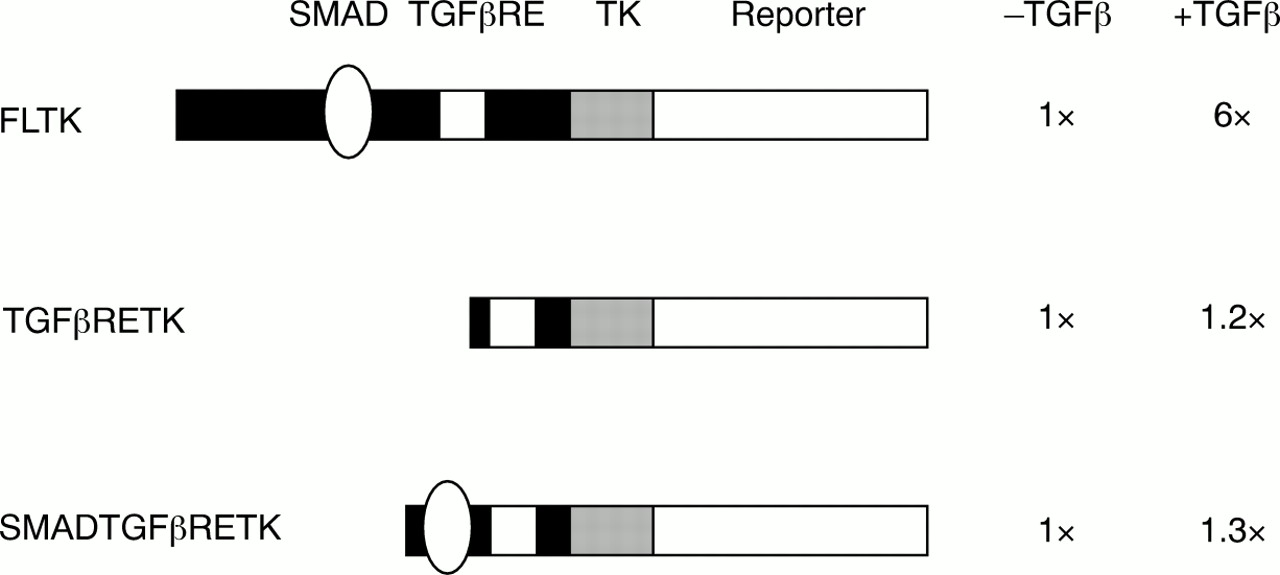
A transforming growth factor β (TGFβ) responsive enhancer (TGFβRE) lies in the ccn2 (ctgf) promoter, but the SMAD binding site and the TGFβRE are insufficient to confer TGFβ responsiveness. Various ccn2 (ctgf) promoter fragments were cloned upstream of a minimal thymidine kinase (TK) promoter–SEAP reporter construct (Clontech, Palo Alto, California, USA). Constructs were transfected into NIH3T3 cells along with a cytomegalovirus (CMV) β galactosidase (Clontech) plasmid to control for transfection efficiencies, and cells were treated for 24 hours with or without TGFβ2.20, 28 Three ccn2 (ctgf) promoter fragments were used: a full length ccn2 (ctgf) promoter fragment (FLTK, containing a fragment between −805 and −50), a 23 bp DNA fragment containing the TGFβRE (TGFβRETK27), and a 55 bp DNA fragment containing the SMAD element and the TGFβRE element (SMADTGFβRETK). Values are expressed relative to the plasmid without TGFβ treatment (N = 6, SEM was less than 25% for all samples). Although the FLTK responds to TGFβ, neither the TGFβRETK nor the SMADTGFβRETK constructs respond to TGFβ, suggesting that there are additional motifs in the ccn2 (ctgf) promoter involved in the TGFβ mediated induction of ccn2 (ctgf).
For this reason, we assessed the ccn2 (ctgf) promoter for additional sequences necessary for the response to TGFβ. TGFβ induction of gene expression has been studied extensively and generally involves the action of members of the SMAD family of proteins.29 Briefly, activation of TGFβ mediated gene expression is frequently mediated through SMAD2, SMAD3, and SMAD4. SMAD2 and SMAD3 are normally present in the cytosol. Once activated by TGFβ, SMAD2 and SMAD3 interact transiently with type I TGFβ receptor kinase and become phosphorylated at the C-terminus. SMAD2 and SMAD3 then form a heteromeric complex with SMAD4, which translocates to the nucleus and activates the expression of target genes. This pathway is negatively regulated by the inhibitory SMADs, SMAD6 and SMAD7. Recent studies have identified a consensus DNA motif, GTCTAGAC, which is a SMAD responsive promoter element to which SMAD3 and SMAD4 bind.30 A sequence similar to the consensus SMAD site is located in the region of the ccn2 (ctgf) promoter immediately upstream of the TGFβRE.28 We found that mutation of the TGFβRE in the context of the full length ccn2 (ctgf) promoter greatly diminishes basal promoter activity but still permits a response to TGFβ.28 Conversely, mutating the putative SMAD binding sequence in the context of an otherwise wild-type, full length CTGF promoter–reporter construct resulted in complete abolition of TGFβ induced gene expression.28 In addition, gel shift analysis showed that SMAD3 and SMAD4 bound to this sequence.28 Cotransfection of SMAD3 and SMAD4 enhanced ccn2 (ctgf) expression significantly, and TGFβ induction of ccn2 (ctgf) did not occur in SMAD3 knockout fibroblasts,28 suggesting that TGFβ induction of ccn2 (ctgf) occurred in a SMAD2 independent fashion. Transfection of the inhibitory SMAD (SMAD7) greatly attenuated the ability of TGFβ, SMAD3, and SMAD4 to increase ccn2 (ctgf) promoter activity, but had little effect on basal expression.28 Conversely, transfecting the inhibitory SMAD (SMAD6) had no effect on the ability of TGFβ to induce ccn2 (ctgf) promoter activity.28 SMAD2/3/4 activity often requires p300 as a transcriptional cofactor.31, 32 However, we found that transfecting either wild-type or dominant negative p300 had no impact on the TGFβ induction of ccn2 (ctgf).28 Thus, ccn2 (ctgf) gene induction requires p300 independent SMAD activity. Intriguingly, the SMAD binding site and the TGFβRE are themselves insufficient to confer TGFβ responsiveness to a heterologous promoter, suggesting that additional factors are required for the TGFβ induction of ccn2 (ctgf) (fig 1).
Because ccn2 (ctgf) gene expression is SMAD dependent and CCN2 (CTGF) is produced spontaneously by scleroderma fibroblasts in culture, we wanted to assess the contribution of SMAD dependent TGFβ signalling to the high degree of CTGF gene expression seen in scleroderma fibroblasts.11 In these studies, we transfected the full length ccn2 (ctgf) promoter fused to a SEAP reporter gene into normal or scleroderma dermal fibroblasts and compared its expression to that of a ccn2 (ctgf) promoter–SEAP reporter construct lacking either a SMAD binding site or a TGFβRE. We found that mutation of the SMAD site had little effect on the increased ccn2 (ctgf) promoter activity seen in scleroderma (fig 2). However, mutation of the TGFβRE site greatly reduced promoter activity both in scleroderma fibroblasts and in normal fibroblasts28 (fig 2). In fact, mutation of the TGFβRE reduced promoter activity in scleroderma fibroblasts to approximately that seen in normal fibroblasts. Thus, SMAD dependent TGFβ signalling seems to have little to do with the maintenance of the scleroderma phenotype, as assessed by constitutive ccn2 (ctgf) gene expression.

{kind=link}
{kind=link}
The transforming growth factor β (TGFβ) responsive enhancer (TGFβRE) of the ccn2 (ctgf) promoter is required for the increased basal ccn2 (ctgf) promoter activity seen in scleroderma fibroblasts. The figure is adapted from data in Holmes et al.30 Three constructs were transfected into normal dermal and scleroderma dermal fibroblasts: a wild-type (nucleotides −805 to +17) ccn2 (ctgf) promoter–SEAP reporter construct (FL), and otherwise identical constructs that contained a mutated TGFβRE element (TGFβRE) or SMAD element (SMAD).30 Values shown are relative to FL construct expression in normal fibroblasts. The standard error in all cases was less than 25%. Mutating the TGFβRE reduces basal ccn2 (ctgf) promoter activity in normal and scleroderma fibroblasts, whereas mutating the SMAD element has no effect on basal expression.
Other regulators of ccn2 (ctgf) expression in fibroblasts
It is obviously important to identify why ccn2 (ctgf) is overexpressed in scleroderma fibroblasts to identify possible targets for drug treatment. Examining the control of ccn2 (ctgf) expression in normal fibroblasts can provide clues to how overexpression can occur in fibrosis. Several cytokines are known to affect ccn2 (ctgf) gene expression in fibroblasts, and the signalling pathways through which these factors act on ccn2 (ctgf) are beginning to be elucidated.
Recently, we found that the proinflammatory cytokine tumour necrosis factor α (TNFα) blocks the TGFβ induction of CCN2 (CTGF) in normal and scleroderma fibroblasts.20 However, TNFα had little or no impact on basal ccn2 (cgtf) expression in scleroderma, suggesting that the increased concentrations of CCN2 (CTGF) found in this disorder may result, in part, from an insensitivity to this cytokine.20 Other regulators of ccn2 (ctgf) gene expression also exist. For example, ccn2 (ctgf) is induced in fibroblasts by the coagulation protease thrombin33 and by dexamethasone,34 and is suppressed by cAMP17 and prostaglandin E2.35. In addition, ccn2 (ctgf) is induced by vascular endothelial growth factor in endothelial cells,36 cortisol in osteoblasts,37 and WT1 (Wilms's tumour suppressor) in kidney mesangial cells.38 Whether these agents are active in fibroblasts or contribute to fibrosis in these tissues is unknown.
Conclusion
The maintenance of the scleroderma phenotype, as assessed by increased CCN2 (CTGF) concentrations, does not appear to result from SMAD dependent TGFβ signalling.28 The TGFβRE27 is required for basal ccn2 (ctgf) promoter activity in normal fibroblasts and also contributes to the increased activity seen in scleroderma fibroblasts.27, 28 The factor(s) binding to this element are currently under investigation, but appear to be independent of AP-1, cAMP responsive element binding protein, or Sp1, factors known to contribute to TGFβ responses in other contexts, because oligonucleotides containing consensus binding sites for these factors do not compete for protein binding to the TGFβRE in gel shift studies (A Leask and S Sa, 1998, unpublished data). Efforts are under way to identify the factors that bind to the TGFβRE. In addition to the promoter motifs, an element in the 3′ untranslated region of ccn2 (ctgf) mRNA has been shown to act as a transcriptional repressor of ccn2 (ctgf) in NIH3T3 fibroblasts.39, 40 The identity of the protein binding to this sequence is unknown, as is its contribution to fibrosis.
Examining the available evidence in the literature, it appears that in scleroderma the initial expansion of the sclerotic lesion may require the activity of TGFβ, although the maintenance of the sclerotic lesion does not result from TGFβ action per se.6–10, 28 Instead, the maintenance of the scleroderma phenotype might result from the failure to suppress downstream responses to TGFβ, such as the induction of CCN2 (CTGF) and collagen synthesis. Thus, although anti-TGFβ/anti-SMAD strategies might be effective in blocking the onset or progression of fibrosis,3 they may not be effective at reversing fibrosis once it has occurred. Given the known profibrotic activity of CCN2 (CTGF), the high degree of constitutive, TGFβ independent ccn2 (ctgf) expression present in the scleroderma lesion may be a cause of the excessive scarring that is seen in patients with scleroderma. Thus, the identification of the molecular mechanism underlying the overexpression of ccn2 (ctgf) in scleroderma might yield effective methods of reversing the fibrotic process in this disorder.
Acknowledgments
This work was supported in part by NIH grant AR45879, Arthritis Research Campaign (UK), the Raynaud's and Scleroderma Association Trust, and the Nightingale Charitable Trust.
References
Supplementary materials
Publisher's CorrectionThe control of ccn2 (ctgf) gene expression in normal and scleroderma fibroblasts
A Leask, S Sa, A Holmes, X Shiwen, C M Black, and D J Abraham
Figure 2 The transforming growth factor ß (TGFß) responsive enhancer (TGFßRE) of the ccn2 (ctgf)
promoter is required for the increased basal ccn2 (ctgf) promoter activity seen in scleroderma fibroblasts
from this article contains an error.A corrected version of the figure is posted here
This error is much regretted
Footnotes
-
↵* According to the recommendation issued by the nomenclature committee of the CCN Society (J Clin Pathol: Mol Pathol 2001;54:M108), the ctgf gene is referred to as ccn2.