


Abstract
Small molecule inhibitors have proven extremely useful for investigating signal transduction pathways and have the potential for development into therapeutics for inhibiting signal transduction pathways whose activities contribute to human diseases. Transforming growth factor β (TGF-β) is a member of a large family of pleiotropic cytokines that are involved in many biological processes, including growth control, differentiation, migration, cell survival, adhesion, and specification of developmental fate, in both normal and diseased states. TGF-β superfamily members signal through a receptor complex comprising a type II and type I receptor, both serine/threonine kinases. Here, we characterize a small molecule inhibitor (SB-431542) that was identified as an inhibitor of activin receptor-like kinase (ALK)5 (the TGF-β type I receptor). We demonstrate that it inhibits ALK5 and also the activin type I receptor ALK4 and the nodal type I receptor ALK7, which are very highly related to ALK5 in their kinase domains. It has no effect on the other, more divergent ALK family members that recognize bone morphogenetic proteins (BMPs). Consistent with this, we demonstrate that SB-431542 is a selective inhibitor of endogenous activin and TGF-β signaling but has no effect on BMP signaling. To demonstrate the specificity of SB-431542, we tested its effect on several other signal transduction pathways whose activities depend on the concerted activation of multiple kinases. SB-431542 has no effect on components of the ERK, JNK, or p38 MAP kinase pathways or on components of the signaling pathways activated in response to serum.
The TGF-β superfamily is a large family of growth and differentiation factors that regulate a wide variety of cellular processes in many different cell types and biological contexts. Different family members regulate cell proliferation (both positively and negatively), migration, extracellular matrix elaboration, adhesion, survival and differentiation, in both developing embryos and adult organisms, ranging from worms to humans (Whitman, 1998; Massagué and Chen, 2000; Massagué et al., 2000). Aberrant signaling by TGF-β, the prototype of the family, has been implicated in a number of human diseases, including cancer, hereditary hemorrhagic telangiectasia, atherosclerosis, and fibrotic disease of the kidney, liver, and lung (Blobe et al., 2000). In addition, low levels of TGF-β signaling have been implicated in compromised wound healing, and inappropriately high levels of TGF-β signaling are associated with excessive scarring (Roberts and Sporn, 1993).
The mechanism of signaling by TGF-β family members is now understood in some detail. The ligands bring together a type II receptor with a type I receptor, both serine/threonine kinases. The type II receptor phosphorylates and activates the type I receptor in the complex. To date, there are five mammalian type II receptors: TβR-II, ActR-II, ActR-IIB, BMPR-II, and AMHR-II and seven type I receptors (ALKs 1–7;Piek et al., 1999). In most cell types, TGF-β signals through the combination of TβR-II and ALK5 (Piek et al., 1999); in endothelial cells, however, ALK1 acts as a TGF-β type I receptor (Oh et al., 2000). Activin and related ligands signal via combinations of ActR-II or ActR-IIB and ALK4, and BMPs signal through combinations of ALK2, ALK3, and ALK6 with ActR-II, ActR-IIB, or BMPR-II (Piek et al., 1999). AMH signals through a complex of AMHR-II with ALK6 (Gouedard et al., 2000), and nodal has been shown recently to signal through a complex of ActR-IIB and ALK7 (Reissmann et al., 2001).
The signals are transduced to the nucleus primarily through activation of complexes of Smads. Upon activation, the type I receptors phosphorylate members of the receptor-regulated subfamily of Smads at two serines in an SSXS motif at their extreme C termini. This activates them and enables them to form complexes with a common mediator Smad, Smad4 (Piek et al., 1999). Smads 1, 5, and 8 are substrates for ALKs 1, 2, 3, and 6, whereas Smads 2 and 3 are substrates for ALKs 4, 5, and 7 (Piek et al., 1999; Jornvall et al., 2001). The activated Smad complexes accumulate in the nucleus, where they are directly involved in the transcription of target genes, usually in association with other specific DNA-binding transcription factors (Massagué and Wotton, 2000). In addition, TGF-β superfamily members can also induce the activation of all three known MAP kinase pathways, although the mechanism underlying this remains unclear (Massagué and Chen, 2000).
Small molecule inhibitors have been invaluable in other systems for dissecting the mechanisms of signal transduction pathways and understanding the role of individual signaling pathways in different biological processes. In addition, they have the potential to be useful for therapeutic applications (Blake et al., 2000 and references therein). Compounds that specifically inhibit receptor kinases for TGF-β superfamily members would be enormously beneficial for furthering our understanding of the mechanism of signaling and determining which biological processes require these signaling pathways. Compounds that selectively inhibit the receptors for TGF-β, in particular, have the potential to be developed for therapeutic applications in the treatment of fibrosis, late-stage carcinogenesis, atherosclerosis, and excessive scarring (i.e., diseases in which the activity of the TGF-β signaling pathway has been implicated).
A potent inhibitor of ALK5 (SB-431542) has recently been developed that acts as a competitive ATP binding site kinase inhibitor and has been shown to inhibit the in vitro phosphorylation of immobilized Smad3 with an IC50 of 94 nM (compound 14; Callahan et al., 2002). We have now investigated the efficiency of SB-431542 as an ALK5 inhibitor and rigorously tested its specificity. We demonstrate that, of the ALKs, it inhibits the activity of ALK5 and also ALK4 and ALK7, which are very similar to ALK5 in their kinase domains. It does not significantly inhibit any of the other ALKs, which have more divergent kinase domains. Consistent with this, SB-431542 inhibits TGF-β- and activin-induced phosphorylation of Smad2, which is mediated by ALK5 and ALK4, respectively, but not BMP-induced phosphorylation of Smad1, which is mediated by ALKs 2, 3, and 6. To demonstrate the specificity of SB-431542 for ALKs 4, 5, and 7, we have tested its effect on several other signaling pathways whose activities depend on the concerted activation of multiple kinases. SB-431542 had no effect on any of these signaling pathways, demonstrating that it is highly selective for these ALKs.
Materials and Methods
Plasmids.
The following plasmids have been described previously: constitutively active human ALK1, ALK3, ALK4, ALK5, ALK6, and rat ALK7 in mammalian expression vectors (Nakao et al., 1997;Macias-Silva et al., 1998; Pierreux et al., 2000; Jornvall et al., 2001), wild-type human ALK4, ALK5, and ALK7 in mammalian expression vectors (ten Dijke et al., 1994; Jornvall et al., 2001), EF-Flag Mixer and EF-Flag XSmad2 (Germain et al., 2000), EF-LacZ (Bardwell and Treisman, 1994), DE-driven luciferase reporter plasmid (Pierreux et al., 2000), Lex-OP-luciferase (Gineitis and Treisman, 2001), mammalian expression plasmids encoding NLex.ElkC (Marais et al., 1993) and NLex.JunN (Price et al., 1996), EF-MEKK1 and EF-RasV12 (Price et al., 1995), and mammalian expression plasmid encoding Flag-tagged constitutively active MKK3 (Raingeaud et al., 1996). The CAGA12-luciferase reporter gene consists of 12 tandem copies of the “CAGA” Smad binding element (Dennler et al., 1998) upstream of the adenovirus major late promoter driving luciferase gene expression. 3D.A-luciferase was constructed by moving the three SRF binding sites and Xenopus laevis minimal γ-actin promoter from 3D.A-Fos (Mohun et al., 1987) into pGL3 (Promega, Madison, WI). EF-Flag XSmad1 was constructed by subcloningX. laevis Smad1 into EF-Flag (Germain et al., 2000). Constitutively active mouse ALK2 was constructed by subcloning the ALK2 coding sequence containing the Q207D mutation (Armes and Smith, 1997) into an EF expression vector.
Cell Culture, Transfections, Inductions, and Inhibitors.
HaCaT, NIH 3T3, C2C12, and T47D cells were all maintained in DMEM containing 10% FCS. NIH 3T3 cells were transfected using LipofectAMINE (Invitrogen, Carlsbad, CA).
Recombinant human TGF-β1 (PeproTech Inc., Rocky Hill, NJ) was dissolved in 4 mM HCl/1 mg/ml BSA at a concentration of 1 μg/ml and was used at a final concentration of 2 ng/ml. Activin was dissolved in 1 mg/ml BSA in phosphate-buffered saline and used at a concentration of 10 to 20 ng/ml. BMP4 (R & D Systems, Minneapolis, MN) was dissolved in 1 mg/ml BSA in phosphate-buffered saline and used at 20 ng/ml. EGF (R & D Systems) was dissolved in 10 mM acetic acid/0.1% BSA and used at 30 ng/ml. Osmotic shock was performed by incubating cells in 0.7 M NaCl in DMEM for 20 min.
Solid anhydrous SB-431542 was dissolved at a concentration of 10 mM in DMSO. Further dilutions of SB-431542 in DMSO were made so that in all cases, SB-431542 was added to cells from a 1000× stock. U0126 (Promega) was dissolved in DMSO and used at a concentration of 25 μM.
Kinase Assays, Whole-Cell Extracts, Western Blotting, and Transcriptional Assays.
Kinase assays were performed as described previously (Laping et al., 2002). For the Western blots shown in Fig.4, extracts were made using 20 mM HEPES, pH 7.5, 10% glycerol, 400 mM KCl, 2 mM EDTA, 1% Triton, 1 mM dithiothreitol, 25 mM NaF, 25 mM sodium-β-glycerophosphate, 1 mM Na3VO4, and protease inhibitors. For all other Western blots, extracts were made using the whole-cell extraction buffer described previously (Khwaja et al., 1998). Western blotting was performed using standard techniques. The following antibodies were used: monoclonal antibody against Smad2 (which also recognizes Smad3; Transduction Laboratories, Lexington, KY); monoclonal antibody against Smad1 [A4 (Santa Cruz Biotechnology, Santa Cruz, CA), Fig. 1, or MADR1 (Upstate, Inc., Lake Placid, NY), Fig. 4]; polyclonal antibodies against phosphorylated Smad1 and Smad2 (a kind gift from Peter ten Dijke) (Faure et al., 2000); monoclonal antibody against GRB2 (Transduction Laboratories); polyclonal antibodies against pan ERK, pan p38, phosphorylated JNK, phosphorylated p38, and phosphorylated ATF2, and a monoclonal antibody against phosphorylated ERK1 and ERK2 (New England Biolabs UK, Hitchin, UK). Transcriptional assays for luciferase reporter genes were performed as described previously (Pierreux et al., 2000).
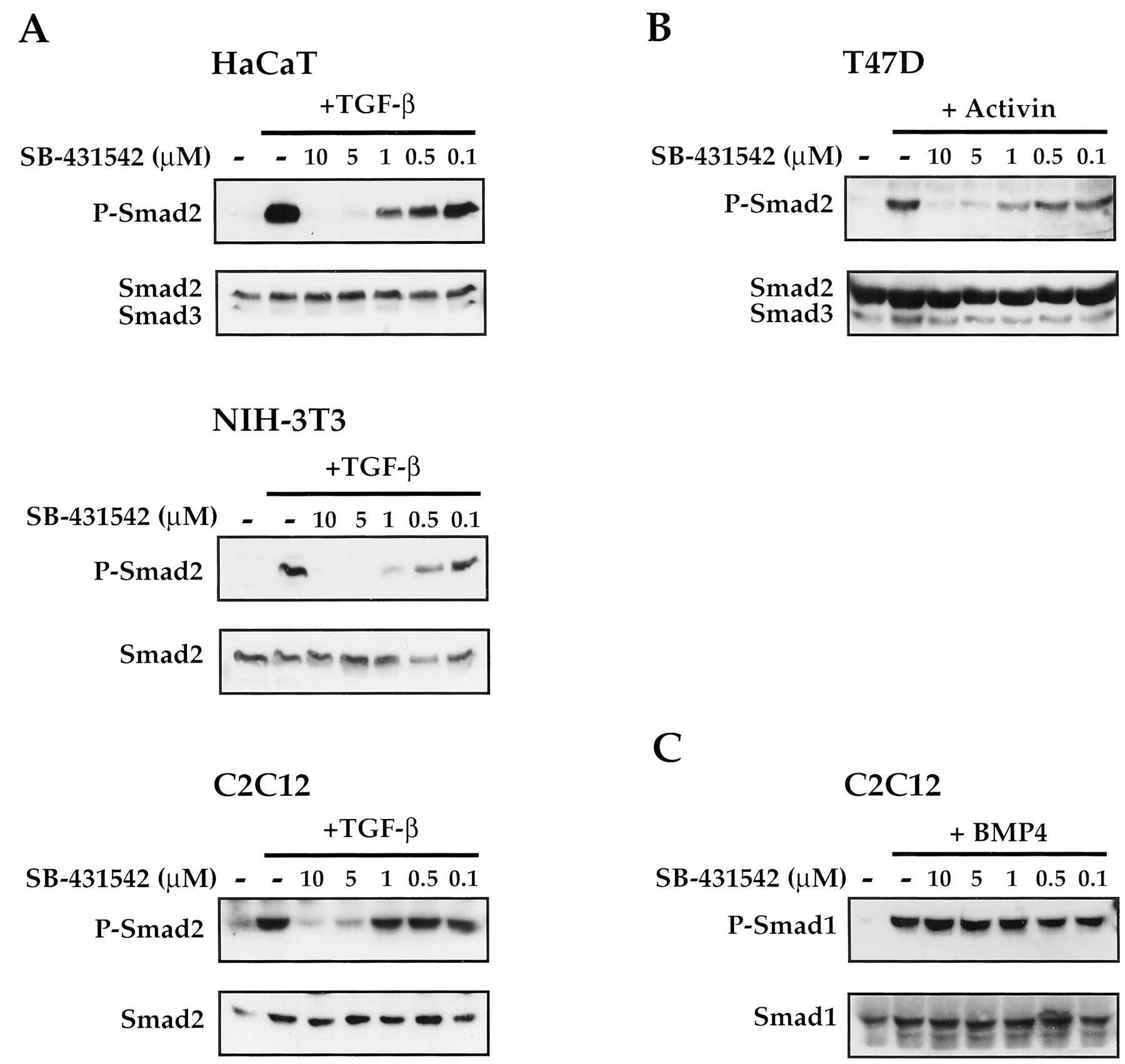
SB-431542 is a potent inhibitor of TGF-β- and activin-induced phosphorylation of Smad2 but not BMP-induced phosphorylation of Smad1. A, HaCaT, NIH 3T3, and C2C12 cells growing in 10% FCS/DMEM were pretreated for 30 min with different concentrations of SB-431542 as indicated and then induced with TGF-β (2 ng/ml) for 1 h. Whole-cell extracts were fractionated by SDS-polyacrylamide gel electrophoresis, and Western blots were performed with an antibody that recognizes activated phosphorylated Smad2 (P-Smad2) or Smad2/Smad3 as a loading control. Note that Smad3 is detectable only in HaCaT cells. B, T47D cells growing in 10% FCS/DMEM were pretreated with SB-431542 as above and then induced with activin (20 ng/ml) for 1 h. Whole-cell extracts were analyzed for phosphorylated Smad2 and Smad2/Smad3 as above. C, C2C12 cells growing in 10% FCS/DMEM were pretreated with SB-431542 as above and then induced with BMP4 (20 ng/ml) for 1 h. Whole-cell extracts were fractionated by SDS-polyacrylamide gel electrophoresis, and Western blots were performed with an antibody that recognizes activated phosphorylated Smad1 (P-Smad1) or Smad1 as a loading control. In A–C, the loading control was performed by stripping the anti-phosphorylated Smad blot and reprobing with an antibody against either Smad2/Smad3 or Smad1, as appropriate, except for the Western blot of extracts from TGF-β-induced C2C12 cells, in which case, duplicate samples were analyzed by Western blotting with the anti-phosphorylated Smad2 antibody or anti-Smad2/3 antibody.

SB-431542 is a potent inhibitor of ALK4, ALK5, and ALK7. A, a phylogenetic tree showing the relationship between human ALK1, ALK2, ALK3, ALK4, ALK5, and ALK6 and rat ALK7 kinase domains (ten Dijke et al., 1994; Jornvall et al., 2001). B, NIH 3T3 cells were transfected with 1.5 μg of plasmids expressing each of the activated versions (ALK*) of ALK1, 2, 3, 4, 5, 6, or 7 together with 1.5 μg of plasmids expressing Smad2 or Smad1. Immediately after the 5-h transfection, SB-431542 was added to the media at a concentration of 10 μM, and cells were incubated for 24 h. Whole-cell extracts were analyzed for phosphorylated Smad2 or phosphorylated Smad1, as appropriate, by Western blotting. Extracts were also blotted for Smad2, Smad1, and GRB2 as loading controls.
Results
SB-431542 Is a Specific Inhibitor of ALK4, ALK5, and ALK7.
SB-431542 was identified as an ALK5 inhibitor (Callahan et al., 2002). Therefore, we first tested whether it was absolutely specific for ALK5 or could inhibit any of the other ALKs, using it initially at a relatively high concentration (10 μM). Seven mammalian type I receptors for TGF-β family members have been cloned (for review, see Piek et al., 1999). Sequence alignment of their kinase domains reveals that they fall into three subclasses: one containing ALK1 and ALK2, one containing ALK4, ALK5, and ALK7, and a third containing ALK3 and ALK6 (Fig. 1A). NIH 3T3 cells were transfected with expression plasmids encoding constitutively activated versions of the ALKs, which have activating point mutations in their glycine- and serine-rich (GS) domains (Wieser et al., 1995). To measure kinase activity of the receptors, cells were cotransfected with either Smad1, which is predominantly phosphorylated by ALK1, ALK2, ALK3, and ALK6, or with Smad2, which is phosphorylated by ALK4, ALK5, and ALK7 (Piek et al., 1999; Jornvall et al., 2001). Smad phosphorylation at the C-terminal “SSXS” motif was detected by Western blotting using antibodies specific for phosphorylated Smad1 or Smad2 (Faure et al., 2000). The levels of Smad1 and Smad2 transfected were monitored with anti-Smad1 or anti-Smad2 antibodies, respectively, and GRB2 was used as a loading control. As expected, activated ALK4, ALK5, and ALK7 all efficiently phosphorylated Smad2 (Fig.1B, top). In all cases, this was inhibited by addition of SB-431542 at a concentration of 10 μM. Constitutively activated ALK1, ALK2, ALK3, and ALK6 efficiently phosphorylated Smad1 in agreement with previous work (reviewed in Piek et al., 1999), and addition of 10 μM SB-431542 did not significantly inhibit their activity except in the case of ALK3, which was weakly affected (Fig. 1B, bottom). We also observed that activated ALK4 and ALK5, but not ALK7, specifically induced phosphorylation of Smad1 (Fig.1B, bottom). This phosphorylation is also clearly dependent on the kinase activities of these receptors, because addition of 10 μM SB-431542 completely abolished this induction of phosphorylated Smad1. We observed no toxic effects of SB-431542 on the cells as measured by cell numbers during the course of the experiment.
The effects of 10 μM SB-431542 were also tested on a selection of the ALKs in vitro (Table 1). In agreement with the results obtained in tissue culture cells, the activity of ALK4 was substantially inhibited (88% inhibition) by this concentration of SB-431542. The BMP-regulated ALKs, however, were either not affected at all (ALK2) or weakly affected (ALK6). We can therefore conclude that SB-431542 is a specific inhibitor for ALK4, ALK5, and ALK7.
Effect of SB-431542 on the activities of protein kinases in vitro
SB-431542 Inhibits the Ability of Activated ALK4, ALK5, and ALK7 to Induce Transcription of Reporter Genes.
Once activated, the R-Smads form complexes with Smad4, which accumulate in the nucleus, where they are directly involved in the transcription of target genes (Massagué and Chen, 2000). We have shown that SB-431542 efficiently inhibits the ability of activated ALK4, ALK5, and ALK7 to phosphorylate Smad2. We next investigated whether it was sufficient to inhibit the ability of these receptors to mediate Smad-dependent transcription. Two different luciferase reporter genes were used for this analysis. One is driven by four copies of the DE from the X. laevis goosecoid promoter. We have shown previously that the paired-like homeodomain transcription factor, Mixer, directly recruits an activated complex of Smad2 and Smad4 to this promoter in response to TGF-β in NIH 3T3 cells (Germain et al., 2000). The other reporter (CAGA12-luciferase) drives luciferase from the adenovirus major late promoter under the control of 12 tandem copies of the CAGA Smad binding element (Dennler et al., 1998). These “CAGA boxes” specifically bind Smad3 and Smad4, and transcription is thought to be mediated by these complexes (Dennler et al., 1998). NIH 3T3 cells were transiently transfected with either DE-luciferase and Mixer or CAGA12-luciferase together with one of the constitutively activated ALKs (ALK4, ALK5, or ALK7). Immediately after transfection, cells were treated with DMSO alone or different concentrations of SB-431542 inhibitor dissolved in DMSO (Fig. 2). For the DE-luciferase assays, substantial activation was seen when DE-luciferase was cotransfected with Mixer and any of the activated receptors (Fig. 2, left), consistent with the ability of Mixer to recruit activated Smad2/Smad4 complexes to the DE to activate transcription (Germain et al., 2000). In all cases, addition of DMSO slightly increased the induced level. All the activated receptors were inhibited by addition of SB-431542. The concentrations of inhibitor required to reduce the induced level of transcription by half (IC50) were 1 μM for activated ALK4, 0.75 μM for activated ALK5, and 2 μM for activated ALK7. Similar results were obtained for the CAGA12-luciferase reporter assays (Fig. 2, right). Coexpression of any of the activated ALKs led to a large increase in transcription mediated by the CAGA sites consistent with the ability of these sites to bind activated complexes of Smad3 and Smad4 (Dennler et al., 1998). Addition of SB-431542 again inhibited all three receptor kinases. The concentrations required to reduce activated transcription by half were 0.75 μM for activated ALK4, 0.5 μM for activated ALK5, and between 1 and 2 μM for activated ALK7. SB-431542 had no inhibitory effects on transcription from the internal control plasmid EF-LacZ, indicating that it was specific for transcription mediated by the activated ALKs (see also below).
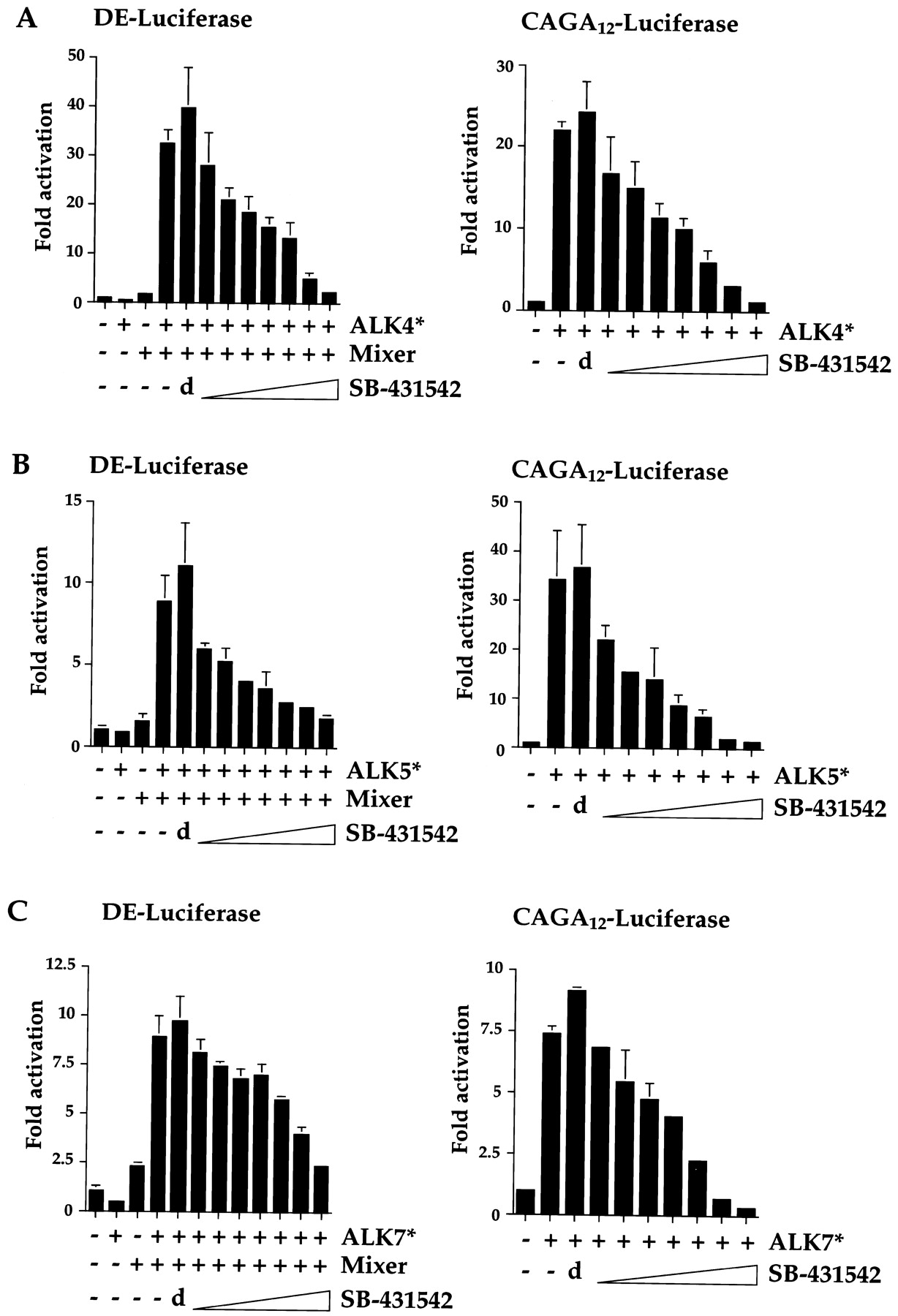
SB-431542 effectively inhibits the ability of ALK4, ALK5, and ALK7 to induce transcriptional activation. NIH 3T3 cells were transiently transfected with 100 ng of DE-luciferase and 50 ng of a plasmid expressing Flag-tagged Mixer (left) or with 100 ng CAGA12-luciferase (right), with 150 ng of plasmids expressing constitutively active ALK4 (ALK4*; A), ALK5 (ALK5*; B), or ALK7 (ALK7*; C) and with 25 ng of EF-LacZ as an internal control. Immediately after transfection, cells were incubated for 24 h in 10% FCS/DMEM containing DMSO alone (d) or 0.25, 0.5, 0.75, 1, 2, 5, or 10 μM SB-431542. Luciferase activity was measured and normalized to β-Gal activity, and fold activations relative to untreated and uninduced samples were determined. The data are representative experiments performed in triplicate and are displayed as mean and S.D.
Thus, SB-431542 specifically inhibits the ability of activated ALK4, ALK5, and ALK7 to induce both Smad2/Smad4- and Smad3/Smad4-dependent transcription. We have demonstrated that it is most effective as an inhibitor of ALK5, because the IC50 for ALK5 is the lowest. The IC50 for ALK4 is slightly higher, and that for ALK7 is higher still. This is consistent with the observation that the ALK4 kinase domain is more closely related to that of ALK5 than the kinase domain of ALK7 is (Fig. 1A).
SB-431542 Effectively Inhibits the Activity of Ligand-Activated ALK4 and ALK5.
Until now, all the assays of the potency and specificity of SB-431542 have made use of constitutively activated ALKs (Figs. 1 and 2; Callahan et al., 2002). For this inhibitor to be useful in vivo, it is clearly important that it efficiently inhibit ligand-induced wild-type ALKs. We therefore investigated its effect on activin-induced ALK4 and on the endogenous TGF-β type I receptor in NIH 3T3 cells, which we presume to be ALK5 because it is the only known TGF-β type I receptor expressed in fibroblasts.
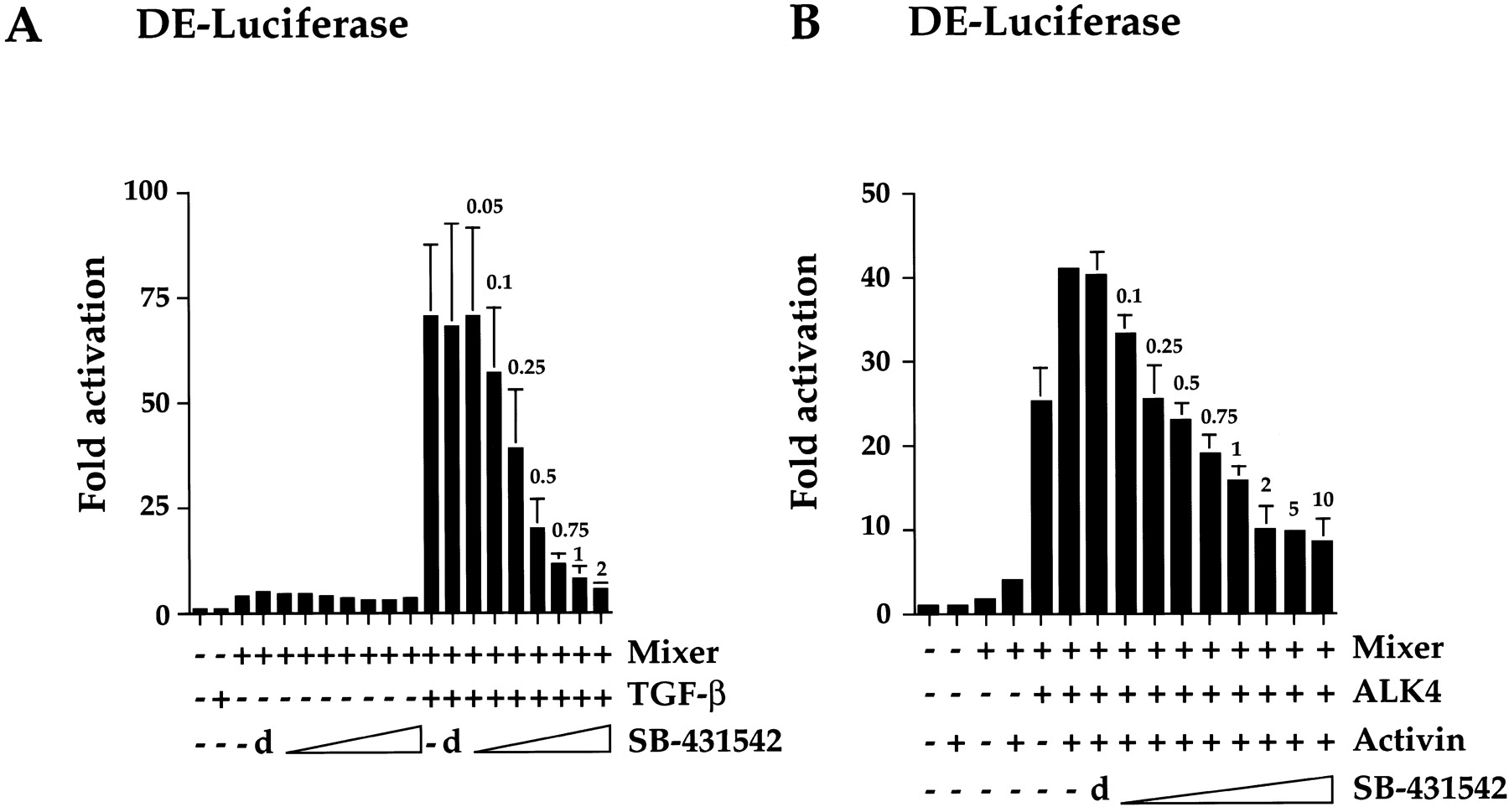
To investigate the effect of SB-431542 on endogenous ALK5, NIH 3T3 cells were transfected with DE-luciferase in the presence or absence of Mixer. Cells were incubated for 24 h then pretreated with either DMSO or different concentrations of SB-431542. After 15 min, cells were either treated or not treated with TGF-β for a further 8 h. In the absence of TGF-β signaling, Mixer induces a very low level of transcriptional activation that is completely unaffected by the inhibitor (Fig. 3A). Upon TGF-β stimulation, Mixer confers very strong transcriptional activation, which is inhibited efficiently by SB-431542. We presume that this is mediated by endogenous ALK5. Half-maximal inhibition occurs at approximately 0.25 μM SB-431542, and total inhibition occurs at a 2-μM SB-431542 concentration (Fig. 3A). This is slightly lower than the amount required to inhibit overexpressed activated ALK5, probably because in this case, the receptor is not overexpressed. We also tested the stability of SB-431542 using this assay and demonstrated that the inhibitor was as effective when added 60 h before TGF-β as it was when added 15 min before (data not shown).

SB-431542 effectively inhibits the activity of ligand-induced ALK4 and ALK5. NIH 3T3 cells were transfected with 100 ng of DE-luciferase reporter gene and 50 ng of a plasmid expressing Flag-tagged Mixer with 100 ng of EF-LacZ as an internal control either alone (A) or together with 250 ng of a plasmid expressing wild-type ALK4 (B) as indicated. Twenty-four hours after transfection, cells were pretreated for 15 min with DMSO (d) or the concentrations of SB-431542 (micromolar) as indicated above the error bars, followed by inductions for 8 h with either TGF-β (A), which signals through endogenous ALK5 receptors, or activin (B), which signals through the transfected ALK4 receptors. Luciferase activity was measured and normalized to β-Gal activity, and fold activations relative to untreated and uninduced samples were determined. A, data are the averages of three independent experiments. B, data are from a representative experiment performed in triplicate and are displayed as mean and S.D.
We tested the effect of the inhibitor on ligand-induced wild-type ALK4 using the same reporter system (Fig. 3B). Overexpression of wild-type ALK4 alone was sufficient to induce transcription of the DE-luciferase reporter gene in the presence of Mixer, probably because of its ability to form ligand-independent complexes with endogenous type II receptors (Attisano et al., 1996). Treatment of the cells with activin enhanced the level of transcriptional activation. SB-431542 efficiently inhibited the activity of activin-induced ALK4, although ALK4-dependent transcription was never completely abolished, even at 10 μM SB-431542. This suggests that the receptor complex may activate weak DE-luciferase reporter gene expression through a mechanism independent of ALK4 kinase activity. Thus, we demonstrate that SB-431542 is a potent inhibitor of ligand-induced ALK4 and ALK5.
SB-431542 Efficiently Inhibits Smad Phosphorylation Induced by TGF-β and Activin But Not BMP4.
Our data indicate that SB-431542 is an effective inhibitor of ALK4, ALK5, and ALK7 but not of ALK1, ALK2, ALK3, or ALK6. Given the ligand specificity of these ALKs, we would expect activin, nodal, and TGF-β responses to be inhibited by SB-431542 but not BMP-induced responses (Massagué et al., 2000;Oh et al., 2000; Reissmann et al., 2001). We investigated this directly by testing the ability of SB-431542 to inhibit TGF-β-, activin-, or BMP4-induced Smad phosphorylation (Fig. 4). In this case, we were assaying the ability of endogenous receptors to phosphorylate endogenous Smads. We tested three different cell lines that respond to TGF-β: the human keratinocyte cell line HaCaT, NIH 3T3 fibroblasts, and the myoblast cell line C2C12 (Hanafusa et al., 1999; Pierreux et al., 2000). In all cases, a high level of phosphorylated Smad2 was detected in response to a 1-h treatment with TGF-β (Fig. 4A). This was completely inhibited in all cases by preincubation with 5 μM SB-431542 and very substantially inhibited, particularly in NIH 3T3 and HaCaT cells, by 1 μM SB-431542. We tested the ability of SB-431542 to inhibit endogenous activin responses using the breast cancer cell line T47D (Cocolakis et al., 2001). These cells respond efficiently to activin as seen by the robust induction of phosphorylated Smad2 (Fig.4B). This was inhibited by pretreatment with SB-431542; a concentration of 5 μM almost completely abolishes the induction. To measure the effect of SB-431542 on an endogenous BMP response, we tested its effect on BMP-induced phosphorylation of Smad1 in C2C12 cells. Levels of phosphorylated Smad1 were dramatically induced upon BMP treatment, and this was not affected by pretreatment of the cells with SB-431542 (Fig.4C). Thus, as predicted from the specificity of SB-431542 for the different ALK family members, SB-431542 is a potent inhibitor of endogenous TGF-β and activin signaling pathways but has no effect on the BMP signaling pathway.
SB-431542 Has No Effect on the Activation of MAP Kinases by Extracellular Signals.
Because SB-431542 is a potent and selective inhibitor of ALK4, ALK5, and ALK7, it has the potential to be a useful inhibitor to dissect these signaling pathways in vivo and enable us to understand the biological processes in which these signaling pathways are involved. For this to be successful, it is obviously important to have a detailed knowledge of the specificity of this compound with regard to other cellular protein kinases. To address this, we first assayed its effects in vitro on a panel of 24 kinases unrelated to the ALKs (Table 1) and demonstrated that the only kinase to be significantly affected in vitro was p38 MAPKα.
We next extended this analysis to a tissue culture system. We assayed the effects of SB-431542 on a selection of well-characterized signal transduction pathways that function through the concerted activation of a series of protein kinases.
The growth factor EGF activates the MAP kinases ERK1 and ERK2 through a signal transduction pathway that depends on the kinase activities of the EGF receptor, the MAP kinase kinase kinase, Raf, and the MAP kinase kinases MEK1 and MEK2 (Bogdan and Klambt, 2001). We tested the effect of SB-431542 on EGF stimulation of ERK1 and ERK2 in NIH 3T3 cells, measuring their activation by the appearance of phosphorylated ERK1 and ERK2 using a Western blot assay. ERK1 and ERK2 are rapidly phosphorylated after 1-min treatment of cells with EGF, and this induction was completely unaffected by pretreatment of the cells with 10 μM SB-431542 (Fig. 5A). It was, however, completely inhibited by addition of the MEK1/2 inhibitor, U0126 (Favata et al., 1998).
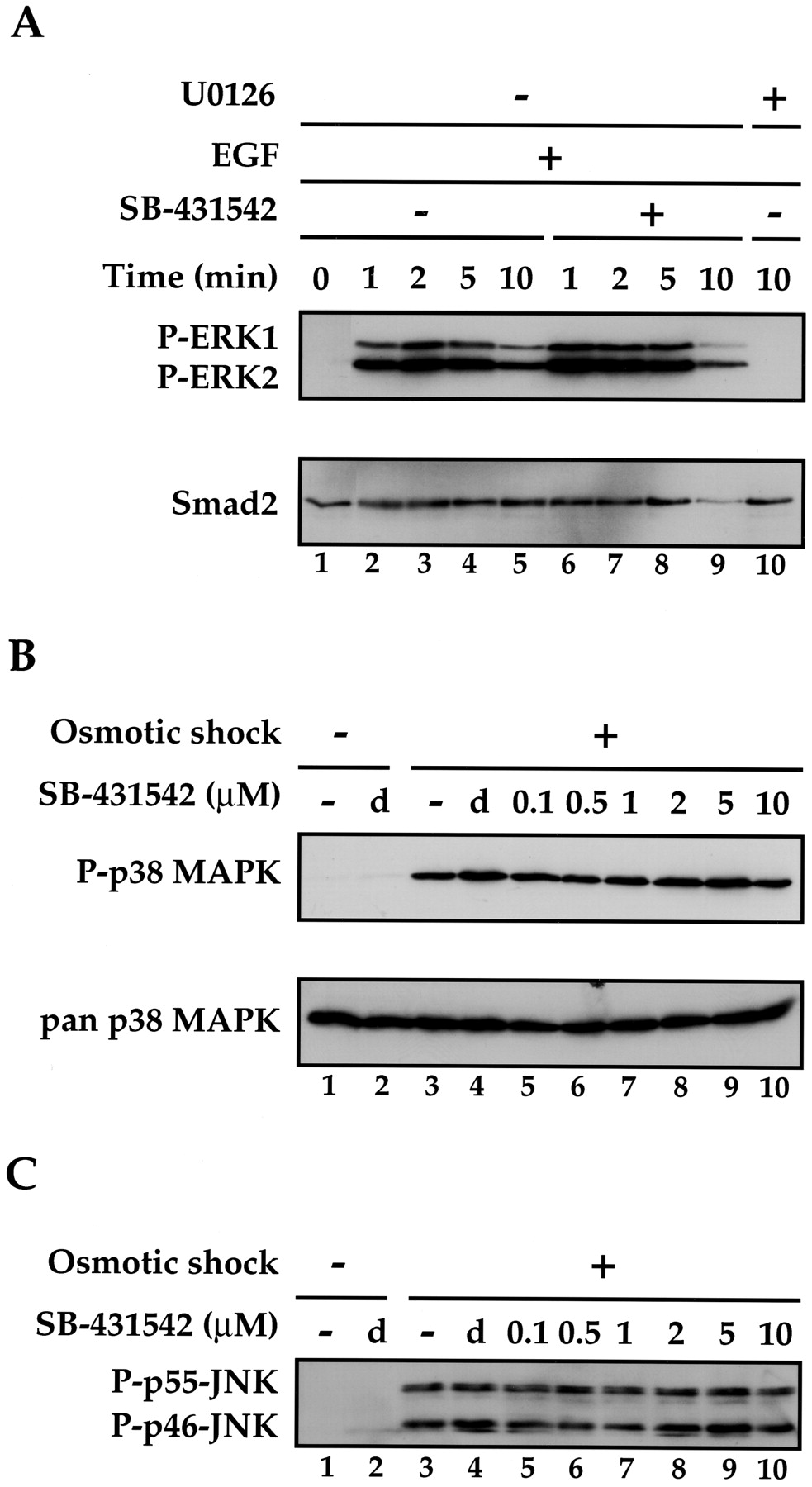
SB-431542 has no effect on the activation of MAP kinases by extracellular signals. A, SB-431542 has no effect on the EGF-induced activation of ERK1 and ERK2. NIH 3T3 cells were serum-starved by incubation in DMEM/0.5% FCS for 24 h. Cells were pretreated with DMSO (lanes 1–5) or 10 μM SB-431542 for 15 min and then induced with EGF (30 ng/ml) for the times shown. The sample in lane 10 was pretreated with the MEK1/2 inhibitor U0126 (25 μM) before addition of EGF. Whole-cell extracts were prepared, which were analyzed for phosphorylated activated ERK1 and ERK2 (P-ERK1 and P-ERK2) and Smad2 as a loading control. B, SB-431542 has no effect on osmotic shock-induced activation of p38 MAP kinase. NIH 3T3 cells were serum-starved by incubation in DMEM/0.5% FCS for 24 h. Cells were pretreated with DMSO (d) or with the concentrations of SB-431542 (micromolar) as indicated for 15 min and then induced with 0.7 M NaCl (osmotic shock) for 20 min. Whole-cell extracts were prepared, which were analyzed for phosphorylated activated p38 (P-p38 MAPK) and pan p38 as a loading control. C, SB-431542 has no effect on osmotic shock-induced activation of JNKs. The Western blot from B (top) was stripped and reprobed with an antibody that recognizes activated phosphorylated JNK (both the p46 and p55 isoforms of both JNK1 and JNK2; Davis, 2000).
The p38 MAP kinases can be activated by osmotic shock. This involves activation of MAP kinase kinase kinases, of which several different families are known: the MEKK group (MEKK1–4), the mixed lineage protein kinase group (MLK1, MLK2, MLK3, DLK, and LZK), the ASK group (ASK1 and ASK2), and TAK1 and TPL2 (Davis, 2000). It is not clear, however, exactly which of these are induced as a result of osmotic stress. These kinases activate MKK3 and MKK6, which in turn activate the four p38 MAP kinase isoforms (p38α, p38β, p38γ, and p38δ; Nebreda and Porras, 2000). NIH 3T3 cells were subjected to osmotic shock by incubation in 0.7 M NaCl for 20 min, which led to a strong induction of phosphorylated p38 MAP kinase as detected by Western blotting with an antibody that specifically recognizes phosphorylated p38 isoforms (Fig. 5B). This induction was completely unaffected by pretreatment of the cells with any concentration of SB-431542 tested (up to 10 μM).
The JNKs are also activated in response to osmotic stress through a pathway that involves some or all of the MAP kinase kinase kinases listed above and the MAP kinase kinase MKK4 (Davis, 2000). To test the effect of SB-431542 on the activity of these kinases, the same Western blot of extracts from osmotically shocked cells that had been probed with the anti-phosphorylated-p38 antibody was stripped and reprobed with an antibody that recognizes the activated phosphorylated p46 and p55 isoforms of both JNK1 and JNK2 (Fig. 5C). The induction of these phosphorylated JNKs in response to osmotic shock was completely unaffected by pretreatment of the cells with up to 10 μM SB-431542. SB-431542, therefore, does not inhibit any kinase on the pathways through which EGF activates ERK1/ERK2 or osmotic shock activates p38 or JNK.
SB-431542 Has No Effect on ATF2 Phosphorylation in Response to Osmotic Shock.
The only kinase unrelated to the ALKs of which activity was found to be inhibited by SB-431542 was the MAP kinase p38α, although the IC50 for p38α in vitro was 10 μM compared with 94 nM for ALK5 (Table 1; Callahan et al., 2002). We therefore tested whether the activity of the p38 isoforms in NIH 3T3 cells was inhibited by doses of SB-431542 up to a concentration of 10 μM. As demonstrated above, p38 and JNK MAP kinases are activated in response to environmental stresses, such as osmotic shock, and both of these kinases phosphorylate the transcription factor ATF2 on threonines 69 and 71 in its N-terminal activation domain (Raingeaud et al., 1996). To examine whether SB-431542 had any inhibitory activity on p38 or JNK, we tested whether it could inhibit the phosphorylation of endogenous ATF2 in response to osmotic shock.
NIH 3T3 cells were pretreated with various concentrations of SB-431542 (0.1–10 μM) and then stimulated by osmotic shock. Extracts were assayed for ATF2 phosphorylation by Western blotting with an antibody specific for the phosphorylated form, and GRB2 was used as a loading control. The phosphorylation of ATF2 by endogenous JNK and p38 MAP kinases was completely unaffected by any concentration of SB-431542 tested (Fig. 6).

SB-431542 has no effect on the phosphorylation of ATF2 in response to osmotic shock. NIH 3T3 cells were serum-starved by incubation in DMEM/0.5% FCS for 24 h. Cells were pretreated with DMSO (d) or with the concentrations of SB-431542 (micromolar) as indicated for 15 min and then induced with 0.7 M NaCl (osmotic shock) for 20 min. Whole-cell extracts were prepared, which were analyzed for phosphorylated activated ATF2 and GRB2 as a loading control.
SB-431542 Has No Effect on the Ability of the Ras-ERK, JNK, or p38 MAP Kinase Pathways or Serum-Induced Signaling Pathways to Activate Transcription.
We then extended our analysis of the specificity of SB-431542 by testing its effect on transcriptional activation mediated by either the Ras-ERK, JNK, or p38 MAP kinase pathways. These MAP kinases phosphorylate and activate transcription factors in the nucleus. Their ability to activate transcription through the appropriate transcription factors is therefore a convenient assay of their activity. The ERK MAP kinases phosphorylate the C-terminal domain of the Ets transcription factor, Elk-1 (Marais et al., 1993), the JNK MAP kinases phosphorylate and activate the N-terminal domain of c-Jun (Derijard et al., 1994), and p38 MAP kinases also phosphorylate and activate Elk-1 in addition to ATF2, which was examined above (Price et al., 1996). Fusions of the relevant regions of Elk-1 and c-Jun with the bacterial LexA repressor (NLex.ElkC and NLex.JunN) mediate transcriptional activation of a reporter gene driven by LexA operators in response to the appropriate signals (Price et al., 1996).
Cotransfection of activated Ras (RasV12) with NLex.ElkC and the Lex-OP-luciferase reporter resulted in extremely high transcriptional activity (Fig. 7A). Addition of SB-431542 up to a 10-μM concentration had no effect on this transcriptional activation, although it was completely inhibited by addition of 25 μM U0126, which is a specific inhibitor of MEK1 and MEK2 (Favata et al., 1998). Activated MEKK1 induced transcription of the Lex-OP-luciferase reporter, mediated by NLex.JunN, through its ability to activate JNK (Price et al., 1996). This was completely unaffected by addition of SB-431542 at concentrations up to 10 μM (Fig. 7B). In addition, activated MKK3 induced transcription of the Lex-OP-luciferase reporter, mediated by NLex.ElkC, through its ability to activate p38 MAP kinase (Raingeaud et al., 1996). This also was completely unaffected by addition of SB-431542 at concentrations up to 10 μM (Fig. 6C).

SB-431542 has no effect on the ability of the Ras-ERK, JNK, or p38 MAP kinase pathways or serum-induced signaling pathways to activate transcription. A, NIH 3T3 cells were transiently transfected with 100 ng of Lex-OP-luciferase, 50 ng of NLex.ElkC, and 200 ng of EF-LacZ as above with or without 100 ng of a plasmid expressing activated Ras. After transfection, cells were incubated in 0.5% FCS/DMEM containing DMSO alone (d) or the indicated concentrations of SB-431542 (micromolar) or the MEK1/2 inhibitor U0126 (25 μM) for 24 h. Luciferase activity was then measured and normalized to β-Gal activity, and fold activations relative to untreated and uninduced samples were determined. B, NIH 3T3 cells were transiently transfected with 100 ng of Lex-OP-luciferase, 20 ng of NLex.JunN, and 50 ng of EF-lacZ with or without 330 ng of a plasmid expressing activated MEKK1. After transfection, cells were incubated in 0.5% FCS/DMEM containing DMSO alone (d) or the indicated SB-431542 concentrations (micromolar) for 24 h. Luciferase assays were performed and quantitated as above. C, NIH 3T3 cells were transiently transfected with 100 ng of Lex-OP-luciferase, 50 ng of NLex.ElkC, and 100 ng of EF-LacZ with or without 350 ng of a plasmid expressing activated MKK3. After transfection, cells were incubated in 0.5% FCS/DMEM containing DMSO alone (d) or the indicated SB-431542 concentrations (micromolar) for 24 h. Luciferase assays were performed and quantitated as above. D, NIH 3T3 cells were transiently transfected with 100 ng of Lex-OP-luciferase together with 50 ng of NLex.ElkC and 350 ng of EF-LacZ as an internal control. After transfection, cells were incubated in 0.5% FCS/DMEM containing DMSO alone (d) or the indicated SB-431542 concentrations (micromolar) or the MEK1/2 inhibitor U0126 (25 μM) for 16 h. Media was then removed and replaced with either 0.5% FCS/DMEM (−) or 10% FCS/DMEM (+ serum) again containing DMSO or concentrations of SB-431542 (micromolar), as indicated above the error bars, or U0126. After a further 8-h incubation, luciferase activity was measured and quantitated as above. E, NIH 3T3 cells were transiently transfected with 450 ng of 3D.A-luciferase, which is driven by three SRF binding sites from the c-fos SRE upstream of a minimal γ-actin promoter (Hill et al., 1995), and 50 ng of EF-LacZ as an internal control. Cells were treated after transfection and induced or not induced with serum exactly as in D. Luciferase assays were performed and quantitated as above. The data are from representative experiments performed in triplicate and are displayed as mean and S.D.
Finally, we tested the effect of SB-431542 on serum-induced signaling pathways. Serum activates transcription at the c-fos serum response element (SRE) via two distinct signaling pathways (Hill et al., 1995). One is mediated by the Ras-ERK MAP kinase pathway and targets the ternary complex factors, such as Elk-1, that are recruited to the SRE by the transcription factor SRF (Hill et al., 1993; Marais et al., 1993). The other signaling pathway targets SRF and requires RhoA and actin cytoskeleton reorganization (Hill et al., 1995;Sotiropoulos et al., 1999). This is thought to involve the activity of both LIM kinase and also the RhoA effector kinase ROCK (Gineitis and Treisman, 2001). We could demonstrate that serum induction of NLex.ElkC-mediated transcription of the Lex-OP-luciferase reporter gene was not affected by pretreatment of cells with any concentration of SB-431542 assayed (Fig. 7D). As expected, this transcriptional activation was completely inhibited, however, by the MEK inhibitor U0126. The serum-induced RhoA-dependent pathway is readily assayed using a reporter gene, which is driven by three SRF binding sites upstream of a minimal γ-actin promoter (Hill et al., 1995). Serum-induced transcription of the reporter gene (3D.A-luciferase) was completely unaffected by pretreatment of cells with SB-431542 (Fig.7E).
Taken together with the data in the previous section, we can demonstrate that SB-431542 has no effect in vivo on any of the kinases unrelated to the ALKs that we have tested. We therefore conclude that it is highly selective for the ALKs.
Discussion
SB-431542 Is a Selective Inhibitor for ALK4, ALK5, and ALK7.
SB-431542 was identified as an inhibitor of ALK5. Here, we demonstrated that in addition to ALK5, SB-431542 also inhibits the activity of ALK4 and ALK7. It has no significant effect, however, on the kinase activity of the other known ALKs (ALKs 1, 2, 3, and 6). These results can easily be rationalized by analysis of the sequences of the kinase domains of these ALKs. ALK4, ALK5, and ALK7 form a subclass of ALKs; the kinase domains of ALK4 and ALK5 are 89.3% identical, and those of ALK5 and ALK7 are 82.4% identical. The kinase domains of the other ALKs, however, are all less than 68% identical to that of ALK5.
Consistent with the selectivity observed for the different ALKs, SB-431542 efficiently inhibits the endogenous TGF-β and activin signaling pathways in vivo and has no effect on BMP signal transduction. Because SB-431542 also effectively inhibits ALK7 activity, we anticipate that it will inhibit endogenous nodal signaling, which has recently been shown to operate through ALK7 (Reissmann et al., 2001).
We have shown herein that inhibition of ALK4 or ALK5 kinase activity is sufficient to inhibit phosphorylation of Smad2 and Smad3 in response to activin or TGF-β. Because SB-431542 is the first soluble ALK4/ALK5 small molecule inhibitor to be described, we have been able to demonstrate here, for the first time, that ALK5 kinase activity is required for Smad2 phosphorylation and Smad-dependent transcriptional activation in vivo without the necessity for overexpression of wild-type or mutant receptors. In addition to the work described here, we have gone on to use this compound to dissect the TGF-β signaling pathway in detail (F. J. Nicolás, G. J. Inman, A. D. Reith, N. J. Laping, and C. S. Hill, manuscript in preparation).
Specificity of SB-431542.
For SB-431542 to be useful in vivo, it is essential to determine whether, in addition to the ALKs, it also inhibits any other cellular kinases. We report its inhibitory activity against a panel of kinases in vitro (Table 1). The only kinase of this panel that SB-431542 weakly inhibits is the MAP kinase p38α, for which it has an IC50 of 10 μM in vitro. Because this compound acts as a competitive ATP-binding inhibitor, we conclude that the ATP-binding pocket of ALK5 and p38α must be similar. Consistent with this idea, several known p38 inhibitors also inhibit ALK5, albeit with higher IC50 values (Laping et al., 2002), and the compounds that were isolated in the screen for ALK5 inhibitors were originally identified as p38 MAP kinase inhibitors (Callahan et al., 2002). The ability of SB-431542 to weakly inhibit p38 MAP kinase isoforms should not compromise the use of this inhibitor in vivo, because the IC50 for ALK5 in vitro is ∼100 times lower than that for p38α (94 nM versus 10 μM, respectively). In fact, we have demonstrated that for the endogenous ALK5 receptors, complete inhibition is observed at 2 μM SB-431542, and no effects on endogenous p38 MAP kinase activity were seen even when the inhibitor was used at a 10-μM concentration.
We have extended this analysis of the specificity of SB-431542 by assaying its effects on a variety of signal transduction pathways that depend on the concerted activation of multiple kinases. In these assays, the inhibitor was used up to a concentration of 10 μM. We demonstrate that SB-431542 has no effect on the ability of growth factors, such as EGF, to induce ERK MAP kinases or stress stimuli to induce JNK or p38 MAP kinases. In addition, we demonstrate that it has no effect on the ability of these three MAP kinase pathways to phosphorylate transcription factors in the nucleus and thereby regulate gene expression. It also has no effect on either of the major signaling pathways by which serum activates transcription via the SRE.
Thus, from this extensive analysis of endogenous serine/threonine kinases, SB-431542 seems to be selective for a subset of the ALKs, although further work is obviously needed to rule out its effects on all other cellular kinases. We anticipate that it will prove an invaluable tool for understanding the role of TGF-β, activin, and nodal signaling in many diverse biological contexts.
Acknowledgments
We thank Roger Davis (University of Massachusetts Medical School, Worcester, MA) for a mammalian expression plasmid encoding activated MKK3, Jon Graff (University of Texas Southwestern Medical Center, Dallas, TX) for XSmad1 and XSmad2, Carlos Ibáñez (Karolinska Institute, Stockholm, Sweden) for wild-type and activated ALK7 expression plasmids, Jim Smith (Wellcome/Cancer Research UK Institute, Cambridge, UK) for an activated ALK2 construct, Peter ten Dijke (The Netherlands Cancer Institute, Amsterdam, the Netherlands) for anti-phospho-Smad2 and anti-phospho-Smad1 antibodies, activated ALK1, -3, -4, -5, and -6 expression plasmids, and CAGA12-MLP-LUC reporter, and Richard Treisman (Cancer Research UK London Research Institute, London, UK) for Lex-OP-luciferase, 3D.A-luciferase, EFRasV12, EFMEKK1, NLex.ELKC, and NLex.JunN. We thank Drs. Stephen P. Davies and Helen Reddy and Professor Sir Philip Cohen (Division of Signal Transduction Therapy and MCR Protein Phosphorylation Unit, University of Dundee, UK) for providing the protein kinase selectivity data for kinases other than ALK2, 4, and 6 (shown in Table 1); we thank Mike Howell for the phylogenetic tree in Fig. 1A and many useful discussions and Karolien De Bosscher, Mike Howell, and Peter Parker for useful comments on the manuscript. We acknowledge the NHPP for providing human recombinant activin A [lot 15365-36(1)].
Footnotes
- Received November 28, 2001.
- Accepted March 21, 2002.
-
This work was funded by Imperial Cancer Research Fund (now Cancer Research UK after the merger of Imperial Cancer Research Fund with the Cancer Research Campaign), GlaxoSmithKline Pharmaceuticals, and a Medical Research Council training fellowship (to F.J.N.).
-
G.J.I. and F.J.N. contributed equally to this work.
Abbreviations
- TGF-β
- transforming growth factor β
- BMP
- bone morphogenetic protein
- AMH
- anti-Müllerian hormone
- ALK
- activin receptor-like kinase
- SB-431542
- 4-(5-benzo[1,3]dioxol-5-yl-4-pyridin-2-yl-1H-imidazol-2-yl)-benzamide
- DE
- distal element
- OP
- operator
- SRF
- serum response factor
- FCS
- fetal calf serum
- BSA
- bovine serum albumin
- EGF
- epidermal growth factor
- DMEM
- Dulbecco's modified Eagle's medium
- DMSO
- dimethyl sulfoxide
- GRB2
- growth-factor receptor-bound protein 2
- ATF
- activating transcription factor
- SRE
- serum response element
- β-Gal
- β-galactosidase
- GS
- glycine- and serine-rich
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}