


Abstract
GS-458967, 6-(4-(Trifluoromethoxy)phenyl)-3-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]pyridine (GS967) is a recently described, novel, sodium channel inhibitor exhibiting potent antiarrhythmic effects in various in vitro and in vivo models. The antiarrhythmic mechanism has been attributed to preferential suppression of late sodium current. However, there has been no reported systematic investigation of the effects of this compound on isolated sodium channels. Here, we examined the effects of GS967 on peak (INaP) and late (INaL) sodium current recorded from cells that heterologously expressed human cardiac voltage-gated sodium channel, the principle cardiac sodium channel. As previously described, we observed that GS967 exerted tonic block of INaL (63%) to a significantly greater extent than INaP (19%). However, GS967 also caused a reduction of INaP in a frequency-dependent manner, consistent with use-dependent block (UDB). GS967 evoked more potent UDB of INaP (IC50 = 0.07 µM) than ranolazine (16 µM) and lidocaine (17 µM). Use-dependent block was best explained by a significant slowing of recovery from fast and slow inactivation with a significant enhancement of slow inactivation in the presence of GS967. Furthermore, GS967 was found to exert these same effects on a prototypical long QT syndrome mutation (delKPQ). An engineered mutation at an interaction site for local anesthetic agents (F1760A) partially attenuated the effect of GS967 on UDB, but had no effect on tonic INaL block. We conclude that GS967 is a preferential inhibitor of INaL, but it also exerts previously unreported strong effects on slow inactivation and recovery from inactivation, resulting in substantial UDB that is not entirely dependent on a known interaction site for local anesthetic agents.
Introduction
Sodium current (INa) in cardiac myocytes carried primarily by voltage-gated sodium channel 1.5 (NaV1.5) channels is responsible for the rapid upstroke of atrial and ventricular action potentials as well as the rapid propagation of depolarization throughout the heart. When NaV1.5 fails to inactivate fully after opening, Na+ influx continues throughout the action potential plateau. The resulting current, referred as late INa (INaL), can promote prolongation of the action potential duration. Late INa is normally small, but its amplitude is greater in certain acquired or heritable conditions, including failing and/or ischemic heart (Le Grand et al., 1995), oxidative stress (Song et al., 2006), or mutations in SCN5A, which encodes NaV1.5 (Bennett et al., 1995; Ruan et al., 2009). SCN5A mutations that cause enhanced INaL produce type 3 long QT syndrome characterized by a high propensity for life-threatening ventricular arrhythmia torsades de pointes, and may also contribute to atrial fibrillation (Antzelevitch et al., 2014).
Preferential inhibition of INaL improves electrical function in myocytes isolated from failing and ischemic hearts or that have been exposed to cardiac glycosides, hydrogen peroxide, enhancers of INaL, or drugs that block the human ether-a-go-go related gene current (IKr) and reduce repolarization reserve (Le Grand et al., 1995; Sicouri et al., 1997; Song et al., 2004, 2006, 2008; Maltsev and Undrovinas, 2008; Sossalla et al., 2010; Wu et al., 2011). Many local anesthetic and antiarrhythmic agents have greater potency to block INaL than peak INa (INaP). Certain compounds, such as ranolazine (Gupta et al., 2015) and F15845 (3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate) (Pignier et al., 2010), are described as preferential INaL blockers.
GS-458967, 6-(4-(Trifluoromethoxy)phenyl)-3-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]pyridine (GS967; a triazolopyridine derivative) (Koltun et al., 2016) is a recently described sodium channel blocker that was originally demonstrated to exert potent antiarrhythmic effects in rabbit ventricular and canine atrial myocytes by a proposed mechanism of action involving preferential INaL block (Belardinelli et al., 2013; Sicouri et al., 2013). More recently, GS967 was shown to suppress arrhythmogenicity evoked by ischemia (Bonatti et al., 2014), hypokalemia (Pezhouman et al., 2014), and catecholamines (Alves Bento et al., 2015) in canine and porcine models. In each of these reports, the effect of GS967 was ascribed to preferential block of INaL. Given these promising effects of GS967, a more comprehensive analysis of its biophysical effects on human cardiac sodium channels is warranted.
In this study, we investigated the molecular pharmacology of GS967 on heterologously expressed human NaV1.5. In addition to preferential block of INaL, we observed a previously unreported and potent use-dependent block of INaP. Use-dependent block of NaV1.5 by GS967 stems from its ability to impair recovery from inactivation and to enhance entry into slow inactivated states. We suggest that these previously unrecognized properties of GS967 may contribute to its antiarrhythmic effects.
Materials and Methods
Plasmid Constructs, Cell Culture, and Transfection.
Plasmids encoding human NaV1.5 and the NaV1.5 mutants delKPQ and F1760A were subcloned into the pRcCMV mammalian expression vector. Channel constructs were then cotransfected with a plasmid encoding green fluorescent protein alone or one with the human sodium channel β1 subunit in series with an internal ribosome entry site (IRES) element and green fluorescent protein as fluorescent markers of transfected cells. The complete coding regions of all plasmid constructs were verified by Sanger sequencing before use in experiments. Cultured cells (tsA201) were transiently transfected with 1.5 μg of pRCCMV-NaV1.5 (or mutants) and 1 μg of pEGFP-IRES or pEGFP-IRES-hβ1 using Lipofectamine (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. The tsA201 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and penicillin (50 units·ml−1)/streptomycin (50 μg·ml−1) at 37°C in 5% CO2. Green or red fluorescent cells were selected for electrophysiological analysis 24–48 hours after transfection. In all experiments, the β1 subunit was coexpressed with NaV1.5 except where mentioned. NaV1.5 was also stably expressed in human embryonic kidney 293 cells and maintained in the same media plus G418 (500 μg/ml).
Electrophysiology.
Sodium currents in transiently transfected tsA201 cells were recorded at room temperature 24–48 hours after transfection using the manual whole-cell patch clamp technique. Patch-clamp pipettes were pulled from thin-wall borosilicate glass (outer diameter: 1.5 mm; Warner Instruments Corp., Hamden, CT) on a P-1000 multistage Flaming/Brown micropipette puller (Sutter Instruments, Novato, CA) and fire polished to a resistance between 1.0 and 2.5 MΩ. To avoid the time-dependent shift of the INa availability curve commonly observed during patch-clamp experiments, voltage-dependent inactivation was always assessed within 2 minutes after cell membrane rupture. The holding potential for all experiments (−120 mV) and specific voltage-clamp protocols are depicted as figure insets. The pipette solution used contained the following (in mM): 10 NaF, 100 CsF, 20 CsCl, 20 1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA), and 10 HEPES, adjusted to pH 7.35 with CsOH. The extracellular (bath) recording solution contained the following (in mM): 145 NaCl, 4 KCl, 1 MgCl2, 10 HEPES, and 1.8 CaCl2, adjusted to pH 7.35 with CsOH. Series resistance was compensated 80%. Data acquisition was carried out using an Axopatch 200B patch-clamp amplifier and pCLAMP 10.0 software (Molecular Devices, Sunnyvale, CA). A Boltzmann function (I = 1/[1 + exp(Vt − V1/2)/k)]) was fitted to the availability and activation curves to determine the membrane potential eliciting half-maximal activation/inactivation (V1/2), where k is the slope factor. All data were analyzed using pCLAMP 10.0 or Excel 2007 (Molecular Devices; Microsoft, Redmond, WA) and plotted using SigmaPlot 10.0 (Systat Software, Inc., San Jose, CA). Experiments examining late INa and ramp currents used tetrodotoxin (TTX; Tocris Bioscience, Bristol, UK) to allow for the determination of TTX-sensitive sodium current. TTX was added to the bath solution from a 3 mM stock solution of TTX (in water) to a final concentration of 10 µM. TTX was applied at the end of the protocol right after GS967. TTX-sensitive current was then determined by offline digital subtraction.
Automated Patch-Clamp Recording.
Automated patch-clamp recording was performed using a Syncropatch 384 PE (Nanion Technologies, München, Germany). Pulse generation and data collection were performed with PatchController384 V.1.3.0 and DataController384 V1.2.1 (Nanion Technologies). Whole-cell currents were filtered at 3 kHz and acquired at 10 kHz. The access resistance and apparent membrane capacitance were estimated using built-in protocols. Series resistance was compensated 95% and leak and capacitance artifacts were subtracted out using the P/4 method. The average seal resistance was 1.01 ± 0.04 GΩ, and cells were excluded from analysis if the current at the holding potential (−120 mV) was >10% of the peak current.
Human embryonic kidney 293 cells stably expressing NaV1.5 were plated into 100-mm culture dishes 48–72 hours prior to the experiment. The day of the experiment, cells (60–80% confluency) were washed once with phosphate-buffered saline (−Mg/Ca), detached with trypsin, and resuspended in culture media and external solution at 180,000 cells/ml. Cells were allowed to recover for at least 30 minutes at 15°C while shaking on a rotating platform. Following equilibration, 10 µl of cell suspension was added to each well of a 384-well, single-hole, low-resistance (2 MΩ) “chip” (Nanion Technologies). Whole-cell currents were recorded at room temperature in the whole-cell configuration. The external solution contained the following (in mM): NaCl 140, KCl 4, CaCl2 2, MgCl2 1, HEPES 10, and glucose 5 (pH = 7.4). The internal solution contained the following (in mM): CsF 110, CsCl 10, NaCl 10, HEPES 10, and EGTA 10 (pH = 7.2).
Data Analysis.
Patch-clamp measurements are presented as the means ± S.E.M. Comparisons were made using Student’s t test, with P < 0.05 considered significant.
Results
Preferential Late INa Block of Human NaV1.5 by GS967.
We examined the effects of 1 µM GS967 on INaP and INaL recorded from tsA201 cells transiently expressing wild-type (WT) human NaV1.5 in the presence of the β1 subunit. As previously reported (Belardinelli et al., 2013), we found that 1 µM GS967 at a slow pulsing rate (0.2 Hz) inhibited INaL to a significantly greater extent than INaP (62 vs. 18.6% inhibition P < 0.001; Fig. 1, A and C). This differential inhibitory effect of GS967 on INaP and INaL was also observed in cells expressing NaV1.5 in the absence of the β1 subunit (Supplemental Fig. S1). GS967 caused a significant 4-mV hyperpolarizing shift in the V1/2 of activation, but it did not modify the inactivation kinetics (Supplemental Fig. S2). GS967 also appeared to cause a significant shift in the V1/2 of steady-state inactivation from −83.2 ± 0.8 to −95.3 ± 1.4 mV (P < 0.001; n = 18). To exclude the confounding effects of time-dependent shifts in steady-state inactivation known to occur with NaV1.5, we examined two separate groups of cells exposed to control (drug-free) conditions or GS967 at a fixed time interval following establishment of the whole-cell seal. We found that GS967 does not modify the voltage dependence of steady-state inactivation (control V1/2: −77.3 ± 1.2 mV vs. GS967: −77.5 ± 1.3 mV; P = 0.45; n = 7 and 10, respectively; Supplemental Fig. S3).
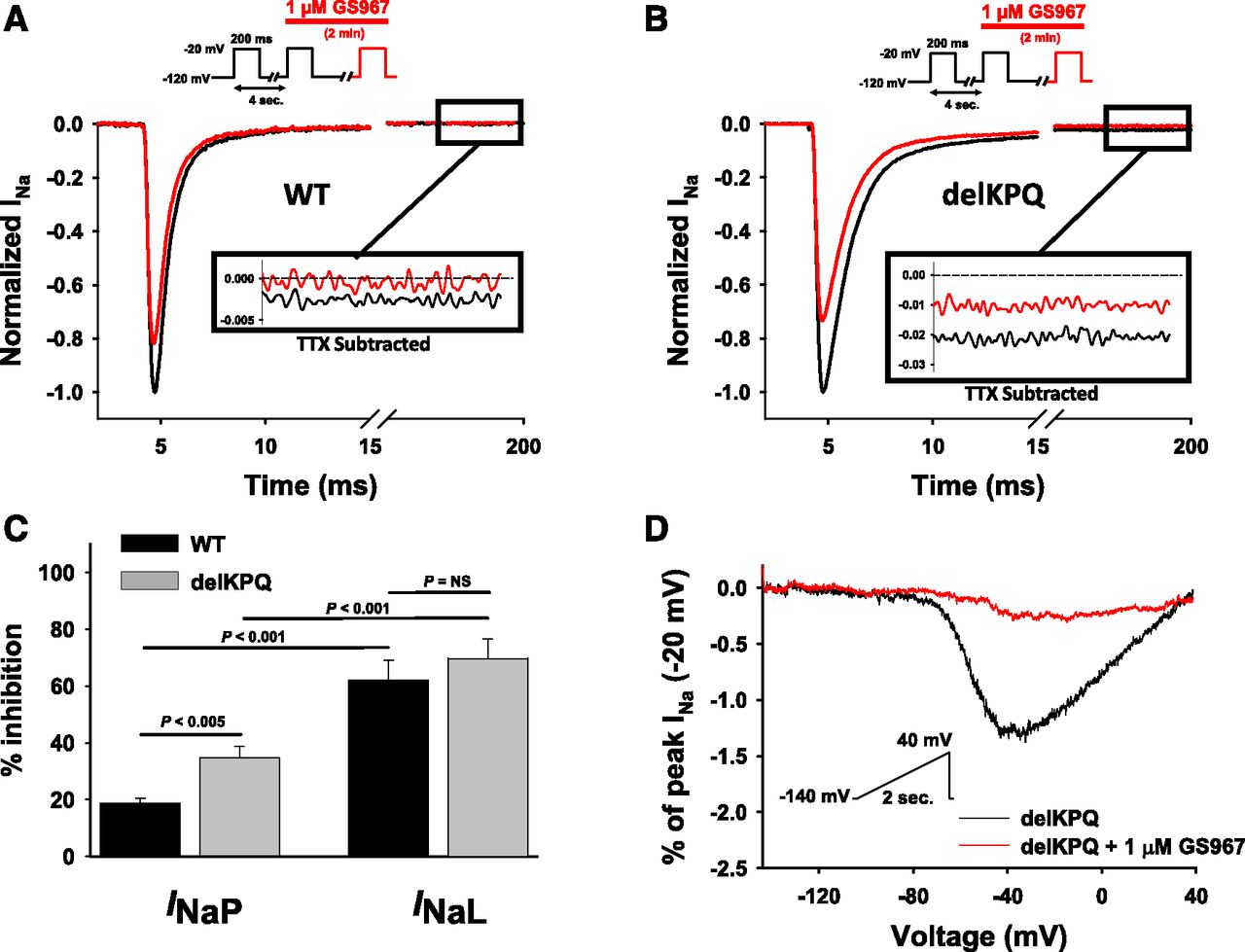
GS967 selectively inhibits NaV1.5 INaL. (A) Representative TTX-subtracted INa from NaV1.5-expressing cells in the absence (black) and presence (red) of 1 µM GS967. Cells were held at a membrane potential of −120 mV and depolarized every 4 seconds to −20 mV for 200 ms. (B) Representative TTX-subtracted INa from cells expressing NaV1.5-delKPQ in the absence (black) and presence (red) of 1 µM GS967. Cells were held at −120 mV and depolarized every 4 seconds to −20 mV for 200 ms. (C) Summary of effects of 1 µM GS967 on WT and delKPQ INaP and INaL. GS967 caused a decrease of INaP and INaL by 18.6 ± 1.8% and 62 ± 6.9%, respectively (n = 16; P < 0.001) for WT, and 34.8 ± 4.0% and 69.7 ± 6.8%, respectively (n = 13; P < 0.001) for delKPQ. INaL was determined from TTX-subtracted current over the final 10 ms of a 200-ms pulse to −20 mV and normalized to peak current. (D) Normalized and averaged TTX-subtracted ramp currents (0.09 mV/ms) recorded from cells expressing NaV1.5-delKPQ in the absence (black) and presence (red) of 1 µM GS967 and expressed as a percentage of INaP. The net charge movement during voltage ramps was 9.2 ± 1.3 pC/nA in the absence and 2.1 ± 0.4 pC/nA in the presence of 1 μM GS967 (n = 12; P < 0.001). All data are presented as the mean ± S.E.M. NS, not significant.
The long QT syndrome (LQTS)–associated NaV1.5 mutation delKPQ exhibits enhanced INaL, providing a more robust target for testing effects of GS967. Figure 1C illustrates that GS967 preferentially blocks delKPQ INaL significantly more than INaP (69.7 vs. 34.8% inhibition, respectively; P < 0.001; Fig. 1, B and C), and the compound nearly eliminates aberrant inward current evoked by slow voltage ramps (Fig. 1D). Notably, GS967 suppresses delKPQ INaP to a greater extent than WT INaP (34.8 vs. 18.6% inhibition; P < 0.005), illustrating a lower degree of selectivity for late over peak current for this LQTS mutation. This difference in INaP between WT and mutant NaV1.5 was not due to differences in the rates of current “rundown.” This observation indicated that peak current block by GS967 was context-specific, and motivated us to examine other effects of GS967 on NaV1.5 channels under different conditions, including a use-dependent block (UDB) paradigm.
Use-Dependent Block of NaV1.5 by GS967.
To test for UDB of NaV1.5 by GS967, we applied a series of 50 short (20 ms) depolarizing pulses to −20 mV at different frequencies (0.5, 1, 2, 10, and 20 Hz). In the absence of GS967, there was no discernable loss of channel availability at stimulation frequencies of 0.5, 1, or 2 Hz, whereas at 10 and 20 Hz, there was an ∼20% reduction in channel availability (Fig. 2B). Following bath application of 1 μM GS967, repetitive pulsing was associated with a progressive reduction of NaV1.5 INaP consistent with UDB of the channel (Fig. 2, A and B). Use-dependent block of NaV1.5 delKPQ INaP by 1 μM GS967 was not significantly different from that observed for WT channels at 2 and 10 Hz. However, at 20 Hz, GS967 showed a greater UDB of delKPQ INaP compared with WT channels (Supplemental Fig. S4). Use-dependent block of WT or mutant cardiac sodium channels by GS967 was not examined previously (Belardinelli et al., 2013; Bonatti et al., 2014; Alves Bento et al., 2015; Burashnikov et al., 2015; Carneiro et al., 2015).

Use-dependent block of human NaV1.5 by GS967. To examine use-dependent block, cells were held at −120 mV and pulsed to −20 mV for 20 ms at five different frequencies (0.5, 1, 2, 10, and 20 Hz), with an interpulse potential of −120 mV (see inset). The peak currents elicited by each pulse were normalized to the peak current of the first pulse and plotted against the pulse number. Black symbols represent the absence of drug, whereas gray symbols represent experiments in the presence of 1 µM GS967. (A) Response of cells expressing the WT channels to 1 µM GS967 at 0.5, 1, 2, 10, and 20 Hz. (B) Relative amplitudes at the last sweep (50th) at 0.5, 1, 2, 10, and 20 Hz were 1.03 ± 0.01, 0.99 ± 0.01, 0.98 ± 0.01, 0.89 ± 0.01, and 0.77 ± 0.02, respectively, in the absence (black) of 1 µM GS967, and 0.92 ± 0.02 (n = 5; P < 0.05), 0.85 ± 0.02 (n = 5; P < 0.05), 0.68 ± 0.02 (n = 11; P < 0.001), 0.25 ± 0.01 (n = 18; P < 0.001), and 0.12 ± 0.01 (n = 18; P < 0.001), respectively, in the presence (gray) of 1 µM GS967. All data are presented as the mean ± S.E.M.
We compared UDB of NaV1.5 by GS967 against lidocaine, a known use-dependent sodium channel blocker (Huang et al., 2012), and another selective INaL blocker, ranolazine (Antzelevitch et al., 2014). Using automated patch-clamp recording, we examined UDB of NaV1.5 across a 1000-fold concentration range for GS967 and lidocaine but only a 100-fold concentration range for ranolazine due to poor solubility at high concentrations. All compounds exhibited UDB of INaP that was potentiated at higher pulsing frequencies (Fig. 3; Supplemental Fig. S5). At all frequencies, GS967 exhibited a similar block as lidocaine but with greater potency (140- to 320-fold lower IC50).

Concentration dependence of NaV1.5 use-dependent block by GS967, ranolazine, and lidocaine. Concentration-response relationships for use-dependent block at three different pulsing frequencies (2, 10, and 20 Hz) were determined as the amplitude of current evoked by the 50th pulse normalized to that of the current evoked by the first pulse plotted as a function of drug concentration. Recordings were performed using an automated planar patch clamp and human embryonic kidney 293 cells stably expressing human NaV1.5. (A) Concentration-response relationships for the use-dependent block at 2 Hz. IC50 = 0.1, 34, and 14 µM for GS967, ranolazine, and lidocaine, respectively. (B) Concentration response for use-dependent block at 10 Hz. IC50 = 0.1, 22, and 32 µM for GS967, ranolazine, and lidocaine, respectively. (C) Concentration response for use-dependent block at 20 Hz. IC50 = 0.07, 17, and 16 µM for GS967, ranolazine, and lidocaine, respectively. Data represent the mean ± S.E.M.; the number of experiments for each concentration is indicated next to the corresponding data points. Values of IC50 are also indicated on each plot.
GS967 Impairs Recovery from Inactivation.
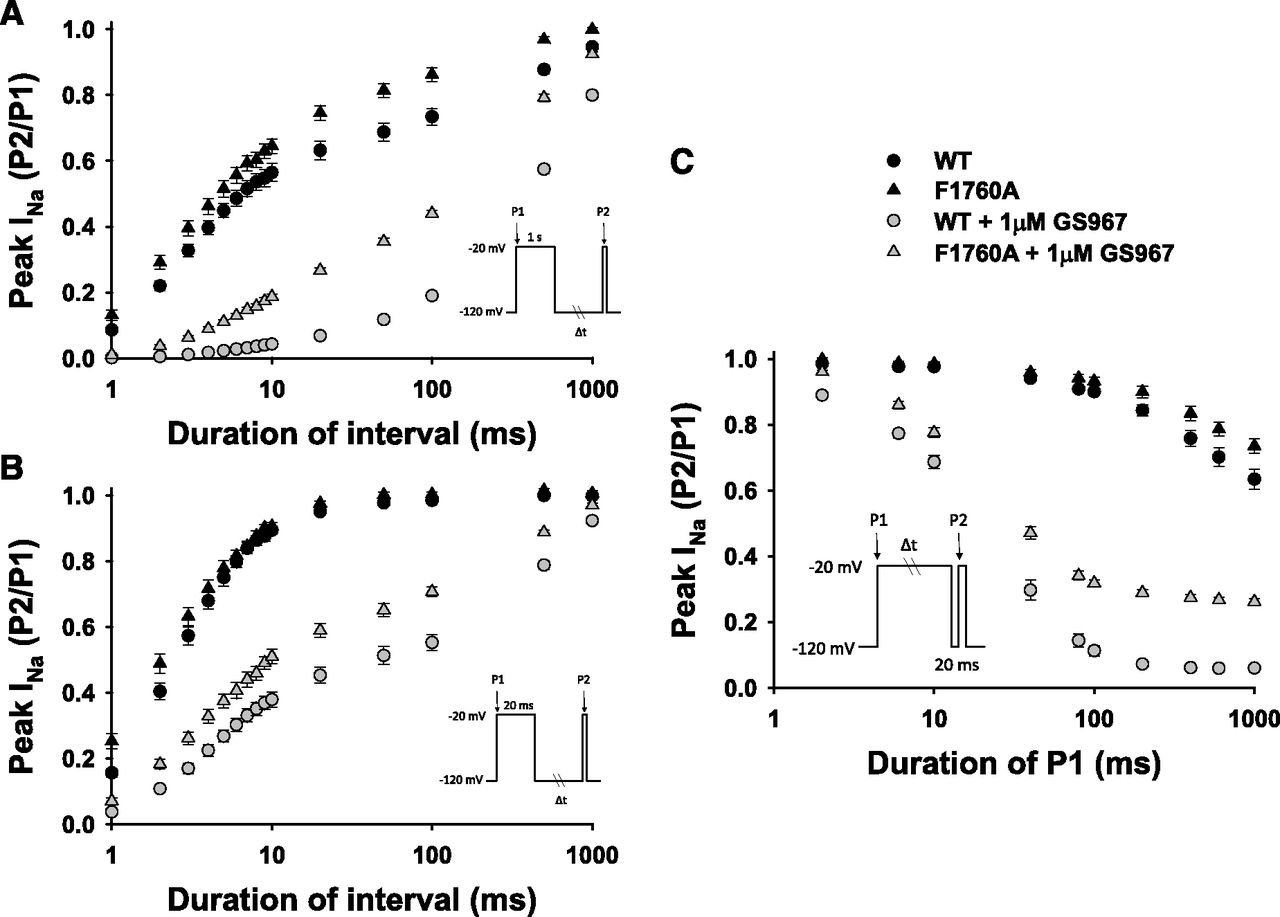
To explore plausible mechanisms to explain the UDB of NaV1.5 by GS967, we first examined recovery from fast and slow inactivation. This was done by utilizing a standard two-pulse protocol consisting of a depolarizing (−20 mV) 20- or 1000-ms pulse to engage fast or slow inactivation, respectively, followed by a variable duration recovery step to −120 mV and a final test pulse (−20 mV, 20 ms). Channel availability after the end of the recovery interval was normalized to initial values and plotted against the recovery time (Fig. 4A).

GS967 modifies NaV1.5 onset of and recovery from inactivation. (A) Recovery from slow (1000-ms step, circles) and fast (20-ms step, triangles) inactivation determined in the absence (black) or presence (gray) of 1 µM GS967. Recovery from slow inactivation exhibited a biexponential time course with fast and slow time constants (time-constant values and relative weights are provided in Table 1). The recovery from fast inactivation was monoexponential in the absence and biexponential in the presence of 1 µM GS967 (time-constant values and relative weights are provided in Table 1). (B) Onset of slow inactivation was determined in the absence (black) and presence (gray) of 1 µM GS967 using a two-pulse protocol (inset). The time constants for onset of slow inactivation are provided in Table 1. All data are presented as the mean ± S.E.M.
The time course of recovery from fast inactivation in the absence of GS967 exhibited a monoexponential time course described by a single time constant (τ = 3.2 ± 0.3 ms), whereas in the presence of GS967, recovery exhibited a biexponential time course with a fast component, which was significantly larger than that observed in the absence of the compound (τfast = 5.2 ± 0.7 ms; P < 0.001), and a prominent slow component (τslow = 602.1 ± 77.5 ms) that represented 57.2 ± 2.8% of the recovering current (Fig. 4A; Table 1). The time course of recovery from slow inactivation was biexponential in the absence or presence of GS967. In the absence of GS967, recovery from slow inactivation was described by fast (τfast = 3.7 ± 0.3 ms) and slow time constants (τslow = 238.5 ± 26.8 ms) with relative weights of 68 ± 2.5% (fast) and 32 ± 2.5% (slow). GS967 significantly affected both fast and slow time constants and increased the relative weight of the slow component to 95 ± 1.0% (Fig. 4A; Table 1). A very similar effect of GS967 on recovery from inactivation of delKPQ channels was also observed (Supplemental Fig. S6; Table 1). These results suggest that GS967 can significantly impair recovery from both fast and slow inactivation.
Effects of GS967 on WT NaV1.5, delKPQ, and F1760A onset and recovery from inactivation
GS967 Enhances Slow Inactivation.
Use-dependent block can also be due to accumulation of channels in a slow inactivated state evoked by prolonged membrane depolarization. We investigated whether kinetic differences in the rate of entry into slow inactivation could account for the UDB of NaV1.5 by GS967. To assess the onset of slow inactivation, cells were held at −120 mV and then depolarized to −20 mV for a variable duration (2–1000 ms) followed by a brief recovery pulse (−120 mV for 20 ms) and a final 20-ms test pulse to −20 mV (voltage protocol illustrated in Fig. 4B). Monoexponential fits were performed to determine the time constants for the onset of slow inactivation in the absence and presence of 1 µM GS967. In the absence of GS967, the time constant was 475.6 ± 29.7 ms, whereas in the presence of 1 µM GS967, the time constant for the onset of slow inactivation was significantly smaller (τ = 28.8 ± 2.6 ms; P < 0.001; Fig. 4B; Table 1), indicating that NaV1.5 channels entered the slow inactivated state more rapidly in the presence of GS967. These kinetic differences likely contribute to UDB by GS967 (Fig. 2). A very similar effect of GS967 on slow inactivation of delKPQ channels was also observed (Supplemental Fig. S6; Table 1).
Using the same protocol as in Fig. 4B, we observed that GS967 had a more potent effect than lidocaine and ranolazine to reduce INaP after long depolarizations (Supplemental Fig. S7). The calculated IC50 for GS967 was 30 nM, 200 and 1300 times lower than for lidocaine (IC50 = 6 µM) and ranolazine (IC50 = 41 µM), respectively (Fig. 5). These differences might be attributed to a greater magnitude of effects on slow inactivation or due to differences in effects on recovery from inactivation (Lenkey et al., 2006).

Concentration dependence of NaV1.5 onset of slow inactivation by GS967, ranolazine, and lidocaine. Concentration-response relationships for the onset of slow inactivation induced by GS967, ranolazine, and lidocaine were determined by automated patch-clamp recording from cells stably expressing human NaV1.5 using the two-pulse protocol illustrated in Fig. 4B. Concentration-response curves for the onset of slow inactivation induced by GS967 (black), ranolazine (blue), and lidocaine (red) were fitted to a four-parameter sigmoidal equation, and the IC50 for each drug or compound was determined. GS967, ranolazine, and lidocaine IC50 values were 0.03, 41, and 6 µM, respectively. Data represent the mean ± S.E.M.; the number of experiments for each concentration is indicated next to the corresponding data point.
GS967 Effect Requires a Local Anesthetic Interaction Site.
Many features of NaV1.5 block by GS967, including suppression of INaL and UDB of INaP, resemble effects of local anesthetic (LA) agents (Nagatomo et al., 2000; Liu et al., 2003). We examined whether the actions of GS967 depend upon a known LA interaction site in the domain 4 (D4)/segment 6 (S6) segment (Catterall, 2014) by testing this compound on NaV1.5 channels with a mutation (F1760A) at this site. Similar to WT channels, GS967 (1 μM) inhibited INaL to a significantly greater extent than INaP (10.9 ± 2 vs. 73.2 ± 4%) at a slow pulsing frequency (0.2 Hz; Fig. 6, A and B). Moreover, the effect of GS967 on INaL was not statistically different between WT and F1760A channels (62.0 ± 7.0 vs. 73.2 ± 4.0% inhibition; P = 0.44). Further, GS967 significantly suppressed ramp currents evoked in F1760A-expressing cells with a 5-fold suppression of net charge movement (Fig. 6C). Together, these results suggest that phenylalanine-1760 is not a critical determinant of INaL block by GS967.

GS967 selectively inhibits NaV1.5-F1760A INaL. (A) Representative TTX-subtracted INa from cells expressing NaV1.5-F1760A in the absence (black) or presence (red) of 1 µM GS967. Cells were held at −120 mV and depolarized every 4 seconds to −20 mV for 200 ms. (B) Summary of effects of 1 µM GS967 on WT and F1760A INaP and INaL. NaV1.5-F1760A INaP and INaL were decreased by 10.9 ± 2.0% and 73.2 ± 4.0%, respectively (n = 14; P < 0.001). (C) Normalized and averaged TTX-subtracted ramp currents (0.09 mV/ms) recorded from cells expressing NaV1.5-F1760A expressed as a percentage of peak INa in the absence (black) or presence of (red) 1 µM GS967. The net charge movement during voltage ramps was 9.4 ± 1.0 pC/nA in the absence and 1.8 ± 0.2 pC/nA in the presence of 1 µM GS967 (n = 14; P < 0.001). All data are presented as mean ± S.E.M. NS, not significant.
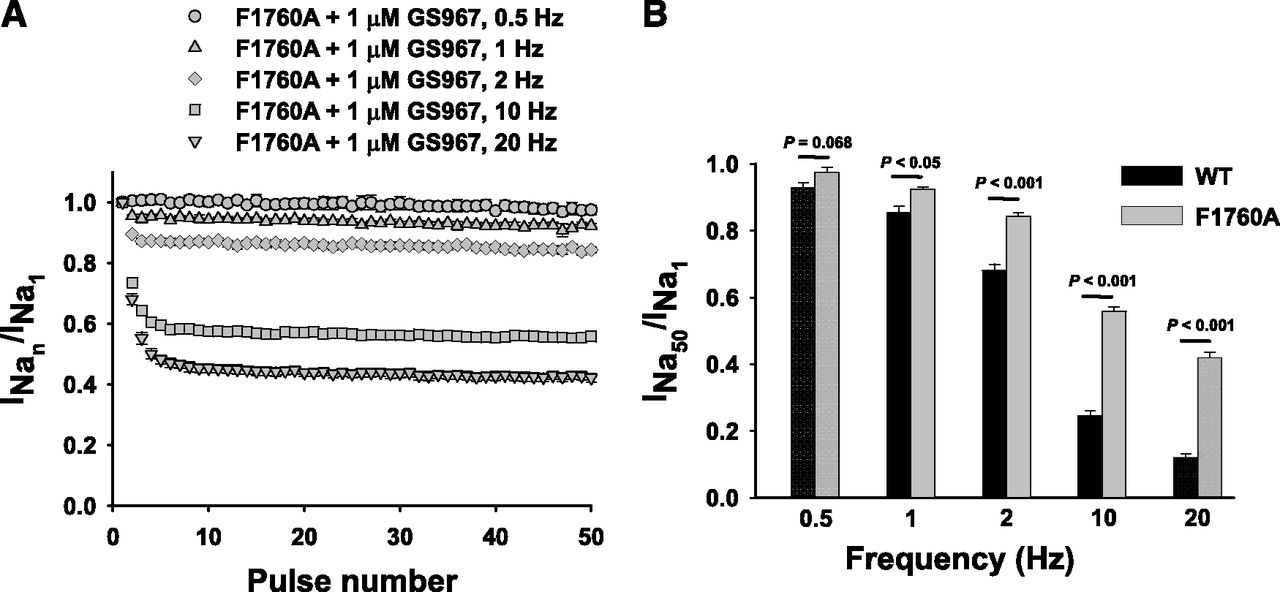
We further assessed whether F1760A modifies UDB of INaP by GS967. First, we compared the intrinsic frequency-dependent loss of channel availability between F1760A and WT NaV1.5 recorded in the absence of the compound. The F1760A mutation exhibits a lower level of intrinsic “run down” at higher frequencies (10 and 20 Hz) than the WT channel (Supplemental Fig. S8). After bath application of 1 μM GS967, repetitive pulsing was associated with a progressive reduction of F1760A INaP, consistent with UDB of the mutant channel at all frequencies (Fig. 7A). However, the degree of UDB of F1760A channels was significantly less than WT channels at 1 Hz or higher pulsing frequencies (Fig. 7B). Collectively, these data suggest that modification of the D4/S6 segment LA interaction site only partially attenuates UDB of INaP by GS967.
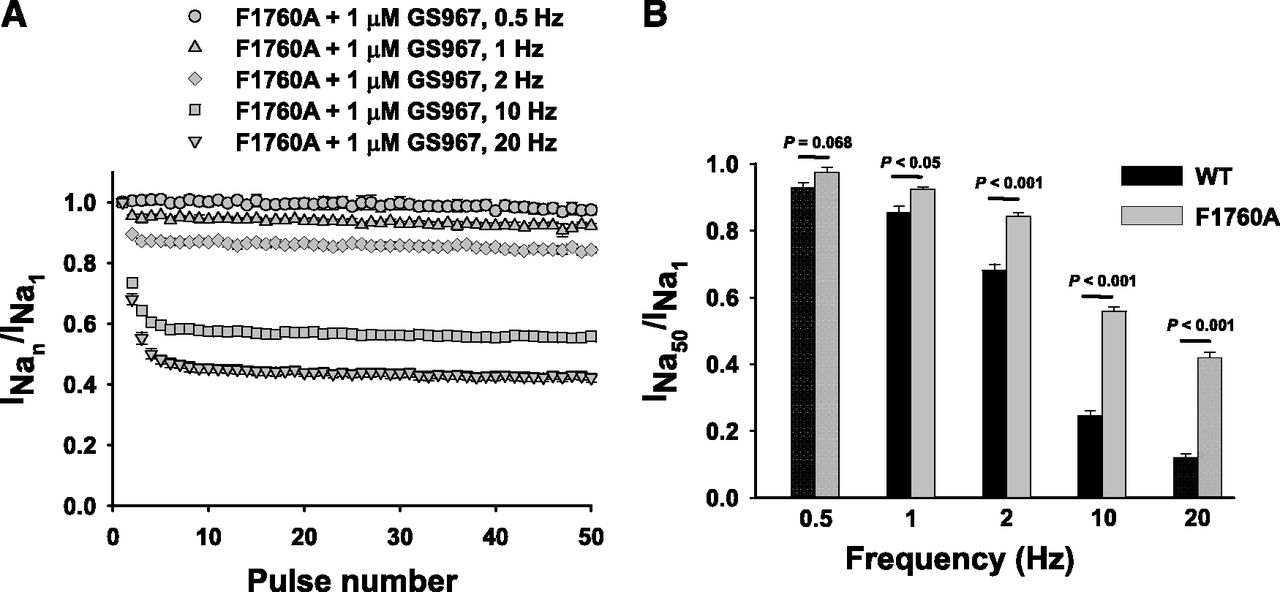
Use-dependent block of NaV1.5-F1760A by GS967. Cells were held at –120 mV and pulsed to −20 mV (20 ms) at five different frequencies (0.5, 1, 2, 10, and 20 Hz), with interpulse potential set at −120 mV (see inset). The peak currents elicited by each pulse were normalized to the first pulse peak current, then plotted against the pulse number. (A) Response of cells expressing NaV1.5-F1760A to 1 µM GS967 at 0.5, 1, 2, 10, and 20 Hz. (B) Comparison of the relative amplitudes at the last sweep (50th) of the use-dependent protocol for the WT (black) and F1760A mutant (gray) at 0.5, 1, 2, 10, and 20 Hz in the presence of 1 µM GS967. Relative amplitudes at the last sweep (50th) at 0.5, 1, 2, 10, and 20 Hz were 0.93 ± 0.02 (n = 5), 0.85 ± 0.02 (n = 5), 0.68 ± 0.02 (n = 11), 0.25 ± 0.01 (n = 18), and 0.12 ± 0.01 (n = 18), respectively, for the WT, and 0.98 ± 0.02 (n = 6; P = 0.068), 0.92 ± 0.01 (n = 6; P < 0.05), 0.84 ± 0.01 (n = 11; P < 0.001), 0.56 ± 0.01 (n = 15; P < 0.001), and 0.42 ± 0.01 (n = 15; P < 0.001), respectively, for F1760A. Data represent the mean ± S.E.M.
Finally, we examined the effect of the F1760A mutation on recovery from fast and slow inactivation. Figure 8 illustrates that F1760A has a slightly faster recovery from slow inactivation compared with the WT in the absence of the compound. GS967 significantly impairs F1760A recovery from slow inactivation but to a lesser extent than for WT NaV1.5 (Fig. 8, A and B; Table 1). This effect could be explained by a reduced slow time constant for recovery from inactivation and a smaller relative weight of this component. Similar to WT channels, GS967 also evoked an additional slow component for recovery from fast inactivation that was not observed in the absence of the compound, but this slow component had a smaller amplitude and faster kinetics than WT channels (Table 1). GS967 dampened the extent of F1760A slow inactivation, but the rate of onset of inactivation was not significantly different from WT NaV1.5 (Fig. 8C; Table 1). These results suggest that mutation of the D4/S6 LA interaction site partially attenuates the effect of GS967 on onset and recovery from inactivation, and this can explain less UDB. However, the effect of GS967 on F1760A was not complete, and therefore, there may be other channel regions important for the full actions of this compound.

GS967 affects NaV1.5-F1760A onset of slow inactivation and recovery from inactivation. Recovery from slow (1000-ms step) (A) and fast (20-ms step) (B) inactivation from cells expressing F1760A or WT NaV1.5 determined in the absence (black) or presence (gray) of 1 µM GS967. The recovery from slow inactivation was biexponential in both the absence and presence of GS967 (see Table 1). F1760A recovery from fast inactivation was monoexponential in the absence and biexponential in the presence of 1 µM GS967 (see Table 1). (C) The onset of slow inactivation from cells expressing WT or F1760A NaV1.5 in the absence (black) and presence (gray) of 1 µM GS967 was assessed using a two-pulse protocol (inset). F1760A onset of slow inactivation time constant was monoexponential (see Table 1). Data represent the mean ± S.E.M.
Discussion
In this study, we investigated the modulation of human NaV1.5 by GS967, a recently described sodium channel blocker with potent antiarrhythmic effects in various in vitro and in vivo models. The antiarrhythmic effect of GS967 was previously attributed to preferential suppression of INaL (Belardinelli et al., 2013; Pezhouman et al., 2014; Burashnikov et al., 2015), but here we offer evidence for other potentially important biophysical effects on the human cardiac sodium channel. Specifically, we demonstrate that GS967 exerts a previously unreported strong effect on slow inactivation and recovery from inactivation, resulting in substantial UDB. These revelations may help explain the pharmacological effects of GS967 in arrhythmia models and other settings.
Initially, we confirmed that GS967 applied to heterologously expressed human NaV1.5 does indeed exhibit significantly greater tonic block of INaL than INaP without modifying inactivation kinetics. This effect was more evident in cells expressing the LQTS mutation delKPQ, which has a greater amplitude of INaL than observed for WT-NaV1.5 channels. Importantly, we also demonstrated that GS967 exerts a strong UDB with IC50 values ranging from 70 to 100 nM, depending on stimulation frequency. GS967 exerts a UDB of NaV1.5 qualitatively similar to that of lidocaine, a prototypic use-dependent blocker of sodium channels, but with significantly greater potency. The basis for UDB by GS967 is most likely a previously unreported strong effect on slow inactivation and recovery from inactivation. The strong UDB exerted by GS967 may contribute to its observed antiarrhythmic efficacy.
It was initially reported that GS967 reduces the proarrhythmic consequences of enhanced INaL in rabbit models of reduced repolarization reserve and ischemia involving ventricular myocardium (Belardinelli et al., 2013). GS967 was also shown to suppress atrial arrhythmogenicity evoked by anemonia sulcata toxin II, E-4031 (N-[4-[1-[2-(6-Methylpyridin-2-yl)ethyl]piperidine-4-carbonyl]phenyl]), or catecholamines (Sicouri et al., 2013). More recent reports highlight the antiarrhythmic effects of GS967 in other models of either ventricular or atrial arrhythmia. GS967 was demonstrated to suppress early after depolarizations and delayed after depolarizations as well as to prevent ventricular tachyarrhythmia in rat hearts exposed to aconitine or hydrogen peroxide (Pezhouman et al., 2014). Bonatti and colleagues (2014) observed prevention of ischemia-induced depolarization and repolarization abnormalities in both atria and ventricles of pigs with acute coronary artery stenosis by GS967, whereas flecainide potentiated the proarrhythmic effects of acute ischemia. GS967 also prevented hypokalemia-induced ventricular fibrillation in rats and rabbits (Pezhouman et al., 2014), catecholamine-induced ventricular tachycardia in pigs (Alves Bento et al., 2015), and chemically induced spontaneous atrial fibrillation in a porcine model (Carneiro et al., 2015). All of these effects were observed with GS967 levels in the range of 0.3–1 μM, which is sufficient to suppress INaL and evoke UDB, allowing us to speculate that antiarrhythmic effects of this compound may be due to the combination of INaL suppression and UDB.
In this study, we also demonstrated that GS967 also suppresses INaL conducted by the LQTS mutation delKPQ. As a result, GS967 nearly eliminates the aberrant inward current evoked by slow voltage ramps. Use-dependent block of NaV1.5 delKPQ INaP by GS967 at low frequencies was not significantly different than that observed for WT channels. The ability of GS967 to target INaL and to produce a significant UDB in the context of type 3 long QT syndrome mutations suggests that compounds sharing these functional properties of GS967 may be useful in the treatment of arrhythmias for LQTS-associated NaV1.5 mutations. Indeed, another selective INaL (GS6615, eleclazine, 4-[(pyrimidin-2-yl)methyl]-7-[4-(trifluoromethoxy)phenyl]-3,4-dihydro-1,4-benzoxazepin-5(2H)-one) is undergoing clinical trials for type 3 LQTS (Gilead Sciences, 2000–2015). Whether GS6615 also exerts effects on slow inactivation and evokes UDB awaits further study.
Slow inactivation influences cellular excitability, particularly in pathophysiological conditions associated with prolonged membrane repolarization, such as epilepsy (Fleidervish et al., 1996), neuronal or cardiac ischemia (Shander et al., 1995), arrhythmias (Veldkamp et al., 2000), and neuromuscular channelopathies (Cannon, 1996). By accelerating the onset of slow inactivation and impairing recovery from inactivation of cardiac sodium channels, GS967 might increase the post-repolarization refractoriness, prevent premature membrane excitation, and enable antiarrhythmic as well as anticonvulsant efficacy. In a separate work, we recently demonstrated antiepileptic effects of GS967 in two mouse models (Anderson et al., 2014), and further work is needed to determine if UDB of neuronal sodium channels contributes to this effect.
Antiarrhythmic drugs with local anesthetic–like properties preferentially target the inactivated state of sodium channels and likely interact with a highly conserved LA drug receptor formed by residues from multiple S6 segments (Catterall, 2014). Whereas many features of NaV1.5 inhibition by GS967, including suppression of INaL and UDB of INaP, resemble effects of LA agents (Liu et al., 2003; Belardinelli et al., 2013), we found that phenylalanine-1760 was not a critical determinant of tonic INaL block by GS967. By contrast, mutation of the D4/S6 LA interaction site attenuates the effect of GS967 on onset and recovery from inactivation and reduces UDB. It has been suggested that phenylalanine-1760 contributes to slow inactivation and links drug action to slow inactivation (Carboni et al., 2005). However, the effect of F1760A on UDB was not complete, and therefore, there may be other channel regions important for the full actions of GS967. We can conclude that the mode of action of GS967 is somewhat distinct from LA, and that the block of INaL and INaP by GS967 does not entirely depend on the known LA drug receptor.
In conclusion, our study demonstrated that GS967 affects multiple biophysical properties of human NaV1.5. Specifically, GS967 exerts strong UDB, most likely because of high-affinity binding of GS967 to NaV1.5-inactivated states, by accelerating entry into slow inactivation and impairing recovery from inactivation. GS967 is a more potent use-dependent blocker than the antiarrhythmic drugs lidocaine and ranolazine. The ability of GS967 to specifically inhibit INaL over INaP and its newly described ability to evoke UDB may contribute to its antiarrhythmic effects.
Acknowledgments
The authors thank Reshma Desai for technical assistance.
Authorship Contributions
Participated in research design: Potet, George.
Conducted experiments: Potet, Vanoye.
Performed data analysis: Potet.
Wrote or contributed to the writing of the manuscript: Potet, Vanoye, George.
Footnotes
- Received January 11, 2016.
- Accepted April 29, 2016.
This work was supported by a Scientist Development Grant [11SDG5330006] from the American Heart Association (F.P.) and a research grant from Gilead Sciences, Inc.
This work was partially supported by a research grant from Gilead Sciences, Inc.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- BAPTA
- 1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid
- D4
- domain 4
- E-4031
- N-[4-[1-[2-(6-Methylpyridin-2-yl)ethyl]piperidine-4-carbonyl]phenyl]
- F15845
- (3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate)
- GS6615
- 4-[(pyrimidin-2-yl)methyl]-7-[4-(trifluoromethoxy)phenyl]-3,4-dihydro-1,4-benzoxazepin-5(2H)-one
- GS967
- GS-458967, 6-(4-(trifluoromethoxy)phenyl)-3-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]pyridine
- INa
- sodium current
- INaL
- late sodium current
- INaP
- peak sodium current
- IRES
- internal ribosome entry site
- LA
- local anesthetics
- LQTS
- long QT syndrome
- NaV1.5
- cardiac voltage-gated sodium channel
- S6
- segment 6
- TTX
- tetrodotoxin
- UDB
- use-dependent block
- WT
- wild type
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}