


Abstract
In response to hypoxia, mammalian cells express multiple gene products [including erythropoietin (EPO) and vascular endothelial growth factor (VEGF)] that serve to increase O2 delivery, as well as glucose transporters and glycolytic enzymes (such as enolase 1) that allow metabolic adaptation to decreased O2availability. Increased transcription of the genes encoding these proteins in hypoxic cells is mediated by hypoxia-inducible factor 1 (HIF-1), a basic helix-loop-helix transcription factor. Expression of HIF-1 and downstream genes can also be induced by exposure of cells to divalent metals (such as CoCl2) or iron chelators [such as desferrioxamine (DFO)]. We report here that the organomercurial compound mersalyl induced expression of VEGF and enolase 1 mRNA, as well as HIF-1 activity, in cultured cells. Expression of reporter genes containing hypoxia response elements from the EPO andVEGF genes was also induced by mersalyl treatment. However, mersalyl inhibited endogenous EPO mRNA expression induced by hypoxia, CoCl2, or DFO. In cells lacking expression of the insulin-like growth factor-1 receptor, mersalyl did not induce HIF-1 activity or VEGF mRNA expression, whereas induction by hypoxia, CoCl2, or DFO was unaffected. The mitogen-activated protein kinase kinase inhibitor PD098059 markedly reduced induction of HIF-1 by mersalyl but not by hypoxia. These results indicate that mersalyl induces expression of HIF-1 and a subset of hypoxia-inducible genes by a mechanism, involving the insulin-like growth factor-1 receptor and mitogen-activated protein kinase activity, that is distinct from mechanisms of induction by hypoxia, CoCl2, or DFO.
Hypoxia plays a fundamental role in the pathophysiological changes in a variety of clinical disorders, including ischemic cardiovascular diseases (for review, see Semenza, 1996). In response to hypoxia, mammalian cells express a variety of gene products (including EPO and VEGF) that serve to increase O2 delivery, as well as glucose transporters and glycolytic enzymes (such as ENO1) that allow metabolic adaptation to decreased O2 availability. Therefore, although ischemia involves substrate deprivation and toxic waste accumulation as well as O2 deprivation, hypoxia is a sufficient stimulus to induce responses that correct all aspects of ischemia by promoting angiogenesis through the action of VEGF. Recent studies have suggested that increases in VEGF levels, resulting from either parenteral VEGF protein administration or gene therapy approaches, may represent a novel approach to the treatment of ischemic disease (for review, see Ferrara and Davis-Smyth, 1997).
The activation of EPO, VEGF, and ENO1gene transcription in response to decreased O2availability is mediated by HIF-1, a basic helix-loop-helix transcription factor composed of HIF-1α and HIF-1β subunits (Wanget al., 1995a; Wang and Semenza, 1995). Expression of HIF-1 increases exponentially as cellular O2concentrations are decreased (Jiang et al., 1996). Expression of the limiting HIF-1α subunit is precisely regulated by O2 concentration and determines the level of HIF-1 DNA-binding activity and transcriptional activity within the cell (Huang et al., 1996; Jiang et al., 1996, 1997b;Semenza et al., 1996; Pugh et al., 1997). In addition to hypoxia, divalent metals (such as CoCl2) and iron chelators (such as DFO) induce HIF-1α expression, HIF-1 DNA-binding activity, andtrans-activation of genes containing HIF-1 binding sites (Wang and Semenza, 1993a,b; Wang et al., 1995a; Jianget al., 1997b; Pugh et al., 1997). The mechanisms of action of these compounds have not been determined but seem to be distinct from the hypoxia signal-transduction pathway (Gleadle et al., 1995b; Ehleben et al., 1997; Fandrey et al., 1997).
In embryonic stem cells, elimination of HIF-1α expression by gene targeting was also associated with loss of hypoxia-inducedVEGF gene transcription (Iyer et al., 1998). Mice lacking HIF-1α expression died at midgestation, with cardiac and vascular defects and extensive cell death throughout the embryo. In fetal sheep subjected to chronic anemia, myocardial hypertrophy and neovascularization were associated with coordinate increases in HIF-1α protein, VEGF mRNA, and VEGF protein (Martin et al., 1998). These studies indicate that HIF-1 is a master regulator of O2 homeostasis that controls both the establishment of essential physiological systems during embryogenesis and their subsequent use in fetal and postnatal life. The modulation of HIF-1 expression may represent a novel therapeutic approach to the treatment of ischemic disorders, because induction of HIF-1 and downstream genes would promote both angiogenesis (via VEGF) and hypoxic adaptation before neovascularization (via glycolytic enzymes). However, increased erythropoiesis mediated by EPO might lead to polycythemia, with the attendant risks of cerebrovascular accidents. Here, we report that the thiol-reactive organomercurial compound mersalyl induces VEGF and ENO1, but not EPO, mRNA expression and thus may represent a lead compound for the development of new pharmacological approaches to the treatment of ischemic disease. Furthermore, mersalyl induces expression of HIF-1 activity and VEGF mRNA by a mechanism involving the IGF-1R, which is distinct from the mechanism of induction in response to hypoxia, CoCl2, or DFO.
Materials and Methods
Chemical reagents.
Mersalyl [o-[(3-hydroxymercuri-2-methoxypropyl)carbamoyl]phenoxyacetic acid], DFO, and CoCl2 were purchased from Sigma Chemical Co. Wortmannin and PD098059 were from RBI (Natick, MA).
Cell lines and tissue culture.
Hep3B cells was cultured in minimal essential medium (Mediatech) with 10% fetal bovine serum and 1% penicillin/streptomycin (Life Technologies, Inc.). W and R− mouse embryo fibroblasts (provided by R. Baserga, Thomas Jefferson University, Philadelphia, PA) and Rat-1A cells (provided by C. Dang, Johns Hopkins University) were cultured in Dulbecco’s modified essential medium with 10% fetal bovine serum and 1% penicillin/streptomycin (Sell et al., 1994). Cells were treated with 50–100 μm mersalyl, 130 μmDFO, or 100 μm CoCl2 for 6–16 hr. Cells were subjected to hypoxia for 6–16 hr in a modular incubator chamber flushed with 1% O2/5% CO2/94% N2.
Nuclear extract preparation and EMSA.
Nuclear extracts were prepared as described previously (Wang and Semenza, 1995). EMSA was performed using oligonucleotide probe W18 (coding strand sequence, 5′-GCCCTACGTGCTGTCTCA-3′), containing the HIF-1 binding site from theEPO gene (Semenza and Wang, 1992).
Immunoblot assays.
Aliquots (15 μg) of nuclear extracts were fractionated by 7% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Blots were incubated with affinity-purified anti-HIF-1α and anti-HIF-1β polyclonal antibodies (Wang et al., 1995a; Jiang et al., 1996), followed by goat anti-rabbit immunoglobulin secondary antibody (1/2000 dilution), with visualization by the enhanced chemiluminescence detection method (Amersham).
RNA blot hybridization.
Total RNA was isolated from cells by acid guanidinium/phenol/chloroform extraction (Chomczynski and Sacchi, 1987). Aliquots (15 μg) of total RNA were fractionated by 1.4% agarose/2.2 m formaldehyde gel electrophoresis and transferred to nylon membranes. Blot hybridization was performed as previously described (Jiang et al., 1997a).
Transient transfection assays.
Plasmid DNA was isolated using a commercial kit (Qiagen). Hep3B cells were electroporated using a Gene Pulser (Bio-Rad), at 260 V and 960 μF. Cells were allowed to recover for 32 hr, the medium was changed, and cells were incubated for an additional 16 hr in the presence or absence of 100 μmmersalyl or 1% O2. Cells were harvested and resuspended in 0.25 m Tris·HCl, pH 8.0, and extracts were prepared by freeze-thaw lysis. β-Galactosidase activity was determined by the hydrolysis ofo-nitrophenyl-β-d-galactopyranoside (Promega), using 25 μg of extract at 37° for 1 hr, followed by spectrophotometric measurement at 420 nm. Luciferase activity was determined using 20 μg of cell extract and 100 μl of assay reagent (Promega). Light emission was measured for 15 sec in a luminometer (Tropix).
Results
The starting point for this study was the report that exposure of Hep3B human hepatoblastoma cells to 100 μm mersalyl inhibited EPO mRNA expression induced by hypoxia, CoCl2, or DFO (Gleadle et al., 1995b). We therefore investigated whether mersalyl also inhibited the expression of other hypoxia-inducible genes. VEGF mRNA was induced in cells exposed to hypoxia, DFO, or CoCl2 (Fig.1), as previously reported (Goldberg and Schneider, 1994; Gleadle et al., 1995a). Mersalyl did not inhibit VEGF mRNA expression induced by hypoxia, DFO, or CoCl2 but, instead, strongly induced VEGF expression in the absence of other stimulating agents. Similar results were obtained for ENO1 mRNA. In contrast, EPO mRNA expression was induced by hypoxia, DFO, or CoCl2 in the absence, but not in the presence, of mersalyl, as previously reported (Gleadleet al., 1995b). Mersalyl therefore induced VEGF and ENO1 mRNA expression but inhibited EPO mRNA expression induced by hypoxia, DFO, or CoCl2.

Effects of mersalyl on VEGF, ENO1, and EPO mRNA expression. Hep3B cells were left untreated (U) or were exposed to hypoxia (H), DFO, or CoCl2 for 6 hr, in the absence (−) or presence (+) of 100 μmmersalyl. Total cellular RNA was isolated and analyzed by blot hybridization, using cDNA probes for VEGF, ENO1, and EPO mRNA and an oligonucleotide complementary to 18 S rRNA.
We next investigated whether the induction of VEGF mRNA or inhibition of EPO mRNA expression was mediated by the HREs of these genes, which are the cis-acting elements required for transcriptional activation under hypoxic conditions and which contain essential binding sites for HIF-1. For this purpose, we used a reporter gene assay that was previously established to define these HREs (Semenza and Wang, 1992; Forsythe et al., 1996). Hep3B cells were transiently transfected with simian virus 40 promoter-luciferase reporter plasmids containing an HRE from the VEGF orEPO gene, located upstream of the transcription unit (Fig.2A). In the presence of mersalyl, expression of these reporter genes was increased 3.1- and 4.0-fold, respectively (Fig. 2B). Mersalyl is thus capable of activating transcription mediated by the HRE from either the VEGF orEPO gene, and the inhibition of EPO mRNA expression must therefore involve some additional mechanism of action that is selective for EPO gene expression. Transcription of the reporter genes was significantly greater in hypoxic cells, compared with mersalyl-treated cells (Fig. 2B), whereas hypoxia and mersalyl induced VEGF mRNA expression to similar degrees (Fig. 1).

Effects of mersalyl on the expression of reporter genes containing HREs. A, Structures of control and reporter plasmids. Reporter plasmids contained one copy of a 47-base pair HRE from theVEGF gene (Forsythe et al., 1996) or two copies of a 33-base pair HRE from the EPO gene (Semenza and Wang, 1992), cloned upstream of the simian virus 40 promoter (SV40 Pro) and luciferase coding sequences.Arrows, HIF-1 binding sites. Hep3B cells were co-transfected with the EPO or VEGF reporter plasmid and a control plasmid containing the simian virus 40 promoter and β-galactosidase (β-gal) coding sequences. B, Analysis of reporter gene expression. Transfected cells were exposed to vehicle (Untreated), 100 μm mersalyl, or 1% O2 (Hypoxia) for 16 hr and harvested, and the luciferase/β-galactosidase activity ratio was determined (relative luciferase activity). For each reporter plasmid tested, the data were normalized to the results obtained with untreated cells. Data represent mean values from three plates of transfected cells.
Transcriptional activation of HRE-containing reporter genes in hypoxic cells is mediated by HIF-1 (Semenza and Wang, 1992; Forsythe et al., 1996). To determine whether mersalyl induced HIF-1, EMSA and immunoblot assays were performed. As in the case of VEGF and ENO1 mRNA expression, induction of HIF-1 DNA-binding activity and HIF-1α protein expression in response to hypoxia, DFO, or CoCl2 was unaffected by mersalyl; in the absence of these agents, mersalyl alone induced HIF-1 expression (Fig.3). The induction of HIF-1 activity and HIF-1α protein expression by mersalyl was somewhat less than the induction by other agents, similar to the results obtained in the reporter gene assay (Fig. 2B).
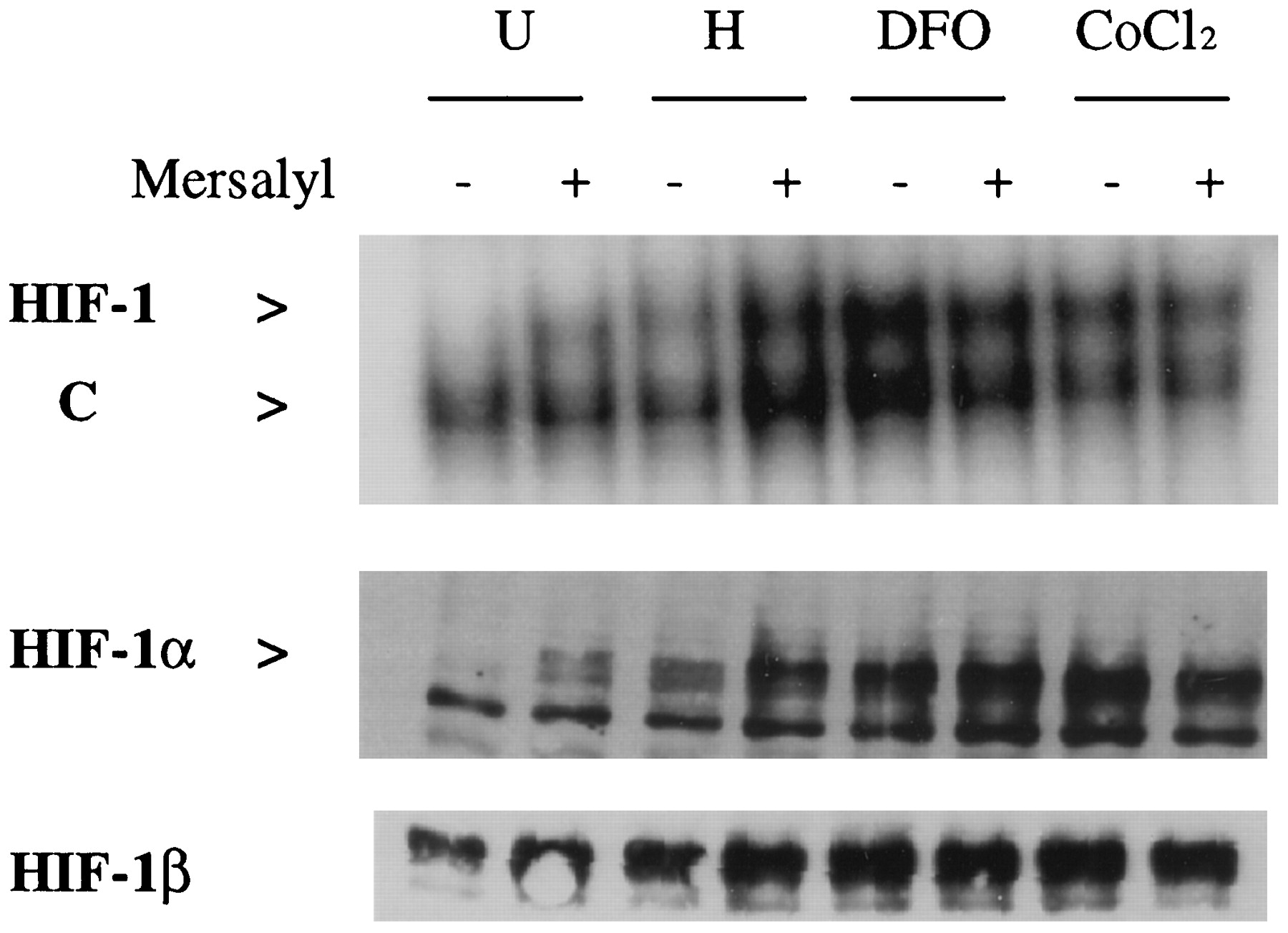
Analysis of HIF-1 expression. Hep3B cells were left untreated (U) or were exposed to hypoxia (H), DFO, or CoCl2 for 6 hr, in the absence (−) or presence (+) of 100 μm mersalyl. Nuclear extracts were prepared and analyzed for HIF-1 DNA-binding activity by EMSA, using a double-stranded oligonucleotide probe containing a HIF-1 binding site that is also recognized by a constitutive factor (C). Nuclear extracts were also analyzed for HIF-1α and HIF-1β protein expression with immunoblot assays.
To determine whether mersalyl induced HIF-1 in other cell types, Rat-1A fibroblasts were analyzed. HIF-1 DNA-binding activity and expression of HIF-1α and HIF-1β protein were induced when these cells were exposed to hypoxia or mersalyl (Fig. 4). Again, hypoxia seemed to stimulate greater HIF-1 expression than did mersalyl, although this difference was more striking in the EMSA than in the immunoblot assays. From these studies (Figs. 1-4), we conclude that 1) mersalyl induces expression of HIF-1 and transcription of downstream genes, 2) mersalyl inhibits EPO mRNA expression by a mechanism that is independent of HIF-1, and 3) mersalyl may also stimulate VEGF mRNA expression by an additional mechanism that is independent of HIF-1.

Induction of HIF-1 by hypoxia and mersalyl in Rat-1A fibroblasts. Cells were left untreated (U) or were exposed to hypoxia (H) or 50 μmmersalyl (M) for 6 hr before nuclear extract preparation, followed by analysis of HIF-1 DNA-binding activity and expression of HIF-1α and HIF-1β protein. C, Constitutive factor.
Among the many biological properties of mersalyl reported in the literature, mersalyl has been shown to induce glycogen deposition (Boot, 1996). Mersalyl is relatively cell-impermeant, and this observation suggests that it might activate a cell surface receptor for insulin. We therefore considered the possibility that the action of mersalyl was mediated by the IGF-1R, which binds both insulin and IGF-1. To test this hypothesis, we assayed VEGF mRNA expression in W cells, a wild-type mouse embryo fibroblast cell line, and in R− cells, which are fibroblasts derived from mouse embryos homozygous for a null allele at the Igf1rlocus (Sell et al., 1994). In W cells, VEGF mRNA expression was induced by hypoxia and strongly induced by mersalyl (Fig.5). In R− cells, VEGF mRNA expression was induced by hypoxia but exposure to mersalyl resulted in little or no induction.

VEGF mRNA expression in wild-type and IGF-1R-deficient mouse embryo fibroblasts. W and R− cells were left untreated (U) or were exposed to hypoxia (H) or 50 μm mersalyl (M) for 14 hr before isolation of total RNA and analysis by blot hybridization, using probes for VEGF mRNA and 18 S rRNA.
Analysis of HIF-1 expression revealed that hypoxia or mersalyl (in either the presence or the absence of serum) induced HIF-1 DNA-binding activity in W cells (Fig. 6). As in Rat-1A cells (Fig. 4), mersalyl was less effective than hypoxia as an inducer of HIF-1 DNA-binding activity but was equally effective as an inducer of HIF-1α and HIF-1β protein expression in W cells (Fig.6). In contrast, neither HIF-1 DNA-binding activity nor HIF-1α and HIF-1β protein expression was induced by mersalyl in R− cells; these cells manifested a response to hypoxia that was similar to that of W cells. In both W and R− cells, HIF-1 expression was elicited by all three previously known inducers (hypoxia, DFO, and CoCl2) (Fig. 7). Thus, R− cells manifested a specific loss of responsiveness to mersalyl as an inducer of HIF-1 expression.

HIF-1 expression in W and R−fibroblasts. Cells were left untreated (U) or were exposed to hypoxia (H) or 100 μm mersalyl in the presence (M) or absence (M*) of 10% fetal bovine serum for 8 hr before nuclear extract preparation and analysis of HIF-1 expression. C, Constitutive factor.

Induction of HIF-1 expression by hypoxia, DFO, or CoCl2 in W and R− fibroblasts. Cells were left untreated (U) or were exposed to hypoxia (H), 130 μm DFO, or 100 μmCoCl2 for 6 hr before nuclear extract preparation and analysis of HIF-1 expression. C, Constitutive factor.
Several cellular responses to insulin and IGF-1 are mediated by MAP kinases (Coolican et al., 1997). These responses can be blocked by exposure of cells to PD098059, a specific inhibitor of the activation of MAP kinase kinase 1 (Alessi et al., 1995). Mersalyl-induced HIF-1 expression was partially or almost completely inhibited by exposure of W cells to 10 or 100 μmPD098059, respectively (Fig. 8A). Several cellular responses to insulin and IGF-1 are mediated by PI-3-kinase and can be inhibited by wortmannin (Cheatham et al., 1994;Coolican et al., 1997). However, wortmannin, at concentrations up to 100 nm, had little effect on the induction of HIF-1 by mersalyl (Fig. 8B). It is noteworthy that insulin stimulation of MAP kinase activity is not blocked by inhibitors of PI-3-kinase (Cheatham et al., 1994). Neither wortmannin nor PD098059 inhibited hypoxia-induced HIF-1 expression (Fig. 8C). Thus, mersalyl induces HIF-1 expression by a unique mechanism that may require IGF-1R and MAP kinase kinase 1 activity.

Effects of PD098059 and wortmannin on the induction of HIF-1 by mersalyl and hypoxia. A and B, W cells were incubated for 6 hr in the absence (−) or presence (+) of mersalyl and 10–100 μm PD098059 (PD) (A) or 1–100 nm wortmannin (W) (B). C, W cells were incubated under nonhypoxic (−) or hypoxic (+) conditions for 6 hr. Where indicated, the cells were exposed to 100 nmwortmannin or 100 μm PD098059 during the incubation. Nuclear extracts were prepared and analyzed for HIF-1 DNA-binding activity (A-C) and HIF-1α protein expression (A and B).C, Constitutive factor.
Discussion
Although hypoxia is the only known physiological stimulus for HIF-1 expression, two different classes of chemical compounds, namely divalent metal ions [such as Co(II), Ni(II), and Mn(II), but not Fe(II)] and iron chelators (including both DFO and the chemically unrelated hydroxypyridinones), have been shown to increase HIF-1α protein levels, HIF-1 DNA-binding activity, and HIF-1-dependent transcriptional activation of hypoxia-inducible genes (Goldberget al., 1988; Wang and Semenza, 1993a,b; Gleadle et al., 1995a; Ho and Bunn, 1996; Huang et al., 1997;Jiang et al., 1997b; Pugh et al., 1997). Recent studies suggested that these two classes of compounds induce HIF-1 expression and downstream gene expression by different molecular mechanisms (Ehleben et al., 1997; Fandrey et al., 1997). In the J1 line of embryonic stem cells, HIF-1α protein, HIF-1 DNA-binding activity, and hypoxia-inducible genes were constitutively expressed, suggesting that HIF-1 expression can be directed by developmental or physiological stimuli other than hypoxia (Iyeret al., 1998). It is not clear whether CoCl2 and DFO activate HIF-1 via the hypoxia signal-transduction pathway or by some other mechanism. However, mersalyl induced HIF-1 by a pathway that is mechanistically distinct from those activated by hypoxia, CoCl2, or DFO. This conclusion is based on the following two observations: 1) HIF-1 was induced by stimuli other than mersalyl in cells lacking IGF-1R tyrosine kinase expression and 2) HIF-1 induction by mersalyl was specifically inhibited by treatment with PD098059.
Tyrosine kinase activity is required for the induction or modulation of HIF-1 and VEGF expression. First, the tyrosine kinase inhibitor genistein blocked hypoxia-induced expression of HIF-1α protein and HIF-1 DNA-binding activity (Wang et al., 1995b) and VEGF mRNA (Mukhopadhyay et al., 1995). Second, cells transfected with the v-src oncogene (which encodes a constitutively active tyrosine kinase) expressed HIF-1α, HIF-1 DNA-binding activity, and ENO1 and VEGF mRNA under nonhypoxic conditions (Jiang et al., 1997a). Remarkably, in contrast to W cells, R− cells were refractory to transformation by all oncogenes tested, with the notable exception of v-src(Valentinis et al., 1997). Third, expression of reporter plasmids containing the VEGF HRE was increased in cells transfected with an activated H-ras oncogene, and this induction required an intact HIF-1 binding site, although the role of HIF-1 was not investigated directly (Mazure et al., 1997). In contrast to the induction mediated by mersalyl, H-ras-induced VEGF transcription was inhibited by wortmannin and dominant-negative mutants of PI-3-kinase (Arbiser et al., 1997; Mazure et al., 1997).
Our analysis of W and R− cells has provided evidence suggesting that the induction of HIF-1 and downstream genes by mersalyl is mediated by IGF-1R, although there is currently no evidence for mersalyl binding to IGF-1R. It is perhaps not coincidental that insulin has been shown to induce expression of mRNAs encoding glucose transporters and glycolytic enzymes (for review, see Pilkis and Granner, 1992), which are also induced by HIF-1 in response to hypoxia (Semenza et al., 1996; Iyer et al., 1998). Insulin may exert its effects on glycolysis at least in part through HIF-1, which would provide a mechanism to integrate the regulation of energy and O2 homeostasis at the transcriptional level. Additional evidence of cross-talk between these systems is provided by the demonstration that expression of IGF-2 and IGF-binding proteins is induced by hypoxia in cultured cells (Kim et al., 1998; Tucci et al., 1998).
Mersalyl is a cell-impermeant organomercurial compound that reacts with free thiol groups of proteins and is known to have diuretic properties when administered to laboratory animals (Amores et al., 1994). Our data indicate that, whereas mersalyl has a positive effect on the VEGF and ENO1 gene expression that occurs at least in part via stimulation of HIF-1 activity, it has a selective negative effect on the expression of EPO mRNA that occurs through an unrelated mechanism. The induction of VEGF mRNA expression under hypoxic conditions is the result of both increased mRNA transcription and decreased mRNA degradation (for review, see Ferrara and Davis-Smyth, 1997). The observation that mersalyl induced VEGF mRNA expression to a greater degree than HIF-1 expression suggests that mersalyl may also induce mRNA stabilization, but we have not performed any experiments to address this question directly. Although pharmacological induction of VEGF and ENO1 gene expression may provide therapeutic benefits under ischemic conditions, by stimulating angiogenesis and glycolysis, respectively, agents that also induce EPO expression and erythropoiesis would not be useful because of the risk of vascular accidents associated with polycythemia (Sokol et al., 1995). The observation that mersalyl selectively induced VEGF and ENO1 mRNA expression suggests that it might represent a reasonable lead compound for the development of novel pharmacological approaches to the treatment of ischemic disorders.
Acknowledgments
We are grateful to Renato Baserga for generously providing W and R− cells and to Chi Dang for the gift of Rat-1A cells.
Footnotes
- Received July 14, 1998.
- Accepted August 7, 1998.
-
Send reprint requests to: Gregg L. Semenza, M.D., Ph.D. The Johns Hopkins Hospital, CMSC-1004, 600 N. Wolfe St., Baltimore, MD 21287-3914. E-mail: gsemenza{at}jhmi.edu
-
G.L.S. is an Established Investigator of the American Heart Association. This work was supported in part by grants from the American Heart Association National Center and the National Institutes of Health (R01-DK39869 and R01-HL55338).
Abbreviations
- EPO
- erythropoietin
- HIF-1
- hypoxia-inducible factor 1
- DFO
- desferrioxamine
- VEGF
- vascular endothelial growth factor
- EMSA
- electrophoretic mobility-shift assay
- ENO1
- enolase 1
- HRE
- hypoxia response element
- IGF-1
- insulin-like growth factor-1
- IGF-1R
- insulin-like growth factor-1 receptor
- MAP
- mitogen-activated protein
- PI-3-kinase
- phosphatidylinositol-3-kinase
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}