


Abstract
The mechanism of action of the metastasis suppressor KiSS1 and its receptor GPR54 is still incompletely characterized. Although the loss of KiSS1 expression by tumor cells has been associated with a metastatic phenotype, the nature of the cellular target of the secreted kisspeptins is unknown. Although an autocrine model of action has been generally assumed, metastasis suppression by KiSS1 has also been shown in cells that do not express GPR54, suggesting a paracrine mechanism in which kisspeptins affect cells in the metastatic niche. Activation of GPR54 was shown to inhibit cell motility and invasion of tumor cells, induce the formation of stress fibers, and reduce the expression of matrix metalloproteinase 9. We showed previously that the activation of GPR54 by kisspeptin-10 suppressed CXCR4-mediated chemotaxis in response to stromal cell-derived factor 1/CXCL12 and abolished the phosphorylation of Akt by CXCR4. We also demonstrated that activation of GPR54 inhibited Akt phosphorylation after the activation of epidermal growth factor receptor and the insulin receptor and triggered apoptosis in epithelial and lymphoid cell lines through a mechanism involving extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase. We show here that the activation of GPR54 induced immediate and profound changes of cell morphology, including cytoplasmic condensation and formation of unpolarized plasma membrane protrusions. These events were dependent on Rho and Rho-Associated Kinase (ROCK) activation. The activation of ROCK also contributed to GPR54-mediated apoptosis in 293 cells, and its effect was additive to and independent of ERK activation. These results suggest that RhoA and ROCK are additional key components of the antimetastatic effect of kisspeptins.
KiSS1 was identified as a metastasis suppressor gene in human malignant melanoma cells (Welch et al., 1994; Lee et al., 1996). As opposed to tumor suppressors, metastasis suppressors do not inhibit the growth of primary tumors but specifically target the spread of tumor cells to distant organs and the establishment of secondary lesions. These metastasis suppressors can interfere with any step of the metastatic cascade, including invasion, angiogenesis, migration and homing, intravasation, survival, and proliferation in a new environment (Stafford et al., 2008). The metastatic phenotype may be conferred by the expression of a limited number of genes, including the chemokine receptor CXCR4, which has been shown to program organ-specific metastatic spread by multiple malignant tumor cell types (Kang et al., 2003; Minn et al., 2005). Other genes that contribute to the metastatic phenotype are involved in establishing a supportive microenvironment (Minn et al., 2005).
KiSS1 was originally identified in experiments in which the transfer of human chromosome 6 to metastatic melanoma cells suppressed metastasis in a mouse xenograft model (Welch et al., 1994; Lee et al., 1996). The gene was identified by subtractive hybridization and differential display and was sufficient to suppress the metastatic phenotype of multiple human cell lines including breast cancer (Lee and Welch, 1997) and ovarian carcinoma (Jiang et al., 2005) in mouse xenograft experiments. In clinical studies, the absence of KiSS1 expression has been linked to poor prognosis in several malignancies, including melanoma (Shirasaki et al., 2001; Martins et al., 2008), carcinoma of the ovary (Prentice et al., 2007), stomach (Dhar et al., 2004), urinary bladder (Sanchez-Carbayo et al., 2003), and esophagus (Ikeguchi et al., 2004).
KiSS1 encodes a secreted protein that is sequentially proteolytically processed to generate multiple polypeptides ranging from 54 to 10 amino acids called kisspeptins (Kp) (Kotani et al., 2001; Muir et al., 2001; Ohtaki et al., 2001; Bilban et al., 2004). Their action is mediated through binding to and activation of GPR54, a G protein-coupled receptor (GPCR), also known as AXOR12 and hOT7T175 and coupled to Gαq(Kotani et al., 2001; Muir et al., 2001; Ohtaki et al., 2001; Bilban et al., 2004). GPR54 activation by kisspeptins was shown to block chemotaxis to fetal bovine serum, activate extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase, induce formation of stress fibers, phosphorylate focal adhesion complex, decrease expression of matrix metalloproteinase 9, and reduce cell proliferation in receptor transfectants (Kotani et al., 2001; Ohtaki et al., 2001; Yan et al., 2001). We have shown previously that activation of GPR54 by kisspeptin-10 (Kp-10) inhibited chemotaxis induced by stromal cell-derived factor 1/CXCL12 and its receptor CXCR4 (Navenot et al., 2005), a mechanism shown in several models to be involved in the migration of CXCR4+ tumor cells to the organs that secrete its ligand such as the lungs, liver, brain, and bones (Müller et al., 2001). Activation of GPR54 suppressed Akt phosphorylation by CXCR4 (Navenot et al., 2005). We recently demonstrated that activation of GPR54 also suppressed the phosphorylation of Akt after activation of their respective receptor tyrosine kinase (RTK) by epidermal growth factor and insulin (Navenot et al., 2009). Considering the critical role of Akt in cell survival, we hypothesized that activation of GPR54 could oppose prosurvival mechanisms. Kp-10 was sufficient for induction of apoptosis in cell lines expressing GPR54. However, the negative cross talk between GPR54 and Akt did not seem to play a significant role in this process. Instead, apoptosis was largely dependent on the activation of ERK1/2 in both HEK-293 and Jurkat cells (Navenot et al., 2009).
Cell migration is a key component of the metastatic process, both for tumor cells and stromal cells recruited in the premetastatic niche such as myofibroblasts, hematopoietic, and endothelial progenitor cells (Kaplan et al., 2005; Psaila et al., 2006; Alison et al., 2009). The nature of the cells expressing Kp and GPR54 in the context of primary tumors or metastatic niches has not been elucidated, but studies based on mRNA expression have shown low levels of GPR54 in multiple normal organs, suggesting that this receptor is expressed in rare normal cell populations (Muir et al., 2001; Ohtaki et al., 2001). In this study, we provide evidence that Kp-10 triggers immediate and profound morphological modifications in cells (both adherent and nonadherent) expressing GPR54. These changes seem to be primarily mediated through the activation of Rho and the Rho-dependent kinase ROCK, a signaling pathway that also contributes to GPR54-mediated apoptosis.
Materials and Methods
Cell Lines and Reagents. The human cell line HEK-293T was modified to express GPR54 with an N-terminal Myc epitope tag by transfection with a pcDNA3.1 construct (Invitrogen, Carlsbad, CA) as described previously (Navenot et al., 2005). Jurkat cells were transfected with an Myc-tagged GPR54 in a pME vector (a gift from Dr. Makio Iwashima, Medical College of Georgia, Augusta, GA). After selection either in G418 (293) or puromycin (Jurkat), transfected cells were selected for expression of the Myc tag by magnetic sorting (Miltenyi Biotec, Auburn, CA) using the 9E10 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Kp-10 (YNWNSF-GLRF-NH2) was synthesized at Kyoto University (Kyoto, Japan). Inhibitors of ROCK (Y-27632), protein kinase C (bis-indolylmaleimide-1), phospholipase C (U73122), MEK1/2 (U0126), and phosphatidylinositol-3 kinase (LY294002) were from Calbiochem (La Jolla, CA). All antibodies used for Western blots were from Cell Signaling Technology (Danvers, MA).
Live Cell Microscopy. 293-GPR54 cells were grown for 48 h on glass-bottomed 35-mm dishes (MatTek, Ashland, MA) coated with poly(l-lysine) (50 μg/ml; Sigma, St Louis, MO). Before the beginning of the experiment, growth medium was replaced with 2 ml of Dulbecco's modified Eagle's medium containing 0.25% BSA (Sigma) and 10 mM HEPES, complemented or not with one of the inhibitors (10 μM Y-27632, 50 μM LY294002, 10 μM U0126, 2 μM U73122, and 10 μM bis-indolylmaleimide-1), and the cells were incubated for 1 h in a CO2 incubator at 37°C. The cells were then placed on an inverted microscope (TE-2000E; Nikon, Melville, NY) equipped with a temperature-regulated stage (20/20 Technology Inc., Wilmington, NC). Image acquisition was done with a 60× Plan Apo 1.4 numerical aperture objective with a slider optimized for high-resolution differential interference contrast (DIC) and a charge-coupled device camera (CoolSnap HQ; Roper, Pleasanton, CA) using Metamorph (Molecular Devices, Sunnyvale, CA). The cells were left to equilibrate for a few minutes, and then one frame was captured every 10 s. After approximately 20 frames (time necessary to observe basal cell movements), 20 μl of Kp-10 was added to a final concentration of 100 nM and left for the entire duration of the experiment. This concentration was chosen because it previously achieved the full biological effect of the ligand in in vitro experiments such as inhibition of chemotaxis (Navenot et al., 2005) or induction of apoptosis (Navenot et al., 2009). Movies were assembled at five frames per second using Metamorph. For Jurkat-GPR54, cells were resuspended in RPMI 1640 medium containing 0.25% BSA and 10 mM HEPES at 1 × 105 cells/ml, and 2 ml was added to a poly(l-lysine)-coated dish. The cells were left to settle on the glass for 1 h in a CO2 incubator at 37°C. That procedure did not change the initial cell morphology but allowed the cells to become immobilized on the coverslip so that the same cells would remain within the microscope field for the entire duration of the experiment. Samples were then processed as described for 293-GPR54 cells.
Analysis of Actin Cytoskeleton. 293-GPR54 cells were processed as described above. After continuous exposure to Kp-10 for times ranging from 30 s to 1 h, the cells were washed rapidly in PBS and fixed in PBS containing 4% paraformaldehyde for 1 h at room temperature. The cells were then permeabilized with PBS containing 0.1% Triton X-100 (Sigma) for 1 min. Detection of F-actin was achieved by incubating the cells for 15 min with phalloidin labeled with Alexa Fluor 594 (Invitrogen). After two washes in PBS, the cells were covered with 1.5 ml of PBS and the same fields were analyzed by DIC and for fluorescence using an inverted microscope (Nikon TE-2000E). At any given time point after exposure to Kp-10, cell morphology was similar in fixed and live cells.
Analysis of Apoptosis by Western Blot. 293 cells were seeded at 5 × 104 cells/35-mm dish in complete growth medium. After 24 h, the medium was replaced with serum-free medium containing 0.25% BSA. For inhibition experiments, the medium contained the inhibitor of MEK1/2 (U0126, 10 μM) or the inhibitor of ROCK (Y-27632, 10 μM). After 1 h at 37°C, Kp-10 (100 nM) was added, and the cells were grown for another 48 to 72 h. The cells in the supernatant were collected and washed in ice-cold PBS. The cells adhering to the dishes were also washed in ice-cold PBS. Detached and adherent cells were pooled for each sample, and lysates were prepared by resuspending the pellets in SDS-reducing sample buffer for 5 min at 100°C. The relative amount of proteins in the samples was determined after SDS-polyacrylamide gel electrophoresis and Coomassie blue staining of 10 μl of each sample and analysis with a LAS-3000 digital imaging system (Fuji, Stamford, CT). Identical amounts of proteins of all samples were then analyzed by Western blotting, the polyvinylidene difluoride membranes (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) being incubated with antibodies specific for caspase 9, cleaved caspase 9, caspase 7, cleaved caspase 7, PARP, and cleaved PARP. After incubation with the appropriate horseradish peroxidase-labeled antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) followed by the ECL Plus substrate (GE Healthcare), blots were analyzed with the LAS-3000 system, and the intensity of the specific bands was quantified using the Multi Gauge software (Fuji).

Changes of cell morphology induced by Kp-10 in 293-GPR54 cells. A, DIC images of 293-GPR54 cells grown for 48 h on poly(l-lysine)-coated glass-bottomed Petri dishes before and after the addition of Kp-10. The same field is shown for all time points. Before stimulation with Kp-10 (t = -220 s and t = 0), the cells only display slow and small movements of the plasma membrane at the periphery. Within 30 s after exposure to Kp-10, the cell cytoplasm retracts around the nucleus, pulling on the membrane and disrupting intercellular junctions and contacts with the support. Numerous cells display highly mobile and randomly positioned membrane protrusions that simultaneously extend and retract for more than 30 min (arrows). B, intense formation of membrane ruffles (arrows) is visible at the edge of the cells in contact with the support. Results are representative of more than 10 identical experiments.
Analysis of Rho, Rac1, and Cdc42 Activation. The determination of activated small GTPases was performed by precipitation of GTP-bound forms of the proteins followed by Western blotting analysis using kits from Millipore Inc. (Billerica, MA). Adherent 293-GPR54 cells were exposed to Kp-10 (or medium for the negative control), rinsed in ice-cold PBS, and lysed in the lysis buffer provided. For pull-down of activated GTPases, cell lysates were incubated with glutathione-agarose beads bound to a GST fusion protein, including a polypeptide from the Rho binding domain of mouse Rhotekin (for Rho analysis) or from the p21-binding domain of human p21-activated kinase-1 (for Rac1 and Cdc42 analysis) according to the manufacturer's protocol. Precipitated proteins were analyzed by Western blotting with antibodies specific for Rho (-A, -B, -C), Rac1, or Cdc42 (Upstate), as described above.
Analysis of Myosin Light Chain 2 Phosphorylation. Adherent 293-GPR54 cells were exposed to Kp-10 for the indicated time, washed in PBS, and lysed in SDS sample buffer. Lysates were analyzed by western blotting as described above with antibodies specific for total myosin light chain 2 (MLC2) or phosphorylated MLC2 (Thr18 and Ser19) (Cell Signaling Technology).
Statistics. Comparison of averages of replicates of experimental data was performed with the Student's t test.
Results
Activation of GPR54 by Kp-10 Results in Immediate Alteration of Cell Shape. The real-time changes induced by exposure to Kp-10 were examined by high-resolution DIC microscopy. Addition of Kp-10 triggered within 20 to 30 s a strong contraction of the cytoplasm in the majority of adherent 293-GPR54 cells. This cytoplasmic contraction around the nucleus resulted in a separation of the plasma membrane from the coverslip at the periphery of the cells and rupture of the intercellular adhesions [Fig. 1A and Supplemental Fig. 1 Video (Supplemental Fig. 1 293-GPR54 Kp-10.wmv)]. This phenomenon was accompanied by an intense formation of membrane ruffles visible at the edge of the cells still in contact with the support (Fig. 1B and Supplemental Fig. 1) and the appearance of numerous short filipodiae. The cells displaying strong cytoplasmic contractions also formed long membrane protrusions that seemed to extend and contract simultaneously (Fig. 1A). Because contraction and membrane protrusions were generalized, there was no evidence of cell polarization. The duration of these changes was greater than 30 min after the addition of Kp-10, although the intensity diminished over time. After 30 to 45 min of continuous exposure to Kp-10, the cytoplasmic contractions progressively subsided, indicating desensitization of the signaling of GPR54 to the cytoskeleton. The cells started spreading over the growth surface again and re-establishing cell-cell contact. None of these morphological changes occurred in 293-GPR54 in the absence of Kp-10 (adding PBS as a control) or in control 293 cells lacking GPR54 after exposure to Kp-10 (data not shown).
The possible cell line specificity of these effects was tested in the nonadherent lymphoid cell line Jurkat. The cells were left to settle onto a glass coverslip coated with poly(l-lysine) before the experiment so that most of the cells would be immobilized. Exposure of Jurkat-GPR54 cells to Kp-10 resulted in the rapid appearance, within seconds of the addition of the ligand, of filipodiae, membrane ruffles, and long membrane protrusions similar to those observed in 293-GPR54 [Supplemental Fig. 2 Video (Suppl Fig. 2 Jurkat-GPR54 Kp-10.wmv)].
Morphological Changes Correlate with Modifications of the Actin Cytoskeleton. HEK-293-GPR54 were exposed to Kp-10, fixed at different times, and stained with fluorescent phalloidin to establish the role of F-actin in the morphological changes observed by time-lapse microscopy. As shown in Fig. 2A, unstimulated cells had limited amounts of F-actin, mostly located in nucleation sites in the cytosol, and no detectable fibers. After the addition of Kp-10, polymerized actin was rapidly formed at the periphery of the contracting cells (Fig. 2B, 30 s) and in membrane ruffles. The highest concentrations were localized in the membrane protrusions (Fig. 2C, 1 min; 2D, 15 min). Stress fibers were not observed before 15 min and were fully formed 30 min after the addition of Kp-10 (Fig. 2, E and F), after cell contraction and synchronous with cell respreading.

Activation of GPR54 induces two waves of actin polymerization. A, staining with fluorescent phalloidin indicates that unstimulated 293-GPR54 cells show little polymerized actin, mostly in the form of nucleation sites distributed in the cytosol. B, Kp-10 first triggers actin polymerization at the periphery of the cells as the cells contract around the nucleus. C, high concentrations of F-actin can be observed below the surface of the membrane protrusions (arrows). The inset shows a different focal plane. D, stress fibers become visible after 15 min of exposure to Kp-10, and F-actin-rich protrusions are still forming (insets shows a different focal plane for DIC and fluorescence of phalloidin). E and F, stress fibers are completely organized after 30 min when the cell movements begin to subside and the cell spreads again on the glass surface. F, magnification of the inset shown in E. Results are representative of three similar experiments.
Cell Morphological Changes Are Dependent of Rho and ROCK Activation. Because the change of cell shape and the remodeling of the actin cytoskeleton suggested the activation of the small G proteins, we investigated whether activation of GPR54 triggered Rho activation and whether this was necessary for the morphological alterations. GST pull-down experiments showed that exposure of 293-GPR54 to Kp-10 resulted in a strong and rapid activation of RhoA (Fig. 3A). A moderate activation of Rac1 was also noted, but no change in the activation state of cdc42 could be detected (Fig. 3B). The contractile response to GPR54 activation temporally coincided with phosphorylation of MLC2, detected by Western blotting with antibodies specific for phosphorylated Thr18 and Ser19 (Fig. 3C). Experiments with pharmacological inhibition were performed to determine whether MLC2 phosphorylation was mediated by the myosin light chain kinase or by the Rho-activated kinase ROCK1. A pretreatment of the cells with the ROCK inhibitor Y-27632 before the addition of Kp-10 blocked the phosphorylation of MLC2 by GPR54, demonstrating the involvement of the Rho-ROCK axis in this procontractile signaling (Fig. 3C).
The role of Rho and ROCK in GPR54-mediated cell shape modifications was further studied by time-lapse microscopy. Pretreatment of 293-GPR54 with Y-27632 induced the formation of thin cytoplasmic extensions, typical of the morphology observed with ROCK inhibition (Fig. 3D). This pretreatment inhibited both cytoplasmic contraction and the formation of membrane protrusions after exposure to Kp-10 [Fig. 3D and Supplemental Fig. 3 Video (Suppl Fig. 3 293-GPR54 Y-27632 Kp-10.wmv)]. However, the cells were still capable of slowly spreading, filling the gaps between the cells, and re-establishing cell-cell contacts, as well as forming membrane ruffles (Fig. 3D). This suggests that GPR54, besides the activation of RhoA through Gαq, is also capable of activating Rac, as shown in pull-down experiments (Fig. 3B). Cells treated with the ROCK inhibitor showed small cytoplasmic fibers of polymerized actin upon stimulation with Kp-10 but were incapable of assembling well organized stress fibers (Fig. 3D).
Pharmacological inhibition of phospholipase C, protein kinase C, or MEK had no effect on the capacity of Kp-10 to elicit in 293-GPR54 cells the observed morphological changes and actin polymerization (data not shown). Whereas inhibition of phosphatidylinositol-3 kinase by LY294002 or wortmannin suppresses the capacity of 293 or Jurkat cells to migrate in response to a chemoattractant like CXCL12, the same treatment did not suppress the morphological changes generated by the activation of GPR54 (data not shown).
Activation of Rho-ROCK by GPR54 Contributes to Apoptosis. ROCK activation has been implicated in apoptosis. Although the membrane protrusions induced by GPR54 activation were not temporally related to membrane blebbing observed in apoptotic cells, experiments were performed to determine the potential role of ROCK activation in GPR54-induced apoptosis. We previously showed that the inhibition of MEK reduced apoptosis in 293-GPR54. Pharmacological inhibition of ROCK suppressed the induction of apoptosis by Kp-10 with a potency similar to MEK inhibition as analyzed by Western blotting experiments analyzing cleavage of caspase 9, caspase 7, and PARP (Fig. 4A). The inhibition by Y-27632 was further enhanced in the presence of the MEK inhibitor U0126 (Fig. 4A). The independence of ERK and ROCK activation was confirmed in Western blotting experiments in which the inhibition of ROCK by Y-27632 had no effect on the phosphorylation of ERK by Kp-10 (Fig. 4B).
Discussion
Despite the dramatic ability of KiSS1 to reverse the metastatic phenotype, our insight into the mechanism of action of Kp and the cognate receptor, GPR54, is limited. Initial studies that focused on the potential autocrine effects of Kp on cells lines transfected with GPR54 showed loss of motility and invasiveness, formation of stress fibers, activation of focal adhesion, and decreased expression of matrix metallo-proteinase 9 (Kotani et al., 2001; Ohtaki et al., 2001; Yan et al., 2001). Whereas some of these signaling events are shared by ligand-receptor pairs that promote cell motility and chemotaxis, the discovery that GPR54 activation negatively regulates Akt and abolishes the activation of Akt by the chemokine receptor CXCR4 in response to CXCL12 (Navenot et al., 2005) introduced a possible mechanism for the suppressive effect of GPR54 on cell motility. We also demonstrated that the same negative cross-talk to Akt extended to the signaling of RTK for epidermal growth factor and insulin, thus inhibiting a signaling component essential to cell survival as well as directed cell migration (Navenot et al., 2009). Activation of GPR54 by Kp-10 was sufficient to induce the apoptosis of 293 and Jurkat cells through a mechanism involving ERK activation (Navenot et al., 2009).

GPR54 activates RhoA and ROCK to induce morphological changes and actin polymerization. A, pull-down experiments using Rhotekin-conjugated agarose beads were performed with duplicates of cell lysates for each time point (1, 2, and 5 min). Proteins eluted from the beads were resolved by SDS-polyacrylamide gel electrophoresis followed by Western blotting with anti-Rho antibody. Results show that activation of GPR54 rapidly generates activated GTP-bound Rho. The histogram shows the quantitative results of two separate experiments that indicate increased activation of Rho at each time point (*, p < 0.05; **, p < 0.01). B, pull-down experiments using p21-activated kinase-1-conjugated agarose followed by Western blotting with anti-Rac1 or anti-cdc42 antibodies show moderate activation of Rac1 but no activation of cdc42 upon GPR54 activation. C, activation of the Rho-ROCK pathway by GPR54 is responsible for the phosphorylation of MLC2 and activation of the contractile machinery. 293-GPR54 cells, pretreated or not with the ROCK inhibitor Y-27632, were stimulated with Kp-10 for the indicated time. Cell lysates were analyzed by Western blotting with antibodies for total or phosphorylated MLC2. Kp-10 induced rapid phosphorylation of MLC2 that was suppressed after inhibition of ROCK. Histograms show quantification of Western blotting results. Results are representative of three similar experiments. D, inhibition of ROCK modified the morphology of 293-GPR54 cells before exposure to Kp-10. After ROCK inhibition, cytoplasmic contraction and emission of membrane protrusions upon the addition of Kp-10 were abolished, but slow cell spreading was preserved, indicating ROCK-independent modifications of the cytoskeleton. In the presence of ROCK inhibition, activation of GPR54 induced the formation of multiple small actin fibers that did not organize in bundles of stress fibers. Bottom, magnifications of the insets. Cell nuclei stained with Hoechst 33342 are shown in the lower magnification image only. Data are representative of four identical experiments.
The data presented here demonstrate that the inhibition of motility resulting from the activation of GPR54 includes profound morphological changes and reorganization of the actin cytoskeleton mediated by the activation of Rho and its effector ROCK. In addition, ROCK activation was involved in apoptosis induced by Kp-10. Cell migration (whether mesenchymal, amoeboid, or collective) is dependent on dynamic changes in the actin cytoskeleton and integrin adhesion complexes that are spatially and temporally regulated by the Rho family members of small GTP-binding proteins Rho, Rac, and Cdc42 (Raftopoulou and Hall, 2004; Vega and Ridley, 2008). The disorganization of this system by the powerful effect of GPR54 activation may be responsible for the decreased motility of cells, both chemotactic and chemokinetic.
GPCRs coupled to Gαq have been shown to affect the activity of small G proteins, and Gαq has been recognized as an activator of RhoA. Constitutively active mutants of Gαq as well as Gα12 and Gα13 are capable of triggering Rho-dependent neurite retraction (Katoh et al., 1998), as observed for LPA4, the receptor for lysophosphatidic acid (Yanagida et al., 2007). The connection between Gαq and RhoA activation has been demonstrated in other systems (Chikumi et al., 2002; Vogt et al., 2003) and involves the guanine nucleotide exchange factor LARG (Vogt et al., 2003), but does not seem to be universal, because the Gαq-coupled angiotensin II receptor (AT1R) did not activate Rho (Takashima et al., 2008). Polymerization of actin, formation of stress fibers, phosphorylation of MLC2 by Rho effector ROCK, and subsequent contraction of actomyosin are the traditional effects of Rho activation and were all observed here after GPR54 activation. It is likely that the strong and sustained cell contraction we observed is responsible for the vasoconstrictive effect reported after activation of GPR54 by Kp in smooth muscle cells in the aorta, coronary arteries, and umbilical vein (Mead et al., 2007). However, the role of Rho has been shown to be even more complex, and RhoA is now recognized as an important factor in the formation of membrane ruffles and membrane protrusions in migrating cells in conjunction to Rac, the traditional effector (Fukata et al., 1999; Palazzo et al., 2001; Pertz et al., 2006). The membrane protrusions we observed in 293 and Jurkat cells expressing GPR54 seemed to involve Rho activation as they were, along with the cytoplasmic contractions, totally abolished after pharmacological inhibition of ROCK. The formation of lamellipodiae and membrane ruffles and the slow cell-spreading induced by Kp-10 in 293-GPR54 after ROCK inhibition may result from both ROCK-independent Rho signaling and Rac activation, because moderate Rac activation was observed (Fig. 3B). This differentiates the signaling of GPR54 from the one recently reported for the sphingosine-1-phosphate receptor 2 (S1P2R) and angiotensin II receptor AT1R (Takashima et al., 2008). Whereas AT1R was incapable of inhibiting migration, S1P2R was shown to inhibit platelet-derived growth factor-induced migration of vascular smooth muscle cells and membrane ruffles formation though the activation of Gαq and Gα12/13. The inhibitory action of S1P2R involved the inhibition of Rac and was dependent on Rho but not on ROCK. These differences in the physiological effects of Gαq-coupled receptor on cell migration illustrate the complexity of the regulation of small GTP-binding proteins (Takashima et al., 2008).
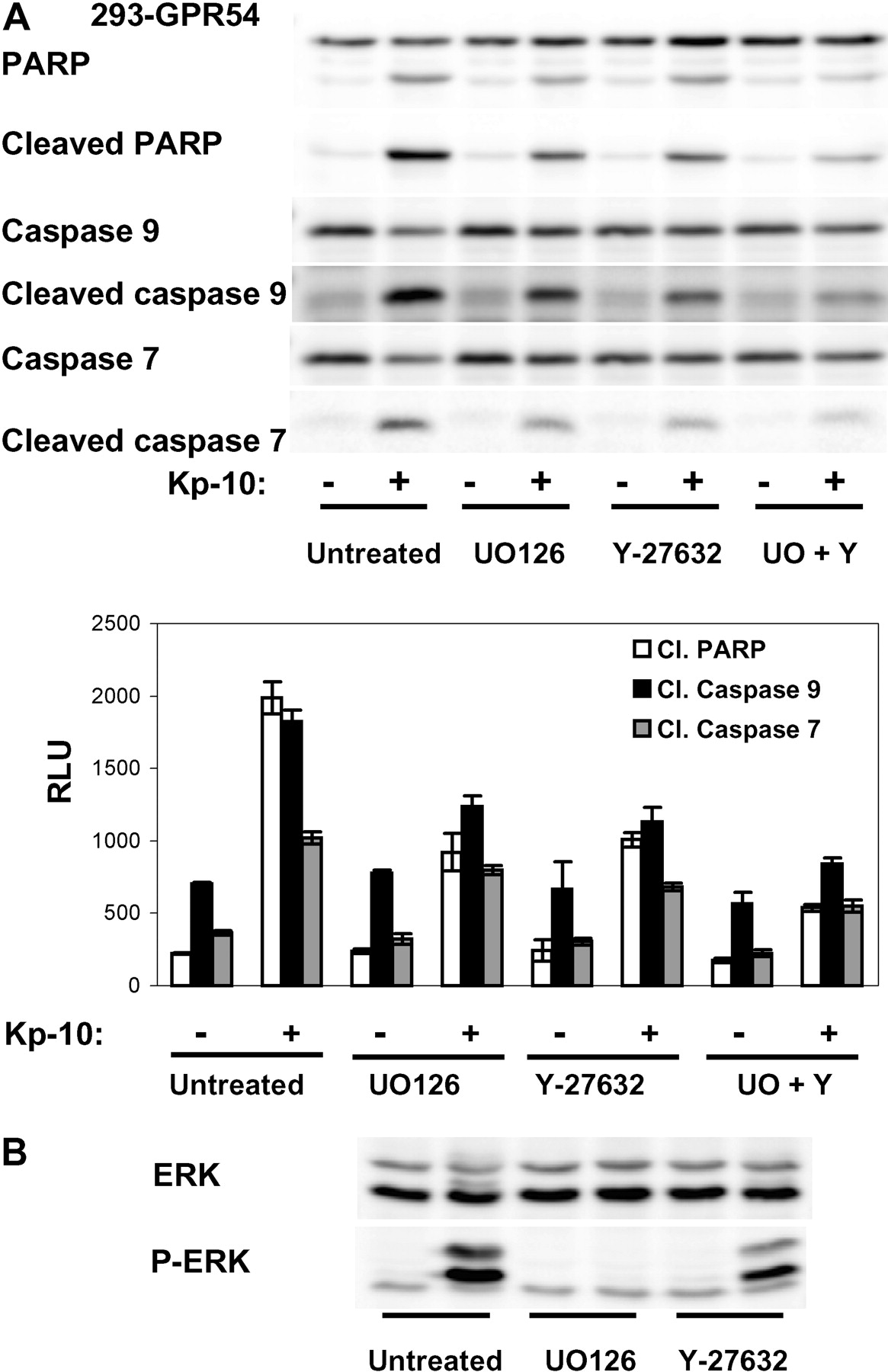
Activation of Rho-ROCK by GPR54 contributes to apoptosis. A, inhibition of ROCK inhibits Kp-10-induced apoptosis in 293-GPR54. Cells were exposed to Kp-10 (100 nM) for 72 h before lysis in SDS sample buffer and analysis by Western blotting for activation of caspases 9 and 7 and cleavage of PARP. For ROCK inhibition, cells were pretreated with Y-27632 for 1 h before the addition of Kp-10. The MEK inhibitor U0126 was used as a control of inhibition of apoptosis. Pretreatment with Y-27632 reduced apoptosis with an efficacy similar to the MEK inhibitor. The combined inhibitions of MEK and ROCK further decreased apoptosis. The histogram represents the quantitative analysis (mean ± S.D.) of two identical experiments. B, inhibition of ROCK had no effect on the ERK activation by GPR54, indicating that the two pathways trigger apoptosis independently. Western blot shows the results of one experiment representative of three similar experiments.
Besides its well recognized role in cell migration, ROCK activation has recently been implicated in apoptosis and, more specifically, the phenomenon of membrane blebbing. In fact, the mechanism of activation of ROCK (change of conformation upon interaction with GTP-bound Rho) was reported to be mimicked by the proteolytic cleavage of its C terminus by activated caspase 3 and granzyme-B (Coleman et al., 2001; Sebbagh et al., 2001, 2005). In this study, we showed that ROCK activation was a contributing factor in GPR54-mediated apoptosis in 293 cells, independent of the role of ERK we established previously. Because activation of Rho and ROCK was also responsible for the morphological changes and the resulting temporary loss of cell-cell contacts and cell adhesion to the support, anoikis is likely to be a contributing factor of GPR54-mediated apoptosis. However, experiments with inhibition of the MEK-ERK pathway by U0126 indicated the existence of an ERK-dependent mechanism that was unrelated to anoikis.
A more complete picture of the potential mechanisms of the antimetastatic signaling of GPR54 begins to emerge. In cells expressing the receptor, exposure to Kp inhibits the Akt activation normally elicited by both GPCR and RTK, thus blocking a pathway critical to both cell survival and directional cell motility. In addition, the activation of Rho and ROCK by GPR54 demonstrated here disturbs the tightly regulated activity of Rho GTPases necessary for dynamic reorganization of the cytoskeleton and cell migration. GPR54-mediated activation of ROCK also contributes to the induction of apoptosis in conjunction with ERK activation. Because these studies were performed in cell lines that over-express GPR54 and do not represent a specific type of tumor cell (e.g., metastatic carcinoma) or stromal cell, each effect observed may not be relevant to a specific cell type or may not be quantitatively representative of a physiological situation. However, it can be rationalized that the strength of GPR54 is to potentially initiate partly independent and partly overlapping signaling mechanisms that collaborate to render GPR54+ cells exposed to Kp largely unresponsive to growth factors and chemokines, depriving them from a nurturing support and from the directional cues necessary for cell migration and homing. These effects not only represent a potential mechanism for the autocrine action of Kp on GPR54+ tumor cells but also support the hypothesis of a paracrine mode of action. Previous studies have shown that, in mouse models, Kp can block metastasis of cells that do not express GPR54 and do not respond to Kp in vitro (Lee and Welch, 1997; Becker et al., 2005; Nash et al., 2007). This raises the possibility that, at least in some types of malignancies, Kp secreted by tumor cells acts on some GPR54+ cellular components of their microenvironment in the metastatic niche such as carcinoma-associated fibroblasts, hematopoietic or endothelial progenitor cells, for instance by affecting their recruitment, survival, or function. The definition of the nature of the cells expressing the ligand and the receptor will provide additional insight into the part of the GPR54 signaling described here that is most relevant in any given type of malignancy.
Footnotes
-
This work was supported by the Georgia Cancer Coalition.
-
ABBREVIATIONS: Kp, kisspeptin; BSA, bovine serum albumin; DIC, differential interference contrast; ERK, extracellular signal regulated kinase; GPCR, G protein-coupled receptor; MLC2, myosin light chain 2; ROCK, Rho-associated kinase; RTK, receptor tyrosine kinase; HEK, human embryonic kidney; PBS, phosphate-buffered saline; PARP, poly(ADP-ribose) polymerase; S1P2R, sphingosine-1-phosphate receptor 2; AT1R, angiotensin II receptor; Kp-10, kisspeptin-10; MEK, mitogen-activated protein kinase kinase; U0126, 1,4-diamino-2,3-dicyano-1,4-bis(methylthio)butadiene; U73122, 1-[6-[[17β-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione; LY294002, 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride; Y-27632, N-(4-pyridyl)-4-(1-aminoethyl)cyclohexanecarboxamide dihydrochloride.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. -
↵1 Current affiliation: Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia, Pennsylvania.
- Received January 27, 2009.
- Accepted March 12, 2009.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}