


Abstract
Hyperpolarization-activated, cyclic nucleotide–gated (HCN) channels are important members of the voltage-gated pore loop channels family. They show unique features: they open at hyperpolarizing potential, carry a mixed Na/K current, and are regulated by cyclic nucleotides. Four different isoforms have been cloned (HCN1–4) that can assemble to form homo- or heterotetramers, characterized by different biophysical properties. These proteins are widely distributed throughout the body and involved in different physiologic processes, the most important being the generation of spontaneous electrical activity in the heart and the regulation of synaptic transmission in the brain. Their role in heart rate, neuronal pacemaking, dendritic integration, learning and memory, and visual and pain perceptions has been extensively studied; these channels have been found also in some peripheral tissues, where their functions still need to be fully elucidated. Genetic defects and altered expression of HCN channels are linked to several pathologies, which makes these proteins attractive targets for translational research; at the moment only one drug (ivabradine), which specifically blocks the hyperpolarization-activated current, is clinically available. This review discusses current knowledge about HCN channels, starting from their biophysical properties, origin, and developmental features, to (patho)physiologic role in different tissues and pharmacological modulation, ending with their present and future relevance as drug targets.
I. Introduction
The family of hyperpolarization-activated, cyclic nucleotide–gated (HCN) channels has attracted increasing attention since the discovery of the coding genes and corresponding proteins, in the late 1990s (Ludwig et al., 1998; Santoro et al., 1998). At that time, the announcement that the molecular fingerprint of the funny current (If) (Brown et al., 1979)—alias hyperpolarization-activated current (Ih) or queer current (Halliwell and Adams, 1982)—was finally sequenced and generated great excitement in a vast audience of scientists belonging to different disciplines, from cardiology to neurosciences, from biophysics to molecular biology and pharmacology. Prompted by the advancement of molecular, electrophysiological, and optical techniques, a mass of information has been accumulating rapidly and exponentially on the biophysical properties and regulatory features of HCN isoforms, including their tissue distribution. Many aspects have been reviewed previously in excellent papers (Accili et al., 2002; DiFrancesco and Borer, 2007; Biel et al., 2009; DiFrancesco and DiFrancesco, 2015). Therefore, this review will recall briefly the main tracts of HCN characteristics, pointing to recent discoveries, and will focus on HCN contribution to cell function and dysfunction, trying to emphasize the potential role of these channels as a target of existing or novel pharmacological approaches.
II. Hyperpolarization-Activated Cyclic Nucleotide–Gated Channels: Basic Facts
The hyperpolarization-activated cyclic nucleotide–modulated proteins are voltage-dependent ion channels, conducting both Na+ and K+, blocked by millimolar concentrations of extracellular Cs+, and modulated by cyclic nucleotides (mainly cAMP) that contribute crucially to the pacemaker activity in cardiac nodal cells and impulse generation and transmission in neurons (DiFrancesco et al., 1986; Pape and McCormick, 1989; DiFrancesco, 1993; Pape, 1996). Their molecular and functional expression has been also detected in human and animal tissues not canonically classified as excitable and in undifferentiated (e.g., stem) or immature cell types. Altogether, these pieces of information allow speculation that HCN channels play a role beyond pacemaking and raise the interest for HCN as targets of therapies, including—but not limited to—ivabradine, the only available drug to date acting as a specific bradycardic agent.
Before starting with the fundamental properties of these channels, Brown et al. (1979) should be acknowledged for the first description of a current activated upon hyperpolarization in the sinoatrial node (SAN); they termed the current funny (If) for its peculiar voltage dependence. However, the most general term hyperpolarization-activated current (Ih) (Yanagihara et al., 1980; Yanagihara and Irisawa, 1980) will be used throughout the text to indicate the current.
A. Genetic and Molecular Characteristics of the HCN Family
The HCN family consists of four isoforms (HCN1–4). The first three full-length cDNAs encoding for HCN1–3 were identified in the mouse brain (Ludwig et al., 1998; Santoro et al., 1998). A fourth isoform (HCN4) was detected by screening a cDNA library from the human heart, and its expression was found remarkably higher throughout human cardiac tissue (atria and ventricles) than in brain (Ludwig et al., 1999). Afterward, HCN4 was detected also in other tissues, such as thalamus and testis (Seifert et al., 1999). A more detailed description of tissue-specific distribution of HCN isoforms will be given in the next section Distribution, Adaptive, and Maladaptive Role of HCN Channels in Mammals.
The general sequence of HCN genes resembles that of six-transmembrane segment, voltage-activated channel subunits; four subunits assemble in homo- or heterotetramers with a stoichiometry that is not completely defined yet (Chen et al., 2001b; Xue et al., 2002; Altomare et al., 2003; Much et al., 2003; Whitaker et al., 2007; Ye and Nerbonne, 2009). However, HCN channels encompass a unique combination of traits typical of different channel families (Fig. 1).

HCN channel topology and associated interacting proteins. Transmembrane and cytoplasmic arrangement of the HCN1 channel subunit (Protein Data Bank code: 5u6p) with distinct domains for interaction with ancillary proteins predicted at the N terminus (caveolin) (Barbuti et al., 2012) and C terminus (Trip8b) (Saponaro et al., 2014); Filamin-A (Gravante et al., 2004); and Nedd4-2 (Wilkars et al., 2014). MIRP-1 (Yu et al., 2001) and KCR1 (Michels et al., 2008) interact also with HCN channels.
First, HCNs have a pore region between S5 and S6, highly conserved in all isoforms and very much like to K+-selective voltage-dependent channels (Kv) in the amino acid sequence (Robinson and Siegelbaum, 2003; Biel et al., 2009). Despite similarity, HCN channels conduct both Na and K ions with a scarce selectivity (DiFrancesco, 1981b; Gauss et al., 1998; Macri et al., 2012). Before defining the structure of HCNs, the evidence that, at variance with K channels, large cations such as Ba2+ or tetraethylammonium do not block Ih (DiFrancesco, 1981a; Ludwig et al., 1998) and that substitution of Thr/Ser residue typical of Kv channels (Zhou and MacKinnon, 2004) with Cys peculiar of HCN has no effect on selectivity (Macri et al., 2012), led to hypothesize the existence of a functionally wider pore. The recent definition of HCN (Lee and MacKinnon, 2017) provides an intriguing explanation: as stated, only two filter sites are present in the HCN pore, at variance with the four sites of Kv channels; moreover, surrounding amino acids reorientate filter amino acids, namely Tyr. Selectivity depends on the limited space allowed by two K+ ions, aligned and bound to the filters in Kv channels. In the presence of a single-bound K+ in HCN, Na+ ions can easily permeate the pore, without binding, thus conducting inward current (Lee and MacKinnon, 2017), with an exceptionally low (∼1 picoSiemens) unitary channel conductance (0.5–1.7 picoSiemens for HCN1 and HCN2, respectively) (DiFrancesco, 1986, Thon et al., 2013; Liu et al., 2016). These properties are essential for the functional role of Ih. In fact, a net inward current flows during the diastolic depolarization of cardiac pacemaker cells (∼−60 mV) resulting from the following: 1) the relative (opposite) contribution of Na+ entry and K+ exit through the open channels, moving along their electrochemical gradients, and 2) the relative permeability, approximately 4:1 for K+ over Na+ (McCormick and Pape 1990b; Ho et al., 1994; Ludwig et al., 1998; Santoro et al., 1998). External K+ concentration greatly influences channel conductance (DiFrancesco, 1982; Maccaferri et al., 1993; Cerbai et al., 1994), i.e., elevation of extracellular K+ due to repetitive firing can amplify Ih and promote depolarization.
Second, HCNs possess a standard voltage sensor in S4, very similar to depolarization-activated channels, i.e., with a regular sequence of positive charged amino acids (Lys and Arg) (Kaupp and Seifert, 2001). A possible explanation of the reverse voltage dependence of these channels comes from the recent structural study by Lee and MacKinnon (2017) on HCN1. They hypothesize that the extraordinary size of S4 and its closeness to the S5–S6 pore, combined with the packed conformation of the S5–S6 helices, compress the pore in a closed state when the membrane is depolarized. Eventually, the inward movement of S4 caused by hyperpolarization displaces the link between S4 and S5, pulling S5 far from S6 and opening the pore like a zipper, instead of closing it as in Kv channels (Männikkö et al., 2002). Such a molecular coupling mechanism between S4, S5, and the C-linker [the region between S6 and the cyclic nucleotide binding domain (CNBD)] was inferred previously on the basis of pioneer studies on gating properties of sea urchin sperm flagellar HCN (SpHCN) by introducing cysteine in the S4–S5 and C-linker and using Cys cross-linking agents such as Cd2+ (Prole and Yellen, 2006).
Third, HCNs possess a distinctive, highly conserved region for cyclic nucleotide binding at the C terminus (CNBD) (Kaupp and Seifert, 2001). A detailed description of CNBD structure and structure–activity relationships for interaction with cyclic nucleotides is given in the section Modulation by Cyclic Nucleotides. The first insight of modulation by direct cAMP binding (and unbinding) as the primary mechanism of autonomic modulation came from the pioneer work of Dario DiFrancesco on f-channels (DiFrancesco and Tortora, 1991). Binding of cAMP is not required to open HCN channels, at variance with retinal and olfactory cyclic nucleotide–gated (CNG) channels (Kaupp and Seifert, 2002; Craven and Zagotta, 2006). However, cAMP promotes channel opening by increasing the open state probability (Thon et al., 2013), accelerating activation, and slowing deactivation (Wicks et al., 2011), thus ultimately modifying voltage dependence and activation kinetics. The CNBD acts as an autoinhibitory mechanism, with cAMP relieving inhibition (Viscomi et al., 2001; Wainger et al., 2001; Wang et al., 2002; Akimoto et al., 2014); it is also implicated, although via a different binding site, in the inhibitory effect of the regulatory subunit tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b) of HCN in neurons (see section The Role of Ancillary Subunit and Regulatory Proteins) (Saponaro et al., 2014). Finally, the C-terminal intracellular region of HCN4 controls—upon cAMP binding—a conformational change, leading to the formation of a tetrameric gating ring (Zagotta et al., 2003; VanSchouwen et al., 2015).
B. Biophysical Features of HCN Isoforms
In patch-clamped cells, upon hyperpolarization below a threshold of −40 to −60 mV, HCN channels activate in a time- and voltage-dependent manner, generating an inward current that does not inactivate (Fig. 2A). Driven by their respective electrochemical gradients, Na+ and K+ flow through the open channels as long as the hyperpolarizing step is maintained, generating a net inward current. Without recapitulating all biophysical features, which have been extensively described elsewhere (Biel et al., 2009), it is anyway necessary to highlight some Ih properties to understand the mechanisms underlying its modulation by exogenous and endogenous substances and the consequences of gain or loss of function in inherited and acquired diseases.

Current properties of HCN homomeric channels. (A) Representative whole-cell current recordings (lower panels) of re-expressed single mHCN1, mHCN2, and hHCN4 isoforms. Each family of time-dependent, inward currents is generated by hyperpolarizing steps to −40/−140 mV, from a holding potential of −30 mV. Activation curves (upper panels), obtained by normalized conductance plotted as a function of test potentials and fitted by a Boltzmann function, exhibit threshold of activation and voltage of half-maximal activation less and most negative for HCN1 and HCN2 isoforms, respectively. (B) Application of cAMP to the cytoplasmic side of the membrane causes a much greater shift in the activation curve of HCN2 compared with HCN1 (modified from Wainger et al., 2001 with permission); (C) schematic representation of the structure of the four HCN isoforms.
Heterologous re-expression of single isoforms, generating homomeric tetramers, demonstrates that HCNs possess different kinetics and voltage-dependent properties. Channel opening upon hyperpolarization generates a time-dependent inward current that can be interpolated by a single- or double-exponential function (see, for example, Wicks et al., 2011; Zong et al., 2012; Kim and Holt, 2013; Nakamura et al., 2013, and, for review of previous literature, Biel et al., 2009). The time constant (tau) of activation differs among isoform subtypes. In native tissue, it depends on the coassembling of different isoforms in the tetrameric channel, the presence of ancillary subunits, post-translational modifications including (de)phosphorylation and N-glycosylation, the effect of ligands, and, finally, experimental conditions such as ionic composition of extracellular milieu (Chen et al., 2001b; Xue et al., 2002; Altomare et al., 2001; Much et al., 2003; Whitaker et al., 2007; Ye and Nerbonne, 2009). When heterologously re-expressed, HCN1 homotetrameric channels exhibit the fastest kinetics of activation and HCN4 the slowest, and the two other isoforms are in between (Ludwig et al., 1999; Seifert et al., 1999; Stieber et al., 2003b; Stieber et al., 2005). Activation kinetics is strongly dependent on voltage: the more negative the step, the faster the activation. This is evident in Fig. 2A, in which a family of HCN1, HCN2, and HCN4 current tracings are plotted as a function of time. The second striking difference relates to the voltage dependence: when the relative amplitude is plotted against the voltage step, HCN1 exhibits the less negative threshold and voltage of half-maximal activation (V1/2) and HCN2 the most negative (Ludwig et al., 1999; Seifert et al., 1999; Stieber et al., 2003b, 2005). Finally, voltage dependence and kinetics of activation of HCN2 and HCN4 are very sensitive to cAMP, at variance with HCN1 (Moroni et al., 2000; Zagotta et al., 2003).
In recombinant systems or native cells, the slow exponential activation of Ih is preceded by a small initial, instantaneous current. The molecular nature of this current is still questioned because variable biophysical and pharmacological properties (e.g., amplitude, sensitivity to Cs+, or organic blockers) have been reported, depending on the frequency of hyperpolarizing pulses, intracellular cAMP or Cl− concentrations, expression of ancillary subunits, and other experimental conditions (Proenza et al., 2002; Mistrik et al., 2006; Proenza and Yellen, 2006). Recent results obtained with mutant HCN1 and HCN2 isoforms in the so-called zipper residues suggest that the two components (instantaneous and slow-activating current) may result from different open states of HCN channels (Wemhoner et al., 2012). At variance with the voltage- and time-dependent HCN current, the physiologic significance of the instantaneous current is unclear and may be dependent on cell types; indeed, a large voltage-independent inward current, blocked by Cs+ and 4-(N-ethyl-N-phenylamino)-1,2 dimethyl-6-(methylamino) pyrimidinium chloride (ZD7288; see Pharmacology, section Ivabradine, Cilobradine, and Other Specific Bradycardic Agents), seems to contribute to neuronal excitability in stellate cells of ventral cochlear nucleus (Rodrigues and Oertel, 2006).
Several models have been proposed in the years to explain HCN behavior, namely voltage dependence, kinetics, and sensitivity to cAMP. Although those basic properties are well described by classic allosteric models (DiFrancesco, 1999), others are not. A more recent four-state model, in which HCN channels transit between two modes, tried to encompass this limitation and explain hysteresis, i.e., the voltage dependence of the HCN channels on their prior activity. In fact, the current–voltage activation curve of Ih is shifted positively after long hyperpolarizing steps (persistent channel opening), and shifted negatively after long depolarizations (closed channels) (Männikkö et al., 2005). The same authors suggest that transition between the two modes, or hysteresis, has important consequences for physiologic (and pathologic) functioning of rhythmic cells. In SAN cells, hysteresis keeps channels closed after an action potential (AP) until complete repolarization (and calcium current recovery), preventing a premature diastolic depolarization; attainment of maximum hyperpolarization (between two APs) stabilizes the open state of HCN channels, pushing the membrane potential toward the threshold for the next AP (Männikkö et al., 2005).
C. Modulation by Cyclic Nucleotides
1. Binding of cAMP and Modification of Gating Properties
HCN channels are primarily activated by hyperpolarization of membrane potential and are regulated by cyclic nucleotides, which interact with a specific site, the CNBD, located in the intracellular C-terminal portion (see section HCN Modulation by Cyclic Nucleotides). cAMP accelerates channel opening and shifts the activation curve to more positive potential (DiFrancesco and Tortora, 1991). The shift in V1/2 depends on the channel subtype and conditions: it varies between 10 and 25 mV for HCN2 and HCN4 (see Wahl-Schott and Biel, 2009 and references cited therein), but only 2–6 mV for HCN1 (Wainger et al., 2001; Wang et al., 2001) (Fig. 2B). cAMP potency (EC50, i.e., the concentration of ligand that produces a half-maximal voltage shift) is in the range 0.06–1.53 μM (Table 1).
Potency of cyclic nucleotides on different HCN channel isoforms
EC50 is the concentration of ligand that produces a half-maximal voltage shift.
According to a generally accepted model, the channel is normally inhibited and the binding of cAMP removes this inhibition, inducing conformational changes and increasing the open probability of channel pore (Wainger et al., 2001). As suggested by Wang et al. (2001) from experiments involving HCN1-HCN2 chimera, the different sensitivity of these two isoforms depends on differences in the sequences involved in the interactions between CNBD and the C-linker. Studies performed on the isolated C-terminal domain (C-linker + CNBD) of HCN2 showed that addition of cAMP changes the proportion between monomer and tetramer in favor of the latter (Zagotta et al., 2003). There is evidence that the isolated C-terminal domain of HCN1 has a higher propensity to tetramerize than those of HCN2 and HCN4 (Lolicato et al., 2011; Chow et al., 2012). Thus, a possible explanation to the lower sensitivity of HCN1 to the natural agonist could be a sort of preactivation of the channel (Chow et al., 2012), probably because it is able to trap endogenous cAMP in the CNBD (Lolicato et al., 2011). The positive effect of cAMP on tetramerization of the isolated intracellular region is translated, in the whole channel, in conformational changes that propagate from the CNBD via the C-linker to the S6 fragment (Lee and MacKinnon, 2017).
Surprisingly, HCN3 is not activated by cAMP; rather, the activation curve is slightly shifted to more negative voltages (Mistrík et al., 2005; Stieber et al., 2005). The lack of sensitivity of HCN3 to cAMP, despite the presence of a functional CNBD in the intracellular region, has been explained by a shorter C-terminal sequence, after the CNBD, which alters the normal autoinhibition of the channel (Stieber et al., 2005).
In recent years, several biophysical techniques, such as isothermal titration calorimetry, surface plasmon resonance, and fluorescence anisotropy, have allowed measurement of the binding affinity of cAMP for the CNBD. Lolicato et al. (2011) used surface plasmon resonance to compare the interaction of cAMP with the C-terminal domain of HCN1, 2, and 4, finding that the three isoforms recognize the natural agonist with similar KD values (respectively, 5, 10, and 11 μM). Xu et al. (2010) used isothermal titration calorimetry and fluorescence anisotropy to measure affinity on the intracellular portion of HCN2 and 4, comparing the results with the EC50 (Table 1); because human HCN4 (hHCN4) does not express in Xenopus laevis oocytes, they used a murine HCN2 (mHCN2)-hHCN4 chimera, in which the C-linker and CNBD of HCN2 were replaced by the same portion of HCN4. cAMP potency, measured in functional studies on the whole channel, was three times higher on HCN2 than on HCN4, but the same difference was not found for affinity (measured on the purified cytosolic domains), giving evidence that binding and gating efficacy might have different requirements.
2. Other Cyclic Nucleotides
As well as cAMP, guanosine-3′,5′-cyclic monophosphate (cGMP) is a full agonist of HCN channels: at saturating concentrations, the shift in V1/2 is in the same range as that of cAMP. However, cGMP displays a 10-fold lower potency for HCN channels (Table 1) (Ludwig et al., 1998). cGMP and cAMP bind to the CNBD in a similar way, only differing in the orientation of the purine ring (Zagotta et al., 2003). On SpHCN, cGMP behaves as a partial agonist showing, when compared with cAMP, an intrinsic activity of 0.5 and about 600-fold lower affinity (Kaupp and Seifert, 2001).
Cytidine 3′,5′-cyclic monophosphate (cCMP) is also able to modulate HCN channels, behaving as partial agonist (DiFrancesco and Tortora, 1991). On HCN2 and HCN4 channels, cCMP shifts the activation curve to more positive potentials, speeds up current activation, and decreases current deactivation, but it has no effect on HCN1 and HCN3. The voltage shift and the maximal increase in the current amplitude were significantly smaller than those observed for cAMP, in agreement with the behavior of a partial agonist (Zong et al., 2012). On HCN2, uridine 3′,5′-cyclic monophosphate (cUMP), purine 3′,5′-cyclic monophosphate (cPMP), and 2-amino-cPMP are all able to shift the activation curve to more positive potentials and to increase channel conductance, with efficacy similar to cAMP and cGMP; on the contrary, inosine 3′,5′-cyclic monophosphate (cIMP), as well as cCMP, behaves as weak activators (Ng et al., 2016).
In addition to cyclic mononucleotides, also cyclic dinucleotides can modulate HCN channels. In mouse SAN myocytes, they behave as antagonists, being able to reduce Ih (Lolicato et al., 2014). Cyclic [guanosine-(2′-5′)-monophosphate-adenosine-(3′-5′)-monophosphate], the cyclic dinucleotide that has been found in mammals, caused a shift of the activation curve toward more negative potentials, and a ∼30% reduction of firing rate. On HCN4 channel expressed in human embryonic kidney (HEK)293 cells, the dose–response curve yielded an IC50 (i.e., the concentration of ligand that produces a half-maximal voltage backshift) of 114 nM. Other cyclic dinucleotides could completely reverse the effect of cAMP on the activation curve, as, for instance, cyclic di-(3′,5′)-GMP, which was ∼16 times less potent than cyclic [guanosine-(2′-5′)-monophosphate-adenosine-(3′-5′)-monophosphate] (IC50 1.8 μM).
D. The Role of Ancillary Subunits and Regulatory Proteins
Different regulatory proteins form macromolecular complexes with HCN channel subunits and define the features of HCN-mediated current in vivo, the regional or subcellular localization of HCN proteins, as well as their susceptibility to modulatory signals.
The number of proteins acting as HCN-binding partners has grown over the last decades; for some proteins, such as Mint2 (Mun18-interacting protein), synaptic scaffolding molecule, and tamalin, only a scaffold function for HCN2 has been identified (Kimura et al., 2004), and a possible role in channel trafficking, distribution, and clustering has been hypothesized.
MinK-related peptide 1 (MiRP1), encoded by kcne2 gene, is a single-transmembrane domain protein, which serves as regulatory subunit of different cardiac ion channels, including HCN channels. It enhances HCN protein and current expression in an isoform-specific manner (Yu et al., 2001; Decher et al., 2003; Qu et al., 2004; Brandt et al., 2009). When coexpressed with HCN channel, current kinetics of activation is accelerated for HCN1 and HCN2, whereas it is slowed down for HCN4, which also displays a shift of the midpoint of activation to more negative voltages. High levels of MiRP1 and HCN subunits (primarily HCN4, and, depending on the species, HCN1 or HCN2) are expressed in the SAN of small and large mammals, including mouse, rabbit, and human (Yu et al., 2001; Accili et al., 2002; Schweizer et al., 2009). A similar interaction among MiRP1 and HCN4, 2, and 1 most likely occurs in atrial and ventricular myocytes, where transcript levels of both subunits are lower compared with SAN (Yu et al., 2001; Decher et al., 2003; Qu et al., 2004; Stillitano et al., 2008, 2013; Sartiani et al., 2010). In these regions, the modification of HCN-mediated current—related to cardiac diseases [atrial fibrillation (AF) and ventricular hypertrophy] or to postnatal maturation—is most likely associated with transcriptional regulation of both HCN channel and MiRP1 subunit.
The K+ channel regulator 1 (KCR1) is a transmembrane protein expressed in cerebellum and heart (Hoshi et al., 1998; Michels et al., 2008). Protein expression analysis in rat and guinea pig revealed an extensive distribution of the protein in the heart with larger amount in the atrioventricular node (AVN), left atrium, and ventricle, followed by right atrium and ventricle, and SAN. KCR1 is a regulatory subunit of diverse native Kv channels, as well as HCN channels. In heterologous expression systems and native ventricular myocytes, KCR1 interacts with HCN2, reducing current size and shifting Ih activation to more negative potentials. These modifications ultimately decrease spontaneous rhythmicity in cultured neonatal cardiac myocytes, suggesting that KCR1 may be an important regulatory subunit of HCN current in vivo.
Caveolin-3 is a lipid raft component of myocyte membrane that colocalizes with and affects the expression and function of HCN channels, as well as their susceptibility to modulating signals. In SAN cells, interaction between caveolin-3 and HCN4 affects channel voltage dependence by shifting V1/2 to negative voltages; it also modifies HCN4 current kinetics by accelerating channel deactivation (Barbuti et al., 2004, 2007, 2012). Because β2-adrenoceptors, but not β1, localize to lipid rafts in the SAN, their activation generates a prominent signal mediating the adrenergic enhancement of HCN current and the rise of heart rate (Barbuti et al., 2007). Following investigations demonstrated that a similar colocalization between caveolin-3 and HCN4 channels occurs in human atrial and embryonic stem cell–derived cardiomyocytes (Bosman et al., 2013; Stillitano et al., 2013). In the latter, the shift of V1/2 is directly related to the increase of caveolin-3 expression during myocyte maturation.
Filamin A is a cytosolic scaffolding protein that exerts a crucial role for the trafficking of numerous ion channels in excitable cells, including neurons and cardiac myocytes. It anchors ion channels to actin cytoskeleton and clusters them in distinct locations on cell surface membrane. In the brain, only HCN1, but not HCN2, 3, or 4, associates with filamin A (Gravante et al., 2004). In heterologous expression systems, this interaction reduces the density of channel expression as well as whole-cell conductance by aggregating HCN1 within restricted regions of the cell membrane. Additionally, filamin A promotes a reversible dynamin-dependent internalization of HCN1 channels and a redistribution of HCN1 channels on cell surface by accumulation of channels in endosomal compartments (Noam et al., 2014). In cultured hippocampal neurons, expression of a dominant-negative filamin A increases the expression of native HCN1, whereas acute abrogation of HCN1–filamin A interaction enhances current size. Whether a similar interaction occurs at cardiac level is unknown, despite filamin A being present in mouse and human atrial myocytes (Rafizadeh et al., 2014).
TRIP8b, also termed Pex5p-related protein (PEX5Rp), and H-channel interacting protein 1 (HIP1), is a brain cytoplasmic protein, member of the Rab family of small GTPase proteins, which are important for vesicle trafficking (Chen et al., 2001a). In neocortical and hippocampal pyramidal neurons, colocalization of TRIP8b to HCN1 promotes an active trafficking of the channels from soma to dendrites that is important to modulate spike firing and synaptic potential (Santoro et al., 2004; Zolles et al., 2009). In TRIP8b-knockout (KO) mice, HCN surface expression in hippocampal pyramidal neurons is dramatically reduced, as well as current size in this region; moreover, normal expression pattern of HCN channels is profoundly altered in pyramidal neuron dendrites (Lewis et al., 2011). Nine isoforms of TRIP8b have been identified that differentially affect HCN channel gating, membrane expression, and trafficking in the nervous system. The overexpression of TRIP8b in cultured hippocampal pyramidal neurons or heterologous expression systems exerts different effects on HCN1 surface expression according to the variant type. TRIP8b (1a–4) and TRIP8b (1a), major splice variants in the hippocampus, enable a correct localization of HCN1 (Lewis et al., 2009; Santoro et al., 2009). Moreover, TRIP8b (1a–4) upregulates HCN1 expression in heterologous systems and promotes its dendritic expression. Conversely, TRIP8b (1a) downregulates HCN1 surface expression in X. laevis oocytes and inhibits the abnormal expression of HCN1 in the axons of pyramidal neurons (Piskorowski et al., 2011). Opposed to TRIP8b, a recent paper shows that ubiquitination by the Nedd 4-2 (neuronal precursor cell–expressed developmentally downregulated four-like) protein decreases HCN1 surface expression and translocation, leading to Ih loss of function (Wilkars et al., 2014).
E. From Transcriptional Control to Post-Translational Modifications
1. microRNAs
Accumulating evidence on sinus node dysfunctions has led to consider the downregulation of HCN channels as possible common cause of different conditions leading to bradycardia (D’Souza et al., 2015). The role of microRNAs (MiR) is emerging as main transcriptional regulator of HCN channels in cardiac pacemaker centers. MiR-1, one of the main muscle-specific microRNA (myomiR), is induced by athletic training in the heart jointly to a downregulation of the transcription factors Tbx3 and NRSF (D’Souza et al., 2014). These changes are consistent with the downregulation of HCN4 and Ih found in SAN cells of trained animals.
On the contrary, in different cardiac pathologies, including myocardial infarction (Suffredini et al., 2012; Yu et al., 2015) or age-related AF (Li et al., 2015b), expression of HCN channels is upregulated, in line with the observed reduction of MiR-1 levels detected in the ventricles and atria. Accordingly, in rats with myocardial infarction, administration of ivabradine, a selective bradycardic agent (Suffredini et al., 2012), or spironolactone, an aldosterone blocker (Yu et al., 2015), counterbalances the overexpression of HCN channels in parallel with upregulation of MiR-1.
In all conditions, the mechanism responsible for the modifications of microRNA levels in the sinus node or in the working myocardium remains unknown.
2. Regulation by Membrane Phosphoinositides
Signaling pathways coupled to membrane phosphoinositide content and downstream derivatives stimulate HCN channels.
Phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] is a membrane constitutive component that increases the opening of recombinant and native HCN channels by shifting the voltage dependence of activation to more positive potentials by 5–20 mV, depending on isoforms. It acts as intracellular allosteric activator that facilitates channel opening (Biel et al., 2009).
In the heart, basal variability of membrane PI(4,5)P2 is likely to contribute to the variations in the voltage dependence of Ih activation in cardiac cells at different maturation degrees (Cerbai et al., 1999b; Qu et al., 2001), derived from different regions, or following stress and pathologic conditions (Suh and Hille, 2007).
In SAN cells, stimulation of bradykinin BK2 receptors coupled to phospholipase C enhances phosphatidylinositol kinase activity that in turn stimulates polyphosphoinositide synthesis, thereby enhancing HCN channel function (Pian et al., 2007). A similar interaction with phosphoinositides occurs in neurons, where PI(4,5)P2 acts as an allosteric modulator of HCN, causing a rightward shift of voltage activation (Zolles et al., 2006; Ying et al., 2011); gating by phosphoinositides may be important to maintain rhythmogenesis when signaling pathways leading to phospholipid degradation reduce channel activation in nerve cells. Recent data in SpHCN suggest that PI(4,5)P2 binds to both the transmembrane core region and the C-linker domain of the channel, with opposite effects (Flynn and Zagotta, 2011). Whether such a dual mechanism also occurs in mammalian HCN is unknown.
Other allosteric modulators of HCN channels are two membrane phosphoinositide derivatives, phosphatidic acid and arachidonic acid, which are products of diacylglycerol kinase and phospholipase A2, respectively. They directly facilitate HCN channel gating by shifting the voltage dependence of activation to more positive values (Fogle et al., 2007).
3. Kinases
Phosphorylation status of HCN channels is an additional regulatory mechanism controlling HCN properties and adapting its activity to the peculiar conditions of different types of cardiac cells and neurons.
Despite the fact that HCN channels are considered end effectors of cAMP, experimental evidence demonstrated a modulatory role of protein kinase A (PKA). In early cardiomyogenesis, activation of PKA has an exclusive role in the stimulation of HCN current following β-adrenergic receptor stimulation (Abi-Gerges et al., 2000). In adult mouse SAN, PKA seems to exert an additive positive shift of the activation curve upon β-adrenergic receptor stimulation (Liao et al., 2010), although the mechanism is most likely mediated by PKA-dependent effects on cAMP production or diffusion rather than phosphorylation of the channel (St Clair et al., 2013).
In hippocampal neurons, phospholipase C–protein kinase C activation, phosphorylating HCN1 isoform, decreases HCN current and HCN1 surface expression (Williams et al., 2015). Similar findings were obtained in respiratory neurons within the pre-Bötzinger complex (Thoby-Brisson et al., 2003).
Tyrosine kinases of the Src family have also a stimulatory function in the mature cardiac cells as well as in different types of neurons (Santoro et al., 1997; Wu and Cohen, 1997; Yu et al., 2004; Zong et al., 2005; Arinsburg et al., 2006). In these cells, the pathway contributes to regulate spontaneous electrogenesis by direct phosphorylation of HCN1, HCN2, and HCN4 and speeding of channel kinetics. Despite the fact that the residue (Y476) conferring such modulation in HCN2 channels is conserved in the other isoforms, a stimulation of kinetics by Src tyrosine kinase has been proven only for HCN4. In addition, the latter isoform undergoes also a positive shift (≅+10 mV) of voltage dependence, most likely because of an additional phosphorylation in a different tyrosine residue (Y531) (Li et al., 2008a). In rat ventricular myocytes, the receptor-like protein-tyrosine phosphatase-α controls the extent of HCN2 channel phosphorylation in this site, shifts the voltage dependence of activation, and decreases channel insertion into the membrane (Huang et al., 2008).
In neuronal cells, HCN channels are also phosphorylated by the serine/threonine kinase, p38 mitogen-activated protein kinase (Poolos et al., 2006), and calcium/calmodulin-dependent protein kinase II (Shin and Chetkovich, 2007).
Modulation by the small ubiquitin-like modifier (SUMO) peptide is one of the mechanisms involved in the regulation of protein–protein interactions. HCN2 SUMOylation occurs in mouse forebrain tissue (Parker et al., 2017); in transfected HEK cells, SUMOylation of HCN2 increased current conductance and surface expression. This finding is of interest in view of HCN dysregulation in central nervous system (CNS) diseases, as discussed later.
III. Origins, Physiology, and Pathophysiology of HCN Channels
A. HCN: Ancient and Early Channels
1. Phylogeny
HCN channels belong to the superfamily of six-transmembrane segment channels and are related to CNG channels and voltage-dependent ether-a-go-go K+ channels Kv10–Kv12 (Lee and MacKinnon, 2017). Being components of the ancestral gene pattern, HCN channels are present across a wide spectrum of invertebrate and vertebrate species. They most likely derive from a single ancestral gene subjected to duplications and diversification events over the species that eventually generated four different isoforms prior to the origin of the vertebrate clade (Jackson et al., 2007). A functional role for HCN in setting heart rhythm has been postulated on the basis of pharmacological studies also in vertebrate ancestors (Wilson and Farrell, 2013). Based on sequence conservation analysis, HCN3, thought to be most similar to the ancestral channel, was the first to diverge as a product of the first duplication. HCN3 was followed by the emergence of HCN4 and then of HCN1 and HCN2, thus composing a group of four variants that collectively shares 80%–90% sequence conservation within the core and transmembrane regions. The latter in all vertebrate and invertebrate contribute to common functional properties of HCN channels. The residual variations in these regions are responsible for subtle isoform-specific differences related to inner selectivity filter, rates of channel opening, and differences in cAMP efficacy to modulate HCN isoforms.
In all four vertebrate isoforms, a region of ∼50 residues upstream to the start of S1 in the NH2 terminus is conserved. In mouse HCN2, this region is involved in intersubunit interactions of tetramer assembly and in the formation of functional channels (Tran et al., 2002). Analogous regions present in the other isoforms are supposed to exert similar functions.
In all vertebrate HCN1, 2, and 4, but not in HCN3, a different block in the COOH terminus is conserved. It represents a PDZ-binding domain enabling channels to interact with PDZ-containing proteins and with the TRIP8b protein (Kimura et al., 2004; Santoro et al., 2004), which regulates channel surface expression.
2. HCN Channels in Stem Cells
The growing number of studies in different types of stem cells has led to identify the presence of several specialized ion channels, including HCN channels (Heubach et al., 2004; Wang et al., 2005; Sartiani et al., 2007). As suggested for other bioelectric signals, mainly K+ or Ca2+ channels, HCN channels also may act as regulators of a wide range of stem cell functions, including proliferation, migration, differentiation, and tissue regeneration of nonexcitable cells (Blackiston et al., 2009; Sundelacruz et al., 2009, 2015; Levin, 2014). Indeed, HCN-mediated mechanisms are most likely involved in the development of a mature phenotype of olfactory sensory neurons, where HCN channels are expressed precociously and drive axon organization (Mobley et al., 2010).
Expression pattern of HCN channels in stem cells is species- and/or origin-dependent, despite the relatively homogenous phenotype of potency markers (Li and Deng, 2011). HCN1 channels are highly expressed in human pluripotent stem cells, but not in mouse cells, where HCN3 largely predominates, suggesting that current phenotype and regulation might diverge among species, as observed in differentiated cells. Differently, human bone marrow–derived stem cells express HCN2 channels. Despite the presence of HCN transcripts and proteins in stem cells, only one study has been performed in mouse pluripotent stem cells, where the channels were found involved in cell proliferation, in particular in cell cycle progression from G0 to G1 phase (Lau et al., 2011).
3. HCN during Organogenesis
a. Cardiogenesis
Occurrence of spontaneous electrical activity is an early event in cardiac morphogenesis. In the mouse, the whole process has been thoroughly dissected, identifying that this pacemaker activity is detectable since embryonic day (E) 7.5 in precardiac mesoderm (cardiac crescent) of the first heart field (Liang et al., 2013; Später et al., 2013; Barbuti and Robinson, 2015). Cells comprised in this early pacemaker region provide efficient peristaltic contractions necessary in the primitive heart tube and express two distinct markers, the transcription factor Nkx2-5 and HCN4 channels (Christoffels et al., 2010). Lineage-tracing experiments show that the Nkx2-5+/HCN4+ cells do not give rise to the cardiac conduction system in the developing heart, but generate atrial and ventricular precursors (Moorman and Christoffels, 2003). At E8, posterior heart field precursors expressing the transcription factor Tbx18 start to proliferate and will form the sinus venosus, a symmetric structure of the heart tube. Following expression of two additional markers (Shox2 and CD166), these cells will become the leading pacemaker (SAN precursors), when at E9.5 a subgroup of cells further expresses the second heart field marker Isl-1 and the transcription repressor Tbx3.
Subsequent increase of Tbx3 and HCN4 and decrease of CD166 expression ultimately form the sinus atrial node in the right atrium, whose typical panel of markers will maintain Isl-1, Tbx18, Shox2, Tbx3, and a high level of HCN4 throughout life. In the left atria, activity of the homeobox factor Pitx2c specifically suppresses the SAN gene program, allowing the correct asymmetric development of the conduction system (Mommersteeg et al., 2007). Despite the fact that HCN4 is functionally expressed ever since the appearance of the pacemaker activity in the precardiac mesoderm, HCN4 starts to play a fundamental function during the formation of the SAN between E9.5 and E11.5, since global and cardiac-specific HCN4-KO mice die in this developmental stage (Stieber et al., 2003a). However, studies in HCN4-deficient embryos show significantly reduced contraction rates and immature AP in deficient embryos compared with wild type (WT), indicating that even before SAN formation HCN4 is critical for normal cardiac development.
Studies with specific deletion of HCN1 and HCN2 confirmed the role of HCN4, because HCN1 (Nolan et al., 2003) or HCN2 (Ludwig et al., 2003; Cao and Oertel, 2011) KO animals do not evidence major cardiac alterations in developing embryos. This also suggests that expression of HCN1 and HCN2 channels in the heart is delayed during cardiogenesis. Indeed, transcription profiling throughout mouse cardiac development indicates that embryonic heart before E9.5 displays abundant HCN4 transcript, whereas other HCN transcripts are almost absent. Toward birth, HCN transcription profile in the atrium and ventricle changes remarkably, because HCN4 is strongly downregulated, whereas HCN1 and HCN2 transcripts slowly emerge. HCN3 isoform shows highest levels at early embryonic stages and then fades to very low levels (Schweizer et al., 2009).
Little information is available on the developmental changes of HCN channels occurring in humans. In a limited time window, i.e., from 11 to 14 weeks of fetal life, a strong protein expression of HCN4 is present in the whole heart that contributes to a remarkable HCN- mediated current in ventricular myocytes (Bosman et al., 2013). At this stage, current density is larger than that retrieved in human healthy adult ventricular cardiomyocytes and close to the values described in ventricular myocytes from ischemic patients (Fig. 3), corroborating the concept of HCN channel expression as a marker of fetal gene reprogramming in cardiac disease (see Altered HCN function and cardiac disease).
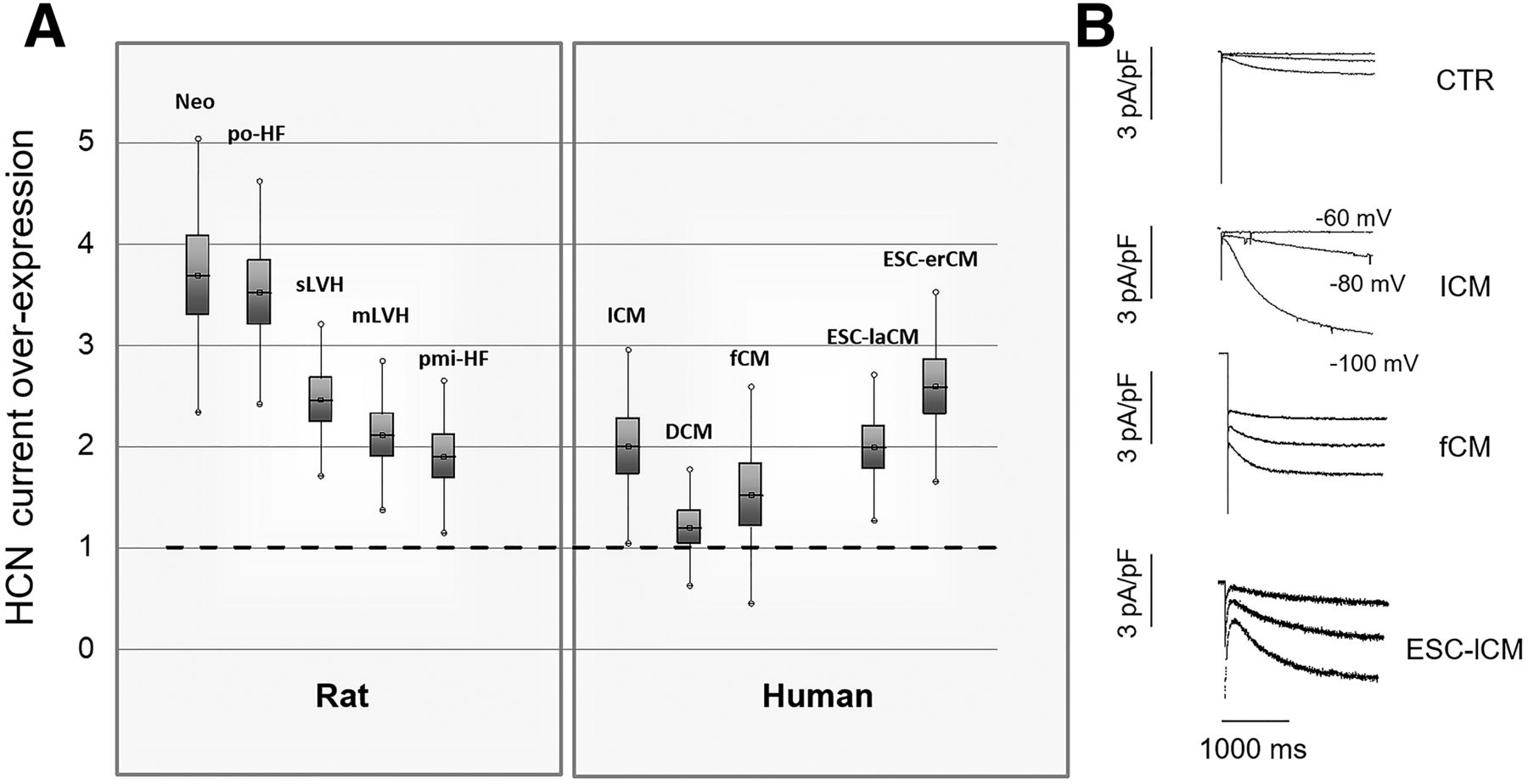
Ih current expression in cardiomyopathies and in cardiac development. (A) Data points represent the ratio between current density measured in ventricular myocytes from diseased hearts and respective control. In neonatal rat cardiomyocytes (Neo), values measured at 2 weeks are compared with those at 2 days after birth. po-HF, pmi-HF: relative increase of Ih in rats with overt heart failure, resulting from pressure overload or following myocardial infarction, respectively. mLVH, sLVH: relative increase of Ih in rats with mild or severe left ventricular hypertrophy caused by aortic banding or long-lasting pressure overload, respectively. DCM, ICM: relative increase of Ih in patients undergoing cardiac transplantation for terminal dilated or ischemic cardiomyopathy, respectively. For all conditions, the relative increase of current density is statistically significant versus controls, that is, normotensive rats, sham-operated rats, or undiseased donor hearts not transplanted for technical reasons, with the exception of DCM patients. fCM, ESC-laCM, and ESC-erCM: relative increase of Ih in human fetal (11–14 weeks) cardiomyocytes and in cardiomyocytes differentiated from human embryonic stem cells in late and early developmental stages, respectively. (B) Representative recordings of Ih in a control (CTR), a hypertrophic (ICM), a fetal (fCM), and a late ESC-derived cardiomyocyte (ESC-laCM).
b. Insights from stem cell–derived cardiomyocytes
A different approach to address the developmental changes during cardiogenesis consists in the use of pluripotent stem cells, whose cardiomyogenic potential in vitro is well established and widely used in the last decades. Following established protocol of differentiation, the in vitro model recapitulates many of the developmental stages described for in vivo cardiogenesis, thus representing a highly valuable tool to investigate the embryonic/fetal modifications of cardiac cells difficult to address in other models. It is also well known that the cardiomyocyte population arising from this model is heterogeneous and composed of atrial-, ventricular-, and SAN-like cells that have been thoroughly characterized for their electrical and structural properties more than 20 years ago (Maltsev et al., 1993). Molecular insights have been reviewed recently (Barbuti and Robinson, 2015). An interesting finding emerged from mouse pluripotent cells is the variable propensity to develop SAN-like or atrial/ventricular-like lineage. The former is characterized by a prominent expression of HCN4/HCN1 channels that are colocalized with caveolin-3 and present together with T-type calcium channels CaV3.1/3.2, thus recapitulating the pattern of channels typically expressed in native SAN (van Kempen et al., 2003; Marionneau et al., 2005; Yanagi et al., 2007; Barbuti et al., 2009). Differently, atrial/ventricular-like lineage is characterized by higher levels of HCN2 and HCN3 compared with HCN4 and HCN1 (White and Claycomb, 2005; Qu et al., 2008).
Phenotype heterogeneity and lineage preference also feature human pluripotent stem cells (He et al., 2003; Mummery et al., 2003), where atrial/ventricular phenotypes associate with HCN2 preponderance over HCN4 (Satin et al., 2004). Consistency of findings among different mammalian species warrants further investigation to better understand whether distinct, still unidentified pluripotency states drive a preferential lineage specification. This hypothesis applies to recent studies on pacemaking activity in early (days 11–21 of differentiation) cardiomyocytes from pluripotent cells (Weisbrod et al., 2013), where HCN current, detectable in most of beating cells, is determined by HCN4 and HCN2 isoforms. The concomitant association with CaV1.3, Na+/Ca2+ exchanger (NCX)-1, and Tbx3 in these cells defines a panel of genes typical of human SAN and perinodal areas (Chandler et al., 2009), suggesting that these cells are early pacemaker or SAN-like cells rather than immature atrial or ventricular cardiomyocytes. Accordingly, cell lines prone to develop atrial/ventricular phenotype exhibit properties resembling those present in adult atrial/ventricular cells, such as expression of NaV1.5, CaV1.2, and HCN2, whereas CaV1.3 and HCN4 are absent (Satin et al., 2004). Differently, studies from our group, performed in a different cell line maintained in long-term culture, evidence the early emergence of immature SAN-like cells, representing the most frequent cardiomyocyte population after 15–30 days of differentiation. This single phenotype diverges into distinct atrial/ventricular myocytes at later maturation stages (55–110 days) (Sartiani et al., 2007; Paci et al., 2012; Bosman et al., 2013). During this developmental period in vitro, HCN channel expression shifts from a SAN-like pattern, mainly composed of HCN1 and HCN4, to an atrial/ventricular pattern, where HCN2 isoform predominates and remains constant. The functional counterpart in the early stage is defined by a robust HCN current, which throughout maturation declines in amplitude and activates much slower, in accordance to a lower expression of HCN1, approaching values like those encountered in native human fetal and adult hypertrophic ventricular cardiomyocytes (Fig. 3). The findings further strengthen the similarities between in vitro and in vivo cardiac differentiation. The advantageous property has been further exploited to investigate the subcellular compartmentation of HCN4 channel in the human setting. During development, HCN4 protein signal shifts from a widespread localization in α-actinin–positive immature cells to restricted sites in mature cardiomyocytes. At this developmental stage, HCN4 increasingly colocalizes with caveolin-3, thus providing a possible explanation for the negative shift of HCN current threshold. A similar modification of caveolin-3/HCN4 interaction occurs in native human cardiomyocytes, as suggested by a similar negative shift of HCN activation threshold characterizing the transition from fetal to adult cardiomyocytes (Bosman et al., 2013).
c. HCN expression in the developing nervous system
At variance with cardiogenesis and stem cell–derived cardiomyocytes, systematic studies investigating isoform- and age-dependent changes in HCN expression levels during pre- and postnatal development of the CNS are missing, with available data referring to selected regions. In CA1 hippocampal pyramidal cell layer of embryonic rats, a robust increase of HCN1 transcript and protein expression occurs during development, with concomitant reduction of HCN4 and relatively stable HCN2 levels. By birth, the contribution of HCN1 to the total HCN channel pool has risen from 30% to 60%. At subcellular level, the proximal-to-distal dendritic gradient of HCN1 is already present at postnatal day 2 (Bender et al., 2001; Surges et al., 2006; Brewster et al., 2007). Similarly, overall Ih density increases nearly sixfold in rat thalamocortical relay neurons during the first 3 months of postnatal life, accompanied by a progressive decrease in cAMP sensitivity. In keeping, quantitative analyses of HCN channel isoforms revealed a steady increase of transcript and protein expression levels of HCN1 and HCN2, with reduced relative abundance of HCN4 (Kanyshkova et al., 2009; Yoshimoto et al., 2015). An interesting observation comes from studies in the brainstem auditory neurons, where HCN channels are crucially involved in the location of sounds (Leao et al., 2006). This function is poorly present at birth in gerbils (as in humans) and undergoes intense postnatal adaption; at this stage, the onset of mature hearing with precise temporal resolution was associated with marked developmental changes in Ih. In particular, the kinetics of activation-deactivation became faster and conductance larger, and activation was shifted rightward. Overall, these changes were attributable in part to an increasing role of HCN1 isoforms, and largely (V1/2 shift) to the maturation of signaling pathways, in particular cAMP- and PI(4,5)P2-dependent modulation (Khurana et al., 2012).
B. Distribution, Adaptive, and Maladaptive Role of HCN Channels in Mammals
1. HCN Current in the Heart: an Ideal Pharmacological Target
Excellent reviews have described the key role of cardiac Ih in automaticity and its interplay with other ion currents in the SAN (Accili et al., 2002; DiFrancesco, 2006, 2010; Biel et al., 2009), as well as in the conduction system (Mangoni and Nargeot, 2008). We refer the reader to these papers for a systematic discussion of physiologic aspects and to recent optical mapping (Torrente et al., 2015) and computational approaches (Fabbri et al., 2017). Overall, HCN channels contribute to two essential features of primary and subsidiary pacemakers: the appearance of a diastolic depolarization and the modulation of its steepness by the sympatho-vagal balance or other endogenous factors (Fig. 4). Indeed, a relevant fraction of the antiarrhythmic and antianginal effect of old and new drugs (from beta-blockers to digoxin and ivabradine) resides in the indirect (antiadrenergic or vagomimetic) or direct (HCN blockade) effect on Ih in pacemaker cells. According to recent computational approach modeling the human SAN AP, HCN current exerts its modulatory role mainly by changing the rate of the diastolic depolarization phase (DDR) over the first 100 ms following the maximum diastolic potential (Fabbri et al., 2017). This might be more relevant in species with low heart rate (e.g., humans versus rodents) because, as elegantly discussed by Zaza (2016), a nonlinear relationship exists between DDR and time: the longer the cycle length, the greater the bradycardic effect of reducing DDR (and vice versa).

Role of HCN channels in cardiac electrophysiological abnormalities. Pathologic implications of dysfunctional HCN channels in cardiac regions arising from transcriptional or post-transcriptional alterations of Ih due to mutations or modulation by endogenous factors. Schematic traces represent the consequences of HCN loss of function (LOF) or gain of function (GOF).
a. Expression and physiologic role of HCN isoforms in cardiac regions
Ih may exhibit different electrophysiological properties (such as activation threshold and amplitude), also depending on the relative expression of different isoforms. Indeed, in the heart, HCN channels exhibit a regional specific distribution (Herrmann et al., 2012; Deng et al., 2015; Li et al., 2015a). A remarkable variation in the total amounts of HCN channels differentiates pacemaker centers from working myocardium, being HCN transcripts and proteins expressed at highest levels in the SAN and in the conduction system (AVN and Purkinje fibers). Additionally, isoform multiplicity differs according to species, except for HCN3, which displays a weak expression, regardless of regions and species. In the SAN of humans, rabbits, mice, and dogs, HCN4 is the main protein isoform compared with the others, the remaining fraction being composed by HCN2 and HCN1 in humans and HCN1 in mouse and rabbits. Differently, rat SAN expresses similar amount of HCN2 and HCN4 (Huang et al., 2016).
In mouse AVN, almost all cells express HCN1 and HCN4, whereas HCN2 is limited to some regions. The bundle of His is particularly enriched of HCN4, whereas bundle branches also display HCN1 and HCN2 (Herrmann et al., 2012). Human, rabbit, and rat AVN largely express HCN4 protein isoform, with HCN1 present in smaller amount (Dobrzynski et al., 2013).
The expression pattern of HCN protein in atria and ventricles also displays isoform variability; however, most mammalians, including humans, exhibit a prevalence of HCN4 and HCN2, followed by smaller amount of HCN1 and negligible levels of HCN3 (Lezoualc’h et al., 2007; Stillitano et al., 2008).
i. HCN in SAN Cells and Its Contribution to Cardiac Pacemaking: Membrane and Calcium Clocks
HCN channels have long been recognized for their primary role in pacemaker impulse generation and regulation. However, the specific contribution of HCN has considerably evolved in recent years. Two main mechanisms, the voltage clock and the Ca2+ clock, mainly sustained by HCN current and Ca2+ release from sarcoplasmic reticulum, respectively, are supposed to contribute to a coordinated system that jointly drives spontaneous electrical activity in SAN. The relative contribution of the two mechanisms to basal rhythm and its adaption to autonomic balance have been the object of an intense debate; to have a taste of different—sometimes conflicting—views, the reader is referred to a lively point–counterpoint exercise by the leading scientists in the field (DiFrancesco and Noble, 2012; Maltsev and Lakatta, 2012) and the enlightening accompanying comment (Rosen et al., 2012).
In brief, in the SAN during the diastolic phase, the fraction of open HCN channels provides a steady-state inward current driving membrane potential (−70/−40 mV, depending on cells) to depolarize toward the threshold required to generate a spontaneous AP (DiFrancesco et al., 1986). A key role of HCN channels, particularly the HCN4 isoform, is suggested—besides other arguments—by bradycardia (or severe bradycardia) consequent to the following: 1) loss of HCN4 function in KO mouse models or patients carrying HCN4 mutations (see later in this section), and 2) the effect of selective HCN blockers (see section Pharmacology of HCN Channels). It is well recognized that the steepness of the diastolic depolarization in pacemaker cells also results from the concerted (simultaneous or sequential) work of other membrane ionic conductances: NCX, L- and T-type Ca2+ channels, Na+/K+ ATPase, voltage-dependent K+ and background Na+ currents—just to mention some relevant ones. The point by DiFrancesco and Noble (2012) is that these conductances concert to set the pacing rate—the membrane clock—with Ih being the conductor, also in force of its exquisite sensitivity to autonomic balance (via intracellular cAMP). Lakatta’s group (Vinogradova et al., 2010) named calcium clock the periodic, spontaneous submembrane calcium release from sarcoplasmic reticulum, triggering Ca2+ extrusion via NCX current, which in turns depolarizes the membrane, activates T- and L-type calcium current, and finally triggers APs. The regular sequence of APs and its adaption to autonomic input might be the result not of a main conductor such as Ih, rather of the interplay between the rate of spontaneous Ca2+ release—roughly periodic—and the fine adjustment operated by the balance of inward/outward membrane conductances, including Ih. At variance with ivabradine, there are not blockers of calcium-clock players specific for SAN cells; in fact, their loss of function impairs atrial and ventricular contractility in vivo. The scientific debate has been also fueled by discrepant results obtained in mouse models undergoing genetic deletion of sinoatrial HCN4, ranging from lethal bradycardia (Baruscotti et al., 2011) to minor rhythm alterations (Herrmann et al., 2007). Although experimental strategies of genetic manipulation as well as the contribution of different HCN isoforms may help explain different results in HCN4-KO mice, it is worth mentioning the following: 1) KO of STIM1, a key regulator of calcium dynamics, also causes severe bradycardia in mice (Zhang et al., 2015), and 2) complete ablation of If by a dominant-negative HCN4 mutant channel expressed in SAN cells alters membrane excitability and calcium cycling (i.e., both clocks), pacemaking and conduction impairment being partially rescued by additional KO of the muscarinic G protein–activated (GIRK4) channels (Mesirca et al., 2014). Interestingly, the latter model confirmed the prominent role of Ih in SAN as sensor of the autonomic nervous system input.
ii. HCN Expression and Function in Subsidiary Pacemakers
In the AVN the function of HCN channels does not diverge substantially; in fact, although the specific role played by HCN channels in this region is less investigated, robust experimental evidence indicates that HCN channels are implicated in AVN pacemaking and conduction (Liu et al., 2008; Marger et al., 2011; Verrier et al., 2014, 2015). Of note, abolition of HCN4 sensitivity through conditional expression of dominant-negative HCN4 channels lacking cAMP sensitivity reduces the spontaneous activity of AVN cells under basal conditions, but does not impair the maximal response to β-adrenergic stimulation, suggesting that HCN4 channels influence AVN basal activity, but are not obligatory for β-adrenergic regulation (Marger et al., 2011).
Transgenic mouse models have further consolidated the functions of HCN channels in SAN and AVN cells. In fact, inducible cardiac ablation of HCN4 in mice leads to progressive severe bradycardia, followed by AVN block, eventually resulting in cardiac arrest and death (Baruscotti et al., 2011).
The physiologic role of HCN channels in the healthy working myocardium remains an issue for which a conclusive function is still unclear. Since the early evidence obtained in human atrial appendage fibers, Ih has been hypothesized to support the spontaneous electrical activity observed in the atrial tissue. Subsequently, a series of studies investigated the specific properties of HCN current in atrial myocytes, showing that voltage dependence, activation kinetics, and ionic selectivity are similar to those retrieved in SAN cells (Carmeliet, 1984). The role of HCN channel in the atria differs from that in SAN because most healthy atrial cardiomyocytes have a stable resting membrane potential and infrequently display spontaneous electrogenesis, in line with a low contribution of HCN current to resting membrane potentials (−80/−70 mV) and absence of spontaneous automaticity. However, when the integrity of the intracellular milieu is preserved, some human atrial myocytes display a clear diastolic depolarization phase, suggesting that HCN current in the atria may overtly influence electrogenesis, in particular when favoring conditions are present, such as reduced repolarizing currents or increased adrenergic tone (Cerbai and Mugelli, 2006).
Similar observations are drawn for HCN channels in the healthy ventricle, where HCN current is readily detectable in most cells displaying a stable resting membrane potential (Cerbai and Mugelli, 2006; Sartiani et al., 2015). Interestingly, recent evidence has uncovered a different function for HCN in the mouse ventricle, where the channel appears involved in the prolongation of AP repolarization. This led to propose that HCN channels, and particularly HCN3, might mediate a depolarizing background current that regulates ventricular resting potential and counteracts the action of hyperpolarizing potassium currents in late repolarization (Fenske et al., 2011). These findings partially agree with a different study in the mouse, where HCN2 and HCN4 isoforms appear to predominate in controlling the late phase of repolarization (Hofmann et al., 2012).
b. Altered HCN function and cardiac disease
i. Altered HCN Properties and Dysfunction of Sinus and Atrioventricular Nodes
The understanding of pacemaker alterations leading to cardiac arrhythmias is rapidly enlarging the genetic basis; currently, the combined efforts of clinical practice and appropriate transgenic models have helped to identify some modifications of HCN channel functions and regulatory proteins associated with dysfunctional cardiac pacemaking and/or conduction (Verkerk and Wilders, 2014).
Screening analysis performed in patients with idiopathic bradycardia has uncovered different loss-of-function mutations of HCN4 gene leading to impaired impulse generation capacity of SAN cells associated or not with conduction dysfunctions (AVN block and altered chronotropic response) (Fig. 4). Deep bradycardia and AVN block are also found in adult transgenic mice with inducible cardiac ablation of HCN4 (Baruscotti et al., 2011), further corroborating the pathophysiological implications of HCN4 reduction in cardiac pacemaker and conduction. Interestingly, HCN4 mutations have been identified in families with bradycardia and left ventricular noncompaction cardiomyopathy, a complex clinical phenotype that associates HCN alterations to cardiac structural abnormalities (Milano et al., 2014). Recently, a novel loss-of-function mutation of HCN4 channel has been identified during a screening in patients with sick sinus and Brugada syndromes (Biel et al., 2016), a finding that further complicates the understanding of proarrhythmic role of HCN channel dysfunctions.
In the healthy heart, a proper sensitivity of HCN4 to cAMP is also important to set basal HCN current magnitude and the contribution of HCN to resting pacemaker automaticity. In fact, transgenic mice expressing human mutated HCN4 gene lacking the cAMP binding site (CNBD) results in basal bradycardia and reduced heart rate sensitivity to adrenergic stimulation (Alig et al., 2009). However, the same model proved that rate adaption during physical activity is preserved, thus indicating that different mechanisms, alone or in combination with HCN channels, are enrolled to increase heart rate following adrenergic stimulation (Alig et al., 2009; Rosen et al., 2012). Of note, adrenergic regulation of heart rate is also preserved, at least partially, in condition of HCN channel blockade, because the state amplifies the effects of any autonomic stimuli on cycle length (Zaza and Lombardi, 2001).
As opposed to the above reports, a recent study in patients with inappropriate sinus tachycardia has identified a gain-of-function mutation (R524Q) in cardiac HCN4 channel. Mutant channels display a higher sensitivity to cAMP and mediate a larger than normal current during the diastolic depolarization, a finding in line with the enhanced cardiac rate detected in these patients (Baruscotti et al., 2016). As a confirmation of the role of Ih in human SAN pacemaking, a recent computational approach shows that all HCN4 mutations associated with a loss of function slow the pacemaker rate down in simulated APs, with negligible effects on other AP parameters, whereas the only gain-of-function mutation described to date has opposite consequences (Fabbri et al., 2017).
Regulatory proteins also have a role in HCN channel dysfunction in pacemaker centers. Currently, a single mutation in the MiRP1/KCNE2 gene has been associated with symptomatic severe sinus bradycardia and suppression of HCN current in vitro, an effect conceivable with an altered interaction between the regulatory subunit and HCN channels (Nawathe et al., 2013).
Sinus bradycardia may also be attributed to nongenetic causes, such as those occurring physiologically with endurance training (Dobrzynski et al., 2013; D’Souza et al., 2014) or ageing (Monfredi and Boyett, 2015). Both conditions are associated with complex and still incompletely defined modifications of SAN and atria that may predispose to develop atrial tachyarrhythmias. On the other side, pathologic conditions such as AF (Jackson et al., 2017) and heart failure (Wang and Hill, 2010) may also lead to sinus node dysfunctions associated with anatomic and electrical changes.
ii. Atrial Remodelling and Fibrillation
HCN current constitutively present in the human atria has since long been proposed to sustain atrial arrhythmias associated with different cardiac pathologies or triggered by various modulatory signals (Opthof, 1998) (Fig. 4).
In diseased conditions, such as cardiac hypertrophy and failure, atrial dysfunction and arrhythmias may occur because of adverse remodeling triggered by chronic impairment of ventricular function. In the context of chronic heart failure, experimental data have documented an altered expression of HCN channels in right atrial tissue, where HCN4 transcript was found significantly increased (Zicha et al., 2005). In line with this study, in atrial tissue from failing human hearts, HCN2 and HCN4 transcripts and proteins were also increased and most likely related to the enhanced HCN current and the positive voltage shift of channel activation (Stillitano et al., 2008). Interestingly, in a different study (Lai et al., 1999), the overexpression of HCN2 in the left atria is positively related with left atrial filling pressure, an indicator of congestive heart failure, further corroborating the link between left ventricular dysfunction and atrial electrical remodeling.
A limited number of studies have investigated the issue in a defined arrhythmic state, such as chronic AF. In this setting, a significant increase of HCN current has been reported in atrial cardiomyocytes from diseased patients compared with control (Stillitano et al., 2013). In particular, at voltage values around myocyte resting membrane potential (≅−70 mV), HCN fractional activation is 10% larger (from 20% to 30%) because of a positive voltage shift in channel activation, suggesting that chronic AF modifies atrial electrogenesis also by enhancing the contribution of HCN current. The molecular counterpart of this modification is not obvious, consisting of unchanged transcript levels for HCN1/HCN2 and reduction of HCN4 and MiR-1; however, protein amounts are preserved, most likely because of post-transcriptional processing. The modifications of HCN transcripts are in agreement with those reported in a previous study performed on patients in chronic AF (Lezoualc’h et al., 2007). The mechanism underlying the increase of HCN current in chronic AF is not clear; it is possible to hypothesize the occurrence of modifications of HCN regulatory subunits that affect channel function. In this regard, the lack of quantitative modifications of caveolin-3 possibly escludes changes of the interaction between caveolin-3 and HCN channels in the diseased atria.
At variance with the above reports, recent studies in a canine model and in humans (Li et al., 2014b, 2015b) have identified an age-related increased expression of HCN2 and HCN4 transcripts and proteins in the atria. These modifications are associated with proarrhythmic alterations in the canine atria and propensity to AF. In humans, similar modifications were found to be associated with reduction of MiR-1 and MiR-133A that were greater in patients with AF (Li et al., 2015b).
The arrhythmogenic potential of adverse cardiac remodeling can be amplified or reduced by different triggers present at local or systemic levels. Some of those having important implications for AF, such as catecholamines, serotonin, atrial natriuretic peptide (ANP), and adenosine, also modulate the function of HCN channels via activation of different types of G protein–coupled receptors (Table 2). In the human atrium, serotonin and catecholamines interact with and activate Gs-coupled receptors, namely subtype-4 serotonin receptor and β1- and β2-adrenoceptors (Pino et al., 1998; Lonardo et al., 2005). Following increase of intracellular cAMP, both serotonin and catecholamines exert a stimulatory effect on HCN amplitude, shifting the activation voltage to positive values. Differently, ANP is synthesized and stored as a prohormone in the atria, and, upon distension, it is released, allowing activation of two receptors (peptide receptors A and B). Both of them stimulate a guanylyl-cyclase activity, thereby increasing intracellular concentration of cGMP; in this case, intracellular cAMP level rises because of cGMP-mediated inhibition of cAMP phosphodiesterases (Lonardo et al., 2004). It has been hypothesized that, in conditions of HCN channel gain of function, as those determined by adverse remodeling, these mediators may promote the function of HCN channels at physiologic potentials, enhancing the propensity to arrhythmias. Among the observations corroborating the hypothesis are the persistence of all mediator activity and associated signaling during AF and the values of affinity constants of serotonin and ANP for the corresponding receptors, which are closer to the physiologic concentrations of serotonin or ANP derived by local release during platelet aggregation or cell stretching. Further investigations are needed to better clarify the pathophysiological implications of these pathways in atrial arrhythmias and to evaluate the interplay of concomitant excitatory effects exerted on HCN channels expressed in atrial myocytes and in SAN cells.
Examples of Ih modulation by endogenous agonists and involved receptors/signaling pathways
iii. Ventricular Hypertrophy and Failure
Over the last decades, much attention has focused on HCN current in nonpacemaker cells and its potential role in triggering ventricular arrhythmias. The first evidences were obtained in the spontaneous hypertensive rat, which develops age-related cardiac hypertrophy associated with several electrophysiological alterations at ventricular level, including an unusual diastolic depolarization phase (Barbieri et al., 1994). Subsequently, this peculiar trait was associated with the occurrence of an atypical Ih that resulted to linearly relate to the severity of cardiac hypertrophy (Cerbai et al., 1994). Ventricular Ih exhibits electrophysiological properties and sensitivity to pharmacological blockade similar to those described in atrial and SAN cells. However, as described for the atrial current, voltage dependence is shifted to more negative potentials compared with SAN cells, being activation threshold at approximately −70 mV. The value is conceivable with a definite contribution of HCN current to ventricular resting membrane potential, ranging from −80 to −90 mV. The atypical voltage dependence of HCN current in the ventricle most likely arises from specific isoform assembling into functional channels, as well as the multiplicity of secondary subunits that influence HCN current properties, analogously to what was reported for nodal and immature cardiomyocytes (Cerbai et al., 1999b; Barbuti et al., 2004, 2007, 2012; Bosman et al., 2013).
HCN current in the ventricle exhibits a typical sensitivity to autonomic transmitters, mimicking that described in the SAN cells (Cerbai et al., 1999a). Upon adrenergic stimulation, the marked positive shift of HCN voltage dependence most likely reflects a major contribution of HCN2 and HCN4 to tetramer assembly, being these isoforms most sensitive to cAMP-mediated effects (Stillitano et al., 2008; Wahl-Schott et al., 2014). The occurrence and density of HCN current in human cardiac ventricular myocytes isolated from explanted hearts are larger compared with control ventricle and related to disease etiology, being more prominent in ischemic than in dilated cardiomyopathy (Cerbai et al., 1997, 2001; Hoppe and Beuckelmann, 1998). In this setting, the gain of function of HCN current relates to increased levels of HCN4 and HCN2 proteins and transcripts (Fernández-Velasco et al., 2003; Stillitano et al., 2008; Suffredini et al., 2012) (Figs. 3 and 4).
Following on these pieces of evidence, a number of studies have extensively documented that increase in HCN channel expression is a common trait of different models of cardiac hypertrophy (Stilli et al., 2001; Fernández-Velasco et al., 2003) and myocardial infarction (Sartiani et al., 2006; Suffredini et al., 2012). Hence, HCN channels most likely have their place in the fetal/embryonic gene pattern re-expressed during functional remodeling of the diseased ventricle (Swynghedauw, 1999). Interestingly, the expression of the HCN channels appears more pronounced in ventricular regions with the greatest overload, suggesting the possibility of mechano‐sensitive mechanism of channel expression (Fernández-Velasco et al., 2003).
Despite the emerging appraisal of HCN channel as representative marker of cardiac remodeling, its arrhythmogenic role in the ventricle has long remained unproven both in experimental and clinical studies. A recent study in a bradycardic model of heart failure has provided insight into the role of HCN channels in the initiation of triggered activity. The model, expressing a dominant-negative form of the transcriptional repressor neuron‐restrictive silencing factor, a regulator of the fetal cardiac gene program, develops heart failure and dilated cardiomyopathy. Along this process, HCN2 and HCN4 channels are upregulated, leading to enhanced HCN current density associated with ventricular tachycardia, premature ventricular contractions, and sudden cardiac death (Kuwabara et al., 2013; Yamada et al., 2014). Accordingly, β‐adrenergic stimulation increased the susceptibility of myocytes to early after‐depolarizations and spontaneous APs. These findings are in agreement with another study (Hofmann et al., 2012) showing that HCN activity increases the arrhythmogenic potential in the failing myocytes through prolongation of the repolarization phase in ventricular APs.
The consolidated knowledge on the pathways involved in cardiac remodeling has led to investigate whether abnormal neurohumoral signals, known to trigger the onset and progression of adverse cardiac modifications, are also involved in HCN channel overexpression. Among the most important signals, adrenergic and renin-angiotensin-aldosterone systems are responsible for increased levels of hormones, which are released systemically and locally (Swynghedauw, 1999; De Mello, 2004) and have a well-established role in the proarrhythmic alterations of the ventricle, such as the reduction of transient outward potassium channel and the prolongation of AP duration. Indeed, in similar experimental context, chronic administration of angiotensin II receptor antagonists, losartan and irbesartan, is effective in restoring normal electrogenesis in the ventricle, including the expression and function of HCN channels (Cerbai et al., 2000, 2003).
More recently, a different pharmacological strategy has emerged from clinical trials demonstrating the efficacy of pacing to modify positively the progression and worsening of cardiac failure (Rao et al., 2007; Fox et al., 2008). That high heart rate was an independent risk factor for cardiovascular mortality and morbidity was demonstrated since the early Framingham studies (Kannel et al., 1987). Accordingly, growing clinical evidence also prompted studies aimed at testing heart-rate–reducing drugs to reverse and/or ameliorate the functional remodeling of the hypertrophic ventricle in animal models (Dedkov et al., 2007). In line with these observations, experimental findings obtained in the postinfarcted rat demonstrate that chronic treatment of ivabradine, the only bradycardic agent in clinical use, is able to ameliorate cardiac function and to counteract the global electrophysiological remodeling of the ventricle, including the overexpression of HCN channels (Ceconi et al., 2011; Suffredini et al., 2012). The mechanism(s) responsible for the benefit of heart-rate–lowering drugs in heart hypertrophy and failure will be further discussed later (section Ivabradine in Angina and Heart Failure).
2. HCN in the Central and Peripheral Nervous System
a. Overview of HCN distribution at regional, cellular, and subcellular level
Data on HCN channel expression at regional level come from in situ hybridization and immunohistochemistry performed in brain-wide sections from both adult and developing rodent brains (Moosmang et al., 1999; Monteggia et al., 2000; Santoro et al., 2000; Bender et al., 2001; Notomi and Shigemoto, 2004). In addition, single-cell reverse-transcription polymerase chain reaction and electron microscopy analysis have provided more detailed information about the expression pattern of HCN genes with cellular and subcellular resolution in selected CNS areas in animal models (Franz et al., 2000; Luján et al., 2005; Bender et al., 2007; Brewster et al., 2007; Huang et al., 2011; Ramakrishnan et al., 2012; Paspalas et al., 2013; Dufour et al., 2014). All four HCN isoforms are expressed in the brain. The relative abundance of each subunit depends on the area, nerve cell type, or subcellular compartment. Despite the different methodologies and animal species, all reports addressing HCN distribution show good agreement. Overall, HCN subunits are strongly expressed in the CNS and peripheral nervous system (PNS), with subunit-specific pattern. In the CNS, HCN1 is highly expressed in the neocortex, hippocampus, cerebellar cortex, brainstem, and spinal cord. HCN2 is nearly ubiquitous across the CNS, but especially abundant in thalamic and brainstem nuclei. Conversely, a limited number of areas exhibits a strong HCN4 expression, such as the olfactory bulb and the thalamus, with a distribution pattern that appears complementary to that of HCN1. The expression of HCN3 is scattered throughout the brain and modest. All isoforms, but HCN3, are present in the retina (Fyk-Kolodziej and Pourcho, 2007). In the PNS, all HCN subunits are expressed. HCN1 is the most abundant in dorsal root ganglia (DRG) (Chaplan et al., 2003), although a prominent function of HCN2 in the transmission of painful stimuli has also been reported (Emery et al., 2011) (see later). The expression of HCN channels has been found also in enteric neurons (Galligan et al., 1990) and in the spiral (Chen, 1997) and trigeminal ganglion neurons (Janigro et al., 1997).
b. Physiologic role of HCN channels in the CNS: general aspects
A large body of literature has accumulated over the past two decades on the role of HCN channels in nervous cells. The basic aspects of HCN channel physiology in the CNS have been the topic of recent reviews (He et al., 2014; Shah, 2016). We will provide a brief overview of HCN channel function in neuronal physiology and then focus on selected contexts of specific relevance for the purpose of this review. HCN current is a major determinant of neuronal excitability in many neuronal types. The action of HCN channels on neuronal excitability arises from four fundamental properties: 1) selectivity for sodium and potassium; 2) activation curve at potentials close to firing threshold; 3) reversal potential at depolarized values (∼−35 mV) with respect to resting membrane potential; and 4) slow gating kinetics. Because of these properties, HCN channels generate a tonic depolarizing current, which drives the membrane potential toward firing threshold. Furthermore, HCN current offsets both negative and positive perturbations in membrane potential by responding with increased activation or deactivation, respectively. Due to the slow kinetics, this filtering action is effective for low-frequency oscillatory inputs (high-pass filtering). Finally, modulation by the action of cAMP, PKA, PI(4,5)P2, or ancillary subunits adds to the complexity and diversity of functions of HCN channels in the CNS.
i. Regulation of Resting Membrane Potential and Intrinsic Excitability
The influence exerted by HCN current on the intrinsic excitability of the neuron has a complex nature. At −65 mV, a fraction of HCN channels is tonically open (Kase and Imoto, 2012). This results in persistent sodium inflow, which depolarizes the membrane and counteracts hyperpolarization. Depolarization causes channel deactivation, and the consequent pause in current flow has a hyperpolarizing effect. Hence, by opposing negative and positive perturbations, tonic HCN current stabilizes membrane potential in the subthreshold range. Therefore, HCN loss of function causes membrane hyperpolarization and reduction of intrinsic excitability. At the same time, however, input resistance at subthreshold potentials increases and the neuron becomes more responsive toward synaptic stimulation. This dual action exerted by HCN current on neuronal function has been clearly established in many diverse brain areas, including the thalamus, the hippocampus, and the cerebellum, and has important repercussions on higher-order brain functions such as learning and memory, control of circadian rhythm, and sleep/wakefulness state (McCormick and Pape, 1990b; Maccaferri et al., 1993; Gasparini and DiFrancesco, 1997; Nolan et al., 2003). Very recently, a novel role for HCN1 in the control of the neuron’s intrinsic excitability has been proposed. In the axon initial segment of medial superior olive principal neurons, HCN1-mediated current reduces spike probability by elevating the firing threshold. The 5-HT1A receptor signaling shifts HCN channel activation curve in the hyperpolarizing direction, thereby relieving the brake on spike probability (Ko et al., 2016). Serotonin-dependent modulation of HCN current is also involved in the control of respiratory rhythm in the retrotrapezoid nucleus. Activation of the 5-HT7 receptor causes a cAMP-dependent depolarizing shift in the activation curve of HCN current, increasing the firing rate of retrotrapezoid nucleus neurons in vitro and respiratory rhythm in vivo (Hawkins et al., 2015).
ii. Rhythmogenesis
In a functional interplay with other conductances, the activation/deactivation cycle of HCN channels sets the pace of subthreshold membrane oscillations, which may eventually generate AP firing. HCN current cooperates with a subthreshold, persistent Na+ current to determine a 4–10 Hz (θ-like) rhythm in layer II grid cells of the entorhinal cortex (EC) (Alonso and Llinas, 1989). Entorhinal grid cells show periodic, hexagonally-patterned firing locations that scale up progressively along the dorsal–ventral axis of medial EC, which are critically involved in spatial navigation (Moser et al., 2015). In this structure, relative HCN1/HCN2 expression ratio goes down, along with oscillation frequency, following a dorsal–ventral gradient (Notomi and Shigemoto, 2004). This frequency gradient is consistent with the distinct time constants of HCN1 and HCN2 isoforms. In HCN1-KO mice, the dorsal–ventral gradient of the grid pattern is preserved, but the size and spacing of the grid fields, as well as the period of the accompanying θ modulation, are expanded (Giocomo and Hasselmo, 2009; Giocomo et al., 2011). In thalamocortical neurons, HCN current mainly results from HCN2 expression. Adrenergic and serotonergic stimulation causes a cAMP-dependent modulation of HCN2-mediated current, which in turn determines the firing mode of thalamocortical neurons. During wakefulness and rapid eye movement sleep, thalamocortical neurons fire in a single-spike, or transmission mode. In this state, information is effectively transmitted to the cortex. During non-rapid eye movement sleep and absence seizures, thalamocortical neurons fire in a burst mode, and transfer of signals to the cortex is believed to be not as effective (McCormick and Pape, 1990a, b). In agreement, global HCN2 deletion causes absence epilepsy with typical spike-and-wave discharges in electroencephalogram (EEG) recordings (Ludwig et al., 2003). In the intergeniculate leaflet neurons, a retino-recipient thalamic structure implicated in orchestrating circadian rhythm, a role for HCN3 subunit has been described. In this work, HCN3-mediated current drives low-threshold burst firing and spontaneous oscillations and is bidirectionally modulated by PI(4,5)P2. Depletion of PI(4,5)P2 or pharmacologic block of HCN current results in a profound inhibition of excitability (Ying et al., 2011).
iii. Synaptic Excitability and Plasticity
Functional HCN channels are strongly expressed along the dendritic arborizations of neocortical and hippocampal neurons with a soma-to-dendrites expression gradient (Bender et al., 2001; Lörincz et al., 2002; Harnett et al., 2015). In these areas, HCN current constitutes a shunt conductance accelerating the membrane’s time constant and the decay phase of excitatory postsynaptic potentials (EPSPs). As a corollary, temporal summation during an EPSP sequence is minimized. This function, discovered and characterized in pyramidal neurons of the hippocampal CA1 region and somatosensory cortex (Magee, 1998, 1999; Williams and Stuart, 2000; Berger et al., 2003), has then been described in subcortical structures (Ying et al., 2007; Engel and Seutin, 2015; Masi et al., 2015). In the dendritic compartment of CA1 pyramidal neurons, the shunting effect exerted by HCN current limits the activation of voltage-dependent calcium entry, with important consequences on synaptic excitability (Tsay et al., 2007). The nonuniform distribution of HCN current is also responsible for a phenomenon termed “site independence” of synaptic potentials, whereby the decay time constant of distally-generated EPSPs is similar to that of proximally-generated EPSPs (Williams and Stuart, 2000). As a rule, HCN current accelerates the kinetics of both forward- and backward-propagating depolarization waves, thus increasing the precision with which the dendrite detects the concurrence of salient events, including EPSP–AP coincidence, an event leading to plasticity (Pavlov et al., 2011). In several brain areas, long-term potentiation and associated cognitive functions may be constrained by normal HCN function. Indeed, HCN1-KO mice show improved hippocampal-dependent learning and memory performance. In these mice, proximal synapses (CA3–CA1 contacts) function normally, whereas they are potentiated at distal locations (EC layer III–CA1 contacts), as predicted by stronger expression of HCN channels in this compartment (Nolan et al., 2004). HCN current affects cortical functions such as cognition, movement planning, and execution by setting the connectivity strength within local cortical microcircuits. HCN current is abundantly expressed in dorsal–lateral prefrontal cortex (PFC) layer III neurons, a population crucially involved in spatial working memory. In these neurons, HCN channels are modulated by the opposite action of α2-adrenergic receptor (α2-AR) and dopaminergic receptor type 1 (D1) stimulation, via cAMP signaling. When α2-AR stimulation inhibits cAMP–HCN channel signaling, neurons are more tightly connected to recurring microcircuit activity and performance is optimal. In contrast, exposure to stress causes a D1-mediated enhancement of cAMP–HCN activity, leading to a functional disconnection from the local network and impairment of spatial working memory (Wang et al., 2007; Arnsten and Jin, 2014). In the mouse primary motor cortex, HCN expression is specifically elevated in corticospinal neurons of layer V. HCN current confers these neurons a 4 Hz–resonance preference and gates synaptic inputs from layer II/III pyramidal neurons, determining the efficacy of signal transmission between the two layers. In this context as well, α2-AR stimulation modulates Ih function (Sheets et al., 2011).
iv. Resonance Properties
HCN current in the dendritic compartment also confers the neuron-specific resonance properties, i.e., the ability to respond preferentially to inputs at a certain frequency, and thus determines to what extent the neuron responds to synchronous network activity. Due to its slow gating kinetics, HCN current has high-pass filtering properties that, combined with the low-pass filtering action exerted by membrane time constant (Bedard et al., 2006), result in a resonance preference of 1–10 Hz (Hutcheon et al., 1996). The increased power of θ oscillation due to HCN1 deletion and potentiation of perforant path–CA1 pyramidal neurons synapse has been proposed as the physiologic background of hippocampal-dependent learning and memory storage (Nolan et al., 2004). The ability to resonate in a HCN-dependent manner has been reported for other areas, but the functional significance at higher-order level has yet to be established (Ulrich, 2002; Wang et al., 2006; Xue et al., 2012b; Borel et al., 2013).
v. Neurotransmitter Release
HCN current at synaptic terminals has been reported to control efficacy of vesicle release. HCN1 and HCN2 isoforms are present at GABAergic terminals of pallidal axon collaterals (Boyes et al., 2007) as well as glutamatergic terminals onto EC layer III pyramidal neurons (Huang et al., 2011). In both settings, pharmacological or genetic inactivation leads to elevation of spontaneous synaptic release. Huang et al. (2011) have suggested that HCN current inhibitory action is exerted by setting the potential to values where Cav3.2 N-type channels are less active. A follow-up study from the same authors shows that HCN1 channels restrict the rate of exocytosis from a subset of cortical synaptic terminals within the EC and constrain non-AP–dependent and AP-dependent spontaneous release, as well as synchronous, evoked release (Huang et al., 2017).
Despite a large mass of information on the cellular effects of HCN channels, our knowledge of their role in more integrate CNS functions and behavior is rather indirect and derived from pathophysiological studies, as described below. An interesting insight comes from recent evidence in Aplysia californica, whose neuroanatomy and physiology are by far less complex than the mammalian brain (Yang et al., 2015). A. californica motor neurons possess only one HCN isoform, acHCN; Ih appears to be involved in classic conditioning upon stimulation by nitric oxide/cGMP signaling and subsequent enhancement of a N-methyl-D-aspartate (NMDA)-like current pathway, similarly to the mammalian hippocampal neurons (Neitz et al., 2014).
c. HCN channels in pathologic states of the CNS
As discussed earlier, HCN current affects the excitability of many neuronal types in a complex manner. Figure 5 exemplifies the major pathophysiological implications of dysfunctional HCN channels in CNS and PNS described to date.

HCN channels in nervous system diseases. Pathologic implications of dysfunctional HCN channels in CNS and PNS arising from preclinical and clinical studies. Alterations in HCN function have been implicated in animal models of major neurologic (gray) and neuropsychiatric disorders (lavender). Neurologic disorders are linked to HCN current dysfunction in specific brain areas. Schematic traces represent the three principal effects of HCN loss of function (LOF) on intrinsic firing (i), stimulated firing (ii), and synaptic input integration (iii).
i. Epilepsy
The majority of studies have reported that increased neuronal excitability, eventually leading to epileptic state, is associated with loss-of-function mutations in HCN1 and HCN2 isoforms (Nava et al., 2014; DiFrancesco and DiFrancesco, 2015). HCN1-null mice are more susceptible to kainic acid–induced seizures, and, in spite of the hyperpolarization in resting membrane potential, synaptic excitability of cortical and hippocampal neurons is increased in these mice (Huang et al., 2009). Global HCN2 deletion results in the appearance of absence seizures (Ludwig et al., 2003). Mice carrying a C terminus–truncated HCN2 protein manifest generalized spike–wave absence seizures. Next-generation sequencing analysis on patients’ cohorts has revealed association with single-point mutations leading to loss-of-function phenotype in HCN2 isoform and idiopathic generalized epilepsy (Tang et al., 2008; DiFrancesco et al., 2011). Another study, however, reported a single-point mutation in HCN2 gene leading to a gain of function, showing a significant association with febrile seizures (Dibbens et al., 2010). In agreement with the hypothesis that epilepsy correlates with a negative functional modulation of HCN current is the evidence that the established anticonvulsants such as lamotrigine (Poolos et al., 2002), gabapentin (Surges et al., 2003), and acetazolamide (Munsch and Pape, 1999) activate HCN current in vitro. However, it is not clear whether HCN current upregulation contributes to the antiepileptic efficacy of these compounds. HCN current loss of function may also result from alterations in the expression of auxiliary subunits, such as TRIP8b. TRIP8b-KO mice feature spontaneous spike–wave discharges on EEG that are the electrographic hallmark of absence seizures, resembling the scenario of a global HCN2 deletion. Compared with global HCN2 KOs, TRIP8b KOs have no cardiac phenotype and less severe seizure phenotype. Mechanistically, TRIP8b-KO mice have significantly reduced HCN channel expression and function in thalamic-projecting cortical layer 5b neurons and thalamic relay neurons, but preserved function in inhibitory neurons of the reticular thalamic nucleus (Heuermann et al., 2016). Finally, a number of studies report a remodelling of HCN1/2 expression following experimental seizure induction, suggesting a reciprocal cause–effect relation between HCN channels and epilepsy (Brewster et al., 2002; Bender et al., 2003). HCN channels are upregulated in rat CA1 pyramidal neurons following hyperthermia-induced seizures. Interestingly, an adaptive increase in GABAergic activity has been reported in this paradigm. GABAergic inhibitory postsynaptic potentials at high frequency (∼50 Hz) tend to summate and to activate HCN channels enough to cause rebound firing at the end of the inhibitory train. This mechanism has been proposed as an explanation for the paradoxical increase in GABAergic inhibition in experimental models of epilepsy (Chen et al., 2001; Dyhrfjeld-Johnsen et al., 2009). In summary, a relatively large number of preclinical and human genetic studies supports the evidence linking HCN channelopathy to epilepsy. These studies do not always hint at a univocal cause–effect relation between HCN alteration and disease. In fact, both up- and downregulation have been reported in association to epileptic states, and HCN expression remodelling is sometimes a consequence rather than the cause of seizures. For these reasons, the hypothesis that HCN channels may serve as new-generation targets for the development of antiepileptic drugs, yet promising, deserves further experimental testing.
ii. Autism Spectrum Disorder
It has been recently established that a reduction in Ih density is one of the alterations determined by Shank3 haploinsufficiency in reprogrammed human neurons (Yi et al., 2016). Shank3 is a ubiquitous scaffolding protein enriched at postsynaptic densities associated with autism spectrum disorder and other neuropsychiatric disorders. Similar reduction in Ih density was found in hippocampal neurons from transgenic mice carrying a similar Shank3 mutation. Finally, chronic treatment of developing mouse neurons with ZD7288 reproduces some of the morphologic abnormalities caused by mutant Shank3.
iii. Schizophrenia
Schizophrenia is characterized by deficits in PFC function and alteration in cAMP signaling pathway. In layer II/III of the PFC, HCN1 and cAMP/phosphodiesterase signaling machinery colocalizes to the neck of thin spines to regulate gating of afferent inputs during the execution of working memory tasks. As discussed earlier, unbalance of cAMP–HCN signaling results in network disconnection and reduced performance in these tasks (Arnsten, 2011). Consistently, D1 stimulation in rat or monkey PFC impairs working memory performance, and HCN channel blockade in PFC prevents this impairment in rats exposed to either stress or D1 receptor stimulation. These findings suggest that D1 stimulation or stress weakens PFC function via opening of HCN channels at network synapses. This effect can be counterbalanced by elevation of cAMP levels with type-4 phosphodiesterase inhibitors (Paspalas et al., 2013; Gamo et al., 2015). Rescue of synaptic connectivity in the PFC seems to be the rationale at the basis of the clinical efficacy of the α2A agonist guanfacine in PFC-related disorders (Arnsten and Jin, 2014).
iv. Substances of Abuse and Addiction
As a conductance involved in the rhythmic activity of the DA reward system, HCN current has been implicated in pathophysiology of addiction. Modulation of HCN current has been associated with exposure to ethanol (Okamoto et al., 2006), cocaine (Arencibia-Albite et al., 2012, 2017; Goertz et al., 2015), and methamphetamine (Gonzalez et al., 2016). It has recently been proposed that the reported impairment of long-term potentiation and spatial memory formation caused by cannabinoids requires activation of cannabinoid receptor type 1 and cGMP-dependent upregulation of HCN current in superficial CA1 pyramidal cells (Maroso et al., 2016).
v. Mood Disorders
Recent studies have suggested a role for HCN channels in animal models of depression. Chetkovich and coworkers (Lewis et al., 2011) have generated a KO mouse lacking the HCN channel auxiliary subunit TRIP8b that significantly regulates the voltage gating and kinetics of Ih. TRIP8b deletion dramatically reduces Ih expression, disrupting subcellular distribution, in hippocampal pyramidal neurons. Behaviorally, mice lacking TRIP8b demonstrate motor-learning deficits and enhanced resistance to multiple tasks of despair with high predictive validity for antidepressant efficacy. Of note, similar resistance to behavioral despair was observed in distinct mutant mice lacking HCN1 or HCN2 (Lewis et al., 2011). Similarly, another study has shown that HCN current–mediated depressant effects are localized in the dorsal hippocampus, as focal infusion of lentiviral particles carrying HCN1 short hairpin RNA has anxiolytic- and antidepressant-like effects. Indeed, knocking down HCN1 channels increases cellular excitability and results in physiologic changes consistent with a reduction of HCN current. Moreover, treated animals display antidepressant- and anxiolytic-like behaviors associated with widespread enhancement of hippocampal activity and upregulation of brain-derived neurotrophic factor–mammalian target ofrapamycin signaling pathways (Kim et al., 2012). HCN-dependent hyperactivity of ventral tegmental area (VTA) DA neurons has been causally associated with susceptibility in social defeat stress paradigms in rats and mice. In the rat study, chronic treatment with the selective serotonin reuptake inhibitor and antidepressant fluoxetine, besides exerting the expected effects, normalizes both HCN current amplitude and VTA firing rate, suggesting a link between the actions at behavioral and cellular levels. The effects of fluoxetine are in part mimicked by intracerebral infusion of ZD7288, thus suggesting a mechanistic involvement of HCN current in the development of depressive state associated with social defeat stress (Cao et al., 2010). In the mouse, upregulated HCN current causes hyperactivity of VTA DA neurons and susceptibility to depression. Paradoxically, further enhancement of HCN function with lamotrigine or optogenetic hyperactivation of VTA discharge rate is able to rescue susceptibility to social defeat stress (Friedman et al., 2014).
vi. Fear and Anxiety
HCN currents control the excitability of principal neurons in the basolateral amygdala by reducing synaptic excitability (Park et al., 2007). In a recent study, HCN1 was identified in one of three loci associated with conditioned fear acquisition and expression in a mouse genetic reference panel. This discovery was validated using behavioral pharmacology and revealed that HCN current in the basolateral amygdala is required for fear acquisition and expression (Knoll et al., 2016). Collectively, these data indicate that HCN channels support negative mood states in animal models in area- and subunit-specific fashion. Direct or indirect negative modulation of HCN current in the brain areas subtending anxiety level, fear, and depression may have a potential therapeutic efficacy.
vii. Neurodegenerative Disorders
Parkinson disease (PD) is caused by the progressive degeneration of nigrostriatal DA neurons and resulting fall of dopamine levels. In midbrain DA neurons, HCN current results from HCN2 and HCN4 expression, and substantia nigra pars compacta (SNc) DA neurons show the highest expression levels (Neuhoff et al., 2002; Dufour et al., 2014). Interestingly, HCN current density is reduced in SNc DA neurons of MitoPark mice, a genetic mouse model in which a DA-targeted mitochondrial defect leads to selective nigrostriatal degeneration and PD-like phenotype (Good et al., 2011; Branch et al., 2016). Of note, this defect marks early, asymptomatic disease stages and is not due to HCN transcript downregulation. In addition, it has been reported that 1-methyl-4-phenylpyridinium, a toxin capable of inducing PD-like selective nigrostriatal degeneration in primates and rodents, is a voltage-dependent inhibitor of HCN channels in SNc DA neurons. The 1-methyl-4-phenylpyridinium–induced HCN block results in increased dendritic excitability and overall responsiveness to excitatory synaptic inputs (Masi et al., 2013). In agreement with relative HCN expression levels, the effect of HCN block on synaptic excitability is significantly greater in SNc compared with VTA DA neurons (Masi et al., 2015). Although preliminary, these findings suggest that altered HCN channel expression or function in experimental PD models deserves consideration in the context of differential vulnerability. A single study has investigated the electrophysiological actions of riluzole, a medication used to retard the onset of respiratory impairment in the treatment of amyotrophic lateral sclerosis, on the firing activity of hypoglossal motor neurons in rat brain slices, and reported that HCN current undergoes a negative shift in the activation curve following drug administration. HCN inactivation contributes to the riluzole-induced reduction in firing rate of hypoglossal motor neurons, the cellular mechanism regarded as the presumed basis of its therapeutic action (Bellingham, 2013). One study has described that HCN1 levels diminished in the temporal lobe of macaques during aging and Alzheimer’s disease patients. The study also suggests a causal role of HCN1 downregulation in the increased susceptibility of Alzheimer’s disease patients to epileptic seizures (Saito et al., 2012).
d. Retinal cells
In situ hybridization studies have shown high levels of HCN1 in retinal photoreceptors, whereas HCN2, 3, and 4 were not initially found in these cells (Moosmang et al., 2001). However, later studies have demonstrated that all four HCN channel isoforms (HCN1–4) were differentially expressed in different classes of retinal bipolar cells particularly clustered at synaptic terminals between bipolar cells and photoreceptors, thus suggesting a HCN possible role in the control of transmitter release (Muller et al., 2003). In particular, it has been suggested that Ih is tonically active in rod bipolar cells in darkness, where it quickens rod bipolar cell responses to dim light stimuli, thus partly explaining the visual side effects of HCN inhibitors (Cangiano et al., 2007).
i. Retinitis Pigmentosa
Although HCN are expressed in the retina, HCN dysfunction was not associated with any retinal disorder until recently. It has been known that patients during prolonged treatment with Ih inhibitor ivabradine experienced visual symptoms such as phosphenes (described as sensations of enhanced brightness in a fully maintained visual field) (Cervetto et al., 2007). However, these symptoms were dose dependent and reverted to normal 1 week after discontinuation of ivabradine without any alteration of retinal morphology and modification of HCN distribution in healthy animals (Della Santina et al., 2010). Moreover, HCN inhibition in dystrophic mice had no effect on either extent or progression of retinal degeneration. However, more recent observations pointed out the importance of HCN1 in a mouse model of CNGB1-linked retinitis pigmentosa in the progression of the disease. Indeed, the absence of HCN1 in CNGB1-KO mice exacerbated photoreceptor degeneration, thus identifying HCN1 as a major modifier of photoreceptor degeneration. This observation suggested that pharmacological inhibition of HCN channels may enhance disease progression in retinitis pigmentosa and achromatopsia (Schön et al., 2016).
e. Dorsal root ganglia
i. Synaptic Transmission in Dorsal Root Ganglion Neurons
Shortly after the first report of a hyperpolarization-activated queer current in hippocampal neurons (Halliwell and Adams, 1982), Ih was described also in neuronal cells of the embryonic mouse DRG (Mayer and Westbrook, 1983). In these neurons, it was noticed the presence of a time- and voltage-dependent conductance activated by membrane hyperpolarization owning similar characteristics of the “pacemaker current,” If, identified in SAN cells a few years before (Brown et al., 1979; Yanagihara and Irisawa, 1980). Its expression was shown to be most prominent in large- and medium-sized neurons, whereas only half of small sized DRG neurons displayed a functional Ih (Scroggs et al., 1994). Of all the four HCN isoforms, HCN1 and HCN2 are predominantly expressed in DRGs, whereas expression of HCN3 and HCN4 is low or undetectable (Acosta et al., 2012). In particular, HCN2 is mainly abundant in small-sized neurons, whereas HCN1 is the predominant subunit expressed in large neurons (Schnorr et al., 2014). Consistently, Ih in large- and medium-sized DRG neurons has a faster activation kinetic and is less cAMP-sensitive as compared with the characteristics of the current in small-diameter neurons (Gao et al., 2012).
HCN1- and HCN2-KO studies largely confirm HCN distribution in different DRG neuronal subtypes. Specifically, HCN1 deletion erased Ih in large DRG neurons, whereas slowly activating cAMP-sensitive Ih was still present in smaller DRG neurons (Momin et al., 2008). In contrast, genetic deletion of HCN2 removed the cAMP-sensitive component of Ih and abolished AP firing caused by an elevation of cAMP in nociceptors (Emery et al., 2011).
ii. HCN as Pacemakers of Pain
The precise role of Ih in DRG both in physiology and in pathology is not perfectly understood. Indeed, healthy sensory neurons seem largely unaffected by HCN blockade. Moreover, clinical studies with Ih blocker ivabradine did not show any adverse drug reaction such as dysesthesias and paresthesias (Herrmann et al., 2015). Therefore, Ih function seems to be mainly affected following pathologic states, such as in neuronal damage or in experimental model of inflammation (Papp et al., 2010; Acosta et al., 2012; Weng et al., 2012; Schnorr et al., 2014). Indeed, in these scenarios, HCN channels have been reported to increase their expression and/or function (Chaplan et al., 2003; Yao et al., 2003; Jiang et al., 2008). These observations suggested an association between HCN overexpression/overfunction and pathologic conditions, such as allodynia and hyperalgesia (Herrmann et al., 2015). Multiple mechanisms most likely underlie the phenomenon, including the following: 1) increased and modified gene expression (Papp et al., 2010; Descoeur et al., 2011; Schnorr et al., 2014); 2) PI(4,5)P2-mediated pathway interaction (Pian et al., 2006, 2007); 3) PKA overactivation (Cheng and Zhou, 2013); and 4) elevated intracellular levels of cAMP triggered by inflammatory substances such as prostaglandin E2, serotonin, and substance P (Jafri and Weinreich, 1998; Momin et al., 2008; Resta et al., 2016).
3. HCN in Other Peripheral Districts
In rat kidney medulla, HCN1, HCN2, and HCN4 isoforms are present at transcript and protein levels. They partially colocalized with the Na+/K+-ATPase at the basolateral membrane, suggesting a potential link to Na+ and K+ homeostasis. However, channels in this tissue mediate a HCN-like cationic current with different properties compared with HCN current described in neuronal and cardiac cells, including lack of blockade by Cs+ and ZD7288 (Bolívar et al., 2008), atypical properties that require further investigation. A recent study has identified the expression of HCN1 and HCN3 channels in rat nephron proximal tubule and that of HCN3 in the thick ascending limb of Henle. In this region, the channel most likely plays a role in the modulation of Na+/K+-ATPase activity and in acid-base homeostasis (Lopez-Gonzalez et al., 2016).
In the upper urinary tract of mice, optical mapping and video microscopy analysis have shown that HCN channel is involved in the pacemaker depolarization that initiates spontaneous peristalsis. Pacemaker potentials appear to originate in cells expressing HCN3 channels located in the pelvis–kidney junction; here, they couple to smooth muscle via gap junctions and trigger ureter peristalsis. Furthermore, HCN3-positive cells coexpress T-type calcium channels that jointly regulate the origin and rate of peristaltic activity (Hurtado et al., 2014; Hashitani et al., 2017).
In rat urinary bladder, all HCN isoforms are present. HCN1, the most prominent isoform, displays an immunolocalization restricted to interstitial cells of Cajal. Based on channel blockade by ZD7288, the function of HCN channels in these cells seems related to muscle relaxation (He et al., 2012). Following studies have confirmed the presence of all HCN isoforms in interstitial cells of Cajal of human bladder (Xue et al., 2012a). Recently, in a model of bladder hyperactivity, a higher expression of all HCN channel isoforms has been reported in association with increased current conductance; this finding suggests that Ih might play an important role in bladder dysfunctions (Deng et al., 2015). Based on the inhibitory effect of ZD7288, HCN channels might be also involved in uterine contractions in term-pregnant rats (Alotaibi et al., 2017).
Similarly to the urinary tract, a functional role has been postulated for HCN channels in smooth muscle cells from various vascular districts, such as the portal vein (Greenwood and Prestwich, 2002) and lymphatic vasa (McCloskey et al., 1999). HCN4 expression has been detected in endothelial populations in the aorta, pulmonary artery, and coronary arteries at specific developmental stages (Liang et al., 2013); however, the role of Ih in endothelial function remains elusive. More defined is the (dys)function of HCN in baroreceptor neurons; in the aortic ganglion, HCN downregulation appears to contribute to blunted excitability in diabetic rats (Li et al., 2008b; Li and Zheng, 2011).
The functional expression of HCN channels in pancreatic β-cells (El-Kholy et al., 2007; Zhang et al., 2009) and α-cells (Zhang et al., 2008) has been demonstrated several years ago by using electrophysiological and pharmacological approaches. In both cell types, channels have been postulated to modulate hormone secretion, although the relevance of HCN blockade in preventing insulin secretion appears only at low extracellular potassium concentration. This observation might be relevant to tone down the risk of adverse effect of Ih blockers. A hyperpolarization-activated current has been described in interstitial cells of Cajal, which act as pacemaker cells in the gastrointestinal tract and transduce mechanical and neurohumoral signals to smooth muscle cells. However, the role of HCN channels is strictly dependent on their regional distribution: Ih blockers inhibit and cAMP-mediated signaling enhance pacemaking in colonic, but not intestinal, Cajal cells (Shahi et al., 2014). The pharmacological relevance of Ih modulation in gut motility abnormalities has not been investigated.
IV. Pharmacology of HCN Channels
A. HCN Modulation by Cyclic Nucleotides
1. Lessons from Crystallography
Up to December 2016, 22 different structures of the intracellular C-terminal domain have been deposited in the Protein Data Bank; of these, 20 were obtained from X-ray diffraction and two from solution nuclear magnetic resonance (NMR) (Table 3). The first crystal structures of the HCN2 C-terminal domain, in complex with cAMP and cGMP, were solved in 2003, allowing a detailed description of this portion (Zagotta et al., 2003). The cytosolic region contains two domains, the C-linker and the CNBD; the former connects the latter to the S6 transmembrane fragment. The C-linker consists of six α-helices (A′-F′) of variable length. The CNBD is formed by four helices (A, P, B, C), with eight β-strands arranged in a jelly roll-like topology between helix A and B; a phosphate-binding cassette is located between β-strands 6 and 7, and include the short P helix. The C-terminal domains assemble as tetramers, with a large subunit–subunit interface almost completely formed by the C-linker region: the first two helices (A′, B′) form a helix-turn-helix motif that interacts with the neighboring subunits (C′ and D′ helices). Cyclic nucleotides establish a series of interactions within the CNBD, involving residues either in β-strands or in helices. The negatively charged cyclic phosphate is engaged in an ionic interaction with R591, whose mutation largely decreased cAMP affinity (Chen et al., 2001b); the two oxygens of the phosphate form H-bond with the NH of C584 (Oeq) and of T592 (Oax). The 2′-OH is H-bonded to E582. This pattern of interactions is common to all cyclic nucleotides crystallized to date in the CNBD of HCN channels. Interestingly, cAMP and cGMP in the binding site assume different conformations, characterized by an orientation anti and syn, respectively, of the purine ring. The NH2 group of adenosine forms an H-bond with the carbonyl oxygen of R632, and some hydrophobic interactions are formed between the aromatic ring and residues in the C-helix and the β-roll. The NH2 group of guanosine is engaged in H-bond with T592, whereas the N1 nitrogen is connected to R632 (C-helix) through a water molecule. cGMP is the only nucleotide that binds in a syn orientation: the purine nucleotides cIMP, cPMP, and 2-NH2-cPMP, as well as the pyrimidine cCMP and cUMP, all bind in an anti-arrangement (Ng et al., 2016).
Structures of HCN channel C-terminal domain deposited in the Protein Data Bank (PDB) up to November 2016
As stated before, HCN subunits display different sensitivity to cAMP; however, the crystal structures of the intracellular region of HCN1, HCN2, and HNC4 with bound cAMP are almost superimposable. The major structural variation was observed in the loop between the β4 and β5 strands, a region that is part of the binding site of cyclic nucleotides: some residues establish contacts with the purine ring of cAMP, and in this region there are localized some sequence differences (Xu et al., 2010, 2012; Lolicato et al., 2011). Xu et al. (2010, 2012) studied the different sensitivity of HCN2 and HCN4 to cAMP (Table 1), suggesting the importance of two substitutions in the β4–β5 loop, M572 and S575 in HCN2, which become Thr and Ala in HCN4. Interestingly, NMR-based evidence suggests an interaction with M572 of the flavonoid fisetin, the only compound structurally unrelated to cyclic nucleotides, which has been reported to activate mHCN2 expressed in Xenopus oocytes, with half-intrinsic activity with respect to cAMP (Carlson et al., 2013).
To understand the conformational rearrangement induced by the binding of cyclic nucleotides, it would be interesting to compare the holo (i.e., cAMP bound) and the apo (i.e., cAMP-free) forms. Unfortunately, nearly all the X-ray structures of the CNBD solved to date are in the holo form and only one apo structure has been obtained for HCN2 (Table 3) (Taraska et al., 2009). The bound and free HCN2 structures show high similarity, possibly due to the presence, in the apo form, of a bromide ion, in a position equivalent to the phosphate group of cAMP in the holo structure, which could induce the same conformational arrangement as the negatively charged cyclic nucleotide. However, comparison of the two structures has revealed a partial uncoiling of F′ helix and of the C-terminal portion of the C-helix, and transition metal ion fluorescence resonance energy transfer has confirmed that the movements triggered by cAMP binding involve this part of the protein (Taraska et al., 2009). A NMR spectroscopy study, performed on HCN4 holo and apo forms, revealed that the conformational changes involve a larger portion, which includes, on one side, helices E′-F′-A (i.e., the N3A motif) surrounding the CNBD, and helices B and C (C-linker) on the other (Akimoto et al., 2014).
Very recently, the structure of hHCN1 has been determined at a 3.5 Å resolution by means of cryoelectron microscopy, with and without cAMP bound (Lee and MacKinnon, 2017), allowing for the first time to compare holo and apo forms under static conditions: the clear change in the position of A, B, and C helices confirms what was found under dynamic conditions. This structure gave a picture, for the first time, of the unique features of this protein, which explain some of its “funny” behavior, as described above (see section Genetic and Molecular Characteristics of the HCN Family).
The crystal structure of the C-terminal domain of HCN4 with cGMP unexpectedly revealed two bound molecules, one in the canonical binding site and the other in a site, called by the authors C-linker pocket located at the interface between the C-linker and the CNBD (Lolicato et al., 2014). This second binding site has been proposed to have an inhibitory effect on cAMP activation: in fact, site-directed mutagenesis studies suggested that this is the interaction site of cyclic dinucleotides, which have been found to have an inhibitory activity (see section Modulation by Cyclic Nucleotides). This finding can be attractive for two reasons: first, the C-linker pocket site is found only in HCN4; therefore, cyclic dinucleotides may represent a mechanism to regulate specifically this isoform. Second, it may be possible to find small molecules that interact with this site, which should be selective for HCN4, as it actually has been proposed (Lolicato et al., 2014) (see section Isoform-Selective HCN Blockers).
2. Structure-Activity Relationships of Cyclic Nucleotides and Analogs
The affinity for the CNBD of cyclic nucleotides has been reported in two different studies (Scott et al., 2007; Möller et al., 2014). In both cases, 8-[[2-[(fluoresceinyl thioureido)amino]ethyl]thio]adenosine-3′,5′-cyclic monophosphate, which binds with high affinity to the intracellular region of HCN channels, was used as fluorescent ligand, and a series of cyclic nucleotides was screened for their ability to displace it from the CNBD. The two studies used different proteins and number of compounds, but reached similar conclusions. In the first one, the affinity of 20 ligands was determined on a dimeric construct, made with the rat HCN2 CNBD and the DNA binding domain of the cAMP receptor protein (Scott et al., 2007). In the more recent one, 47 analogs were screened on the C-terminal domain (C-linker and CNBD) of mHCN1, mHCN2, and hHCN4 channels, kept as monomers by means of a maltose-binding protein tag (Möller et al., 2014).
The 47 tested compounds did not display selectivity for the three isoforms, but structural modifications greatly affected affinity. Among the naturally occurring cyclic nucleotides, cAMP was, as expected, the most potent, whereas others (cGMP, cCMP, cIMP, cPMP, cUMP) interacted with lower affinity. The pivotal role of the ionic bond with R591 has been confirmed by the drop-in affinity found for the cAMP and cGMP analogs having the equatorial phosphate oxygen atom substituted with sulfur; a similar replacement on the axial oxygen atom, involved in H-bond with Thr, reduced affinity too, although to a lesser extent. The 2′-hydroxyl group forms an H-bond as donor with a key glutamate residue (Zhou and Siegelbaum, 2007); this interaction is necessary because methylation strongly reduced or abolished binding. Substitution in position 8 with Br, fluorescein, or other groups gave compounds with improved affinity, indicating some space available for bulky substituents. On the contrary, modifications on the NH2 groups of cAMP and cGMP were detrimental, suggesting that these groups too are involved in binding. Unexpectedly, the replacement of the nitrogen atom in position 7 with CH on cAMP, cGMP, and other analogs improved affinity: the EC50 for 7CH-cAMP were 100-, 64-, and 108-fold lower than those of cAMP on, respectively, HCN1, HCN2, and HCN4 C-terminal domains. The reason for this increase in affinity was clear from the X-ray structure of the complex of 7CH-cAMP with the HCN4 intracellular region; although cAMP and its CH analog interact in the same way, the hydrophobic environment surrounding the seven position of the purine ring stabilizes a lipophilic CH group better than a hydrophilic N atom. On human HCN4 channel, expressed in HEK293 cells, 7CH-cAMP behaves as an activator with potency four times higher than cAMP; moreover, it displayed some selectivity for HCN channels with respect to PKA and to exchange protein directly activated by cAMP.
B. Pharmacology of HCN Blockade by Exogenous Ligands
1. Ivabradine, Cilobradine, and Other Specific Bradycardic Agents
The definition “specific bradycardic agent” (SBA) refers to a group of structurally diverse substances, discovered in the second half of 1900. These compounds belong to two different chemical classes: N-alkyl-aminoimidazolines, exemplified by alinidine (2-[N-allyl-N-(2,6-dichlorophenyl)amino]imidazoline) and phenylalkylamines structurally related to falipamil (AQ-A-39) (Fig. 6). The word “specific” meant that these molecules, and their congeners, were able to reduce heart rate at concentrations at which other hemodynamic parameters were not affected; it was proposed that these compounds exerted their activity directly on the SAN, blocking the funny current, just discovered at that time (Kobinger and Lillie, 1987). Later on, it was found that the activity of SBAs was not confined to cardiac tissue because these compounds could block the hyperpolarization-activated current also in neurons (Pape, 1994).

Chemical structure of specific bradycardic agents.
The development of alinidine and analogs was stopped due to the concern about the formation of clonidine as metabolite, and by the observation of visual side effects in phase I clinical trials (Kobinger, 1987). On the contrary, the research on phenylalkylamines yielded some interesting compounds. Increasing the size of the five-membered pyrrolidone ring of falipamil into a seven-membered benzazepinone moiety gave a more potent and safer compound, zatebradine (UL-FS49), which entered clinical trials; the development of this molecule too was halted due to adverse effects. The benzazepinone moiety is found also on other two phenylalkylamines: cilobradine (DH-AK269) and ivabradine (S16257). Both compounds are reduced-flexibility analogs of zatebradine: in cilobradine, the three-methylene chain became part of a six-membered piperidine ring, whereas in ivabradine the phenethyl group was incorporated into a bicyclo[4.2.0]octa-1(6),2,4-trien-7-yl methyl moiety. In both cases, a chiral center was introduced into the molecules, and the eutomer is the S enantiomer (Figure 6).
The three drugs have a similar mode of action: they reduce spontaneous AP firing in SAN preparation, mainly through a reduction in the rate of diastolic depolarization (Thollon et al., 1994). On sheep Purkinje fibers, these drugs reduced Ih amplitude, without modifying the voltage dependence (Van Bogaert and Pittoors, 2003). Ih blockade is use-dependent (Goethals et al., 1993) and occurs on the open channel (Goethals et al., 1993; DiFrancesco, 1994; Bois et al., 2007). No effect on contractility is produced (Lillie and Kobinger, 1986; Bois et al., 1996). Cilobradine is reported to be more potent than zatebradine in sheep Purkinjie fibers and mouse DRG (Raes et al., 1998). At variance with the other two drugs, zatebradine prolonged APs in some cardiac preparations (Thollon et al., 1994; Perez et al., 1995b), showing class III antiarrhythmic properties (Valenzuela et al., 1996). This could be one of the reasons for which the development of zatebradine has stopped; another was, again, the observation of visual side effects, coming from zatebradine blockade of Ih involved in the recovery of photoresponse in retinal rods (Satoh and Yamada, 2002). Site-directed mutagenesis, electrophysiology, and molecular modeling studies suggest that cilobradine and ivabradine share a common binding site, located in the internal mouth of the channel pore (Cheng et al., 2007; Bucchi et al., 2013); the proposed mechanism is that these drugs enter the binding site from the intracellular side of the channel (Bucchi et al., 2006).
ZD7288 (Fig. 6) is another blocker of the hyperpolarization-activated current, which is structurally unrelated to the other SBAs. Its pharmacological characterization showed that it behaves as specific bradycardic agent acting on the SAN without having any other direct effects on the heart (BoSmith et al., 1993). Different from phenylalkylamines, the Ih blockade by ZD7288 is not use-dependent, but there is evidence from site-directed mutagenesis that the binding site is the same of ivabradine (Bucchi et al., 2013). ZD7288 has not been developed as drug, but it is widely used as a tool to study Ih.
The word “specific” does not imply that SBAs are completely selective for HCN channels; all compounds reported in this section are able to interact also with other targets. For instance, alinidine is a K+ channel blocker (McPherson and Angus, 1989; Jonas et al., 1992); ZD7288 interacts with Na+ and Ca2+ channels (Sanchez-Alonso et al., 2008; Wu et al., 2012). Several papers have reported that the interaction of ivabradine with other ion channels occurs at concentration higher than those active on HCN channels (Perez et al., 1995a; Bois et al., 1996; Delpon et al., 1996; Koncz et al., 2011); however, more recently, two independent studies showed that ivabradine blocks human ether a-go-go (hERG) K+ channels and HCN4 channels with similar potency (Lees-Miller et al., 2015; Melgari et al., 2015), raising concern on the safety of this drug (Hancox et al., 2015).
The SBAs discussed in this section were disclosed before discovering the heterogeneity of HCN channel family; their ability to discriminate among HCN channel isoforms has been checked only for zatebradine, cilobradine, and ivabradine (see next section).
2. Isoform-Selective HCN Blockers
The ability of ivabradine, cilobradine, and zatebradine to discriminate among HCN channel isoforms has been tested on human homomeric HCN1–4 channels expressed in HEK293 cells (Stieber et al., 2006). The three compounds did not display selectivity or preference for one isoform over the others; the IC50 were in all cases in the low micromolar range. The lack of selectivity may not surprise, if one considers that the four isoforms share a high degree (80%–90%) of sequence identity in the region comprising the six transmembrane segments (S1–S6) and the CNBD (Kaupp and Seifert, 2001); this region harbors the proposed binding site for ivabradine, cilobradine, and ZD7288. However, sequence alignment of the S5-P-S6 region of HCN1, HCN2, and HCN4 isoforms—where site-directed mutagenesis studies locate the residues important for binding (Shin et al., 2001; Cheng et al., 2007; Chan et al., 2009; Bucchi et al., 2013)—reveals some differences in the sequence, that, in principle, could pave the way to selectivity for compounds’ interaction at this site.
Indeed, structural modifications of the nonselective blocker zatebradine gave compounds (Fig. 7) showing different activity on homomeric HCN1, HCN2, and HCN4 channels expressed in HEK293 cells. The inclusion of the three-methylene chain of zatebradine into a cyclohexane ring gave EC18, showing a sixfold preference for HCN4 over HCN1 and HCN2 channels (Del Lungo et al., 2012). The replacement of the three-methylene chain with a cis-butene moiety, combined with the introduction of a chiral center on the phenethyl group, gave MEL55A [labeled in the original paper as (R)-5], which was fourfold and 11-fold more potent on HCN2 than on HCN1 and HCN4, respectively (Melchiorre et al., 2010). MEL57A, serendipitously discovered while synthesizing MEL55A, displayed 30- and 170-fold selectivity for HCN1 over HCN2 and HCN4, respectively. Notably, the selectivity found in recombinant systems was maintained in tissues expressing different HCN isoforms. On guinea pig SAN cells, the reduction of Ih produced by EC18 was threefold higher than that induced by MEL57A; both compounds were tested at concentrations close to the IC50 values. In contrast, in mouse DRG neurons, MEL57A, but not EC18, significantly reduced Ih. In dog cardiac Purkinje fibers, EC18 reduced the amplitude and slowed the slope of the spontaneous diastolic depolarization, whereas MEL57A was ineffective (Del Lungo et al., 2012). In cultured DRG, MEL55A reduced Ih amplitude and cell excitability, its effect being quantitatively similar to that of ivabradine (unpublished data). The utility of isoform-selective and HCN-channel–specific compounds appears evident in studying Ih from tissues expressing different isoforms: for instance, the application of the HCN4-preferring blocker EC18 suggested a functional role of this isoform in mediating Ih in GABAergic interneurons of the dorsal part of the lateral geniculate nucleus (Leist et al., 2016).

Chemical structure of isoform-selective HCN blockers.
Researchers at Johnson & Johnson (San Diego, CA) disclosed a HCN1-selective compound. Starting from the high throughput screening of a proprietary library, and after optimization of the initial lead, piperazine 1 was obtained (Fig. 7); this compound was 12-, 7-, and 10-fold more potent on homomeric HCN1 channels than on HCN2, HCN3, and HCN4, respectively (McClure et al., 2011). Differently from the nonselective compound ZD7288, 1 displayed antihyperalgesic activity in a spared nerve injury model at doses at which heart rate was not affected.
Among the compounds described in the next section, amiodarone shows some isoform selectivity: its IC50 value for blockade of homomeric channels expressed in Xenopus oocytes is 4- and 22-fold lower on HCN4 than on HCN1 and HCN2, respectively (Fan et al., 2011). However, multi-ion channels blocking properties may obscure the selectivity of this compound.
Although it is reasonable to hypothesize that EC18, MEL55A, and MEL57A, being structural analogs of ivabradine and cilobradine, bind to the same interaction site, nothing is known about the binding site of compound 1 and amiodarone. As reported in the previous section (Lessons from Crystallography), the X-ray structure of the complex cGMP-HCN4 revealed the existence of a pocket in the C-linker, proposed to be the binding site for cyclic dinucleotides, but available also for synthetic modulators (Lolicato et al., 2014). Molecules binding at this site should display selectivity because this pocket is not present in other isoforms. Indeed, by means of virtual screening, a compound has been disclosed (N′-biphenyl-2-yl-N-[1-(3-cyanobenzyl)piperidin-4-yl]-N-(pyridin-3-ylmethyl)urea, 2; Fig. 7), which was found able to antagonize cAMP activation on HCN4, but not on HCN2 channels. This pocket may represent a new opportunity for delivering HCN4-selective compounds.
3. HCN Blockade by Other Drugs
Several drugs have been shown to block HCN channel, a property that can either contribute to their pharmacological activity or, alternatively, produce side effects. Some of the compounds mentioned in this section are G protein–coupled receptor modulators, controlling cAMP concentration through adenylyl cyclase–coupled Gα proteins; therefore, they could be endowed with mixed direct–indirect activity. In the following discussion, only compounds used in the clinic are included; for a more comprehensive list, see Romanelli et al. (2016). The compounds have been divided into subgroups according to their pharmacological activity.
a. Analgesic/Antihyperalgesic drugs
Dexmedetomidine and clonidine are α2-AR agonists commonly used for several indications, including analgesia. They share some structural similarities, carrying a five-membered imidazole or imidazoline ring. In addition, as discussed in section Ivabradine, Cilobradine, and Other Specific Bradycardic Agents, clonidine is a metabolite of the SBA alinidine. Both compounds have been shown to block recombinant HCN channels expressed in HEK293 cells (Knaus et al., 2007; Yang et al., 2014), and in vivo studies suggested that the analgesia by dexmedetomidine can be produced by an α2-independent mechanism (Brummett et al., 2011; Yang et al., 2014). For both drugs, bradycardia is a common side effect (Anger, 2013; Isbister et al., 2017), although only clonidine has been tested on SAN (Knaus et al., 2007). The analgesic activity of both compounds can also be due to a receptor-mediated mechanism: α2-AR activation by these drugs is reported to decrease Ih in several neuronal preparations (Parkis and Berger, 1997; Carr et al., 2007; Shirasaka et al., 2007; Inyushin et al., 2010), including some involved in pain perception (Yagi and Sumino, 1998; Takeda et al., 2002). In some instances, it appears that the receptor-mediated inhibition of HCN channels does not occur through the classic Gα,i-linked reduction of cAMP concentration, rather through activation of protein kinase C via Gβγ modulation (Carr et al., 2007; Inyushin et al., 2010).
Loperamide and tramadol are opioid receptor agonists, which have been reported to block Ih by opioid receptor–independent mechanisms. Tramadol was tested on GH3 cells, where it inhibited Ih with micromolar potency; the effect was not affected by preincubation with naloxone (Liu et al., 2009). Loperamide is mainly used as antidiarrheal drug, but it showed analgesic activity and antihyperalgesic properties in animal models of neuropathic pain; in some instances, this activity was not dependent of opioid receptors (Ringkamp et al., 2012). Loperamide was able to block HCN1 and HCN4 channels expressed in HEK293 cells (Vasilyev et al., 2009), and to inhibit Ih in rat DRG, acting from the extracellular side of the channel (Vasilyev et al., 2007); the authors suggested that this effect could contribute to the analgesic activity of the drug.
The antihyperalgesic activity of minocycline has been tested in several models of neuropathic pain, and it has been associated with inhibition of microglia activation (Mika, 2008; Möller et al., 2016); the molecular mechanism is unknown, but could be related also to the ion channel–blocking properties of this drug. On substantia gelatinosa neurons of rat spinal cord, minocycline reduced Ih and decreased the rate of AP firing; the maximal inhibition was 40% and was produced by a direct interaction with the channel from the extracellular side (Liu et al., 2015). Potency was in the micromolar range, but IC50 value was two orders of magnitude higher than that for Na+ channel blockade in DRG (Kim et al., 2011).
Eugenol, a dental analgesic obtained from clove oil, was found able to block hyperpolarization-activated current and decrease firing of APs in trigeminal ganglion neurons. After rat infraorbital nerve chronic constriction injury, it reversed mechanical allodynia at lower doses with respect to those active in thermal hyperalgesia. The Ih-blocking properties may contribute to the analgesic and antihyperalgesic activity of this molecule (Yeon et al., 2011).
b. General anesthetics
Propofol is a general anesthetic, with additional antiemetic and antiepileptic properties (Kotani et al., 2008; Lundström et al., 2010). Propofol is a small lipophilic molecule, which interacts with multiple targets, including several ion channels (Kojima et al., 2015). Ih blockade by propofol has been studied in several tissues: hippocampal neurons (Funahashi et al., 2001; Higuchi et al., 2003), area postrema (Funahashi et al., 2004), thalamocortical neurons (Ying et al., 2006; Chen et al., 2009a), and EC (Li et al., 2016). Overall, these studies suggest that Ih blockade can contribute substantially to the pharmacological actions of the drug, which seems to involve mainly HCN1 and HCN2 channel isoforms (Ying et al., 2006; Chen et al., 2009a). In X. laevis oocytes expressing homomeric HCN1, HCN2, and HCN4, and heteromeric HCN1-HCN2 channels, HCN1 was the most sensitive isoform (Cacheaux et al., 2005; Chen et al., 2005). Consistent with HCN channel blockade are the findings that propofol reduces mechanical and thermal hyperalgesia in a peripheral nerve ligation model of neuropathic pain (Tibbs et al., 2013), and that bradycardia is a common side effect after administration of this drug (Lundström et al., 2010).
Ketamine is another general anesthetic with multiple action sites, among which HCN channels: in HEK293 cells the drug strongly inhibited homomeric HCN1 and heteromeric HCN1–HCN2 channels, but not homomeric HCN2 channels. This activity is likely to contribute to the hypnotic action of ketamine, as demonstrated in HCN1-KO mice (Chen et al., 2009a). Interestingly HCN1 inhibition alone can theoretically explain the changes in EEG produced by ketamine or propofol, without taking into account interaction with NMDA or GABA receptors (Bojak et al., 2013).
Other volatile general anesthetics have been shown to reduce Ih; for instance, halotane and isoflurane interact with HCN1 and HCN2 channels, producing a hyperpolarizing shift in voltage dependence of activation and a decrease in maximal available current at clinically relevant concentrations (Chen et al., 2005, 2009b). Studies using global and forebrain-selective HCN1-KO mice showed that interaction with this isoform in the forebrain contributes to hypnotic and amnestic effects of isoflurane and sevoflurane, but not to their immobilizing action (Zhou et al., 2015b). In thalamocortical neurons, xenon reduced signal propagation by interacting with HCN2 channels, the predominant isoform expressed in this tissue (Ludwig et al., 1998); this effect was not seen in HCN2-KO animals. Accordingly, xenon produced a sedative effect in WT mice, but not in mice lacking HCN2 channels (Mattusch et al., 2015).
c. Local anesthetic and antiarrhythmic drugs
The Ih-blocking activity of the local anesthetic and antiarrhythmic drug lidocaine has been measured in recombinant systems and on neuronal and cardiac tissues. Lidocaine was tested on homomeric mHCN1, mHCN2, and mHCN4, and heteromeric mHCN1–HCN2 channels expressed in X. laevis oocytes and HEK293 cells (Meng et al., 2011): on all isoforms the drug caused a decrease in both tonic and maximal current and slowed current activation kinetics; only for HCN1-containing channels it produced a hyperpolarizing shift in voltage dependence of activation. On rat DRG (Bischoff et al., 2003; Putrenko and Schwarz, 2011) and in substantia gelatinosa neurons (Hu et al., 2016), the blockade was reversible; the dose–response curves yielded similar IC50 values (∼80–99 μM). In a mice model of sciatic nerve block, lidocaine was more potent in WT mice that in HCN1−/− mice, and the duration of lidocaine anesthesia was longer in WT than in HCN1−/− mice (Zhou et al., 2015a). It was suggested that HCN channel blockade might substantially contribute to the drug action during epidural and spinal anesthesia. When tested on rat ventrobasal thalamocortical relay neurons, lidocaine blocked Ih with potency similar to that found in DRG (Putrenko and Schwarz, 2011); the authors suggest that this activity could contribute to lidocaine’s systemic analgesic actions and may partially explain neurotoxicity.
In addition to lidocaine, bupivacaine and mepivacaine also have been tested on rat DRG neurons (Bischoff et al., 2003). The mechanism of Ih blockade was similar to that of lidocaine; bupivacaine was about twice and four times as potent as lidocaine and mepivacaine, respectively.
In cardiac tissues, the Ih-blocking properties of lidocaine have been demonstrated more than 30 years ago in sheep cardiac Purkinje fibers (Carmeliet and Saikawa, 1982), and more recently on rabbit SAN myocytes (Rocchetti et al., 1999). In the latter study, the IC50 value (38 μM, twofold lower than in neuronal tissues) suggested some concern on a possible interference of the drug on normal cardiac automaticity. Indeed, when tested on recombinant mHCN4 channel, Ih blockade was found significant also at physiologic potentials (−70 mV) (Meng et al., 2011).
However, under different experimental conditions, the effect produced by lidocaine on recombinant rabbit HCN4 channels, at −70 mV, was considered clinically not relevant (Tamura et al., 2009). This study was performed on a large series of antiarrhythmic drugs, to gain information about their effects on the pacemaker current for a more rational clinical use of these compounds. To simulate physiologic conditions, i.e., the tonic stimulation by the sympathetic nerve and the presence in the cytosol of cyclic nucleotides, the blocking activity was measured in presence of 0.3 mM intracellular cAMP, and the calculated IC50 values were compared with therapeutic concentrations. For several compounds (quinidine, disopyramide, cibenzoline, mexiletine, aprindine, propafenone, flecainide, propranolol, verapamil, sotalol, as well as lidocaine), the effect was considered negligible; on the contrary, the IC50 values of amiodarone and bepridil were close to their therapeutic concentrations.
The effect of amiodarone on Ih was further characterized on human HCN channel isoforms (HCN1, HCN2, and HCN4) expressed in X. laevis oocytes (Fan et al., 2011): on all isoforms the blockade was concentration- and use-dependent, but the activation curves were not modified. Interestingly, amiodarone was found to be 4 and 20 times more potent on HCN4 that on HCN2 and HCN1, respectively. Amiodarone was found to block Ih also in rabbit SAN preparations (Satoh, 1991), ventricular myocytes from spontaneously hypertensive and normal rats (Li et al., 2014a), but not in isolated human atrial myocytes (Hoppe and Beuckelmann, 1998). In ventricular myocytes from spontaneously hypertensive rats, amiodarone significantly suppressed Ih density and downregulated HCN2 and HCN4 expression (Li et al., 2014a).
Dronedarone is a structural analog of amiodarone, used in the treatment of AF. On hHCN4 channel expressed in Chinese hamster ovary cells, dronedarone was equipotent with amiodarone and ivabradine in blocking Ih (Bogdan et al., 2011). Clinical trials have evidenced a mild bradycardic effect of this drug (Singh et al., 2007), which has been shown to be mainly due to HCN channel inhibition (Sobrado et al., 2013). In a porcine model, dronedarone induced a significant reduction in ventricular rate during AF, owing to Ih inhibition in AVN, which slowed conduction (Verrier et al., 2014).
C. In Vivo Studies and Clinical Pharmacology
1. Ivabradine in Angina and Heart Failure
The current therapeutic indication for ivabradine stems from its primary development as selective HCN blocker: ivabradine is the only commercially available specific bradycardic agent. To understand why bradycardia represents an attractive target in angina, it is worth to recall briefly the mechanisms underlying myocardial ischemia. Ischemic cardiomyopathy arises from an imbalance between oxygen supply to and demand by the myocardium. Several factors contribute to this disequilibrium, namely flow reduction through epicardial coronary arteries, due to plaques, vasospasm, or—as more recently acquired—microcirculatory dysfunction (for a review, see Camici et al., 2016). Myocardial perfusion and oxygen extraction occur mainly during the diastolic phase, in either healthy or diseased heart, but allowing a sufficient rest time between two consecutive beats is more crucial for the latter condition, due to the uneven blood flow. At the same time, heart rate is the primary determinant of oxygen demand. Therefore, decreasing heart rate is a primary therapeutic goal in ischemic cardiomyopathy, traditionally achieved by using beta-blockers, which attenuate angina symptoms. Nevertheless, a significant percentage of patients do not achieve the optimal beta-blocker dosage because of adverse effects or contraindications. Thus, recent European Society of Cardiology guidelines recommend ivabradine as a second-line therapy (class IIa, level B) in patients with ischemic cardiomyopathy intolerant to or poorly controlled by beta-blockers (Montalescot et al., 2013). This recommendation is based on clinical evidence: in the BEAUTIFUL trial, ivabradine reduced severe cardiovascular events in patients with angina and a heart rate ≥70 beats per minute (bpm) (Fox et al., 2008). Alone or combined with beta-blockers, ivabradine reduces SAN rhythm, both at rest and during exercise, without (further) depressing contractility.
However, recent work suggests that the interplay between heart rate and cardiac protection might be more complex than a plain cardiac cycle/oxygen balance scheme. In animal models of ischemic cardiomyopathy, ivabradine protects against risk factors, such as atherosclerotic plaque formation (Custodis et al., 2008), energetic imbalance, and electrophysiological remodeling (Ceconi et al., 2011). These results led to new hypothesis on the relationship between heart rate reduction and improved coronary perfusion, because of collateral artery growth and/or recovery of endothelial cell function during bradycardia (Schirmer et al., 2012). Thus, the distinction between classic and pleiotropic (ancillary) mechanisms of action is tenue, because—based on present knowledge—all can be largely ascribed to HCN blockade in SAN cells. Indeed, at variance with beta-blockers, ivabradine does not possess intrinsic vasoconstrictive activity, which may explain a gain of function during exercise, i.e., by preserving coronary dilation mediated by catecholamines (Camici et al., 2016). In the same line, the reduction of heart rate, but not the dosage, was significantly related to the survival in patients with heart failure treated with beta-blockers (McAlister et al., 2009).
In the SHIFT Trial (Bohm et al., 2010), adding ivabradine to current standard therapy in patients with heart failure safely lowered heart rate below 70 bpm, which is considered a desirable target to improve outcome in patients with ventricular dysfunction not often achieved with beta-blockers due to side effects. More recently, the SIGNIFY study in patients with preserved ejection fraction and coronary artery disease, in which ivabradine was added to standard background therapy, showed a significant increase in cardiovascular events concomitant to, or despite, heart rate reduction (about 60 bpm versus 70 bpm) (Fox et al., 2014). Based on the results of these three large studies and their post hoc analysis, ivabradine should be used as a second-line therapy to reduce the risk of hospitalization in patients with symptomatic chronic heart failure and resting heart rate >70 bpm despite maximally tolerated dosage of beta-blockers (or contraindications to them) (Ponikowski et al., 2016). Adverse effects reported in these large trials also reflect ivabradine cardiac and extracardiac actions: the most common are bradycardia and visual disturbances (phosphenes) due to blockade of—respectively—cardiac and retinal HCN channels.
a. HCN blockade and arrhythmogenesis: an open issue
Far less defined and consequently exploitable are the potential antiarrhythmic profile of ivabradine and in general HCN blockade. Verrier et al. (2013) showed that the capability to control ventricular rate by dronedarone, the de-iodinated analogous of amiodarone, resides in its HCN-blocking effect on AVN conduction in a pig model of AF. This is not surprising because, as described above, HCN4 channels are functionally relevant in nodal conduction (Liu et al., 2008). Subsequent work by the same group reported a similar effect in pigs treated with ivabradine alone (Verrier et al., 2014) or with ranolazine (Verrier et al., 2015): the association reduced AVN conduction and AF dominant, leaving ventricular contractility and duration of the QT interval (an ECG marker of repolarization velocity) unaltered. Finally, recent reports in AF patients showed improved rate control with ivabradine added to beta-blockade (Caminiti et al., 2016; Kosiuk et al., 2016), although these sporadic observations are obviously too weak for drawing conclusions.
As mentioned above, a subgroup analysis of the SIGNIFY study unraveled increased cardiac events in patients with preserved contractile function treated with ivabradine, but neither bradycardia nor emergent AF appear to have an impact (Fox et al., 2015). A proarrhythmic potential of HCN blockade has been inferred based on telemetric recording of ECGs in mice treated with different specific bradycardic agents (Stieber et al., 2004). Periodic fluctuations of the R-R interval appeared in the ECG, depending on dosage and drug type, the order of potency being cilobradine > zatebradine >> ivabradine, without significant alterations in AVN conduction. These fluctuations resemble a well-known phenomenon termed Wenckebach’s periodicity, typical of the AVN. In SAN cells, the mechanism underlying the sinoatrial Wenckebach’s periodicity may reside in the different HCN expression and sensitivity to bradycardic agents in the central versus peripheral sinus node (Stieber et al., 2004; Mangoni and Nargeot, 2008). Finally, the bradycardic effect of ivabradine might stimulate the sympathetic drive, which is per se a proarrhythmic stimulus (Dias da Silva et al., 2015).
However, extrapolation of data obtained in rodents to the clinical setting may be unwarranted, and indeed the usefulness of ivabradine has been postulated in AF, although limited clinical evidence exists at present (Turley et al., 2016). Of note, Ih seems to undergo significant remodeling in chronic AF, as described above (Stillitano et al., 2013). Such a gain of function in the working atrial myocardium parallels that observed by several groups, including some of us, in hypertrophied and failing ventricular cardiomyocytes [reviewed in Sartiani et al. (2015)]. It is interesting to observe that chronic treatment with ivabradine exerts a reverse remodeling action in animal models of heart failure (Ceconi et al., 2011), by counteracting electrophysiological abnormalities, including HCN overexpression and function in atrial and ventricular cardiomyocytes (Suffredini et al., 2012). Finally, off-label uses of ivabradine have been reported in other settings (Oliphant et al., 2016), although evidence from clinical trials is lacking.
2. Treatment of Pain
a. HCN channel modulation in neuropathic pain
The analgesic action of the HCN channel blocker ZD7288 in different models of neuropathic pain has been largely demonstrated (Dalle and Eisenach, 2005; Jiang et al., 2008; Takasu et al., 2010; Nava et al., 2014). In particular, tactile allodynia and thermal hyperalgesia were largely decreased by ZD7288 application. This is not surprising due to the wide expression of HCN channels along the peripheral pain neuraxis (Papp et al., 2006; Papp et al., 2010). Interesting results derived from genetically engineered HCN mutants underlined the important role played by HCN2 in neuropathic pain maintenance (Schnorr et al., 2014). Emery et al. (2011, 2012) demonstrated that conditional deletion of HCN2 abolished pain behavior in the chronic constriction injury model. These results were largely confirmed in a different model of neuropathy using a virtually identical HCN2 mutant (snsHCN2KO) (Herrmann et al., 2015), thus pointing out HCN2 as the most important subunit modulating neuropathic pain.
However, in chemotherapy-induced periphery neuropathic models, HCN1 modification seems to play a prominent role compared with HCN2 in modulating pain sensitization. Recently, Zhang and Dougherty (2014) demonstrated an increased expression of HCN1 in DRG neurons in a rat model of paclitaxel-induced peripheral neuropathy. They also observed an increased frequency of spontaneous discharging neurons in both myelinated and unmyelinated neurons. The hyperpolarization-activated current mediated by HCN1 may facilitate the rise of spontaneous activity in these neurons (Zhang and Dougherty, 2014). Furthermore, Descoeur et al. (2011) observed a significant increase in HCN1 mRNA levels in DRG from oxaliplatin-treated mice. Moreover, they observed that ivabradine exerted analgesic effect on cold allodynia, but not on mechanical hyperalgesia. These two examples provide evidence of a prominent role for HCN1 channel in chemotherapy-induced neuropathy.
Recently, the role of HCN channels in CNS for neuropathic pain has also been investigated. Interestingly, sciatic nerve injury caused an activity-dependent dysfunction of HCN channels in the dendrites of layer 5 pyramidal neurons, resulting in an increased neuronal activity in cortical areas, which has been postulated to be one of the key manifestations of chronic pain (Kuner, 2010; Saab, 2012). It was demonstrated that specific activation of 5-HT7 receptor increased HCN channel function, thus restoring normal dendritic integration and reducing mechanical pain hypersensitivity in nerve-injured animals in vivo (Santello and Nevian, 2015).
b. Participation of HCN channels in inflammatory pain behavior
HCN appears to play a role also in modulation of inflammatory pain. Both nonselective Ih blockers and genetic deletion of HCN display a significant analgesic effect in different animal models of inflammation (Dunlop et al., 2009; Emery et al., 2011). The cAMP-sensitive isoform HCN2 seems to be particularly involved in heat hyperalgesia, as inferred from experiments using conditional HCN2-KO mice challenged with carrageenan or prostaglandin E2–mediated inflammatory stimuli (Emery et al., 2011). The inflammatory processes, which activate signaling transduction pathways, cause an increase of cAMP in pain-sensing neurons responsible for peripheral HCN2 channel activation, which in turn controls the acute phase of neuronal hypersensitivity. Indeed, cAMP shifts the resting membrane potential to depolarized values and increases AP firing in isolated DRG neurons, whereas these modifications do not occur in HCN2 KO or by using nonselective HCN channel blockers (Emery et al., 2011; Schnorr et al., 2014; Young et al., 2014). On the contrary, models of inflammation not implying Gs activation are insensitive to HCN2 deletion or HCN channel blockade (Schnorr et al., 2014), thus demonstrating the absence of HCN influence on cAMP-independent nociceptive behavior.
Similarly, chronic inflammatory states, in which transcriptional regulatory processes are more important compared with intracellular cAMP increase for HCN-mediated sensitization, have been observed to be partially HCN-independent. Indeed, it appears that HCN channels only modulate tactile hypersensitivity, being heat hyperalgesia not affected by either HCN2 deletion or HCN channel block in chronic inflammatory models (Luo et al., 2007; Weng et al., 2012; Schnorr et al., 2014). These results suggest that distinct neuronal pathways convey mechanical and thermal sensitization in chronic inflammatory conditions (Abrahamsen et al., 2008; Cavanaugh et al., 2009; Herrmann et al., 2015). Nonetheless, pharmacological targets modulating Gi-coupled receptors have been reported to interact via HCN modulation to a decreased nociceptive response (Resta et al., 2016).
V. Conclusions
Despite the mass of information that has been accumulating on the biophysical and molecular properties of HCN channels and their (patho)physiologic role in excitable (and not-excitable) cells, we are now seeing the beginning of pharmacological and therapeutic exploitation. Indeed, the unique properties of this channel family required a long-standing effort for clarifying its structural features and modulatory pathways. It is not surprising that the first (and single for the moment) HCN-related drug in the medical armamentarium was a bradycardic agent, targeting the earliest physiologic function ascribed to Ih.
Still, clues emerge for opportunities of future investigation aimed at identifying pharmacological strategies: the prevalence of different HCN subtypes in organs and tissues, the possibility to target HCN gain or loss of function associated with disease, the feasibility of novel isoform-selective drugs, as well as the discovery of HCN-mediated effects for old medicines. The modulation of neuropathic pain could be likely the next in line precisely because of a combination of these favorable factors. However, the impressive evidence emerging from genetic and molecular studies involving HCN channels in the CNS could pave the way to unforeseen therapeutic innovation.
Acknowledgments
We thank Alessandro Mugelli for invaluable support and insightful contribution to authors’ research in this field.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Sartiani, Masi, Mannaioni, Romanelli, Cerbai.
Footnotes
- Received April 12, 2017.
- Accepted July 7, 2017.
A.M. is supported by a fellowship from the Minister of Health (Bando Ricerca Finalizzata e Giovani Ricercatori 2011–2012, GR-201102346829). This work was supported by Ente Cassa di Risparmio di Firenze (Grants 2013.0683A2202.2677 to L.S. and 2013.0102 to G.M.).
L.S. and G.M. are co-first authors.
Abbreviations
- 5-HT
- 5-hydroxytryptamine (serotonin)
- α2-AR
- α2-adrenergic receptor
- AF
- atrial fibrillation
- ANP
- atrial natriuretic peptide
- AP
- action potential
- AVN
- atrioventricular node
- bpm
- beats per minute
- cCMP
- cytidine-3′,5′-cyclic monophosphate
- cGMP
- guanosine-3′,5′-cyclic monophosphate
- cIMP
- inosine 3′,5′-cyclic monophosphate
- CNBD
- cyclic nucleotide binding domain
- CNG
- cyclic nucleotide gated
- CNS
- central nervous system
- cPMP
- purine 3′,5′-cyclic monophosphate
- cUMP
- uridine-3′,5′-cyclic monophosphate
- D1
- dopaminergic receptor type 1
- DA
- dopaminergic
- DDR
- diastolic depolarization rate
- DRG
- dorsal root ganglia
- E
- embryonic day
- EC
- entorhinal cortex
- EEG
- electroencephalogram
- EPSP
- excitatory postsynaptic potential
- HCN
- hyperpolarization-activated cyclic nucleotide–gated channel
- HEK
- human embryonic kidney
- hHCN
- human HCN
- If
- funny current
- Ih
- hyperpolarization-activated current
- KCR1
- K+ channel regulator 1
- KO
- knockout
- mHCN
- murine HCN
- MiR
- microRNA
- MiRP1
- MinK-related peptide 1
- NCX
- Na+/Ca2+ exchanger; NMDA, N-methyl-D-aspartate
- NMR
- nuclear magnetic resonance
- PD
- Parkinson disease
- PFC
- prefrontal cortex
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PKA
- protein kinase A
- PNS
- peripheral nervous system
- SAN
- sinoatrial node
- SBA
- specific bradycardic agent
- SNc
- substantia nigra pars compacta
- spHCN
- sea urchin sperm HCN channel
- SUMO
- small ubiquitin-like modifier
- TRIP8b
- tetratricopeptide repeat-containing Rab8b-interacting protein
- V1/2
- voltage of half-maximal activation
- VTA
- ventral tegmental area
- WT
- wild type
- ZD7288
- 4-(N-ethyl-N-phenylamino)-1,2 dimethyl-6-(methylamino) pyrimidinium chloride
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}