Article Text

Abstract
Background Clinical classification of rare sequence changes identified in the breast cancer susceptibility genes BRCA1 and BRCA2 is essential for appropriate genetic counselling of individuals carrying these variants. We previously showed that variant BRCA1 c.5096G>A p.Arg1699Gln in the BRCA1 transcriptional transactivation domain demonstrated equivocal results from a series of functional assays, and proposed that this variant may confer low to moderate risk of cancer.
Methods Measures of genetic risk (report of family history, segregation) were assessed for 68 BRCA1 c.5096G>A p.Arg1699Gln (R1699Q) families recruited through family cancer clinics, comparing results with 34 families carrying the previously classified pathogenic BRCA1 c.5095C>T p.Arg1699Trp (R1699W) mutation at the same residue, and to 243 breast cancer families with no BRCA1 pathogenic mutation (BRCA-X).
Results Comparison of BRCA1 carrier prediction scores of probands using the BOADICEA risk prediction tool revealed that BRCA1 c.5096G>A p.Arg1699Gln variant carriers had family histories that were less ‘BRCA1-like’ than BRCA1 c.5095C>T p.Arg1699Trp mutation carriers (p<0.00001), but more ‘BRCA1-like’ than BRCA-X families (p=0.0004). Further, modified segregation analysis of the subset of 30 families with additional genotyping showed that BRCA1 c.5096G >A p.Arg1699Gln had reduced penetrance compared with the average truncating BRCA1 mutation penetrance (p=0.0002), with estimated cumulative risks to age 70 of breast or ovarian cancer of 24%.
Conclusions Our results provide substantial evidence that the BRCA1 c.5096G>A p.Arg1699Gln (R1699Q) variant, demonstrating ambiguous functional deficiency across multiple assays, is associated with intermediate risk of breast and ovarian cancer, highlighting challenges for risk modelling and clinical management of patients of this and other potential moderate-risk variants.
- Cancer: breast
- Genetics
- Genetic epidemiology
Statistics from Altmetric.com
Introduction
The clinical classification of rare sequence changes identified in the high-risk breast cancer susceptibility genes BRCA1 and BRCA2 is essential for appropriate genetic counselling of individuals carrying these variants. Classification of BRCA1 and BRCA2 variants was facilitated by the development of a multifactorial likelihood model,1 which provides a quantitative estimate of pathogenicity by assessing measures of genetic and other features of variant carriers relative to characteristics observed for classical high-risk mutations. Moreover, this quantitative assessment of risk has been linked to clinical management guidelines to provide a basis for standardised variant reporting, variant classification and management of families with such variants.2 The multifactorial likelihood methodology has been applied in multiple studies,1 ,3–15 with more than 200 BRCA1 or BRCA2 variants now classified using this approach.16 However, the multifactorial approach is designed to distinguish high-risk mutations from variants with no or little clinical significance, and it is likely that additional methods are required to detect and validate BRCA1 or BRCA2 rare variants associated with more modest risks than the average penetrance reported for classical mutations in these genes, that is, 65% risk of breast cancer and 39% risk of ovarian cancer to age 70 years for BRCA1 mutations, and 45% risk of breast cancer and 11% risk of ovarian cancer to age 70 years for BRCA2 mutations.17
We previously showed that the variant BRCA1 R1699Q (c.5096G>A p.Arg1699Gln) located in the BRCA1 carboxyl terminal (BRCT) regions of the transcriptional transactivation domain (TAD) demonstrated equivocal results from a series of functional assays, when compared with wild-type control and known pathogenic missense mutation BRCA1 A1708E (c.5123C>A p.Ala1708Glu) which was null in all assays.8 In particular, this variant displayed intermediate transcriptional transactivation activity in human 293T and T47D cell lines and wild-type centrosome amplification function, but behaved as a deleterious mutation when assayed for formation of nuclear foci and trypsin sensitivity. There is also inconsistency in assay results from other functional studies, including discrepancies between yeast and mammalian transcriptional transactivation assay results in a single report,18 and categorisation of R1699Q as a variant with strong functional effect due to compromised peptide binding activity and specificity, and compromised transcriptional activity in yet another study.19 Most recently, Chang et al20 performed an extensive study of the R1699Q substitution using mouse embryonic stem (ES) cell-based functional assays, and demonstrated that this variant affected mouse ES cell survival and differentiation, and was unable to rescue embryonic lethality of Brca1-null mice. However, this study also demonstrated that the variant did not cause significant cell-cycle defects, and had no effect on genomic stability, but it was suggested that abrogated repression of oncomir miR-155 was the underlying mechanism for BRCA1-mediated tumour suppression. The equivocal behaviour of this variant can be explained at a protein level, as demonstrated by protein modelling predictions shown in supplementary figure S1. R1699 is located in the linker connecting the BRCT repeat domain, and participates in a salt bridge between the BRCT repeats.21 The loss of salt-bridging interactions and steric strain associated with accommodating a tryptophan substitution contributes to conformational instability of the R1699W (c.5095C>T p.Arg1699Trp) pathogenic mutation and, subsequently, disrupts transcriptional transactivation function. By contrast, substitutions with little or no effect on structures, such as R1699Q, may be fully or partially active in these assays. Moreover, R1699 lies in a conserved phosphopeptide-binding groove of the BRCA1 repeat, and plays an important role in phosphopeptide recognition through its interaction. Specifically, our protein modelling results directly comparing R1699Q and R1699W show that the volume of R1699W is likely to cause steric clashes with the phosphopeptide, whereas, the smaller surface and volume presentation of R1699Q will not cause steric clashes, but may modestly alter phosphopeptide recognition (see supplementary figure S1). These modelling predictions explain the experimental results from biophysical assays of BACH1 binding affinity which demonstrated that R1699W leads to a significant 160-fold reduction in affinity compared with wild-type, whereas, the reduction is only 24-fold for R1699Q.22
We previously proposed that the R1699Q variant has partial abrogation of BRCA1 functions, and may confer low to moderate risk of cancer that would be better measured using pooled family studies.8 In a study assessing pathogenicity of 1433 variants based on family history, co-occurrence and cosegregation data from a large dataset derived from clinical testing at Myriad Genetic Laboratories, the combined odds that BRCA1 R1699Q was a pathogenic variant compared with neutral/no clinical significance was 2.5:1, based on a sample of 16 family histories with cosegregation data on only three of these.11 By contrast, BRCA1 R1699W at the same residue was classified as pathogenic, with odds in favour of pathogenicity of 39 978:1.11 Bioinformatic analysis shows that the arginine at position 1699 is conserved through tunicate, but the severity of the amino acid substitution is much less marked for glutamine (Grantham deviation 43) compared with tryptophan (Grantham deviation 101). Accordingly, the Align-GVGD algorithm (http://brca.iarc.fr) classifies R1699Q as a C35, while R1699W falls in the most severe C65 category for missense alterations. Based on an analysis of the same Myriad dataset, C35 variants were estimated to have a prior probability of pathogenicity of 0.66, while C65 variants were associated with a prior probability of 0.81.11 ,23
Mohammadi et al24 assessed the likelihood of causality by cosegregation analysis of a single family, and reported a likelihood ratio (LR) of 1.4 for R1699Q. In another genetic study of several BRCA1/2 sequence variants, Gomez Garcia et al25 examined the R1699Q and R1699W variants as part of a model-building exercise that incorporated family history, and estimated the probability of pathogenicity to be 0.87 for R1699Q and >0.99 for R1699W. Although this model classified both variants as pathogenic mutations, the authors noted that R1699Q did not cosegregate completely with disease in one of three of the families in which such data were available.
In summary, a number of different studies to date indicate that the R1699Q variant demonstrates inconsistent or inconclusive results at the functional and genetic level. In an extension of our previous study,8 we confirmed the intermediate transcriptional transactivation activity of BRCA1 R1699Q in the 293T cell line relative to pathogenic variant R1699W at the same residue, and then initiated large-scale genetic studies to assess if this intermediate function might translate to the lower risk of breast and ovarian cancer in families for R1699Q compared with R1699W.
Methods
Confirmation of transcriptional transactivation activity
Using methods previously described,8 we first compared transcriptional transactivation activity of BRCA1 R1699Q in the 293T cell line with that of pathogenic variant, R1699W, at the same residue, and also to pathogenic control, A1708E, and confirmed our original findings that this variant displayed intermediate function compared with wild-type sequence and known pathogenic TAD variants (see supplementary figure S2).
Genetic analyses
With ethical approval from the relevant institutional review boards, we then initiated large-scale genetic studies to assess if this intermediate function might translate to the risk of breast and ovarian cancer in families. Informed consent was obtained from all participants. Through collaboration facilitated in part by the ENIGMA consortium,26 we ascertained sufficient information from multiple clinical cancer genetics centres around the world (table 1) to compare family history and risk profiles of families in which the R1699Q variant had been identified, with families with the known pathogenic mutation R1699W at the same residue. For an additional reference group, we also collected a set of pedigrees that had been clinically tested for BRCA1 and BRCA2 mutations from the same centres within the same time frame as the R1699Q and R1699W families, but for which no pathogenic mutation or any other unclassified variant had been found (BRCA-X). The time frame was determined by the centres to ensure that a similar criterion for testing was used. The proband in each instance was defined as the individual initially screened for BRCA1/2 mutations.
Number of families included in the family history and penetrance analyses
Family history analysis
As a measure of how each family fit the characteristics of a BRCA1 mutation-positive family, we used the Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm (BOADICEA) risk-prediction algorithm27–30 to calculate the probability that the proband from each family was a carrier of a BRCA1 mutation based on the pedigree structure and the phenotypes of individuals in the pedigree. BOADICEA uses a full pedigree likelihood approach, and incorporates ages at diagnosis of breast and ovarian cancer, presence of pancreatic and prostate cancer, the age at last follow-up for unaffected individuals, and the year of birth to account for cohort effects in penetrance. The model estimates the simultaneous effects of the high-risk genes BRCA1 and BRCA2 using the age-specific penetrance estimates derived from 22 population-based studies,17 while allowing for unknown genetic effects that explain the residual familial clustering of breast cancer. The residual familial clustering is explained by a polygenic component with variance that decreases linearly with age.
The estimated probabilities of the proband carrying a pathogenic BRCA1 mutation based on the BOADICEA prediction model, Bi, were then transformed in order to better fit a Gaussian distribution using a logit transformation bi = logit(Bi)=ln(Bi/(1−Bi), so that standard statistical methods could be used. For each centre that contributed R1699Q/W families, we calculated the mean and SD of the probabilities calculated for the BRCA-X families from this centre. This distribution was used to create z-scores as Zij = (bij−Xj)/Sj, where bij is the logit of the BOADICEA-predicted probability of a BRCA1 mutation in the ith BRCA-X family in the jth centre, Xj and Sj are the sample mean and SD of the logit-transformed Bi from the jth centre. For families with the sequence variants of interest, R1699Q and R1699W, these Zij thus represent the position of family histories of probands carrying an R1699Q or R1699W variant within the distribution of families tested negative for BRCA1/2 mutations in the same centres and time frame. Letting ZQi be the standardised logit score of the ith R1699Q family- and ZWi represent the corresponding score for the ith R1699W family, and assuming further that the ZQi and ZWi are Normally distributed with means μQ and μW and variances σ2Q and σ2W respectively, these scores can then be used to test the following two hypotheses:
-
The family histories of R1699Q probands are more BRCA1-like than those of matched BRCA-X. That is, we test the null hypothesis μQ = 0 versus the alternative μQ>0 with a one-sample t test. Rejection of the null hypothesis indicates that the R1699Q families have proband/family histories more compatible with a pathogenic BRCA1 mutation than the centre-matched BRCA-X families.
-
The family histories of R1699Q are less ‘BRCA1-like’ than those of R1699W mutations. This is tested by a two-sample t test of the null hypothesis μQ = μW against the one-sided alternative μQ < μW .
If both these null hypotheses are rejected, this indicates that R1699Q variants are, in some sense, intermediate in terms of their BRCA1 family history profile compared with BRCA-X and BRCA1 R1699W families.
Segregation analyses
Risk was analysed more directly through analysis of cosegregation of the R1699Q/W genotypes in the relatives of probands presenting with R1699Q/W variants.31 Analyses included 30 R1699Q informative families with 111 total tested individuals and 19 R1699W families with 80 tested individuals. Risks were estimated by examining the likelihood of the genotypes of the family members (both, women affected with breast or ovarian cancer, and healthy women) as a function of BRCA1 penetrance, conditional on the proband's genotype and all pedigree phenotypes. The conditioning is needed to account for the fact that families were ascertained on the basis of the cancer phenotypes in the entire family, and the fact that the proband carried the variant. In this situation, most information about penetrance derives from the distribution of variant genotypes among unaffected women. Because there was insufficient additional genotyping in these families to reliably estimate age-specific risk ratios for each age group, we examined the risk associated with the R1699Q/W variants relative to those associated with the ‘average pathogenic BRCA1 mutation’, as found in much larger studies of predominantly truncating mutations.17 In these analyses, the age-specific HR (by decade) was assumed to be a constant multiple of the estimate of Antoniou et al,17 with cumulative penetrances re-estimated at each trial value of the multiplier. This allowed for a similar pattern of age-specific effects, as in BRCA1, but only required estimation of a single parameter. We also repeated the analyses allowing for separate penetrance multipliers for breast cancer and ovarian cancer to allow for the possibility that the functional effects of R1699Q or R1699W might be more relevant to cancer risk for one but not both these cancers. We varied the multiplier of the assumed standard penetrance of BRCA1 from 0.05 to 2, in increments of 0.01, in order to find the value that maximised the likelihood of the observed data (and to obtain CIs). If under a particular model, a given value of the penetrance implied risks of cancer in carriers lower in a given age group than in non-carriers, these were constrained to be the same as the non-carrier rates.
The analysis of penetrance was done using the LINKAGE package of programs32 to calculate pedigree likelihoods, and the other statistical analyses were performed using STATA V.11.0 (StatCorp, College Station, Texas, USA).
Results
Table 2 shows the results of the analyses comparing family history scores of probands from R1699Q families, R1699W families, and families with no BRCA1 pathogenic mutation. Of note is the ordered progression of the BOADICEA raw scores showing clear differences between all three groups of families, and the tests of significance between groups. The Z-scores for R1699Q (adjusted for the mean and SD of the BRCA-X families from the same centres) were significantly greater than 0 (p=0.0004), indicating that carriers of the R1699Q variant have more ‘BRCA1-like’ family histories than families that test negative for both genes, and that they have some of the characteristics of family history (eg, ovarian cancer) of BRCA1. However, they are also clearly less ‘BRCA1-like’ than family histories of probands carrying the previously classified pathogenic R1699W mutation (p<0.00001).
Analysis of family history scores
Although the above analyses indicate that families carrying R1699Q are different in terms of their personal and family history from both BRCA-X families, and families carrying the R1699W variant, these analyses do not directly address the question of cancer risks conferred by these mutations. They also do not provide a level of evidence that the variant is pathogenic, as in the typical assessment of cosegregation within the framework of the multifactorial model.1 ,33 Segregation analyses were thus undertaken. For R1699W, the maximum likelihood estimate of the relative proportion of the standard BRCA1 penetrance was 0.24 (95% CI 0.06 to 1.10), which was not significantly different from 1.0 (LR X12 = 3.44; p=0.06). The odds in favour of pathogenicity at this value of the penetrance multiplier were 314 100 : 1. When we allowed the possibility that there were different multipliers for breast and ovarian cancer, the estimates were 0.11 for breast cancer and 2.35 for ovarian cancer, with corresponding odds of 2 420 000 : 1 in favour of pathogenicity. The LR test provided some evidence for difference from a single value (X12=4.08; p=0.043) and for a difference from standard penetrance (X22=7.53; p=0.023).
For R1699Q, the maximum likelihood estimate of the penetrance multiplier parameter was 0.20 (95% CI 0.09 to 0.45), was significantly reduced compared with the standard model (X12=14.2; p=0.0002). The odds in favour of pathogenicity were 6226 : 1 for R1699Q at this value of the multiplier, whereas, they were only 5 : 1 under the standard model. In contrast with R1699W, allowing separate multipliers for breast and ovarian cancer did not result in a big difference in likelihood, with estimated parameters of 0.18 for breast cancer and 0.30 for ovarian cancer (odds of 6787:1), which was not significantly different from a single value of 0.20 (p=0.7) for breast and ovarian cancer.
Clearly, there is a reduced penetrance for this variant, compared with the standard penetrance of BRCA1 as estimated by Antoniou et al.17 To represent these estimated parameters in terms of absolute risks which are perhaps more clinically relevant, we can translate the penetrance multipliers into age-specific relative risks of breast and ovarian cancer, and use these to obtain cumulative risks of breast and/or ovarian cancer by age, based on the age-specific relative risks in Antoniou et al.17 Figure 1 shows the predicted cumulative risks of developing either breast or ovarian cancer based on the maximum likelihood parameter estimates of the breast and ovarian relative risk multiplier parameters for R1699Q and R1699W, compared with the standard model and population rates. Similar figures for breast cancer risk and ovarian cancer risk, individually, are provided in the supplementary figure S3. If our model is correct, the risk of breast or ovarian cancer to age 70 is 24% (95% CI 10% to 40%) for carriers of BRCA1 R1699Q, and 58% (95% CI 7% to 72%) for carriers of BRCA1 R1699W, assuming the best fitting model of separate risk multipliers for breast and ovarian cancers. This compares with 4.6% for women in the general population, and 68% for the carriers of an average pathogenic mutation. The risks for R1699Q are higher than that conferred by family history alone, but still lower than those conferred by BRCA2 and PALB2 mutations.
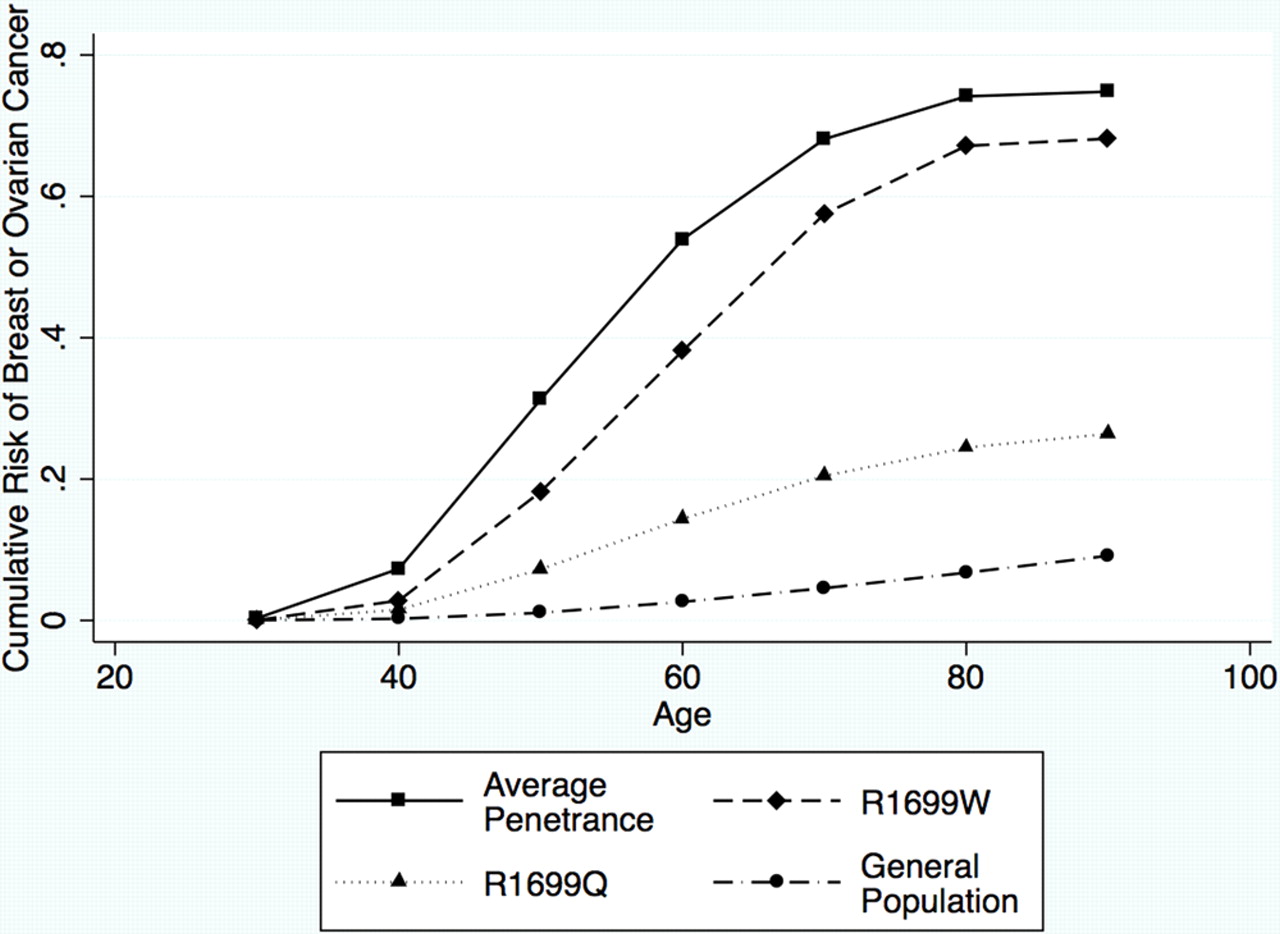
{kind=link}
Cumulative risk of breast or ovarian cancer by age, assuming the best fitting models of penetrance for R1699Q (dotted line) and R1699W (dashed line). The corresponding curves for the standard penetrance (solid line) and for the general population (dash-dotted line) are also shown.
Discussion
Although we have presented results of analyses examining risk, our goal was not to estimate penetrance per se, but rather to compare these two specific variants with the penetrance of the ‘average’ BRCA1 mutation (the vast majority of which are truncating), both in terms of family histories of probands carrying these variants and in terms of cosegregation of the variants within families. Here we provide, for the first time, significant evidence that a BRCA1 variant can be associated with reduced risks of breast cancer compared with the ‘average’ pathogenic mutation. It is of particular relevance and consequence for future studies, since the variant R1699Q was selected for study due to its behaviour in a variety of functional assays. Depending on the assay, this missense variant has demonstrated either wild-type function, abrogated function akin to known pathogenic mutations, or functional activity intermediate between that observed for wild-type BRCA1 and known truncating pathogenic and missense pathogenic mutations.
Interestingly, there was also evidence that the R1699W variant was associated with significantly lower breast cancer risk and a markedly increased risk of ovarian cancer. We recognise that the estimation of breast and ovarian cancer parameters, separately, is somewhat difficult given the necessity of conditioning the data on all pedigree phenotypes, but the results, nevertheless, raise the question that differences in risk of breast versus ovarian cancer may be a characteristic of some missense mutations in the BRCT repeat domains. In this regard, we note that in the 34 R1699W families, there were an average of 2.24 breast cancers and 1.48 ovarian cancers, while in the 68 R1699Q families, there were 2.35 breast cancers and 0.85 ovarian cancers per family, consistent with the higher estimated risk of ovarian cancer in these families. Further study of a large number of such variants will be necessary to address such an intriguing possibility that would have clear clinical implications.
Using the standard multifactorial model, the posterior probability for R1699Q is calculated to be 0.79 from the available data, namely: prior probability of pathogenicity of 0.66 based on the A-GVGD class C3523; segregation odds of 5:1 in favour of pathogenicity from this study of 30 families; LRs from Easton et al11 of 8:1 against pathogenicity for family history, and 3:1 in favour of pathogenicity for co-occurrence data. That is, using the model developed based on the characteristics of BRCA1 pathogenic mutations of ‘average’ penetrance, R1699Q would be classified as International Agency for Research on Cancer (IARC) Class 3 ‘uncertain’.
Our conclusive finding that BRCA1 c.5096G>A R1699Q can be shown to have both intermediate functional deficiency in several assays, and is associated with breast and ovarian cancer risk at significantly lower levels than truncating BRCA1 mutations, has a number of consequences. Our findings suggest that results from a battery of functional assays may highlight other variants with intermediate or equivocal results for investigation as potential moderate risk variants. Indeed, the variant BRCA1 A1708V showed abrogated centrosome amplification, but normal nuclear foci formation and trypsin sensitivity equivocal results from a series of functional assays in our original report,8 and is a candidate for further investigation as a potential moderate risk variant.
If this observation of intermediate function translating to intermediate risk is a general finding, it is likely that there will be a subset of variants that are difficult to classify using the standard multifactorial likelihood approaches that are based on comparing data for a particular variant under the hypothesis that it is a fully penetrant pathogenic BRCA1 mutation, against the hypothesis that it is neutral, or of no clinical significance, with respect to risk. As shown for the R1699Q variant with more families available for analysis than will likely be achieved for most other rare variants, the standard cosegregation analysis yielded odds of only 5:1 in favour of the variant being pathogenic compared with the >6000:1 odds when a lower penetrance was allowed. Further, and more importantly, we must now face the question of how these women should be counselled in terms of cancer risk and the management of that risk. We do not propose that counselling be any different for R1699W, although results from the two parameter analyses suggest that particular attention should perhaps be paid to ovarian cancer for this known pathogenic variant. We emphasise, however, that the CI are wide, particularly for cancer site-specific risks, and future studies are necessary to confirm the markedly increased ovarian cancer risk observed in our dataset. While there is certainly significant evidence that R1699Q carriers are at increased risk over population rates, this risk is markedly lower than that observed for the average BRCA1 mutation. The findings presented here are likely to provide impetus for research studies considering approaches to clinical management of patients with cancer risks intermediate to those conferred by BRCA1/2 mutations, and those from family history alone. In the case of R1699Q, counselling could be similar to that for other moderate-penetrance genes such as PALB2, CHEK2 and RAD51C, although that may change if ovarian cancer screening improves given the increased rate of ovarian cancer over the general population. In all these cases, the incorporation of the now 30+ common breast cancer susceptibility alleles into comprehensive risk prediction models will be of great value in allowing women and their providers to make informed management decisions. In addition, it would be interesting to specifically explore if BRCA1 haplotypes altering promoter activity,34 ,35 or potentially altering 3′ untranslated region (UTR) microRNA binding,36 influence the level of function of R1699Q in vivo, and explain in part the variable presentation of families.
In summary, we provide evidence that a BRCA1 variant demonstrating equivocal functional deficiency across multiple assays is associated with intermediate risk of breast and ovarian cancer, highlighting challenges for risk modelling and clinical management of patients of this and other potential moderate-risk variants.
Acknowledgments
We thank the many families who participated in this study. This work is supported by the efforts of laboratory and clinical staff from many centres around the world. In particular, we would like to acknowledge the efforts of the individuals named in appendix A for their contribution to this specific study. kConFaB thanks Heather Thorne, Eveline Niedermayr, kConFab research nurses and staff, heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study for their contributions to kConFab, and the many families who contribute to kConFab.
Appendix
ENIGMA collaborators (excluding those named in the author list)
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
Footnotes
▸ Additional supplementary figures are published online only. To view these files please visit the journal online (http://jmg.bmj.com)
-
Funding This work was supported in part by project grants from The National Health and Medical Research Council (NHMRC) to ABS. ABS is supported by an NHMRC Senior Research Fellowship. kConFab is supported by grants from the National Breast Cancer Foundation, the NHMRC and by the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. The kConFab Clinical Follow Up Study was funded by NHMRC grants (145684 and 288704). BJF is supported by the Canadian Institutes of Health Research Team Grant in Familial Risks of Breast Cancer CRN-87521. AL thanks the Swedish Cancer Society for support. The work of the German Consortium GC-HBOC is supported by a grant of the German Cancer Aid (grant 107364, RKS) and by the Centre for Molecular Medicine Cologne, Cologne, Germany (RKS, BW). The French Consortium thanks the Association d'Aide à la Recherche Cancérologique de Saint Cloud (ARCs) and the Ligue 92 contre le Cancer for their financial support. FJC and DEG are supported by NIH grant CA116167, an NIH Recovery Act supplement (CA116167Z), and an NIH Specialised Programme of Research Excellence (SPORE) in Breast Cancer (CA116201). LG is supported by a Komen Race for the Cure Fellowship. Research by TvOH was supported by the NEYE Foundation. SMD is supported by funding from the Komen Foundation for the Cure. Ohio State University CCG is supported by the OSU Comprehensive Cancer Center (AET). EJVR is funded by grants from the Cancer Association of South Africa. The research coordinated by MPGV was supported by Dutch Cancer Society grants 2001-2471 and 2006-3677. DEG is supported by NIH grant CA116167. Coordination of ENIGMA is funded by The National Institutes of Health Recovery Act supplement award (CA116167Z).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.