Article Text

Abstract
Objective We aimed to delineate the neurodevelopmental spectrum associated with SYNGAP1 mutations and to investigate genotype–phenotype correlations.
Methods We sequenced the exome or screened the exons of SYNGAP1 in a total of 251 patients with neurodevelopmental disorders. Molecular and clinical data from patients with SYNGAP1 mutations from other centres were also collected, focusing on developmental aspects and the associated epilepsy phenotype. A review of SYNGAP1 mutations published in the literature was also performed.
Results We describe 17 unrelated affected individuals carrying 13 different novel loss-of-function SYNGAP1 mutations. Developmental delay was the first manifestation of SYNGAP1-related encephalopathy; intellectual disability became progressively obvious and was associated with autistic behaviours in eight patients. Hypotonia and unstable gait were frequent associated neurological features. With the exception of one patient who experienced a single seizure, all patients had epilepsy, characterised by falls or head drops due to atonic or myoclonic seizures, (myoclonic) absences and/or eyelid myoclonia. Triggers of seizures were frequent (n=7). Seizures were pharmacoresistant in half of the patients. The severity of the epilepsy did not correlate with the presence of autistic features or with the severity of cognitive impairment. Mutations were distributed throughout the gene, but spared spliced 3′ and 5′ exons. Seizures in patients with mutations in exons 4–5 were more pharmacoresponsive than in patients with mutations in exons 8–15.
Conclusions SYNGAP1 encephalopathy is characterised by early neurodevelopmental delay typically preceding the onset of a relatively recognisable epilepsy comprising generalised seizures (absences, myoclonic jerks) and frequent triggers.
- Epilepsy and seizures
- Genetics
- Neurology
Statistics from Altmetric.com
Introduction
The human SYNGAP1 gene on chromosome 6p21.3 encodes the synaptic RAS-GTPase-activating protein 1, a protein of the post-synaptic density (PSD) of glutamatergic neurons.1 ,2 SYNGAP1 interacts with PSD95 (DLG4) and SAP102 (DLG3), and is able to positively or negatively regulate the density of N-Methyl-D-aspartic acid (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors at the glutamatergic synapses and mediate signalling downstream of glutamate receptor activation.3 ,4 While complete Syngap1 deficiency in mice is lethal at early postnatal stages, heterozygous syngap1+/- mice are viable but show behavioural and cognitive disturbances.5–8 Syngap1 haploinsufficiency disrupts the excitatory/inhibitory balance in the developing hippocampus and cortex and results in accelerated glutamatergic synapse maturation. When this process occurs during critical developmental windows, it alters the synaptic plasticity necessary for the refinement of connections that ultimately shape cognitive and behavioural modalities.4 ,9 Different SYNGAP1 protein isoforms exist and are generated through alternative splicing and alternative promoter usage, in a process regulated by synaptic activity and postnatal age in mice. Two of the main SYNGAP1 mouse isoforms that differ in their N-terminal and C- terminal sequences have opposite effects on glutamate activation pathway.10 Although several isoforms have also been described in humans, their specific role has not yet been established.
Recently, several groups have independently reported de novo SYNGAP1 mutations in patients with intellectual disability (ID), epileptic encephalopathy (EE) or autism spectrum disorders (ASD) identified by exome sequencing11–15 or direct sequencing of the SYNGAP1 gene through a candidate gene approach.16–24 Recently, seven SYNGAP1 mutations were identified by exome sequencing in a series of 1133 patients, 83% of whom had ID, indicating a frequency of SYNGAP1 mutation of ∼0.74% in patients with ID.25 One patient with a chromosomal translocation interrupting SYNGAP126 and five patients with 6p21.3 deletions encompassing SYNGAP123 ,27–30 have also been reported. Thus, to date, SYNGAP1 appears one of the most relevant ID-causing genes, with mutations possibly explaining 0.7 to 1% of ID. Genotype–phenotype correlations have not been clearly established. Moreover, because most patients with SYNGAP1 mutation were identified in large-scale exome or panel studies, the clinical features and the natural history of the SYNGAP1-associated ID and epilepsy remain to be precisely described. Here, we have gathered the molecular and clinical data of 15 unreported and two previously reported patients to investigate in more detail the SYNGAP1 mutational and neurodevelopmental spectra.
Methods
Patients
We analysed 251 patients with variable neurodevelopmental phenotypes including ID, EE and ASD (see online supplementary methods for details) by exome sequencing (n=59) or direct sequencing of genes encoding synaptic proteins (n=192). One additional patient had an intragenic SYNGAP1 deletion identified by microarray-based comparative genomic hybridisation (array-CGH). Clinical and molecular data of 13 additional patients with SYNGAP1 mutation, identified in 12 other centres, were collected: all patients with a mutation introducing a premature termination codon or occurring de novo (ie, proven pathogenic), with the exception of patients with genomic deletions encompassing other genes than SYNGAP1, were eligible for inclusion. Patients #2 and #10 have been previously reported.12 ,24 Each patient’s referring physician filled out a table with detailed developmental, neurological, behavioural and epilepsy history, including EEG and imaging data if available. Most patients were evaluated according to developmental scales routinely used in enrolled centres by clinicians trained in neurodevelopment or neuropsychologists (eg, Brunet-Lezine, HAWIK-IV or SON-R2 scales). The sex ratio was eight males/nine females. Mean age at the time of the study was 10.3 years (range 3–29 years).
Supplemental material
Exome sequencing
The exome of index cases or parent–offspring trios was sequenced by IntegraGen (Evry, France) or by the Genotypic and sequencing facility of ICM.31 Exons were captured from fragmented genomic DNA samples using the SureSelect Human All Exon 50 Mb exome kit (Agilent Technologies) or the SeqCap EZ Solution-Based Enrichment V.3.0 (Roche), and paired-end 150-base massive parallel sequencing was carried out on an Illumina HiSeq2500 or a NextSeq500, according to manufacturers’ protocols. Bioinformatics analyses were respectively done using the in-house pipeline developed by Integragen SA, as previously described,31 or by the iCONICS ICM facility platform as follows: sequencing reads passing quality filtering were aligned to the human reference genome (hg19) with Burrows–Wheeler aligner (BWA);32 GATK33 was used to recalibrate base quality scores, realign around indels and mark duplicate reads. Variants were filtered based on their impact on the gene (missense, nonsense, frameshift, splice site-altering variants) and a minor allele frequency <1% in databases (Exome Variant Server, 1000 Genomes, HapMap, Exome Aggregation Consortium and in-house databases). Calling of de novo variants in trios was done using the Eris interface (Integragen SA) or Polyweb (University Paris-Descartes).
SYNGAP1 screening and Sanger sequencing
All exons and intron–exon junctions of SYNGAP1 (NM_006772.2) and 18 other synaptic genes were amplified using the Fluidigm Access Array technology (IFC Controller AX, FC1 Cycler, 48×48 Access Arrays) and sequenced on a MiSeq Illumina sequencer as paired-end 2×250 bp reads. Alignment of reads on the human reference was performed with BWA and GATK, and additional bioinformatics steps including filtering for novel coding variants were done using an in-house pipeline. Mutations identified by next-generation sequencing (exome or panel) were validated by Sanger sequencing. De novo occurrence was tested by analysing available parents. The predicted effect of mutations was interpreted with Alamut 2.2 (Interactive Biosoftware).
SYNGAP1 isoforms and genotype–phenotype correlations
Human SYNGAP1 cDNA and protein sequences were retrieved from NCBI and Uniprot, aligned using Clustalw2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) and compared with mouse and rat isoforms.10 We first assessed genotype–phenotype correlations in the 17 affected individuals from our cohort.
Review of individuals with previously published SYNGAP1 mutations
The terms ‘SYNGAP1’ and ‘mutation’ were used to search for articles reporting patients with SYNGAP1 mutation in PubMed. In addition, SYNGAP1 mutations and variants present in the HGMD professional (Biobase) and Exac databases were retrieved, listed and visualised on the schematic representation of the SYNGAP1 gene. Statistical analysis was done using the Fisher exact test.
Results
Genetic analyses and review of SYNGAP1 mutations
In our cohort of 251 patients with neurodevelopmental disorders, we identified 3 patients (1.2%) with novel de novo pathogenic heterozygous mutations of SYNGAP1 using exome or panel sequencing. One additional patient had a SYNGAP1 deletion of 16.6 kb encompassing exons 2–9, identified by array-CGH. We collected additional phenotypic information for 2 cases published previously12 ,24 and 11 additional patients with SYNGAP1 mutations identified in other centres (see table 1 and online supplementary table S2).
Molecular and clinical data from the 17 patients with SYNGAP1 mutations*
Supplementary table 2
SYNGAP1 mutations occurred de novo in all 12 patients for whom DNA of both parents was available and, with the exception of one de novo missense mutation, all of them introduced a premature termination codon in the protein sequence (table 1 and figure 1). None of the mutations were reported in control databases (Exome Variant Server, 1000Genomes, HapMap, Exome Aggregation Consortium). The single missense mutation of this study (c.1685C>T, p.Pro562Leu, rs397514670), also identified in a previously reported patient,20 altered a highly conserved amino acid of the RasGap/GTPase domain of the protein (up to yeast) and was predicted damaging by SIFT and PolyPhen-2.

Summary of SYNGAP1 mutations identified in this study and the literature. (A) Location of mutations on the different SYNGAP1 isoforms. Mutations in red correspond to the patients identified in this study. Mutations in black correspond to previously published patients. Recurrent mutations are underlined. Isoform 1 corresponds to the longest isoform (NM_006772.2, N-terminus: SYNGAP A, C-terminus: SYNGAP α2); isoform 2 is obtained through alternative splicing of exons 18 and 19 and differs in its C-terminus (SYNGAP β: 1265–1343: RLMLVEEELR…NGEFRNTADH →SPSLQADAGGGGAAPGPPRHG); isoform 3 is obtained through alternative transcription start site usage involving an additional exon and differs in its N-terminus (SYNGAP B: 1–98: MSRSRASIHR…PVEGRPHGEH→MGLRPPTPSP…RRCSSCCFPG); isoform 4 is obtained through alternative splicing of exon 19 and differs in its C-terminus (SYNGAP γ: 1296–1343: ERQLPPLGPTNPRV…LQITENGEFRNTADH→LLIR). Isoform 5 corresponds to a rat isoform obtained through transcription start site usage (SYNGAP C); its existence in humans has not been demonstrated and therefore remains putative. Note that other isoforms, not represented on this schematic, have been described in rodents but not yet in humans, in particular isoform α 1, which differs in the C-terminus (QTRV). (B) Schematic representation of the mutations (above) and the variants present in the Exome Aggregation (ExAc) database (below) on the longest SYNGAP1 isoform (NM_006772.2) and corresponding protein domains.
In total, 47 patients (including two monozygotic twins23) carrying 43 different point mutation or indels limited to the SYNGAP1 gene have been described to date (figure 1 and online supplementary table S3). Three recurrent mutations (c.321_324del, c.427C>T/p.Arg143*, c.1685C>T/p.Pro562Leu) were found in two patients each. Pathogenic mutations in SYNGAP1 are distributed throughout the gene, especially in exons 5, 8 and 15, which are among the largest exons of SYNGAP1. Interestingly, the two first and two last exons, which are alternatively spliced and included in 3 out of 5 SYNGAP1 isoforms, but also exons 9 and 16, present in all known isoforms seem to be spared (figure 1).
Supplementary table 3
Clinical and neurodevelopmental features of SYNGAP1-related encephalopathy
All patients with SYNGAP1 anomalies of our series had ID, which was evaluated as severe in 10 patients, moderate in 5 and mild in 2 (see table 1 and online supplementary table S1). The mean age of sitting unsupported was 12 months (median age 10 months, n=15) and of walking 27.7 months (median age 24 months, n=15). Also, 10/17 patients could walk by age 2 years and 14/17 by age 3 years. All patients had speech delay: 12 of them spoke first words at a mean age of 2.5 years and 5 patients did not speak at age 10 years or older. In most patients, both receptive and expressive languages were affected. Two patients had mild ID, including one without motor delay. In those, mild, progressive language delay and behavioural anomalies were the most prominent features.
Supplementary table 1
In total, 8 out of 16 patients (50%) older than 3 years old were diagnosed with ASD. Patients with ASD had remarkably poor verbal and non-verbal communication abilities as well as impaired social interactions (see online supplementary table S1). Half of the patients (n=4/8) with severe ID, 1/5 with moderate ID and 2/2 with mild ID were diagnosed with ASD. Independent from a formal diagnosis of ASD, many of the patients exhibited stereotypies (n=10), temper tantrums, aggressiveness, self-injurious behaviour and/or restlessness (n=7).
Neurological examination, performed at a mean age of 8.9 years, was considered normal in two patients. Gait was clumsy or unsteady in five patients and ataxic in five others. Truncal hypotonia was reported in 10 patients and facial hypotonia in 4. Some patients had orthopaedic problems, such as pes planus and rotation of the hips.
Brain MRI performed in all 17 patients (mean age 5.4 years) was either normal or revealed nonspecific features (arachnoid cysts in two patients, mild myelination delay in one and signal abnormities in another).
Epilepsy was diagnosed in 16/17 patients (table 2). The only patient without epilepsy, who was aged 5 at the time of this study, had a single afebrile seizure at the age of 3.5 years. Excluding this patient, first seizures occurred at a mean age of 3 years (range 1–8 years) and consisted of drop attacks, massive myoclonic jerks, atonic seizures, myoclonic absences or absences. A diagnosis of myoclonic astatic epilepsy (MAE, ie, Doose syndrome) and epilepsy with myoclonic absences (EMAs) was made in three and one patients, respectively. The others were diagnosed with unclassified genetic generalised epilepsy (GGE). None had a diagnosis of Lennox–Gastaut syndrome (LGS).
Epilepsy features in SYNGAP1-related encephalopathy
The epilepsy responded to a single antiepileptic drug (AED), mostly sodium valproate, in seven patients and was pharmacoresistant in nine (list of AEDs is reported in table 2). During the active phases of epilepsy, seizures occurred daily in five patients, 10 times per day or more in two and 100 times daily or more in two others. Seizures were of short duration, and the most frequent seizure types were typical or atypical absences (n=9), massive myoclonic jerks with or without falls (n=7), eyelid myoclonia (n=3), clonic or tonic clonic seizures (n=3), myoclonic absences (n=3) and atonic seizures (n=2). Head drops or falls were relatively frequent (n=5) and reported as myoclonic astatic, atonic seizures or drop attacks. Eight patients had several seizure types. No patient had status epilepticus, and exacerbation by fever was mentioned in four. We found no correlations between the diagnosis of ASD and the age at epilepsy onset. The proportion of patients with ASD was identical among those with pharmacoresistant (n=5/10) and pharmacosensitive epilepsy (n=3/6).
The most frequent anomalies reported on EEG traces (figure 2) from 16 patients were ictal or interictal bursts of spikes, spike waves or slow waves that were either generalised (n=13), generalised with a posterior predominance or posterior only (n=5). Paroxysmal anomalies were localised to central regions in six instances. Triggers of seizures were identified in seven patients, including photosensitivity (PS, n=5), fixation-off sensitivity (FOS, n=1), PS and FOS (n=1) and chewing (n=1).
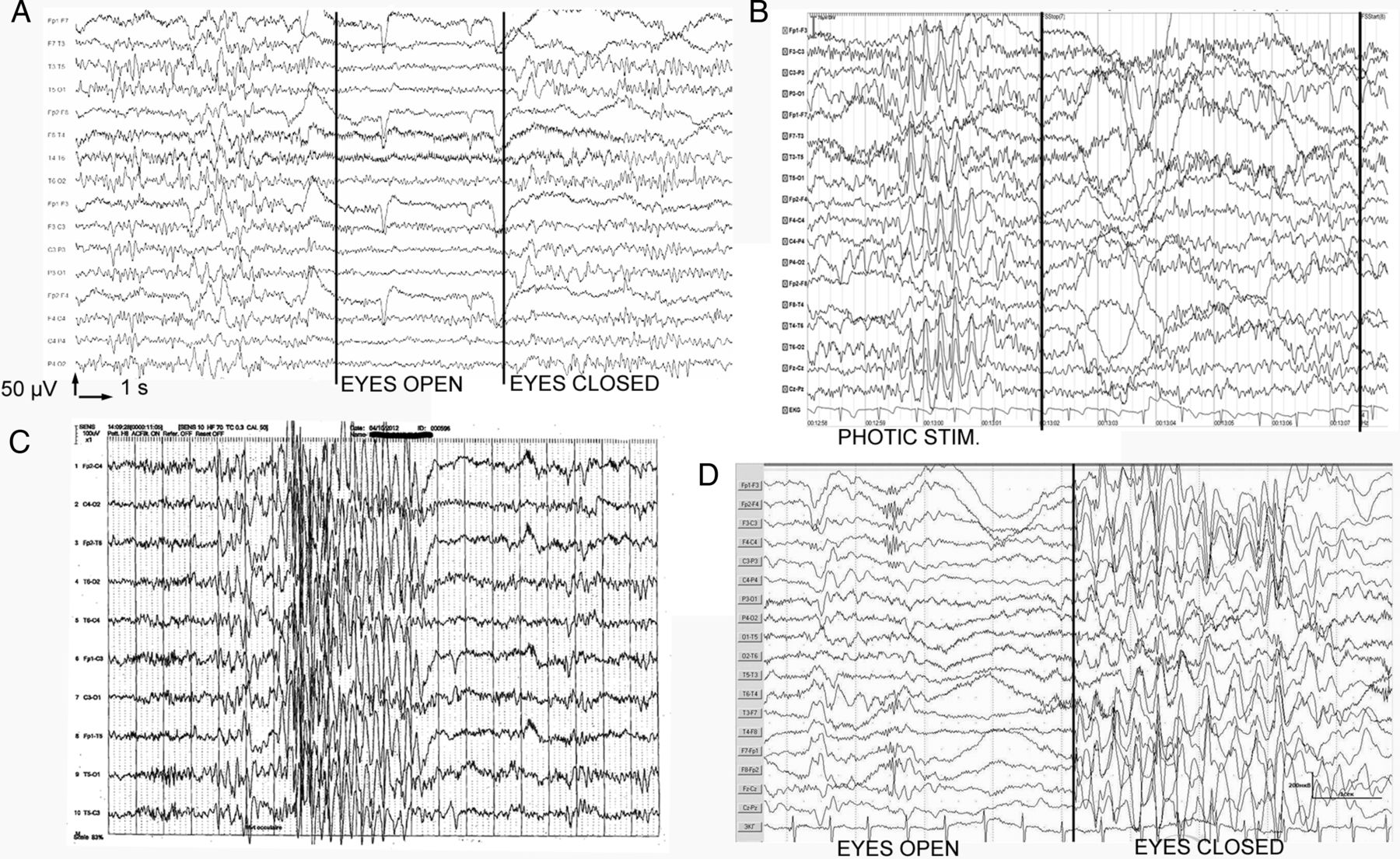
EEG samples from patients exemplifying electroencephalographic findings in SYNGAP1-related encephalopathy. (A) Sample demonstrating normalisation of paroxysmal activity by eye opening, that is, fixation-off sensitivity, in patient #2. (B) Sample showing paroxysmal activity under photic stimulation, that is, photosensitivity, in patient #2. (C) Sample from patient #1: burst of generalised spikes concomitant of a rapid eye deviation (fast rhythms are due to benzodiazepine therapy). (D) Sample from patient #12 showing the appearance of generalised spike wave complexes with a low degree of bilateral synchronisation after eye closure (fixation-off phenomenon).
Genotype/phenotype correlations
We observed no definite correlation between the location of the mutation on the gene and the severity of ID or ASD diagnosis. However, schematic representation of the clinical features of our 17 patients, ordered by the position of the mutation on the gene (figure 3), revealed that the epilepsy of patients with mutations in exons 4–5 was mainly pharmacosensitive (5/6 patients), whereas that of patients with mutations in exons 8–15 was mainly pharmacoresistant (8/9, p=0.01).

{kind=link}
{kind=link}
{kind=link}
Graphical representation of clinical data (age at epilepsy onset, level of intellectual disability (ID) and pharmacoresistance or pharmacosensitivity) in our patients series. X-axis indicates the number of the patient, ordered by the position of the mutation on the gene, except patient 1, who corresponds to the patient with the intragenic SYNGAP1 deletion. Y-axis indicates the age at seizure onset (in months). The proportion of patients with mild (circles), moderate (triangles) and severe (squares) ID is not different in the pharmacoresistant (red) and in the pharmacosensitive (green) groups. One patient (black square, patient 10), who had a single afebrile seizure and was thus not considered strictly as having epilepsy, was not considered for this analysis. The age at the first seizure is neither related to the resistance or sensitivity of the epilepsy to antiepileptic drug nor to the position on the gene. The age at seizure onset is not correlated with the level of ID. The mutations of most patients with pharmacosensitive epilepsy cluster in exons 4–5, whereas those of most patients with pharmacoresistant epilepsy spread over exons 8–15 (p=0.001).
Discussion
In this study, we collected the comprehensive molecular and clinical data of the largest series of patients with SYNGAP1 mutation so far in order to describe more accurately the neurodevelopmental and epilepsy phenotype and to address genotype–phenotype correlations. Delineation of the phenotype from 36 patients with SYNGAP1 mutations showed that it includes mild to severe ID in all, generalised epilepsy in most and autistic behaviour in a half of them (see online supplementary table S3). In the present study, we describe the phenotype of 17 cases with SYNGAP1-associated encephalopathy, bringing the total number of reported patients with SYNGAP1 mutations to 47.
Neurological examination in SYNGAP1-associated encephalopathy
Truncal hypotonia, sometimes in association with facial hypotonia, was the main recurrent feature in our patients, in line with previous series.20 ,23 Likewise, ataxia, with a broad-based or clumsy gait, was frequent in our patients and recurrently mentioned in others.20 ,23 Gait abnormalities are probably due to a combination of hypotonia, lack of global coordination, poor motor control, inattentiveness and orthopaedic issues.
Occipitofrontal circumference was normal in 78% of patients from the literature and in 100% of ours. Though microcephaly has been mentioned in some cases,17 ,20 ,23 it seems to be not a common aspect in patients with SYNGAP1 mutations.
As with previously reported patients, MRI in our patients showed either no or nonspecific features, implying that brain imaging is not helpful in the diagnosis of SYNGAP1-related disorders.
The neurodevelopmental phenotype in SYNGAP1-associated encephalopathy
In our series as well as in the literature, early motor delay with severe language impairment is the first manifestation of SYNGAP1 encephalopathy. Fourteen patients of our series acquired a few words between 1 and 4 years old but only three patients were able to speak simple sentences, which is likely related to ID. These data highlight that language acquisition in most patients with SYNGAP1 mutation rapidly reaches a plateau. It may even be subjected to regression since seven of our patients acquired a few words but eventually lost them again during the first years of life.
Slowing of global development and seizures appeared to occur concurrently in some patients, suggesting that SYNGAP1 mutation might be a cause of EE, as previously suggested.18 By definition, EE is an epilepsy disorder in which the “epileptic activity itself may contribute to severe cognitive and behavioral impairments above and beyond what might be expected from the underlying pathology alone”.34 The concept of EE may apply to specific syndromes (West syndrome and LGS) usually associated with ID or to epileptic individuals with an encephalopathic course.34 West syndrome and LGS were not diagnosed in our patients. However, retrospective analysis of the clinical history of some of them may illustrate an ‘encephalopathic course’ apparently related to frequent daily seizures. As an example, patient #14 in whom first seizures occurred up to 100 times a day had increasing behavioural disturbances and a concomitant stagnation of cognitive acquisition; her language and communication skills significantly improved once the epilepsy was controlled. On the contrary, the epilepsy of patient #4 responded to sodium valproate alone at 4 years old but her cognitive evolution was very poor at 10 years. Beyond these particular clinical histories, a global view of the epilepsy and neurodevelopmental disorder in our series shows that the level of ID is not related to the resistance or sensitivity of the epilepsy to AED (figure 3). In addition, the age at first seizure does not correlate with the resistance to AED and is not clearly linked to the severity of ID. Finally, among the eight patients with language regression reported here, two of them only had a concomitant first seizure. Epilepsy in the others started several months or years after language regression. The contribution of interictal EEG abnormalities to cognitive regression is theoretically possible but cannot be demonstrated since EEG were recorded after the first seizure. Consequently, while the concept of EE may possibly correspond to the encephalopathic course of a subgroup of patients with pharmacoresistant epilepsy in our series, evidence to extend this concept to SYNGAP1-related neurodevelopmental disorder in general is lacking.
Epilepsy in SYNGAP1-associated encephalopathy
SYNGAP1 mutation rate was 0.74% in a large series of 940 patients with ID25 and up to 1% (5/500) in another large series of patients with EE.18 Overall, about 85% patients with SYNGAP1 mutations had seizures. This suggests that epilepsy is extremely common in the SYNGAP1-associated encephalopathy and that SYNGAP1 is one of the most frequently mutated genes in patients with ID and epilepsy. All patients in our series had generalised seizures, like those reported in a previous study,20 only a few of them also experienced focal clonic or tonic clonic seizures. Generalised bursts of spikes, spike waves and slow waves, sometimes with an occipital predominance, were the main recurrent EEG features in our patients. Thus, falls and myoclonic jerks, (typical or atypical) absences, sometimes in combination, define the most common seizures types that, together with the finding of interictal generalised and/or occipital anomalies on EEG, may guide towards the diagnosis of SYNGAP1 mutation in patients with ID.
Though most of our patients with SYNGAP1 mutations had a diagnosis of unclassified GGE, seizure types were suggestive of epilepsy syndromes associated with ID, particularly EMA and MAE, whose diagnosis has been suggested in three and one patient(s), respectively. To our knowledge, two other patients with EMA were found to carry a de novo genetic anomaly affecting SYNGAP1: one with a frameshift mutation20 and another with a gene interruption due to a balanced translocation.26 However, the sequencing of SYNGAP1 in four other patients with EMA and in another one with MAE failed to reveal any mutations. This result is in agreement with a previous work in which a single SYNGAP1 mutation was identified in three patients with EMA, 10 with MAE and 2 with LGS.20 This suggests that SYNGAP1 mutations are relatively uncommon causes of these epilepsy syndromes.
Photosensitivity has been mentioned in previously reported SYNGAP1 patients,17 ,23 but has not been emphasised. The fixation-off phenomenon has been described once.24 In our series, PS as a trigger for seizure was found in half of the patients. Parents or caregivers of four patients noticed it as sensitivity to sunlight, artificial light or the television. This high rate of PS is significant since clinical PS is found in only 10% of patients with epilepsy in the 7–19-year-old group.35 We assume that PS may have not been detected in some of our patients because it is an age-dependent phenomenon with a peak around puberty; it could therefore still appear in some of them; or because of the poor cooperation of patients during the recording. These data suggest that PS, when present, might be a diagnostic clue from the EEG of an underlying SYNGAP1 mutation.
Genotype/phenotype correlations
Although patients with SYNGAP1 mutations show a common core clinical picture, the phenotype is relatively variable, particularly regarding the severity of ID, pharmacoresistance and the presence of ASD. Since SYNGAP1 is a complex gene, giving rise to several protein isoforms with opposite effects on the glutamate activation pathway, via alternative splicing and transcription start sites,10 it was tempting to speculate that the location of the mutation on the gene could correlate to the clinical outcome. However, we found little correlation between the location of the mutation and the severity of ID, epilepsy and/or ASD. Yet, the epilepsy of patients with mutations in exons 4–5 appeared more pharmacosensitive than that of patients with mutations in exons 8–15. Interestingly, exons 4 and 5 are not present in SYNGAP C, an isoform obtained through alternative promoter usage, whose existence has been demonstrated in mice and rats. Although this isoform has not been shown to exist in humans as well, our results suggest that it could also exist and have a different function, as already proven for isoforms α1 and α2, which differ in their C-terminus. Further study is necessary to confirm this finding and decrypt the precise function of each human SYNGAP1 isoform and its relationship with the human pathology characteristics.
Nevertheless, the comparison of the clinical features of patients with identical mutations revealed significant clinical differences (see online supplementary tables S2 and S3), confirming that there is a real variability of the phenotype that depends on other factors than the mutation itself. On the contrary, monozygotic twins had strikingly similar phenotypes, suggesting that these modifier factors could be of genetic origin.23
ASD in SYNGAP1-associated encephalopathy and hypothetical consequences of SYNGAP1 mutations on brain development
Although all patients with validated pathogenic SYNGAP1 mutations reported to date had ID, only half of them had a diagnosis of ASD (including data from the literature and our series). In our series, the presence of autistic traits was neither limited to patients with moderate or severe ID, nor to those with pharmacoresistant or early-onset epilepsy. Thus, ASD, like epilepsy, could be considered as an additional feature of the SYNGAP1-related phenotype in the context of ID, irrespectively of its severity, rather than an ‘isolated’ diagnosis.
This observation is in agreement with previous studies showing that many neurodevelopmental disorders are caused by mutations in genes encoding synaptic proteins, and more specifically constituents of the PSD.36 The fact that a subset of patients with SYNGAP1 mutations exhibit autistic behaviours suggests that a single mutation in a synaptic gene is not sufficient to cause ASD and that the genetic or epigenetic background of the patient probably plays an important role in the occurrence of autistic features in a context of intellectual development impairment. Many genes mutated in patients with ASD and ID are linked with neuronal signalling pathways and may alter the synaptic plasticity underlying the building, refinement and consolidation of neuronal networks associated with learning and adaptive behaviours, with the balance between inhibitory and excitatory signals being determinant in this process.37 ,38 ,39 Given the function of the SYNGAP1 protein in regulating excitatory inputs downstream of NMDA receptors, the SYNGAP1-associated encephalopathy is likely a manifestation of the disruption of this balance. ASD as well other neurodevelopmental disorders could in many cases result from the interruption or impairment of the maturation processes of neuronal networks that are driven by neuronal activity during a critical period of brain development.39 This scenario is particularly relevant to the fact that the clinical and morphological consequences of SYNGAP1 haploinsufficiency in mice, that is, behavioural disturbances and premature dendrite elongation, are restricted to gene disruption during a given period of brain development.4 ,9 Following this hypothesis, SYNGAP1 encephalopathy may be regarded as an example of premature closing of the time window for cognitive development in humans. In the SYNGAP1-associated encephalopathy, disruption of the excitatory/inhibitory balance, which is also a cause of epilepsy, may therefore prematurely end the maturation process of synapses and lead to ID, ASD and epilepsy by a common pathophysiological mechanism.
URLs/resources
NCBI PubMed: http://www.ncbi.nlm.nih.gov/pubmed
Uniprot: http://www.uniprot.org/
Exome Variant Server: http://evs.gs.washington.edu/EVS/;
ExAC Browser (Beta)|Exome Aggregation Consortium: http://exac.broadinstitute.org/
BIOBASE HGMD Professional: http://www.biobase- international.com/product/hgmd
Acknowledgments
The authors thank the families for their participation in this study, the iCONICS facility, especially Ivan Moszer and Justine Guegan, for bioinformatic analysis of exome and panel sequencing data, Patrick Nitschké and the Paris-Descartes Bioinformatics Platform for access to the Polyweb interface, and the DNA and cell bank of the U1127 for DNA extraction and collection.
References
Footnotes
Collaborators List of EuroEPINOMICS-RES MAE working group coinvestigators: Dana Craiu (Pediatric Neurology Clinic II, Department of Neurology, Pediatric Neurology, Psychiatry, Neurosurgery, “Carol Davila” University of Medicine, Bucharest, Romania; Pediatric Neurology Clinic, “Professor Doctor Alexandru Obregia” Clinical Hospital, Bucharest, Romania), Peter De Jonghe (Neurogenetics Group, Department of Molecular Genetics, VIB, Antwerp, Belgium), Ingo Helbig (Division of Neurology, The Children's Hospital of Philadelphia, Philadelphia, Pennsylvania; Department of Neuropediatrics, University Medical Center Schleswig-Holstein, Christian Albrechts University, Kiel, Germany), Renzo Guerrini (Pediatric Neurology Unit and Laboratories, Children's Hospital A. Meyer, University of Florence, Florence, Italy), Anna-Elina Lehesjoki (Folkhälsan Institute of Genetics, Helsinki, Finland; Research Programs Unit, Molecular Neurology and Neuroscience Center, University of Helsinki, Helsinki, Finland), Carla Marini (Pediatric Neurology Unit and Laboratories, Children's Hospital A. Meyer, University of Florence, Florence, Italy), Hiltrud Muhle (Department of Neuropediatrics, University Medical Center Schleswig-Holstein, Christian Albrechts University, Kiel, Germany), Rikke S Møller (Danish Epilepsy Centre, Dianalund, Denmark), Bernd Neubauer (Department of Neuropediatrics, University Medical Faculty Giessen and Marburg, Giessen, Germany), Deb Pal (Department of Clinical Neuroscience, Institute of Psychiatry, King's College London, London, UK), Kaja Selmer (Department of Medical Genetics, Oslo University Hospital, Oslo, Norway), Ulrich Stephani (Department of Neuropediatrics, University Medical Center Schleswig-Holstein, Christian Albrechts University, Kiel, Germany), Katalin Sterbova (Child Neurology Department, University Hospital Motol, Prague, Czech Republic), Pasquale Striano (Pediatric Neurology and Muscular Diseases Unit, Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, ‘G Gaslini Institute’, Genova, Italy), Tiina Talvik (Department of Pediatrics, University of Tartu, Tartu, Estonia; Department of Neurology and Neurorehabilitation, Children's Clinic, Tartu University Hospital, Tartu, Estonia), Sarah von Spiczak (Department of Neuropediatrics, University Medical Center Schleswig-Holstein, Christian Albrechts University, Kiel, Germany).
Contributors CM, CvS, and CN contributed equally. Design and coordination of the study: CM, CvS, CN, GK, CD. Contributing genetic and/or phenotypic data: CM, CvS, CN, DV, DS, GL, AR, BG, YM, CK, IB, DH-Z, ES, MR-D, UY, HÇ, AI, IM, EP, CK, ER, AR, SB-W, AR, CZ, JH, AR, MM, MB, KM, LH-H, BM, SS, SW, CTM, HCM, KH, SB, JL, DH, GK, CD. Writing of the manuscript: CM, CvS, CN, GK, CD. Revision of the manuscript: CM, CvS, CN, GL, CZ, SS, SW, GK, CD.
Funding This study was financially supported by INSERM, Fondation de France (FdF—Engt n°15144 to D. Héron), Agence Nationale de la Recherche (ANR SAMENTA SynDivAutism), Assistance Publique des Hôpitaux de Paris (APHP) and the “Investissements d'Avenir” programme ANR-10-IAIHU-06 (IHU-A-ICM). C D and CN are members of the Bio-Psy Labex.
Competing interests None declared.
Patient consent Obtained.
Ethics approval This study was approved by INSERM (RBM C12-06) and the ethical CCPRB committee from La Pitié-Salpêtrière (Paris, France).
Provenance and peer review Not commissioned; externally peer reviewed.